

Catalytic protocols that generate a-branched amines efficiently and enantioselectively facilitate the preparation of many important biologically active molecules.[1]Among such entities are homopropargyl amines, used in the total synthesis of a number of natural products.[2] Several investigations have adopted the chiral auxiliary strategy; the desired products are obtained in high diastereoselectivity as trimethylsilyl-substituted alkynes.[3] In contrast, the corresponding catalytic protocols are scarce. The first relevant report included three examples of reactions of allenyl stannanes with a glyoxylate-derived tosylimine,[4] affording homopropargyl sulfonamides in 34–96% yield and 55:45–93:7 enantiomeric ratio (e.r.).[5] A notable recent advance entails enantioselective additions of a readily available allenylboron to tosylimines catalyzed by a Ag-phosphine catalyst to furnish a wider range of products and higher enantioselectivity (87:13 to more than 98:2 e.r.). Nonetheless, reactions of substrates that do not bear an aryl substituent proved to be less efficient, those of enolizable alkyl-substituted tosylimines were not reported and, as with the aforementioned initial development, removal of the tosyl unit requires strong reducing conditions.[6-8]
Herein, we present a broadly applicable and efficient catalytic method for enantioselective synthesis of homopropargyl amides (Scheme 1); acid hydrolysis generates the parent amines. Transformations are performed with 0.25–2.0 mol% of a chiral N-heterocyclic carbene (NHC) complex of copper, derived from a readily available chiral imidazolinium salt and CuCl or the more robust CuCl2·2H2O, both of which are commercially available. Aryl-, heteroaryl-, alkenyl-, as well as alkyl-substituted N-phosphinoyl imines can serve as substrates. Additions proceed to completion in seven hours or less, delivering homopropargyl amides in 65% to more than 98% yield and 92:8 to more than 98:2 e.r. The Cu-catalyzed process is amenable to gram-scale operations, and can be performed in a common fume hood without the need for strictly anhydrous and/or oxygen-free conditions.
Scheme 1.
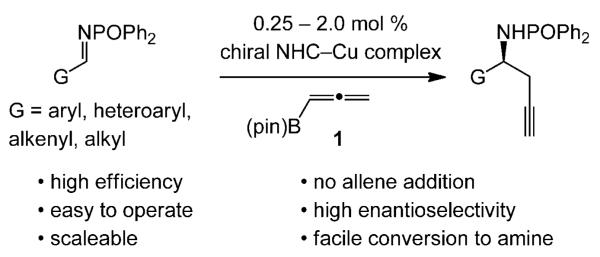
A practical and broadly applicable, efficient, and highly enantioselective method for synthesis of homopropargyl amines.
We began by probing the capacity of chiral C1-symmetric imidazolinium salt[9] 3a (Scheme 2), effective for reactions of allylborons with N-phosphinoyl imines,[10] in serving as the catalyst precursor. In the presence of the NHC–Cu complex prepared in situ with CuCl and NaOtBu, more than 98% conversion is achieved within six hours, and the desired amine 4a is formed in 98% yield and 98:2 e.r (Scheme 2, top). None of the allene addition product is formed (less than 2%, as judged by 400 MHz 1H NMR analysis). When CuCl2·2H2O, more stable to air and moisture than CuCl, is utilized, the reaction is complete in 30 min and similarly enantioselective (Scheme 2). The practicality of the catalytic process is under-lined by the gram-scale transformation shown in Scheme 3. The reaction is performed with 0.25 mol% of the catalyst in a standard fume hood; treatment of the product mixture with aqueous HCl affords the homopropargyl amine (5) in 86% overall yield and 96:4 e.r. The desired product is isolated in analytically pure form after routine aqueous wash without the need for costly silica gel chromatography and the associated-solvents. The amide can be obtained through simple trituration; such ease of isolation is due to the strong tendency of N-phosphinoyl amides to be crystalline.[11,12]
Scheme 2.

NHC–Cu-catalyzed propargyl addition can be performed with CuCl or the air-stable CuCl2·2H2O with exceptional efficiency and high enantioselectivity. Mes=2,4,6-(Me)3C6H2.
Scheme 3.
