

Abstract
Although exposure to major psychological trauma is unfortunately common, risk for related neuropsychiatric conditions, such as post-traumatic stress disorder (PTSD), varies greatly among individuals. Fear extinction offers a tractable and translatable behavioral readout of individual differences in learned recovery from trauma. Studies in rodent substrains and subpopulations are providing new insights into neural system dysfunctions associated with impaired fear extinction. Rapid progress is also being made in identifying key molecular circuits, epigenetic mechanisms, and gene variants associated with differences in fear extinction. Here, we discuss how this research is informing understanding of the etiology and pathophysiology of individual differences in risk for trauma-related anxiety disorders, and how future work can help identify novel diagnostic biomarkers and pharmacotherapeutics for these disorders.
Keywords: extinction, anxiety, amygdala, prefrontal cortex, PTSD, cross-species translation
Introduction
Exposure to severe psychological traumas can result in debilitating anxiety disorders, such as PTSD [1]. However, although the proportion of the US population exposed to at least one severe trauma might be as high as 75% [2], the lifetime prevalence of PTSD is only approximately 7% [3]. Thus, population wide, the number of people exposed to trauma and stress far exceeds the number developing a clinical condition such as PTSD (i.e., resilience is the statistical norm in environments where major traumas are relatively rare). Nonetheless, there is still a significant population of individuals who are susceptible and, for groups exposed to severe traumatic events (e.g., military combat, terrorist acts, or severe accidents), rates of PTSD are 20–30% [4].
A major step towards advancing the ability to screen and treat effectively individuals at elevated risk for trauma-related anxiety disorders will be identifying key pathophysiological factors that influence relative risk. In recent years, the field has seen rapid advances in elucidating neural systems and circuits that are dysfunctional in patients with anxiety disorders, including PTSD [5], and recruited in rodent and human subjects during impaired processes, such as fear extinction. There have also been new insights into some of the molecular and genetic factors that moderate PTSD risk via the formation and function of these neural circuits. Here, we discuss how a growing body of evidence from studies in rodent and human subjects that utilize fear extinction as a readout of recovery from learned trauma is rapidly informing understanding of the etiology and pathophysiology of individual differences in risk for PTSD and other trauma-related conditions.
Fear extinction as a tractable, translational assay for modeling individual differences in recovery from trauma
Most individuals exposed to trauma exhibit some of the symptoms of PTSD in the immediate aftermath [6], but PTSD is distinguished from the normal reaction to trauma by the persistence of these symptoms (defined by [1] as at least 1 month post-trauma) and can therefore be conceptualized to some extent as a failure in the process of recovery [7]. Preclinical assays that measure the capacity to recover from some of the effects of a traumatic exposure therefore have good face validity for modeling PTSD.
One assay that meets this criterion is fear extinction (for illustration, see Box 1). Extinction, recognized by Pavlov almost 100 years ago [8], occurs when ‘a fear conditioned organism exposed to a fear-eliciting cue in the absence of any aversive event. . .result[ing] in a decline in conditioned fear responses’ [9]. Under most conditions (with exceptions, such as juvenile rodents [10]), the behavioral manifestation of fear extinction likely reflects the emergence of a new, inhibitory form of learning that successfully competes with the trauma memory to reduce fear expression. Fear extinction is an evolutionarily conserved process that can be readily measured across species, making it a tractable preclinical assay for modeling trauma recovery in animals. Indeed, because patients with PTSD and certain other anxiety disorders exhibit impaired fear extinction [11], probably as a result rather than an antecedent of the trauma [12] (but see [13]), this measure has clear translational relevance to human experimental studies and the clinical condition itself.
Box 1. Population differences in fear extinction and environmental risk factors.
Psychological traumas that lead to anxiety disorders, such as PTSD, are by definition extreme stressors, but a history of stress before a trauma can act as an additional risk factor. In rodents that exhibit good extinction under non-stressed conditions (Figure Ia), stress before fear conditioning can impair the ability to extinguish fear subsequently (Figure Ib). An early example in mice found a relatively modest regimen of three exposures to forced swim stress, given before conditioning, impaired extinction learning in the absence of any increases in fear [32]. In fact, as little as a single 30-min (elevated platform) or 2-h restraint stressor, given either before or after conditioning or after partial extinction learning, is sufficient to impair subsequent extinction learning in mice and rats [46,74-76], and only 10 min of predator odor was enough to disrupt extinction in a subset of the Lewis rat strain [77]. More prolonged stressors involving daily exposure to restraint or corticosterone treatment for a week or more also impair extinction acquisition or retrieval [34,78-82].
Chronic exposure to alcohol also impairs extinction in rodents [48,83,84] (Figure Ib). This is intriguing from a clinical perspective because alcoholism and anxiety disorders frequently co-occur in the same individuals [85] and excessive drinking increases exposure to traumas (e.g., automobile accidents or domestic violence; http://www.who.int/violence_injury_prevention/violence/world_report/factsheets/ft_intimate.pdf).
Stress and alcohol might impair fear extinction by impacting the same cortico-amygdala circuits that are dysfunctional in extinction-impaired populations. Both stress- and alcohol-induced extinction deficits are associated with dysmorphology in medial prefrontal cortex (mPFC) pyramidal neurons [32,48], decreased mPFC NMDAR expression [48,79], and a reduction in cue-evoked IL firing [34,48]. Stress also produces sustained neuronal hyperexcitability and dendritic hypertrophy in BLA pyramidal neurons [86,87]. Thus, it appears that exposure to stress and alcohol shifts the balance of cortico-amygdala function to favor fear over extinction.
Exposure to these environmental risk factors in individuals already at risk (e.g., from their genetic constitution) would be predicted to combine to impact deleteriously extinction and associated risk for PTSD, as is the case in other affective disorders [14]. On a brighter note, recent studies demonstrate that stress- and alcohol-induced impairments in fear extinction are reversible. For example, DCS [76] or a putative BDNF-TrkB agonist [82] can reverse stress-induced extinction deficits. In addition, both neuropeptide S infused into the LA around the time of stress [46] or systemically administrated diazepam given before stress [74] prevent stress-impaired extinction and some of the associated neural changes (e.g., amygdala hyperexcitability [46]).
In humans, individual differences in fear extinction likely arise from a combination of life history, prior exposure to stress and trauma, and predisposing genetic and biological factors. The complexity of these differences makes the task of parsing the relative contribution of specific influences challenging [14]. The experimental control afforded by rodent models, combined with the greater capacity for making precise delineation of neural systems, molecules, and genes in rodents compared with humans, makes them an essential compliment to human studies. It also facilitates the study of how certain environmental risk factors, including a prior history of stress or alcohol exposure, exert extinction-impairing effects (Box 1).
