

Abstract
Background
Neutrophils are the predominant phagocytes that provide protection against bacterial and fungal infections. Genetically determined neutrophil disorders confer a predisposition to severe infections and reveal novel mechanisms that control vesicular trafficking, hematopoiesis, and innate immunity.
Methods
We clinically evaluated seven children from five families who had neutropenia, neutrophil dysfunction, bone marrow fibrosis, and nephromegaly. To identify the causative gene, we performed homozygosity mapping using single-nucleotide polymorphism arrays, whole-exome sequencing, immunoblotting, immunofluorescence, electron microscopy, a real-time quantitative polymerase–chain-reaction assay, immunohistochemistry, flow cytometry, fibroblast motility assays, measurements of apoptosis, and zebrafish models. Correction experiments were performed by transfecting mutant fibroblasts with the nonmutated gene.
Results
All seven affected children had homozygous mutations (Thr224Asn or Glu238Lys, depending on the child's ethnic origin) in VPS45, which encodes a protein that regulates membrane trafficking through the endosomal system. The level of VPS45 protein was reduced, as were the VPS45 binding partners rabenosyn-5 and syntaxin-16. The level of β1 integrin was reduced on the surface of VPS45-deficient neutrophils and fibroblasts. VPS45-deficient fibroblasts were characterized by impaired motility and increased apoptosis. A zebrafish model of vps45 deficiency showed a marked paucity of myeloperoxidase-positive cells (i.e., neutrophils). Transfection of patient cells with nonmutated VPS45 corrected the migration defect and decreased apoptosis.
Conclusions
Defective endosomal intracellular protein trafficking due to biallelic mutations in VPS45 underlies a new immunodeficiency syndrome involving impaired neutrophil function. (Funded by the National Human Genome Research Institute and others.)
Rare hematopoietic disorders often reveal basic mechanisms of immune processes. One component of this system, the neutrophil, plays a particularly prominent role in the defense against bacterial and fungal infections.1 Functional defects in neutrophils can involve impaired granule content (e.g., neutrophil-specific granule deficiency), secretion of inflammatory proteins, impaired cell adherence (e.g., leukocyte adhesion deficiency), cell migration (e.g., as a result of CXCR4 mutations), chemotaxis, endocytosis, and oxygen-dependent killing (e.g., chronic granulomatous disease).2 Other disorders related to neutrophils are manifested as severe neutropenia, which may result from impaired differentiation, as in mutations in the gene encoding the transcription-repressor protein growth factor independent 1, or from increased susceptibility to apoptosis due to mutations in ELANE (encoding elastase, neutrophil expressed) or HAX1 (encoding HCLS1-associated protein X1).3
A fundamental feature of neutrophils is their capacity to secrete proteins stored in specialized vesicles and to fuse membrane compartments in order to kill ingested microbes in phagolysosomes. These functions are influenced by the processes of exocytosis and endocytosis, which also govern internalization of nutrients and pathogens, intracellular signaling, and recycling of membrane proteins. Impaired membrane trafficking occurs in cases of AP3B1 deficiency4 and LAMTOR2 (p14) deficiency,5 resulting in melanocyte dysfunction (hypopigmentation) and infections due to neutropenia.
One critical component of the endocytic pathway in neutrophils involves vacuolar protein sorting (VPS).6 Proteins involved in vacuolar sorting are required for endosomal trafficking and protein recycling through the trans-Golgi network. Several monogenic diseases testify to the critical role of VPS in normal organ function. Defects in VPS13A, VPS13B, VPS33B, and VPS35 cause cho-reoacanthocytosis, Cohen's syndrome (hypotonia, obesity, and prominent incisors), the arthrogryposis–renal dysfunction–cholestasis syndrome, and Parkinson's disease type 17 (autosomal dominant, adult-onset form), respectively.7-10
We describe a new immunodeficiency syndrome due to biallelic mutations in VPS45. The syndrome is characterized by a severe defect in neutrophils, bone marrow fibrosis, nephromegaly, and life-threatening infections.