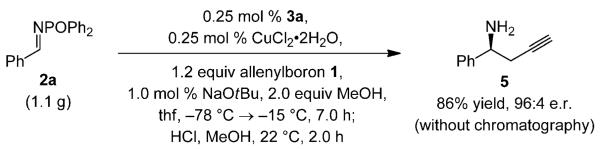
Gram-scale NHC–Cu-catalyzed homopropargyl amine synthesis performed in a standard fume hood.
Enantioselective homopropargyl amine synthesis can be performed with a range of aryl-substituted imines; transformations proceed to completion with 1.0 mol% of the chiral catalyst (Table 1). Regardless of whether the substrate carries an electron-withdrawing (entries 2, 5 and 6), electron-donating (entries 4 and 7), or a sterically demanding aryl group (entries 1 and 3), products are isolated in 88–98% yield and 93:7–98:2 e.r. Reaction times span from one to six hours; either CuCl or the more robust CuCl2·2H2O can be used. Homopropargyl amides are obtained in high yield regardless of the metal salt employed.
Table 1.
NHC−Cu-catalyzed enantioselective propargyl additions to aryl-substituted aldimines.[a]

| Entry | Substrate (Ar) | 3 | With CuCl | With CuCl2.2 H2O | ||
|---|---|---|---|---|---|---|
| yield [%][b] | e.r.[c] | yield [%][b] | e.r.[c] | |||
| 1 | 2b (2-naphthyl) | 3b | 97 | 98:2 | 95 | 94:6 |
| 2 | 2c (0-FC6H4) | 3a | 96 | 97.5:2.5 | 96 | 95:5 |
| 3 | 2d (o-MeC6H4) | 3a | 93 | 97.5:2.5 | 92 | 94:6 |
| 4 | 2e (o-MeOC6H4) | 3a | 97 | 97:3 | 98 | 94:6 |
| 5 | 2f (m-BrC6H4) | 3a | 97 | 96:4 | 93 | 93:7 |
| 6 | 2g (p-ClC6H4 | 3a | 92 | 97:3 | 88 | 96:4 |
| 7 | 2h (p-MeOC6H4) | 3a | 98 | 97:3 | 92 | 96:4 |
Reactions were performed in an N2 atmosphere. Addition reactions with CuCl: 3.0 mol % NaOtBu at −50°C for 6.0 h; addition reactions with CuCl2.2H2O: 5.0 mol% NaOtBu at −78→−15°C for 1.0 h; 1.4 equiv of 1 in all cases. Less than 2% allene addition in all cases.
Yields of isolated purified products (±5%).
Determined by HPLC analysis (±2%). See the Supporting Information for all experimental details and analytical data.
The occasional use of structurally modified imidazolinium salts, such as 3b (Table 1, entry 1), is not because the catalyst derived from 3a is ineffective. Rather, in some cases, slightly higher efficiency and/or enantioselectivity can be attained with modified NHC–Cu complexes 3b and 3c. For example, with 3a, formation of 4b proceeds to more than 98% conversion, affording 4b in 96% yield and 95:5 e.r. (vs. 97% yield and 98:2 e.r. with 3b).

Heterocycle-, alkenyl-, and alkyl-substituted N-phosphinoyl imines serve as substrates (Scheme 4). Enantioselective additions proceed readily with O-, as well as S- or N-substituted heterocyclic aldimines without detectable catalyst inhibition (6–8, Scheme 4); homopropargyl amides are isolated in 94% to more than 98% yield and 94:6–97:3 e.r. Allylamide 9 and the more sterically hindered 10 (Scheme 4) are obtained in similarly high yields and enantiomeric purities (81% and 98% yield, and 96:4 and 96.5:3.5 e.r., respectively). The catalytic protocol can be readily extended to substrates that bear a linear, β- or α-branched alkyl group (11–13, Scheme 4); products are isolated in 65–80% yield and in 92:8-97:3 e.r. Optimal results for synthesis of 13 are obtained with the catalyst derived from imidazolinium salt 3c; 90.5:9.5 and 90:10 e.r. and 76% and 83% yield are obtained, respectively, with 3a and 3b. In all transformations shown in Scheme 3, conditions involve the more robust CuII salt (1.0 mol% loading, 1.0 h). As with aryl-substituted imines (Scheme 2 and Table 1), the additions proceed with similar efficiency and slightly higher enantioselectivity when CuCl is used; for example, thienyl 7 and pyridyl 8 are generated in 91% yield and 95% yield and in more than 98:2 and 97:3 e.r., respectively (with 3a as catalyst precursor; more than 98% conversion and less than 2% allene addition).[13]
Scheme 4.