One useful approach in rodent studies has been to divide, a posteriori, a sample of rats or mice into subgroups exhibiting good versus poor extinction (for a pioneering example, see [15]) and then to examine whether the groups differed in an extinction-related neural phenotype [16]. Another valuable method is to selectively breed for fear [17] or anxiety-like behavior [18] and then test for differences in fear extinction. A third approach utilizes inbred strains of rats and mice. Within an inbred strain, each individual is essentially genetically identical, which allows for the testing of an unlimited sample of clones within and across experiments. Thus, because any given inbred strain differs genetically to other strains [19], two inbred strains can be treated as two genetic populations each comprising genetically homogeneous individuals. Where strains differ in a fear phenotype of interest, such as fear extinction, associated neurobiological factors can be studied in genetically stable populations that, unlike subdivided outbred or selectively bred lines, can be reproduced ad infinitum. For example, using this approach, the C57BL/6J (‘B6’) and 129S1/SvImJ (‘S1’) inbred mouse strains were identified as exhibiting reliably intact and impaired fear extinction, respectively [20,21].
Cortico-amygdala circuitry associated with variation in extinction
The brain regions mediating fear extinction have been increasingly well mapped by studying the impairing effects of permanently lesioning or temporarily inactivating specific brain areas in rodent populations normally exhibiting good fear extinction. This work has centered on the amygdala, mPFC, and hippocampus [16,22-26]. An important extension to this approach has been to measure patterns of endogenous activation, simultaneously in multiple brain regions, associated with population and/or strain differences in extinction. Neuronal correlates of individual differences in extinction have been examined using imaging techniques, including metabolic mapping (via 2-deoxyglucose uptake [27]) and, in particular, quantification of immediate-early gene (IEG) expression, which is a marker of neuronal activation (reviewed in [28]). These studies reveal how impaired extinction is associated with patterns of aberrant neuronal activation in relevant brain circuits.
One key regional node within the extinction circuitry is the infralimbic cortex (IL). Impaired extinction has been associated with relatively low levels of IL IEG expression in various models. These models include the aforementioned S1 mouse strain [20], a poor-extinction subpopulation of B6 mice [29], B6 mice treated with a cannabinoid CB1 receptor antagonist [30], and rats either extinguished immediately after conditioning [31] or bred for high anxiety-like behavior [18]. This highly consistent observation of IL hypoactivity concurs with other data, such as the finding that stress-induced extinction impairment is associated with IL dendritic hypotrophy in mice [32] and the observation that low in vivo IL neuronal activity predicts poor extinction in rats [16]. Importantly, it also extends to humans, in whom functional magnetic resonance imaging (fMRI) studies indicate that hypoactivation and lesser thickness of a functionally analogous ventromedial PFC (vmPFC) region correlates with poor extinction in healthy humans and patients with PTSD (reviewed in [16,33]).
Emerging evidence suggests that a region of the vmPFC neighboring the IL [the prelimbic cortex (PL) in rodents and dorsal anterior cingulate (dACC) in humans] has an opposite role to IL in regulating fear and extinction. Consistent with lesion and in vivo electrophysiology data suggesting a fear-promoting role of PL [34-37], PL IEG activity is significantly elevated in the extinction-deficient S1 mouse strain [38] and in rats showing high fear [39]. Furthermore, the dACC is hyperactive in patients with PTSD [16]. These various lines of evidence suggest that variation in the relative recruitment of PL and IL is a significant predictor of individual differences in the ability to extinguish a fear memory both in animals and humans.
Another major hub of the extinction circuit is the amygdala [23,26]. IEG analysis of extinction-impaired mice (S1 strain, B6 subpopulation, and protease nexin-1 knockout) indicates reduced recruitment of the lateral (LA), basal (BA), and central lateral (CeL) nuclei of the amygdala [20,29,38,40]. The medial (ventrally positioned) intercalated cell mass (ITC), part of a GABAergic feedforward relay station interconnecting amygdala nuclei [26,40,41], is also hypoactive [38,42]. To some degree, the attenuated extinction-related activation of BA and CeL may reflect a failure to engage specialized extinction-encoding neurons recently identified in both of these regions [43,44], but this remains to be determined. It is currently unclear how these functional aberrations might be related to the pyramidal dendritic hypertrophy found in amygdala neurons in some models of impaired extinction [21], although it is interesting that stress produces a similar morphological effect [45] in tandem with impaired extinction [32,34,46].
In contrast to the extinction impairment-associated attenuation of LA–BA–ITC–CeL recruitment, impaired extinction is associated with elevated IEG activity in the central medial nucleus (CeM) of the amygdala (and PL and insular cortex) [20,38]. The CeM is the major output station of the amygdala that drives fear via its connections to the hypothalamus and brainstem regions [44]. Although its small size and location deep in the brain hampers the imaging of specific subregions of the amygdala in humans, the amygdala as a whole also exhibits hyperactivation in patients with anxiety disorders (reviewed in [16,33]). These data in rodents and humans are consistent with sustained amygdala fear drive in extinction-impaired individuals, possibly resulting from a failure to engage proextinction circuits in cortical and subregions of the amygdala (Figure 1).
Figure 1.

Variation in cortico-amygdala function underlying individual differences in effective extinction versus a bias towards sustained fear. (a) Effective extinction is associated with recruitment of (i) a pathway from the infralimbic cortex (IL), via certain intercalated cell masses (ITC) (and possibly an area known as the capsular IL target zone or intramedullary gray [41,42], not shown), and including the medial central amygdala (CeM) [88]; (ii) connections from a subpopulation of cells in the basal amygdala (BA) [43], some via ITC, to the CeM; and (iii) an inhibitory drive from some cells in the lateral central amygdala (CeL) to CeM [44,89]. (b) Poor extinction and persistent fear is associated with inadequate engagement of extinction circuits and dominance of pro-fear pathways from the lateral amygdala (LA) to BA to CeM [44], as well as reciprocal connections between prelimbic cortex (PL) and BA [16,36]. Note: for simplicity, this model does not include all connections or other brain regions (e.g., hippocampus or thalamus) that likely contribute to individual differences in fear processing and extinction [16,22,23,25]. (c) The rescue of impaired extinction has been tightly coupled to the normalization of many of the corticolimbic abnormalities associated with sustained fear in extinction-impaired populations. An illustrative example is provided by immediate-early gene analysis of neuronal activation in the S1 mouse strain model given the multitarget extinction treatment of dietary zinc restriction [38]. Under basal non-extinction (CS−) conditions, the good-extinguishing B6 (black bars) and poor-extinguishing S1 (red bars) mouse strains do not differ in the number of Zif268-positive cells, in any region, regardless of zinc restriction (ZnR). Following extinction training (CS+), treatment increased activation in previously hypoactive regions (green shading) in the S1 strain that subserve the formation of extinction memories, including the IL, LA, BA, ITC, and CeL, to levels equivalent to baseline activity seen in a good-extinguishing (B6) mouse strain. Conversely, neuronal activity in the previously hyperactive (pink shading), pro-fear regions of the CeM and PL decreased after treatment. This shows how neural abnormalities in extinction-deficient individuals are not permanent and can be effectively reversed by effective drug treatments. Note: for simplicity, neither the insular cortex (hyperactivity in S1 normalized by treatment) nor regions showing no differential activation are shown). Data in bar graphs are redrawn, with permission, from [38].