Methods
Patients
We evaluated children at the Edmond and Lily Safra Children's Hospital, Sheba Medical Center, in Tel Hashomer, Israel; Universitair Ziekenhuis Brussel, in Brussels; and the Dr. von Hauner, Children's Hospital, Ludwig-Maximilians-University, in Munich, Germany. In addition, samples from these children and healthy controls were examined at the National Institutes of Health, in Bethesda, Maryland. Parents of patients and parents of healthy controls gave written informed consent for their children to participate in the study, which was approved by the local institutional review boards.
Single-Nucleotide Polymorphism Arrays
Single-nucleotide polymorphism (SNP) genotyping was performed on genomic DNA with the use of the HumanOmniExpress DNA Analysis Bead-Chip (Illumina). SNPs were analyzed with the use of GenomeStudio software (Illumina).11,12
Sequencing
An in-solution hybridization method of exome capture was carried out with the use of the SureSelect Human All-Exon System version 1.0 (Agilent Technologies). Sequencing was performed with the use of the Genome Analyzer IIx sequencer (Illumina).13 VPS45 variants were examined by means of dideoxy sequencing, with primers covering exon 7 and its flanking intronic region (for sequencing details and protein-modeling techniques see the Methods section in the Supplementary Appendix, available with the full text of this article at NEJM.org; sequences are available on request).
Cell Studies
Primary fibroblasts were cultured from forearm skin-biopsy specimens in the affected children and from foreskin from healthy human adults (American Type Culture Collection) in the controls, as previously described.14 For the cell-migration assay, fibroblasts were grown to 80% confluence in 48 hours in 6-well or 24-well plates coated with fibronectin. A scratch was made in the confluent layer of cells with a pipette tip. The migration of the cells into the wound area was imaged with the use of a Nikon Digital Sight DS-Fi1 microscope and camera and was measured in triplicate. Methods for immunofluorescence, immunoblotting, electron microscopy, real-time quantitative polymerase-chain-reaction (PCR) assay, and evaluation of apoptosis with the CaspACE FITC-VAD-FMK In Situ Marker (Promega) are described in the Methods section in the Supplementary Appendix.
Plasmids and Transfections
For gene correction studies, human VPS45 complementary DNA (cDNA) clones tagged with Myc-DDK (inserted into a pCMV6 vector with gentamicin B1 resistance) were purchased from OriGene Technologies. VPS45 and empty pCMV6-myc plasmids were overexpressed in patient and control fibroblasts with the use of the Nucleofector 2b Device with a human dermal fibroblast transfection agent (Lonza). Transfection efficiency was more than 70%, as measured by immunofluorescence analysis of living cells cotransfected with pmaxGFP plasmid.
Neutrophil Function
Neutrophils were isolated (98% purity and viability) from heparinized blood by dextran sedimentation, centrifugation with the use of Ficoll histopaque gradients, and erythrocyte lysis. Superoxide production, hydrogen peroxide production, analysis of the NADPH-oxidase subunits, and chemo-taxis were assayed as described in the Methods section in the Supplementary Appendix.
Flow Cytometry
Samples of peripheral blood obtained from patients and healthy controls were incubated with anti-CD45 PerCP antibodies (Dako) and anti-CD29 FITC antibodies (Serotec). Lymphocytes, granulocytes, and monocytes were gated. The cell-surface expression of both markers was measured with the use of flow cytometry (Epics V, Coulter Electronics).
Studies In Zebrafish
Zebrafish (Danio rerio) were maintained under an approved National Human Genome Research Institute protocol for studies in animals, in accordance with the Zebrafish Book.15 Gene-specific morpholinos were purchased from Gene Tools and microinjected into the yolk sac of wild-type zebrafish embryos at the 1-cell to 4-cell stage. The embryos were of the Ekkwill (EK) strain. Embryos were maintained at 28°C in E3 embryo medium15 and scored for survival 24 hours after fertilization. Whole-mount in situ hybridization was performed as previously described,16 with the use of riboprobes for mpx.17 Staining with Sudan black B was performed as previously described18 (see the Methods section in the Supplementary Appendix for details).