NHC–Cu-catalyzed enantioselective synthesis of heteroaryl-, alkenyl-, and alkyl-substituted homopropargyl amines. (Conditions as in Table 1 with 3a and CuCl2·2H2O, except 3c used for 13. See the Supporting Information for details.)
The NHC–Cu-catalyzed processes likely involve allenyl-copper species, such as A (Scheme 5).[14] Complex A originates from the corresponding Cu–alkoxide, either by s-bond metathesis with the allenylboron 1, or through ligand exchange via allenylboronate derived from addition of a metal alkoxide to 1M. [15] Computational studies[16] point out that the Cu–allene is energetically favored as opposed to the alternative, and probably more nucleophilic, Cu–propargyl (A is 9.9 kcalmol−1 lower in energy compared to B; Scheme 5); the alkynyl complex would produce the unobserved allenyl amides. Examination of the geometry-optimized structures of C and D indicates that the mode of reaction C, with the substrate coordinating from the most accessible quadrant of the chiral complex is preferred (vs. D);[16] such a scenario accounts for the identity of the observed major enantiomers.[17]
Scheme 5.

Theoretical studies point to a strong energetic preference for the NHC–Cu–allenyl A versus the derived propargyl complex B and a model that accounts for the observed stereochemical outcomes.
The strong preference for the intermediacy of allenyl–copper A (vs. the propargyl derivative B) might be the reason that the processes presented here are operationally more robust than the corresponding allyl additions.[10] Although the CuII salt can be used in either case, unless stringently controlled conditions are adopted, there is significant diminution in e.r. when allylcopper intermediates are involved. For example, when the reaction with (pinacolato)allylboron and 2a is carried out under the conditions shown in Scheme 2 (bottom reaction), the desired homoallyl amide is generated in 84:16 e.r. (vs. 97:3 e.r. under strictly inert (glove box) conditions). Such disparity may originate from the higher sensitivity of the more nucleophilic allylmetals (Cu–Csp3 vs. Cu–Csp2 in allenylmetals; see Scheme 6). Moreover, when CuCl2·2H2O is used, transformations are likely catalyzed by an NHC–CuI complex,[18] a scenario supported by the close similarity of the stereochemical outcomes compared to processes involving CuCl. The in situ reduction of the transition metal thus occurs under the reaction conditions.[19]
Scheme 6.

Representative examples demonstrate the utility of the NHC–Cu-catalyzed method for enantioselective synthesis of homopropargyl amines; o-NBSH= o-nitrobenzenesulfonylhydrazide.
The two instances depicted in Scheme 6 demonstrate utility. As illustrated by the formation of 14, the homopropargyl amides can be transformed to the derived carboxylic acids through an efficient and mild Ru-catalyzed transformation.[20] The corresponding β-amino acids, building blocks in the preparation of biologically active molecules,[21] can be accessed by removal of the phosphinoyl group (see Scheme 3).
The second application is connected to the preparation of the amine segment of the anticancer agent aza-epothilone A (Scheme 6).[22] Catalytic enantioselective addition to heterocyclic aldimine 15 proceeds in 98% yield and more than 98:2 e.r. Homopropargyl amide 16 is then converted to the derived iodoalkyne and subsequently reduced[23] with complete Z selectivity to deliver 17 in 76% overall yield and exceptional enantiomeric purity. The Z-vinyl iodide (17) can be utilized in enantioselective synthesis of the biologically active target or other members of the same family through a formerly reported strategy based on catalytic cross-coupling.[24]
In summary, we introduce the first practical, general, and efficient method for catalytic enantioselective preparation of homopropargyl amines. The protocol requires a catalyst composed of an inexpensive metal salt, a chiral ligand that can be prepared in four or five steps, and the allenylboron reagent, which can be purchased; product isolation is straightforward and inexpensive. Design and development of additional chiral catalysts for efficient and enantioselective C–C bond forming processes with readily accessible organoboron reagents is in progress.