From variation in extinction systems to molecular mechanisms
Does IEG activation tell more about the extinction process than whether certain brain regions are over or under recruited? It seems that it likely does because IEGs, such as c-Fos and Zif268, are activated as part of a broader set of signaling pathways involved in neuroplasticity [25]. Thus, aberrant IEG activity in extinction-impaired rodents may, at least in part, reflect an inability to engage effectively mechanisms necessary for the long-term changes in circuit functions that subserve extinction memory formation. The precise molecular mechanisms regulating extinction-related synaptic plasticity are the subject of intense investigation and the reader is referred to excellent comprehensive literature reviews on the topic [9,25].
One key mechanism worth noting in the context of individual differences is glutamatergic signaling and synaptic plasticity mediated through the NMDA receptor (NMDAR). Pyramidal neurons in the vmPFC and BLA are glutamatergic and express NMDARs. GABAergic interneurons in these regions also express NMDARs, but presynaptically. The functional importance of NMDARs to extinction has been demonstrated in several ways. First, IL neurons exhibit a pattern of burst firing during extinction that is NMDAR (GluN2B subunit) dependent, and NMDAR antagonists infused into the vmPFC impair extinction consolidation [47,48]. Second, NMDAR or GluN2B-specific antagonists injected into the BLA impede extinction learning [49,50], whereas, conversely, either systemic or intra-BLA administration of D-cycloserine (DCS; a drug that acts as a partial agonist at NMDARs at low concentrations) facilitates extinction learning in rodents and human phobics [51].
Interestingly, however, the extinction-promoting effects of DCS appear to vary as a function of the amount of extant extinction learning. A pro-extinction effect of DCS was only seen in those rats that showed some evidence of extinction learning [52] or when a sufficient number of trials were given to initiate extinction learning [53]. Similarly, although DCS facilitates extinction in a mouse strain (B6) that exhibits extinction learning under untreated conditions [54], the drug is ineffective in an extinction-deficient strain such as S1 [20]) unless some extinction learning is elicited (for example after weak conditioning) [54].
Mechanistically, this suggests that NMDAR-mediated plasticity is necessary but not sufficient for extinction memory formation, and that recruitment of other molecular pathways is required to initiate extinction learning and gate the facilitatory action of DCS. The requirement for extant extinction learning is not surprising given DCS likely promotes extinction via enhancing consolidation [55], but it could have important implications for how the drug is to be most effectively used in patients with PTSD. The preclinical data predict that activating NMDARs would have relatively poor efficacy in individuals that are severely extinction impaired or fail to respond to exposure therapy. There is some initial support for this: randomized, placebo-controlled studies showed DCS to be ineffective in combat-related PTSD [56] unless patients completed extensive exposure therapy [57]. Further studies along these lines would be valuable.
Future strategies for rescuing impaired extinction
A deeper understanding of the neural, molecular, and genetic basis of impaired extinction opens up novel, mechanistically based avenues to potential therapies to reverse these impairments. The plethora of extinction-related neurotransmitters, molecular signaling pathways, and epigenetic mechanisms (Box 2) that are being revealed bodes well for the identification of new, targetable mechanisms [9,25]. Excitingly, therapeutic mechanisms may not be limited to the pharmacological, but extend to manipulations, including deep-brain stimulation [54,58,59] and purely behavioral interventions that exploit recall-induced memory lability and reconsolidation [60,61].
Box 2. Epigenetic changes associated with fear extinction.
There is increasing evidence that epigenetic mechanisms, including histone modifications and DNA methylation, contribute critically to the regulation of synaptic plasticity and learning [90]. This extends to fear extinction, which is known to depend on coordinated gene expression and protein synthesis that, in turn, is modulated by epigenetic mechanisms [91]. Indeed, histone acetylation is dynamically regulated in the rodent brain following successful extinction learning [92-97]. These changes are functionally relevant because pharmacological or genetic silencing of specific HDAC isoenzymes facilitates fear extinction [91,97-102]. Extending these data, it was recently found that treatment with a HDAC1,2,3-selective inhibitor rescued impaired extinction in a mouse model (i.e., the S1 strain) by enabling consolidation of extinction learning [54]. Dietary zinc deficiency, which among its various neural targets, inhibits zinc-dependent HDACs, also rescues extinction in the same model [54].
It is not yet clear which specific HDAC isoform(s) mediate extinction. Pharmacological or genetic silencing of HDAC1 or HDAC2 isoforms enhances fear or extinction consolidation [100,103]. Valproic acid (VPA), an anticonvulsant that acts as a robust pan-HDAC inhibitor (in addition to promoting GABA signaling), has been shown to facilitate extinction learning in healthy humans [104]. VPA rescues extinction learning and retrieval in the S1 strain [105], which is a broader behavioral effect than the consolidation-specific effect of HDAC1,2,3 inhibition [105]. Interestingly in this context, the extinction-promoting effects of VPA have been attributed specifically to histone H4 acetylation, acting on the BDNF promoter to enhance BDNF gene expression in the mPFC [91,98]. These preliminary findings raise the possibility that HDAC isoforms differentially contribute to fear extinction by targeting specific regional nodes within the neural circuitry.
The aforementioned work on DCS provides an exemplar of how a pharmacological approach developed from hypothesis-driven studies on rodent extinction has successfully translated to therapeutic application in anxiety disorders, including phobia and PTSD [51,62,63]. Pharmacotherapeutic intervention strategies such as this are built around the idea that, by targeting known extinction-mediating mechanisms, extinction can be strengthened by treatments given as adjuncts before, during, or just after exposure therapy [9,64]. In fact, in some cases, the lasting fear-reducing effects of extinction training can be mimicked by pharmacological treatments (e.g., brain-derived neurotrophic factor gene, BDNF) in the absence of any training [65]. Given that extinction is context [66] and time [8] dependent, the goal of therapy is to produce an extinction memory that is robust across environments and the passage of time.
To date, several drugs have been shown to rescue deficient extinction in rodent models of impaired extinction (Table 1). These include the metabotropic glutamate receptor subtype-7 agonist, AM082 [54], the α2-adrenoceptor antagonist, yohimbine [20], and the serotonin reuptake inhibitor fluoxetine [21]. Fluoxetine is noteworthy among these examples by virtue of being a first-line treatment for anxiety disorders (although its utility as an effective adjunct has been questioned [67,68]. Two other examples from animal studies are illustrative of some general issues. The first involves an experimental multi-target approach, in the form of dietary zinc restriction. Depleting zinc fully rescued impaired extinction behavior in the S1 mouse strain and further facilitated extinction over untreated baseline in the B6 strain [38]. As in the case of fluoxetine [21], the effect was produced when animals were treated after conditioning, mimicking the clinical situation in which treatment typically commences after trauma. However, perhaps the most interesting aspect of the zinc depletion rescue was that it normalized cortico-amygdala activation abnormalities in parallel in parallel with the rescue of extinction. As demonstrated by IEG mapping, depletion restored activity in various pro-extinction brain regions (e.g., IL, BA, ITCv, and CeL) that were under-recruited in the absence of treatment, and suppressed pro-fear regions (e.g., PL, CeM, and insular cortex) [38] (illustrated in Figure 1d). The reduction in amygdala output (and insular activity [69,70]) is reminiscent of the reversal of amygdala hyperactivity seen in patients with anxiety after successful antidepressant [71] or exposure therapy [70]. These findings suggest that clinically relevant neural correlates of treatment effects can be identified in tandem with the rescue of extinction. These translatable neural ‘biomarkers’ could prove useful for tracking the efficacy of other treatments in rodent models and patient populations.