Results
Clinical Studies
Our original cohort consisted of five affected children, in Families A through D (Fig. 1A), who were products of four marriages of consanguineous self-reported Palestinians. Families B and C were distantly related. All the children presented in infancy with poor weight gain, hepatomegaly, splenomegaly, and severe infections or deep-seated abscesses, apparent on imaging (Fig. S1A, S1B, and S1C in the Supplementary Appendix).
Figure 1. Clinical Findings.
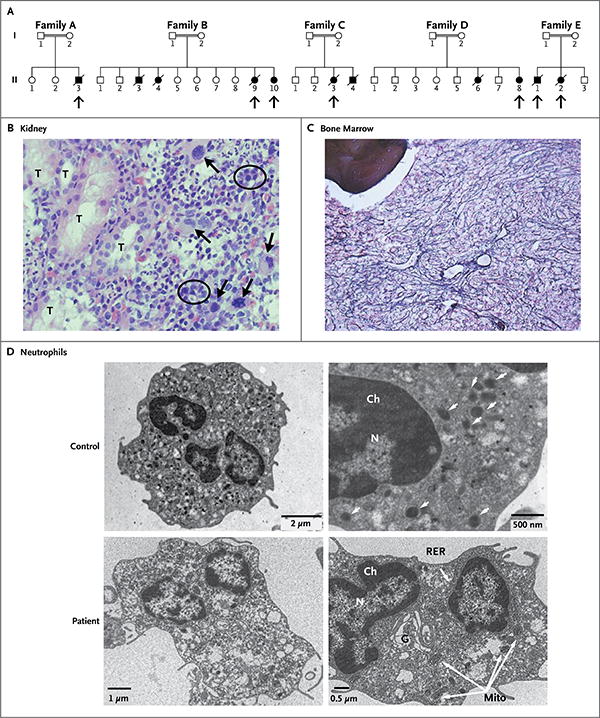
Panel A shows the pedigrees of five affected families. Double lines indicate consanguineous unions. Squares denote male family members, circles female family members, solid symbols affected family members, open symbols unaffected family members, and slashes deceased family members. Arrows point to the children who were studied. Panel B (hematoxylin and eosin) shows extramedullary hematopoiesis in a renal-biopsy specimen from one of the affected children. Megakaryocytes have large, lobulated, hyperchromatic nuclei (arrows) and are the hallmark of extramedullary hematopoietic tissue. Normoblastic red cells are circled. T denotes renal tubule. In Panel C (reticulin stain), a bone marrow–biopsy specimen from one of the children shows prominent, diffuse, intersecting reticulin fibers, representing fibrosis (grade 3 of 4). Bone is shown in the upper left corner of the image. Panel D shows electron-microscopical images of peripheral-blood neutrophils from a child with a mutation in VPS45 and an age-matched control. The micrographs on the left are at lower magnification than those on the right. In the control neutrophils, the number and structure of the mitochondria are normal and there are multiple granules (arrows). In comparison, the patient's neutrophils contain abundant mitochondria (Mito) and far fewer cytoplasmic granules (arrows); also, the Golgi apparatus (G) and rough endoplasmic reticulum (RER) are more developed, and the chromatin (Ch) and cytoplasm are less condensed. N denotes nucleus.