Supplementary Material
Footnotes
Financial support was provided by the NIH (GM-57212). We are grateful to Dr. B. Li for securing X-ray structures and to Frontier Scientific, Inc. for gifts of the allenylboron reagent. We thank Boston College Research Services for providing access to computational facilities.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201202694.
References
- [1].a) Nugent TC, El-Shazly M. Adv. Synth. Catal. 2010;352:753–819. [Google Scholar]; b) Nugent TC, editor. Chiral Amine Synthesis. Wiley-VCH; Weinheim: 2010. [Google Scholar]
- [2].For example, see: indolizidine alkaloid family: Song Y, Okamoto S, Sato F. Tetrahedron Lett. 2002;43:8635–8637. subcosine II: Cui L, Li C, Zhang L. Angew. Chem. 2010;122:9364–9367. Angew. Chem. Int. Ed. 2010;49:9178–9181. doi: 10.1002/anie.201004712. l- forosamine: Zacuto MJ, Tomita D, Pirzada Z, Xu F. Org. Lett. 2010;12:684–687. doi: 10.1021/ol9026667. hederacine B: Yamashita M, Yamashita T, Aoyagi S. Org. Lett. 2011;13:2204–2207. doi: 10.1021/ol2004353.
- [3].For representative cases, see: Voituriez A, P rez-Luna A, Ferreira F, Botuha C, Chemla F. Org. Lett. 2009;11:931–934. doi: 10.1021/ol802912f. Fandrick DR, Johnson CS, Fandrick KR, Reeves JT, Tan Z, Lee H, Song JJ, Yee NK, Senanayake CH. Org. Lett. 2010;12:748–751. doi: 10.1021/ol9028258. Cyklinsky M, Botuha C, Chemla F, Ferreira F, P rez-Luna A. Synlett. 2011:2681–2684.
- [4].For a review on catalytic enantioselective propargyl additions, see: Ding C-H, Hou X-L. Chem. Rev. 2011;111:1914–1937. doi: 10.1021/cr100284m.
- [5].Kagoshima H, Uzawa T, Akiyama T. Chem. Lett. 2002:298–299. [Google Scholar]
- [6].Wisniewska HM, Jarvo ER. Chem. Sci. 2011;2:807–810. [Google Scholar]
- [7].A single case has been reported involving an acylhydrazone as the substrate and the relatively moisture-sensitive trichlorosilyl-allene as the reagent; the phenyl-substituted homopropargyl amine was obtained in 53% yield and 89:11 e.r. See: Chen J, Captain B, Takenaka N. Org. Lett. 2011;13:1654–1657. doi: 10.1021/ol200102c. Furthermore, in a recent study, homopropargyl amines were obtained as minor byproducts; see: Huang Y-Y, Chakra-barti A, Morita N, Schneider U, Kobayashi S. Angew. Chem. 2011;123:11317–11320. Angew. Chem. Int. Ed. 2011;50:11121–11124. doi: 10.1002/anie.201105182.
- [8].For an example of catalytic enantioselective synthesis of propargyl glycine derivatives through the use of chiral-phase transfer catalysts, see: Castle SL, Srikanth GSC. Org. Lett. 2003;5:3611–3614. doi: 10.1021/ol035236x.
- [9].For the use of C1-symmetric NHC–Cu complexes in enantioselective synthesis of C–C, C–Si, and C–B bond formation, respectively, see: Lee K.-s., Hoveyda AH. J. Org. Chem. 2009;74:4455–4462. doi: 10.1021/jo900589x. Lee K.-s., Hoveyda AH. J. Am. Chem. Soc. 2010;132:2898–2900. doi: 10.1021/ja910989n. O Brien JM, Lee K.-s., Hoveyda AH. J. Am. Chem. Soc. 2010;132:10630–10633. doi: 10.1021/ja104777u.
- [10].Vieira EM, Snapper ML, Hoveyda AH. J. Am. Chem. Soc. 2011;133:3332–3335. doi: 10.1021/ja200311n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].The addition can be performed on gram-scale with the catalyst derived from CuCl (0.25 mol% loading, 10 h, under otherwise the same conditions as shown in Scheme 2), affording 4a in 81% yield and more than 98:2 e.r (without chromatography).