Table 1.
Examples of effect of extinction facilitating and/or rescuing treatments in animal models of impaired extinction and human anxiety disordersa
| Agent class | Treatment | Animal studiesb | Human studies | |||
|---|---|---|---|---|---|---|
| FC | Model | Short-term extinction |
Extinction retrieval |
Efficacy as adjunct to exposure therapy |
||
| Adrenergic α2 R antagonist | Yohimbine | Cue | Genetic [20] | Nd | + | + Claustrophobia [106] |
| NMDAR agonist | DCS | Context | Stress [76] | Nd | + | + Acrophobia, social phobia |
| DCS | Cue | Genetic [20] | − | − | Panic disorder ([51,62,63]) | |
| DCS | Cue | Adolescent [107] | Post | + | −/(+) PTSD | |
| DCS | Cue* | Genetic [38] | Post | + | PTSD, +/− OCD ([51,56,57,62,63]) | |
| AMPAR agonist | PEPA | Cue | Genetic [38] | − | − | Nd |
| mGLUR7 agonist | AMN082 | Cue | Genetic [38] | + | + | Nd |
| FAAH inhibitor | AM3506 | Cue | Genetic [73] | + | + | Nd |
| SSRI | Fluoxetine | Cue | Genetic [54] | − | + | + PTSD [68], − social phobia [67] |
| HDAC inhibitor | MS-275 | Cue | Genetic [54] | − | − | Nd |
| Cue* | Genetic [54] | Post | + | Nd | ||
| Neurotrophic factors | BDNF | Cue | Stress [82] | + | ? | Nd |
| Neuropeptides | NPS | Cue | Stress [46] | + | ? | Nd |
| Multitarget (GABAergic, GC, GLUergic, HDAC inhibitor) |
ZnR | Cue | Genetic [54] | + | + | Nd |
| Multitarget (GABAeric, HDAC inhibitor) |
VPA | Cue | Genetic [54] | + | + | Nd |
| Other approaches | DBS | Cue | Genetic [54] | − | + | Nd |
DBS, accumbal deep brain stimulation; FC, fear conditioning; GC, glucocorticoid; GLU, glutamate; HDAC, histone deacetylase; Nd, not determined; post, given post extinction training (all other treatments given before or during extinction training); R, receptor; SSRI, selective serotonin reuptake inhibitor; VPA, valproic acid; ZnR, dietary zinc restriction.
+ effect, − no effect, +/− mixed results
weak fear conditioning.
Preclinical drug studies can also provide leads to genetic biomarkers for individual differences in risk for trauma-related anxiety disorders. A recent illustration of this approach stems from basic research implicating the endocannabinoid system in fear extinction [72]. Pharmacological inhibition of fatty acid amide hydrolase (FAAH), an enzyme controlling levels of the endocannabinoid anandamide, was found to rescue impaired fear extinction in the S1 mouse strain by augmenting endocannabinoid signaling and synaptic plasticity in the amygdala [73]. This observation generated the hypothesis that variation in the gene encoding FAAH in humans would associate with differences in anxiety-related behavior, possibly via moderation of amygdala functions. In support of this hypothesis, a lesser-functioning SNP in FAAH was associated with low scores on a trait (i.e., stress reactivity) that predicts reduced risk for PTSD [73]. This trait was also associated with more rapid habituation of amygdala responses to a threatening stimulus (i.e., fearful faces) [73]. This work and others like it (see Box 3 for some excellent examples) provides proof-of-principle of how rodents models of impaired extinction can scale up to predict individual differences in human brain functions and behaviors, even beyond fear extinction.
Box 3. Gene variants moderating fear extinction, PTSD and cortico-amygdala function.
Studies in identical twins (the gold standard for gauging the genetic contribution to disease) estimate the heritability component of PTSD to be 25–35% [108,109]. Experimentally induced learned fear also has a strong genetic contribution [110]. Although extinction is impaired in PTSD, what is less clear is whether deficient extinction is an endophenotype for PTSD in the true genetic sense. This would be demonstrated by evidence that poor fear extinction precedes exposure to a PTSD-causing trauma, or if poor extinction was found to occur at higher than normal rates in unaffected relatives of patients with PTSD.
The structure and function of cortico-amygdala circuits subserving fear regulation is moderated by genetics in humans [111] and rodents [112]. Healthy identical twins of patients with combat-related PTSD exhibit higher activation of the dACC, relative to unrelated, combat-exposed, control subjects without PTSD [113]. Furthermore, smaller hippocampal volumes have been reported in combat-PTSD and noncombat twins than in combat-experienced non-PTSD non-relatives [114], although this may depend upon the subjects’ co-morbid history of alcoholism (Box 1) [115]. These data suggest possible pre-existing neural antecedents of PTSD. By contrast, combat-exposed PTSD individuals have less gray matter in the pregenual ACC than do their combat-exposed twins without PTSD, which is more consistent with an effect resulting from, rather than being antecedent to, PTSD [116].
There is intense interest in identifying specific gene variants underlying risk for PTSD [117]. It is fair to say that, as with other neuropsychiatric conditions, research aimed at identifying specific genes associated with PTSD has been inconclusive. Nevertheless, several potential candidates have emerged. Some of the most compelling studies connect a gene variant with differences in extinction or PTSD prevalence and provide parallel evidence, from human or preclinical work, of the related neurobiological correlates. For example, variation in the gene encoding the serotonin transporter, SLC6A4, is associated with increased PTSD rates in high-trauma populations [118] and impaired extinction in healthy volunteers [119]. Deleting the same gene in mice also impairs extinction and causes dendritic dysmorphology in the vmPFC and amygdala [120], suggesting a translationally relevant neural correlate of the behavioral effect. Along similar lines, a ‘val(66)met’ SNP in the BDNF gene [121] is associated with impaired extinction in healthy subjects, in transgenic mice with a humanized version of the variant [122], and in rats with low levels of endogenous hippocampal BDNF [65]. Likewise, a variant in the gene encoding human prodynorphin, PDYN, predicts amygdala hyperactivity and diminished vmPFC coupling during extinction, while dynorphin-knockout mice show impaired extinction and aberrant vmPFC and/or amygdala activation [123]. In another excellent example of cross-species translational evidence, variation in the genes encoding the pituitary adenylate cyclase-activating polypeptide gene and its receptor (ADCYAP1 and ADCYAPR1, respectively) associates with fear learning and PTSD prevalence in females and amygdala ADCYAPR1 mRNA expression increases with fear learning in mice [124].