Cultures grew mainly Staphylococcus aureus and aspergillus. Clinical findings (Table 1) included neutrophil counts of 100 to 520 per cubic millimeter and lack of a response to high-dose (20 μg per kilogram of body weight) recombinant granulocyte colony-stimulating factor (G-CSF). All the children had hypergammaglobulinemia and nephromegaly (Fig. S1D and S1E in the Supplementary Appendix); examination of kidney-biopsy specimens in Patients A-II-3, B-II-9, and C-II-3 showed extramedullary hematopoiesis (Fig. 1B). Nucleated red cells, indicating substantial bone marrow damage or stress, were present in the peripheral blood of the children (Table 1). Results of evaluations of T cells, including lymphocyte subsets and lymphocyte response to mitogenic stimulation, were normal in all the children, as was production of T-cell–receptor excision circles (TRECs) (data not shown). Molecular and genetic evaluations were negative for known congenital neutrophil defects. Two of the affected children underwent hematopoietic stem-cell transplantation (HSCT) from matched related donors; both died of infection after failed engraftment. A third child died of infection before HSCT could be performed, and the other two children are awaiting the procedure.
Table 1. Characteristics of the Seven Affected Children*.
| Variable | Patient No. | Reference Range | ||||||
|---|---|---|---|---|---|---|---|---|
| A-II-3 | B-II-9 | B-II-10 | C-II-3 | D-II-8 | E-II-1 | E-II-2 | ||
| Age at diagnosis (mo) | 6 | 3 | 3 | 2 | 12 | 0 | 0 | |
| Sex | M | F | F | F | F | M | F | |
| Hemoglobin (g/dl) | 7.6 | 7.8 | 8.1 | 9.3 | 10.5 | 8.8 | 7.7 | 11–15 |
| White cells (per mm3) | 5640 | 4600 | 4500 | 4400 | 2800 | 3500 | 7200 | 5000–15,000 |
| Neutrophils (per mm3) | 520 | 300 | 510 | 100 | 240 | 477 | 560 | 1500–8500 |
| Lymphocytes (per mm3) | 4717 | 4910 | 3320 | 1900 | 2250 | 1183 | 2380 | 2000–8000 |
| Platelets (per mm3) | 131 | 145 | 118 | 111 | 73 | 37 | 46 | 150–500 |
| Nucleated red cells (per 100 cells) | 3 | 7 | 4 | 0 | 1 | 2 | 3 | 0 |
| IgG (mg/dl) | 1140 | 2500 | ND | 1740 | 2280 | 865 | 1660 | 300–1000 |
| Superoxide production (nmol/106 neutrophils/min)† | 0 | ND | ND | 0 | 0.1 | ND | ND | 2–10 |
| Outcome | Died after HSCT | Died | Awaiting HSCT | Died after HSCT | Awaiting HSCT | Died | Died | |
HSCT denotes hematopoietic stem-cell transplantation, and ND not determined.
Superoxide production in neutrophils was stimulated by phorbol myristate acetate (PMA).
A Moroccan family (Family E) had two children in whom the syndrome was diagnosed immediately after birth (Fig. 1A). The clinical manifestations in these two children were similar to those seen in the affected children of Families A through D, including recurrent infections and diarrhea, poor weight gain, hepatomegaly, splenomegaly, and nephromegaly. Hematologic evaluation showed persistent neutropenia, a poor response to G-CSF, and repeated requirements for blood and platelet transfusions (Table 1). In addition, these children had substantial neurologic abnormalities manifested as delayed development, cortical blindness, hearing loss, thin corpus callosum on magnetic resonance imaging, and dysrhythmia on electroencephalography. Both children died of infections before transplantation could be performed.
The parents of the affected children from all five families were clinically healthy. Blood counts and killing functions of neutrophils, assessed by examination of superoxide production and chemotactic activity, were normal in the parents and healthy siblings from Families A, C, and E (data not shown).