- [12].For an overview regarding the utility of N-phosphinoylimines in chemical synthesis, see: Weinreb SM, Orr RK. Synthesis. 2005:1205–1227.
- [13].Additionally, 6 is obtained in more than 98% yield and 96.5:3.5 e.r., 9 is isolated in 88% yield and 98:2 e.r., and 12 is formed in 75% yield and 98:2 e.r. (> 98% conv. and < 2% allene addition in all cases). See the Supporting Information for additional details.
- [14].For the role of MeOH in the catalytic cycle, see Ref. [10].
- [15].Jung B, Hoveyda AH. J. Am. Chem. Soc. 2012;134:1490–1493. doi: 10.1021/ja211269w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].See the Supporting Information for details.
- [17].Since protonation of the intermediate NHC–Cu–allene by MeOH does not constitute a major pathway, it is likely that N-phosphinoylimines are associated to the Lewis acidic transition metal when an NHC–Cu species is generated, leading to an intramolecular allyl transfer. Such considerations, together with the high levels of enantioselectivity, suggest that open transition structures are likely not involved. For a related discussion, see: Vrancken E, G rard H, Linder D, Ouizem S, Alouane N, Roubineau E, Bentayeb K, Marrot J, Mangeney P. J. Am. Chem. Soc. 2011;133:10790–10802. doi: 10.1021/ja200702a.
- [18].For catalytic processes that involve the use of CuII salts and bis(pinacolato)boron, see: Chea H, Sim H-S, Yun J. Bull. Korean Chem. Soc. 2010;31:551–552. Guzman-Martinez A, Hoveyda AH. J. Am. Chem. Soc. 2010:132, 10634–10637. doi: 10.1021/ja104254d. Ibrahem I, Breistein P, Cordova A. Angew. Chem. 2011;123:12242–12247. doi: 10.1002/anie.201105458. Angew. Chem. Int. Ed. 2011;50:12036–12041. doi: 10.1002/anie.201105458.
- [19].A possible route for reduction of CuII involves reaction of NHC–CuIICl(OtBu) with 1 to generate (pin)B-OtBu and NHC–CuIICl(allene). Subsequent homolytic Cu–C bond cleavage affords NHC-CuICl and the product from allene dimerization.
- [20].Yang D, Chen F, Dong Z-M, Zhang D-W. J. Org. Chem. 2004;69:2221–2223. doi: 10.1021/jo0357925. [DOI] [PubMed] [Google Scholar]
- [21].For a review on the significance of β-amino acids, see: Seebach D, Beck AK, Bierbaum DJ. Chem. Biodiversity. 2004;1:1111–1239. doi: 10.1002/cbdv.200490087.
- [22].For synthesis and biological activity of aza-epothilones, see: Schinzer D, Altmann K-H, Stuhlmann F, Bauer A, Wart-mann M. ChemBioChem. 2000;1:67–70. doi: 10.1002/1439-7633(20000703)1:1<67::AID-CBIC67>3.0.CO;2-I.
- [23].For examples of Z-selective reduction of iodoalkynes with o-nitrobenzenesulfonylhydrazide (o-NBSH), see: Rouden J, Seitz T, Lemoucheux L, Lasne M-C. J. Org. Chem. 2004;69:3787–3793. doi: 10.1021/jo0498157. Zhu W, Jim nez M, Jung W-H, Camarco DP, Balachandran R, Vogt A, Day BW, Curran DP. J. Am. Chem. Soc. 2010;132:9175–9187. doi: 10.1021/ja103537u.
- [24].a) Borzilleri RM, Zheng X, Schmidt RJ, Johnson JA, Kim S-H, DiMarco JD, Fairchild CR, Gougoutas JZ, Lee FYF, Long BH, Vite GD. J. Am. Chem. Soc. 2000;122:8890–8897. [Google Scholar]; b) Stachel SJ, Lee CB, Spassova M, Chappell MD, Bornmann WG, Danishefsky SJ, Chou T-C, Guan Y. J. Org. Chem. 2001;66:4369–4378. doi: 10.1021/jo010275c. Chakra. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.