Concluding remarks
Our goal here was to further underscore the utility of considering individual differences in risk for trauma-related disorders and, as a consequence, the potential for developing animal models of a translatable, disorder-associated endophenotype, such as deficient Pavlovian fear extinction. Basic research to date using rodent models of impaired extinction has provided new insights into the cortico-amygdala systems and gene variants underlying differences in fear extinction. In an important extension of this work, recent studies have demonstrated how treatments that effectively rescue impaired extinction produce concomitant functional normalization in aberrant cortico-amygdala recruitment. As we schematize in Figure 2, the emerging preclinical research on individual differences in fear extinction offers a tractable strategy for identifying novel therapeutic approaches and diagnostic risk biomarkers for PTSD and other trauma-related anxiety disorders.
Figure 2.

A strategy for advancing the neurobiological understanding and future treatment of post-traumatic stress disorder (PTSD) by screening for individual differences in fear extinction. The ideal subject pool is a population of individuals with trauma-related anxiety disorders, or fear-conditioned animals, ideally with detailed biographical and/or experimental information on exposure to predisposing environmental risk factors, such as stressful life events and alcohol abuse. Following screening for fear extinction, individuals are selected on the basis of impaired versus intact extinction and compared for aberrant versus effective neural circuit function, respectively. This provides a basis for identifying specific molecules and gene variants underlying these circuit variations, which in turn can lead to pro-extinction therapeutic target development to benefit existing patients, as well as novel biomarkers for the future screening of individuals at elevated risk of trauma-related anxiety disorders.
Figure I.
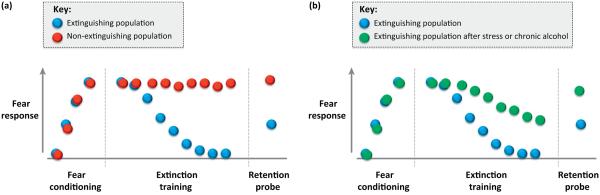
Extinguishing and non-extinguishing populations, and effects of stress or chronic alcohol in extinguishing populations. (a) A similarly conditioned fear response to a previously neutral stimulus (e.g., tone) repeatedly paired with an inherently aversive stimulus (e.g., electric shock) can be extinguished by repeated presentation of the stimulus in the absence of an aversive outcome in some populations of humans and rodents, but not others. (b) Even in populations that normally extinguish, impairments in either extinction learning and/or retention can be produced by stress or chronic alcohol exposure.
Acknowledgments
A.H. is supported by the NIAAA Intramural Research Program. N.S. is supported by the Austrian Science Fund FWF (SFB F4410-B19 and W 1206-B18 SPIN).
References
- 1.American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders. 4th APA Press; 1994. [Google Scholar]
- 2.Breslau N, Kessler RC. The stressor criterion in DSM-IV posttraumatic stress disorder: an empirical investigation. Biol. Psychiatry. 2001;50:699–704. doi: 10.1016/s0006-3223(01)01167-2. [DOI] [PubMed] [Google Scholar]
- 3.Kessler RC, et al. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wisnivesky JP, et al. Persistence of multiple illnesses in World Trade Center rescue and recovery workers: a cohort study. Lancet. 2011;378:888–897. doi: 10.1016/S0140-6736(11)61180-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lissek S, et al. Classical fear conditioning in the anxiety disorders: a meta-analysis. Behav. Res. Ther. 2005;43:1391–1424. doi: 10.1016/j.brat.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 6.McFarlane AC. Posttraumatic stress disorder: a model of the longitudinal course and the role of risk factors. J. Clin. Psychiatry. 2000;61(Suppl. 5):15–20. discussion 21–13. [PubMed] [Google Scholar]
- 7.Yehuda R, LeDoux J. Response variation following trauma: a translational neuroscience approach to understanding PTSD. Neuron. 2007;56:19–32. doi: 10.1016/j.neuron.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Pavlov IP. Conditioned Reflexes. Oxford University Press; 1927. [Google Scholar]
- 9.Myers KM, Davis M. Mechanisms of fear extinction. Mol. Psychiatry. 2007;12:120–150. doi: 10.1038/sj.mp.4001939. [DOI] [PubMed] [Google Scholar]
- 10.Kim JH, Richardson R. New findings on extinction of conditioned fear early in development: theoretical and clinical implications. Biol. Psychiatry. 2010;67:297–303. doi: 10.1016/j.biopsych.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Milad MR, et al. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol. Psychiatry. 2009;66:1075–1082. doi: 10.1016/j.biopsych.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milad MR, et al. Presence and acquired origin of reduced recall for fear extinction in PTSD: results of a twin study. J. Psychiatr. Res. 2008;42:515–520. doi: 10.1016/j.jpsychires.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guthrie RM, Bryant RA. Extinction learning before trauma and subsequent posttraumatic stress. Psychosom. Med. 2006;68:307–311. doi: 10.1097/01.psy.0000208629.67653.cc. [DOI] [PubMed] [Google Scholar]
- 14.Caspi A, et al. Genetic sensitivity to the environment: the case of the serotonin transporter gene and its implications for studying complex diseases and traits. Am. J. Psychiatry. 2010;167:509–527. doi: 10.1176/appi.ajp.2010.09101452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bush DE, et al. Individual differences in fear: isolating fear reactivity and fear recovery phenotypes. J. Traumat. Stress. 2007;20:413–422. doi: 10.1002/jts.20261. [DOI] [PubMed] [Google Scholar]
- 16.Milad MR, Quirk GJ. Fear extinction as a model for translational neuroscience: ten years of progress. Annu. Rev. Psychol. 2012;63:129–151. doi: 10.1146/annurev.psych.121208.131631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sokoloff G, et al. Anxiety and fear in a cross of C57BL/6J and DBA/2J mice: mapping overlapping and independent QTL for related traits. Genes Brain Behav. 2011;10:604–614. doi: 10.1111/j.1601-183X.2011.00699.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muigg P, et al. Impaired extinction of learned fear in rats selectively bred for high anxiety: evidence of altered neuronal processing in prefrontal-amygdala pathways. Eur. J. Neurosci. 2008;28:2299–2309. doi: 10.1111/j.1460-9568.2008.06511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yalcin B, et al. Sequence-based characterization of structural variation in the mouse genome. Nature. 2011;477:326–329. doi: 10.1038/nature10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hefner K, et al. Impaired fear extinction learning and cortico-amygdala circuit abnormalities in a common genetic mouse strain. J. Neurosci. 2008;28:8074–8085. doi: 10.1523/JNEUROSCI.4904-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Camp MC, et al. Genetic strain differences in learned fear inhibition associated with variation in neuroendocrine, autonomic, and amygdala dendritic phenotypes. Neuropsychopharmacology. 2012;37:1534–1547. doi: 10.1038/npp.2011.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pape HC, Pare D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol. Rev. 2010;90:419–463. doi: 10.1152/physrev.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ehrlich I, et al. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 24.Herry C, et al. Neuronal circuits of fear extinction. Eur. J. Neurosci. 2010;31:599–612. doi: 10.1111/j.1460-9568.2010.07101.x. [DOI] [PubMed] [Google Scholar]