Bone marrow–biopsy specimens from the children, obtained before G-CSF therapy, revealed hypercellularity, distortion of hematopoietic tissue, prominent reticulin fibrosis (grade 3 of 4), and collagenous fibrosis (Fig. 1C). Leukocytes were predominant. There was an increased number of mature neutrophils, many of which extended from their normal central intertrabecular location into the paratrabecular spaces. In the fibrotic areas, the neutrophils were characterized primarily by nuclear hypolobulation and pale cytoplasm. Numerous apoptotic nuclei were also seen (Fig. S2 in the Supplementary Appendix). Normoblastic red cells were slightly hypoplastic; megakaryocytes were normal in number and morphologic features. Fibrosis was noted in all biopsy specimens but without chromosomal aberrations or features of myelodysplasia.
Electron microscopy of peripheral-blood neutrophils from Patients B-II-10 and D-II-8 revealed immature cells suggestive of abnormal myeloid differentiation19 (Fig. 1D). As compared with neutrophils from age-matched healthy controls, the neutrophils from the two affected children contained fewer granules (azurophils) and more abundant mitochondria; the Golgi apparatus and rough endoplasmic reticulum were more developed, and the chromatin and cytoplasm were less condensed. Quantitative morphometric studies20 confirmed that these differences were significant (Fig. S3 in the Supplementary Appendix).
VPS45 Mutations and Expression
Consanguinity suggested autosomal recessive inheritance, and analysis of SNP arrays in Families A, B, and C identified excessive homozygosity. One homozygous region, from 149.3 to 154.3 Mb on chromosome 1 (hg19 in the human genome reference sequence of the National Center for Biotechnology Information), was observed in all the affected children and only in the affected children (Fig. S4A in the Supplementary Appendix). Whole-exome sequencing in Patient B-II-9 yielded 62,235 variants, but only 569 were in the candidate region; 92 were not in the dbSNP database. Fourteen variants affected protein sequence (Table S1 in the Supplementary Appendix), including 12 variants in HRNR (encoding hornerin), 1 in NUP210L (encoding nucleoporin 210-kD–like protein), and 1 in VPS45. VPS45 was pursued because of its role in intracellular trafficking; dideoxy sequencing (Fig. S4B in the Supplementary Appendix) confirmed a missense mutation (c.671C→A; p.Thr224Asn) in exon 7 in all five affected children. The mutation segregated with the disease, was present in both peripheral-blood mononuclear cells and fibroblasts, and was absent from normal genome and exome databases and from 250 ethnically matched control alleles (data not shown). The affected siblings in Family E also had a homozygous mutation (c.712G→A; p.Glu238Lys); again, the mutation segregated with the phenotype. Both the p.Thr224 and p.Glu238 residues are highly evolutionarily conserved (Fig. S5A in the Supplementary Appendix).
We constructed a complete three-dimensional representation of VPS45; the structure of UNC-18 protein was used as a template, producing only a few misaligned loop regions and a root-mean-square error of 0.72 Å. VPS45 is a globular protein (Fig. S5B in the Supplementary Appendix) belonging to the alpha-plus-beta class. The two mutant residues (p.Thr224 and p.Glu238) are central to the protein matrix (SARIG program21) (Fig. S5C in the Supplementary Appendix) and are predicted to alter the conformation of several residues (Fig. S5D and S5E in the Supplementary Appendix). Seventeen computational programs were applied to predict changes in thermostability and function; for both Thr224Asn and Glu238Lys, 16 programs predicted thermodynamic destabilization or impaired function (Table S2 in the Supplementary Appendix).
Fibroblast extracts from Patient B-II-9 contained a normal amount of VPS45 messenger RNA (data not shown). However, the VPS45 protein level in peripheral blood was 20% of the level in an unaffected control, and the level in fibroblasts was approximately 50% of the normal level (Fig. 2A). Immunoblots also showed reduced amounts of the VPS45-interacting proteins rabenosyn-5 and syntaxin-16 (Fig. 2A). Immunofluorescence studies showed that VPS45 bearing the Thr224Asn mutation was reduced in amount and was diffusely present throughout the cytoplasm in the patient's neutrophils and fibroblasts, in contrast to the strong perinuclear localization of the non-mutant VPS45 in normal cells (Fig. S6 in the Supplementary Appendix).