- 25.Orsini CA, Maren S. Neural and cellular mechanisms of fear and extinction memory formation. Neurosci. Biobehav. Rev. 2012;36:1773–1802. doi: 10.1016/j.neubiorev.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pare D, Duvarci S. Amygdala microcircuits mediating fear expression and extinction. Curr. Opin. Neurobiol. 2012;22:717–723. doi: 10.1016/j.conb.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toledo-Rodriguez M, et al. Stress during puberty boosts metabolic activation associated with fear-extinction learning in hippocampus, basal amygdala and cingulate cortex. Neurobiol. Learn. Mem. 2012;98:93–101. doi: 10.1016/j.nlm.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 28.Singewald N. Altered brain activity processing in high-anxiety rodents revealed by challenge paradigms and functional mapping. Neurosci. Biobehav. Rev. 2007;31:18–40. doi: 10.1016/j.neubiorev.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 29.Herry C, Mons N. Resistance to extinction is associated with impaired immediate early gene induction in medial prefrontal cortex and amygdala. Eur. J. Neurosci. 2004;20:781–790. doi: 10.1111/j.1460-9568.2004.03542.x. [DOI] [PubMed] [Google Scholar]
- 30.Plendl W, Wotjak CT. Dissociation of within-and between-session extinction of conditioned fear. J. Neurosci. 2010;30:4990–4998. doi: 10.1523/JNEUROSCI.6038-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SC, et al. Lack of medial prefrontal cortex activation underlies the immediate extinction deficit. J. Neurosci. 2010;30:832–837. doi: 10.1523/JNEUROSCI.4145-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Izquierdo A, et al. Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J. Neurosci. 2006;26:5733–5738. doi: 10.1523/JNEUROSCI.0474-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin LM, Liberzon I. The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology. 2010;35:169–191. doi: 10.1038/npp.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilber AA, et al. Chronic stress alters neural activity in medial prefrontal cortex during retrieval of extinction. Neuroscience. 2011;174:115–131. doi: 10.1016/j.neuroscience.2010.10.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sierra-Mercado D, et al. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology. 2011;36:529–538. doi: 10.1038/npp.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burgos-Robles A, et al. Sustained conditioned responses in prelimbic prefrontal neurons are correlated with fear expression and extinction failure. J. Neurosci. 2009;29:8474–8482. doi: 10.1523/JNEUROSCI.0378-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li S, et al. Differential involvement of the medial prefrontal cortex in the expression of learned fear across development. Behav. Neurosci. 2012;126:217–225. doi: 10.1037/a0027151. [DOI] [PubMed] [Google Scholar]
- 38.Whittle N, et al. Rescue of impaired fear extinction and normalization of cortico-amygdala circuit dysfunction in a genetic mouse model by dietary zinc restriction. J. Neurosci. 2010;30:13586–13596. doi: 10.1523/JNEUROSCI.0849-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knapska E, Maren S. Reciprocal patterns of c-Fos expression in the medial prefrontal cortex and amygdala after extinction and renewal of conditioned fear. Learn. Mem. 2009;16:486–493. doi: 10.1101/lm.1463909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meins M, et al. Impaired fear extinction in mice lacking protease nexin-1. Eur. J. Neurosci. 2010;31:2033–2042. doi: 10.1111/j.1460-9568.2010.07221.x. [DOI] [PubMed] [Google Scholar]
- 41.Pinard CR, et al. Medial prefrontal cortical innervation of the intercalated nuclear region of the amygdala. Neuroscience. 2012;205:112–124. doi: 10.1016/j.neuroscience.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Busti D, et al. Different fear states engage distinct networks within the intercalated cell clusters of the amygdala. J. Neurosci. 2011;31:5131–5144. doi: 10.1523/JNEUROSCI.6100-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herry C, et al. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- 44.Ciocchi S, et al. Encoding of conditioned fear in central amygdala inhibitory circuits. Nature. 2010;468:277–282. doi: 10.1038/nature09559. [DOI] [PubMed] [Google Scholar]
- 45.Vyas A, et al. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J. Neurosci. 2002;22:6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chauveau F, et al. Prevention of stress-impaired fear extinction through neuropeptide S action in the lateral amygdala. Neuropsychopharmacology. 2012;37:1588–1599. doi: 10.1038/npp.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burgos-Robles A, et al. Consolidation of fear extinction requires NMDA receptor-dependent bursting in the ventromedial prefrontal cortex. Neuron. 2007;53:871–880. doi: 10.1016/j.neuron.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 48.Holmes A, et al. Chronic alcohol remodels prefrontal neurons and disrupts NMDAR-mediated fear extinction encoding. Nat. Neurosci. 2012;15:1359–1361. doi: 10.1038/nn.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sotres-Bayon F, et al. Acquisition of fear extinction requires activation of NR2B-containing NMDA receptors in the lateral amygdala. Neuropsychopharmacology. 2007;32:1929–1940. doi: 10.1038/sj.npp.1301316. [DOI] [PubMed] [Google Scholar]
- 50.Parkes SL, Westbrook RF. The basolateral amygdala is critical for the acquisition and extinction of associations between a neutral stimulus and a learned danger signal but not between two neutral stimuli. J. Neurosci. 2010;30:12608–12618. doi: 10.1523/JNEUROSCI.2949-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davis M, et al. Effects of D-cycloserine on extinction: translation from preclinical to clinical work. Biol. Psychiatry. 2006;60:369–375. doi: 10.1016/j.biopsych.2006.03.084. [DOI] [PubMed] [Google Scholar]
- 52.Weber M, et al. Effects of D-cycloserine on extinction of learned fear to an olfactory cue. Neurobiol. Learn. Mem. 2007;87:476–482. doi: 10.1016/j.nlm.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 53.Bouton ME, et al. D-cycloserine facilitates context-specific fear extinction learning. Neurobiol. Learn. Mem. 2008;90:504–510. doi: 10.1016/j.nlm.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whittle N, et al. Deep brain stimulation, histone deacetylase inhibitors and glutamatergic drugs rescue resistance to fear extinction in a genetic mouse model. Neuropharmacology. 2012;64:414–423. doi: 10.1016/j.neuropharm.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ledgerwood L, et al. Effects of D-cycloserine on extinction of conditioned freezing. Behav. Neurosci. 2003;117:341–349. doi: 10.1037/0735-7044.117.2.341. [DOI] [PubMed] [Google Scholar]
- 56.Litz BT, et al. A randomized placebo-controlled trial of D-cycloserine and exposure therapy for posttraumatic stress disorder. J. Psychiatr. Res. 2012;46:1184–1190. doi: 10.1016/j.jpsychires.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 57.de Kleine RA, et al. A randomized placebo-controlled trial of D-cycloserine to enhance exposure therapy for posttraumatic stress disorder. Biol. Psychiatry. 2012;71:962–968. doi: 10.1016/j.biopsych.2012.02.033. [DOI] [PubMed] [Google Scholar]
- 58.Rodriguez-Romaguera J, et al. Deep brain stimulation of the ventral striatum enhances extinction of conditioned fear. Proc. Natl. Acad. Sci. U.S.A. 2012;109:8764–8769. doi: 10.1073/pnas.1200782109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maroun M, et al. Enhanced extinction of aversive memories by high-frequency stimulation of the rat infralimbic cortex. PLoS ONE. 2012;7:e35853. doi: 10.1371/journal.pone.0035853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schiller D, et al. Preventing the return of fear in humans using reconsolidation update mechanisms. Nature. 2009;463:49–53. doi: 10.1038/nature08637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Monfils MH, et al. Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science. 2009;324:951–955. doi: 10.1126/science.1167975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Norberg MM, et al. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol. Psychiatry. 2008;63:1118–1126. doi: 10.1016/j.biopsych.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 63.Bontempo A, et al. D-cycloserine augmentation of behavioral therapy for the treatment of anxiety disorders: a meta-analysis. J. Clin. Psychiatry. 2012;73:533–537. doi: 10.4088/JCP.11r07356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Holmes A, Quirk GJ. Pharmacological facilitation of fear extinction and tthe search for adjunct treatments for anxiety disorders: the case of yohimbine. Trends Pharmacol. Sci. 2010;31:2–7. doi: 10.1016/j.tips.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peters J, et al. Induction of fear extinction with hippocampal-infralimbic BDNF. Science. 2010;328:1288–1290. doi: 10.1126/science.1186909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bouton ME. Context, ambiguity, and unlearning: sources of relapse after behavioral extinction. Biol. Psychiatry. 2002;52:976–986. doi: 10.1016/s0006-3223(02)01546-9. [DOI] [PubMed] [Google Scholar]
- 67.Davidson JR, et al. Fluoxetine, comprehensive cognitive behavioral therapy, and placebo in generalized social phobia. Arch. Gen. Psychiatry. 2004;61:1005–1013. doi: 10.1001/archpsyc.61.10.1005. [DOI] [PubMed] [Google Scholar]
- 68.Schneier FR, et al. Combined prolonged exposure therapy and paroxetine for PTSD related to the World Trade Center attack: a randomized controlled trial. Am. J. Psychiatry. 2012;169:80–88. doi: 10.1176/appi.ajp.2011.11020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hauner KK, et al. Exposure therapy triggers lasting reorganization of neural fear processing. Proc. Natl. Acad. Sci. U.S.A. 2012;109:9203–9208. doi: 10.1073/pnas.1205242109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schienle A, et al. Symptom provocation and reduction in patients suffering from spider phobia: an fMRI study on exposure therapy. Eur. Arch. Psychiatry Clin. Neurosci. 2007;257:486–493. doi: 10.1007/s00406-007-0754-y. [DOI] [PubMed] [Google Scholar]
- 71.Harmer CJ, et al. Antidepressant drug treatment modifies the neural processing of nonconscious threat cues. Biol. Psychiatry. 2006;59:816–820. doi: 10.1016/j.biopsych.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 72.Riebe CJ, et al. Fear relief-toward a new conceptual frame work and what endocannabinoids gotta do with it. Neuroscience. 2012;2004:159–185. doi: 10.1016/j.neuroscience.2011.11.057. [DOI] [PubMed] [Google Scholar]
- 73.Gunduz-Cinar O, et al. Convergent translational evidence of a role for anandamide in amygdala-mediated fear extinction, threat processing and stress-reactivity. Mol. Psychiatry. 2012 doi: 10.1038/mp.2012.72. http://dx.doi.org/10.1038/mp.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Akirav I, Maroun M. The role of the medial prefrontal cortex-amygdala circuit in stress effects on the extinction of fear. Neural Plast. 2007 doi: 10.1155/2007/30873. 2007, 30873 Article ID 30873, 11 pages. ( http://dx.doi.org/10.1155/2007/30873) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baratta MV, et al. Controllable versus uncontrollable stressors bi-directionally modulate conditioned but not innate fear. Neuroscience. 2007;146:1495–1503. doi: 10.1016/j.neuroscience.2007.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yamamoto S, et al. Effects of single prolonged stress and D-cycloserine on contextual fear extinction and hippocampal NMDA receptor expression in a rat model of PTSD. Neuropsychopharmacology. 2008;33:2108–2116. doi: 10.1038/sj.npp.1301605. [DOI] [PubMed] [Google Scholar]
- 77.Goswami S, et al. Impact of predatory threat on fear extinction in Lewis rats. Learn. Mem. 2010;17:494–501. doi: 10.1101/lm.1948910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Miracle AD, et al. Chronic stress impairs recall of extinction of conditioned fear. Neurobiol. Learn. Mem. 2006;85:213–218. doi: 10.1016/j.nlm.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 79.Gourley SL, et al. A history of corticosterone exposure regulates fear extinction and cortical NR2B, GluR2/3, and BDNF. Neuropsychopharmacology. 2009;34:707–716. doi: 10.1038/npp.2008.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Baran SE, et al. Chronic stress and sex differences on the recall of fear conditioning and extinction. Neurobiol. Learn. Mem. 2009;91:323–332. doi: 10.1016/j.nlm.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Garcia R, et al. Hippocampal low-frequency stimulation and chronic mild stress similarly disrupt fear extinction memory in rats. Neurobiol. Learn. Mem. 2008;89:560–566. doi: 10.1016/j.nlm.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 82.Andero R, et al. Effect of 7,8-dihydroxyflavone, a small-molecule TrkB agonist, on emotional learning. Am. J. Psychiatry. 2011;168:163–172. doi: 10.1176/appi.ajp.2010.10030326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ripley TL, et al. Repeated withdrawal from ethanol impairs acquisition but not expression of conditioned fear. Eur. J. Neurosci. 2003;18:441–448. doi: 10.1046/j.1460-9568.2003.02759.x. [DOI] [PubMed] [Google Scholar]
- 84.Bertotto ME, et al. Influence of ethanol withdrawal on fear memory: effect of D-cycloserine. Neuroscience. 2006;142:979–990. doi: 10.1016/j.neuroscience.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 85.Thomas JL, et al. Prevalence of mental health problems and functional impairment among active component and National Guard soldiers 3 and 12 months following combat in Iraq. Arch. Gen. Psychiatry. 2010;67:614–623. doi: 10.1001/archgenpsychiatry.2010.54. [DOI] [PubMed] [Google Scholar]
- 86.Vyas A, et al. Effects of chronic stress on dendritic arborization in the central and extended amygdala. Brain Res. 2003;965:290–294. doi: 10.1016/s0006-8993(02)04162-8. [DOI] [PubMed] [Google Scholar]
- 87.Rodriguez Manzanares PA, et al. Previous stress facilitates fear memory, attenuates GABAergic inhibition, and increases synaptic plasticity in the rat basolateral amygdala. J. Neurosci. 2005;25:8725–8734. doi: 10.1523/JNEUROSCI.2260-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pare D, et al. New vistas on amygdala networks in conditioned fear. J. Neurophysiol. 2004;92:1–9. doi: 10.1152/jn.00153.2004. [DOI] [PubMed] [Google Scholar]