Figure 2. Cellular Phenotypes.
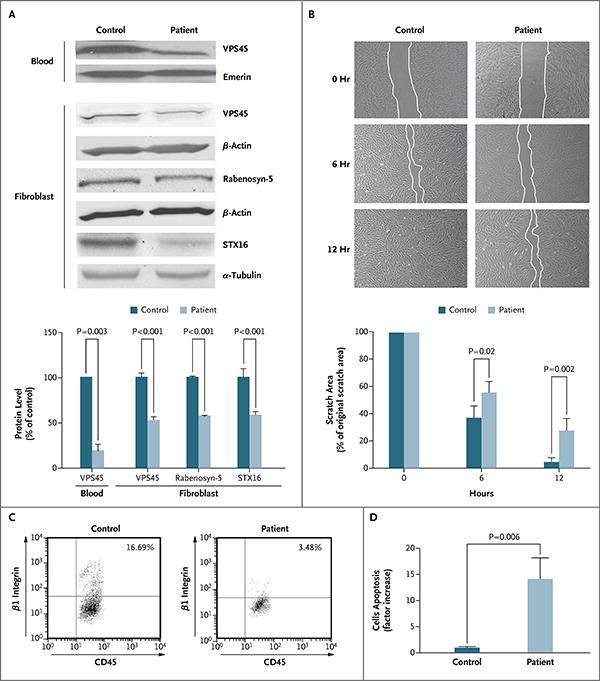
In Panel A, immunoblot analysis and graphic representation of densitometry show reduced expression of VPS45 in peripheral blood and fibroblasts and reduced expression of the interacting partners of VPS45 — rabenosyn-5 and syntaxin-16 (STX16) — in fibroblasts from one of the affected patients as compared with a control. Loading was controlled by immunoblotting the same membrane for emerin, β-actin, or α-tubulin. Densitometry was performed with the use of the ImageJ 1.45s Gel Analysis tool (National Institutes of Health). Panel B shows impaired cell motility. Fibroblasts were grown on coverslips coated with fibronectin. The cultures were “wounded” by introducing a scratch (approximately 800 μm in width) across the field of cells; the same field of cells was imaged immediately, after 6 hours, and after 12 hours. The fibroblasts from Patient B-II-9 migrated significantly more slowly toward the wounded area than did control cells (similar results were obtained in fibroblasts from Patient E-II-1). Each experiment was performed in triplicate, and similar results were obtained in four independent experiments. The histogram shows the quantification of the area of the scratch at 0, 6, and 12 hours. Panel C shows flow-cytometric analysis for CD45 and β1 integrin that was performed on peripheral blood in a patient and a control. The patient's blood (with VPS45-mutant neutrophils) has far fewer cells that are positive for β1 integrin. Panel D shows apoptosis in fibroblasts from a patient and a control. Staining for activated caspase 3, an in situ fluorescein isothiocyanate marker and indicator of apoptosis, was increased by a factor of 14 in fibroblasts from the patient as compared with control cells. In Panels A, B, and D, T bars indicate the standard deviation.
Functional Characterization of Mutant VPS45
Immunoblotting of the NADPH-oxidase subunits (e.g., gp91phox, p22pho, p47phox, and p67phox) was normal (data not shown). In contrast, neutrophil chemotaxis, assessed by stimulation with N-formyl-methionyl-leucyl-phenylalanine (fMLP), was markedly reduced in cells obtained from Patients A-II-3 and C-II-3 (0 to 4 cells per field) as compared with cells from healthy controls (42 to 57 cells per field). In addition, the cells from Patient B-II-9 that were transformed by Epstein–Barr virus did not respond normally to stimulation by fMLP, interleukin-8, or zymosan-activated serum (data not shown). As an indication of NADPH-oxidase dysfunction, superoxide production, assessed by stimulation with phorbol myristate acetate (PMA), was lacking in the neutrophils from Patients A-II-3, C-II-3, and D-II-8 (Table 1).
Markers of cell-migrating capacity were also deficient in VPS45-mutant cells. In neutrophils, cell motility requires β1 integrin on the plasma membrane.22 Analysis by means of fluorescence-activated cell sorting (FACS) showed that VPS45-mutant neutrophils (from Patients B-II-10 and D-II-8) expressed much lower levels of β1 integrin than did the neutrophils from a healthy control (Fig. 2C). VPS45-mutant neutrophils and fibroblasts also showed reduced β1 integrin on immunofluorescence staining (Fig. S7 in the Supplementary Appendix). In neutrophils, β1 integrin was missing from the cell surface. In addition, fibroblasts from Patients B-II-9 and E-II-1, grown as a confluent monolayer on fibronectin-coated cover slides, had poor recovery from a scratch, as compared with healthy foreskin fibroblasts (Fig. 2B).
Fibroblasts and bone marrow with mutant VPS45 also showed evidence of increased apoptosis. Staining with CaspACE FITC-VAD-FMK, which binds to activated caspase and serves as a marker for apoptosis, was 14 times as high in the VPS45-mutant fibroblasts as in the control cells (mean [±SD] proportion of total cells that were stained, 6.6±2% vs. 0.5±0.1%) (Fig. 2D, and Fig. S8A in the Supplementary Appendix). This result was reinforced by the increased number of caspase 3–positive cells in bone marrow–biopsy specimens from the patient, indicating enhanced apoptosis (Fig. S8B in the Supplementary Appendix). Measurement of RNA expression and protein levels of BiP, a marker of stress-induced apoptosis in the endoplasmic reticulum that is elevated in some congenital neutropenias,23 showed no significant difference between fibroblasts from the patients and those from the controls (Fig. S9 in the Supplementary Appendix).
Correction of Vps45-Mutant Phenotypes
On transient transfection of mutant fibroblasts with a plasmid containing nonmutated VPS45 cDNA, the levels of the VPS45-interacting proteins rabenosyn-5 (Fig. 3A) and syntaxin-16 (not shown) returned to normal. In addition, on scratch testing, the VPS45-transfected fibroblasts from Patient B-II-9 migrated significantly more rapidly toward the wounded area than did cells from the patient that were transfected with empty plasmid (Fig. 3B). Transfection with the nonmutated VPS45 vector decreased the amount of fibroblast apoptosis by 50%, but not to the levels observed in normal fibroblasts transfected with empty vector (Fig. 3C). However, given that the transfection efficiency was approximately 70%, the correction of the apoptotic phenotype in fibroblasts from the patient was nearly complete.
Figure 3. Correction of the VPS45 Mutant Phenotype.
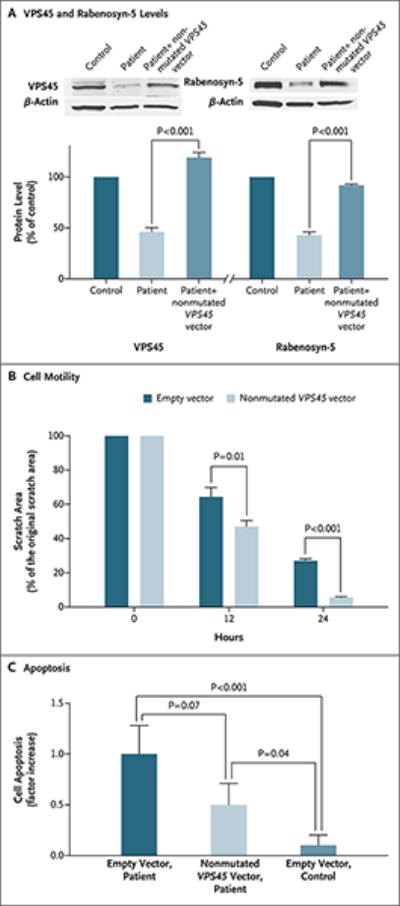
Panel A shows that in VPS45-deficient fibroblasts, which contain approximately half the normal amount of both VPS45 and its interacting protein, rabenosyn-5, levels of both proteins are restored after transfection of the fibroblasts with a plasmid containing nonmutated VPS45. β-Actin was used as the control protein. Panel B shows cell motility after fibroblasts from a patient were transfected with either empty vector or a vector containing nonmutated VPS45 complimentary DNA (cDNA) and then grown on coverslips coated with fibronectin. The cultures were “wounded” by introducing scratches. The histogram shows the quantification of the area of the scratch at various time points. The VPS45-transfected fibroblasts migrated significantly more rapidly toward the wounded area than did cells transfected with empty plasmid. Panel C shows apoptosis, as assessed by staining for activated caspase 3, in patient fibroblasts transfected with either empty vector or a vector containing non-mutated VPS45 cDNA, as compared with control fibroblasts transfected with empty vector. Transfection with the nonmutated VPS45 vector reduced apoptosis by 50%.