- 89.Haubensak W, et al. Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature. 2012;468:270–276. doi: 10.1038/nature09553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Day JJ, Sweatt JD. Epigenetic mechanisms in cognition. Neuron. 2011;70:813–829. doi: 10.1016/j.neuron.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bredy TW, et al. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn. Mem. 2007;14:268–276. doi: 10.1101/lm.500907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stafford JM, et al. Increasing histone acetylation in the hippocampus-infralimbic network enhances fear extinction. Biol. Psychiatry. 2012;72:25–33. doi: 10.1016/j.biopsych.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Levenson JM, et al. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- 94.Peleg S, et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science. 2010;328:753–756. doi: 10.1126/science.1186088. [DOI] [PubMed] [Google Scholar]
- 95.Fontan-Lozano A, et al. Histone deacetylase inhibitors improve learning consolidation in young and in KA-induced-neurodegeneration and SAMP-8-mutant mice. Mol. Cell. Neurosci. 2008;39:193–201. doi: 10.1016/j.mcn.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 96.Bousiges O, et al. Spatial memory consolidation is associated with induction of several lysine-acetyltransferase (histone acetyltransferase) expression levels and H2B/H4 acetylation-dependent transcriptional events in the rat hippocampus. Neuropsychopharmacology. 2010;35:2521–2537. doi: 10.1038/npp.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Monsey MS, et al. Epigenetic alterations are critical for fear memory consolidation and synaptic plasticity in the lateral amygdala. PLoS ONE. 2011;6:e19958. doi: 10.1371/journal.pone.0019958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bredy TW, Barad M. The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learn. Mem. 2008;15:39–45. doi: 10.1101/lm.801108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lattal KM, et al. Systemic or intrahippocampal delivery of histone deacetylase inhibitors facilitates fear extinction. Behav. Neurosci. 2007;121:1125–1131. doi: 10.1037/0735-7044.121.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guan JS, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fujita Y, et al. Vorinostat, a histone deacetylase inhibitor, facilitates fear extinction and enhances expression of the hippocampal NR2B-containing NMDA receptor gene. J. Psychiatr. Res. 2012;46:635–643. doi: 10.1016/j.jpsychires.2012.01.026. [DOI] [PubMed] [Google Scholar]
- 102.Itzhak Y, et al. Histone acetylation rescues contextual fear conditioning in nNOS KO mice and accelerates extinction of cued fear conditioning in wild type mice. Neurobiol. Learn. Mem. 2012;97:409–417. doi: 10.1016/j.nlm.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bahari-Javan S, et al. HDAC1 regulates fear extinction in mice. J. Neurosci. 2012;32:5062–5073. doi: 10.1523/JNEUROSCI.0079-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kuriyama K, et al. Psychopharmacology. 2011;218:589–597. doi: 10.1007/s00213-011-2353-x. [DOI] [PubMed] [Google Scholar]
- 105.Whittle N, et al. Deep brain stimulation, histone deacetylase inhibitors and glutamatergic drugs rescue resistance to fear extinction in agenetic mouse model. Neuropharmacology. 2013;64:414–423. doi: 10.1016/j.neuropharm.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Powers MB, et al. Facilitation of fear extinction in phobic participants with a novel cognitive enhancer: a randomized placebo controlled trial of yohimbine augmentation. J. Anxiety Disord. 2009;23:350–356. doi: 10.1016/j.janxdis.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 107.McCallum J, et al. Impaired extinction retention in adolescent rats: effects of D-cycloserine. Neuropsychopharmacology. 2010;35:2134–2142. doi: 10.1038/npp.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tambs K, et al. Structure of genetic and environmental risk factors for dimensional representations of DSM-IV anxiety disorders. Br. J. Psychiatry. 2009;195:301–307. doi: 10.1192/bjp.bp.108.059485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kremen WS, et al. Twin studies of posttraumatic stress disorder: differentiating vulnerability factors from sequelae. Neuropharmacology. 2012;62:647–653. doi: 10.1016/j.neuropharm.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hettema JM, et al. A twin study of the genetics of fear conditioning. Arch. Gen. Psychiatry. 2003;60:702–708. doi: 10.1001/archpsyc.60.7.702. [DOI] [PubMed] [Google Scholar]
- 111.Hariri AR, Holmes A. Genetics of emotional regulation: the role of the serotonin transporter in neural function. Trends Cogn. Sci. 2006;10:182–191. doi: 10.1016/j.tics.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 112.Yang RJ, et al. Variation in mouse basolateral amygdala volume is associated with differences in stress reactivity and fear learning. Neuropsychopharmacology. 2008;33:2595–2604. doi: 10.1038/sj.npp.1301665. [DOI] [PubMed] [Google Scholar]
- 113.Shin LM, et al. Exaggerated activation of dorsal anterior cingulate cortex during cognitive interference: a monozygotic twin study of posttraumatic stress disorder. Am. J. Psychiatry. 2011;168:979–985. doi: 10.1176/appi.ajp.2011.09121812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gilbertson MW, et al. Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nat. Neurosci. 2002;5:1242–1247. doi: 10.1038/nn958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Woodward SH, et al. Hippocampal volume, PTSD, and alcoholism in combat veterans. Am. J. Psychiatry. 2006;163:674–681. doi: 10.1176/ajp.2006.163.4.674. [DOI] [PubMed] [Google Scholar]
- 116.Kasai K, et al. Evidence for acquired pregenual anterior cingulate gray matter loss from a twin study of combat-related posttraumatic stress disorder. Biol. Psychiatry. 2008;63:550–556. doi: 10.1016/j.biopsych.2007.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Norrholm SD, Ressler KJ. Genetics of anxiety and trauma-related disorders. Neuroscience. 2009;164:272–287. doi: 10.1016/j.neuroscience.2009.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang Z, et al. The relationship between combat-related posttraumatic stress disorder and the 5-HTTLPR/rs25531 polymorphism. Depress. Anxiety. 2011;28:1067–1073. doi: 10.1002/da.20872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hartley CA, et al. Serotonin transporter polyadenylation polymorphism modulates the retention of fear extinction memory. Proc. Natl. Acad. Sci. U.S.A. 2012;109:5493–5498. doi: 10.1073/pnas.1202044109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wellman CL, et al. Impaired stress-coping and fear extinction and abnormal corticolimbic morphology in serotonin transporter knock-out mice. J. Neurosci. 2007;27:684–691. doi: 10.1523/JNEUROSCI.4595-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Andero R, Ressler KJ. Fear extinction and BDNF: translating animal models of PTSD to the clinic. Genes Brain Behav. 2012;11:503–512. doi: 10.1111/j.1601-183X.2012.00801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Soliman F, et al. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science. 2010;327:863–866. doi: 10.1126/science.1181886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bilkei-Gorzo A, et al. Dynorphins regulate fear memory: from mice to men. J. Neurosci. 2012;32:9335–9343. doi: 10.1523/JNEUROSCI.1034-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ressler KJ, et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature. 2011;470:492–497. doi: 10.1038/nature09856. [DOI] [PMC free article] [PubMed] [Google Scholar]