Knockdown of VPS45 Expression in Zebrafish Embryos
VPS45 function was investigated in vivo with the use of zebrafish embryos injected with one of two vps45-specific morpholino oligonucleotides (MOs) (I2E3-MO, targeting the splice acceptor site of intron 2, or ATG-MO, targeting the translation initiation site) (Fig. 4A). The optimal dose of each morpholino was determined by its toxic effects on injected embryos 24 hours after fertilization (Fig. S10 in the Supplementary Appendix); no obvious morphologic defects were observed. Real-time quantitative PCR analysis of injected embryos showed that I2E3-MO caused skipping of exon 3, resulting in a frame shift and premature truncation of the protein. Immunoblotting confirmed decreased protein levels for both MOs (Fig. 4B). As compared with untreated embryos, the embryos treated with vps45 morpholino had a large decrease in the number of neutrophils, assessed by means of both myeloperoxidase staining24 (Fig. 4C) and Sudan black B staining18 (Fig. S11 in the Supplementary Appendix).
Figure 4. Targeted Knockdown of Vps45 with the Use of Morpholino Oligonucleotides (MOs) That Block Splicing and Translation in Zebrafish Embryos.
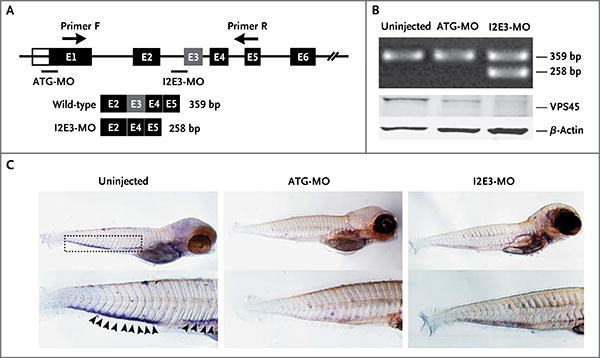
Panel A is a diagram of morpholino design showing the zebrafish gene vps45 and morpholino antisense strategies to block either the translation of the zebrafish vps45 messenger RNA (ATG-MO) or the splice acceptor site of exon 3 (I2E3-MO). Primers F and R validate exon skipping, including the expected size of amplicons that result from loss of exon 3. Below the diagram is a schematic depiction of the spliced transcript in the I2E3-MO–injected embryos (258 bp) as compared with the uninjected and ATG-MO–injected embryos (359 bp). In Panel B, the top immunoblot shows the results of reverse-transcriptase–polymerase-chain-reaction analysis of vps45 transcript from uninjected and morpholino-injected embryos 5 days after fertilization. The bottom immunoblot shows the results of Western blot analysis of whole-protein lysates from uninjected and morpholino-injected embryos 5 days after fertilization. Lysates from embryos injected with ATG-MO and from those injected with I2E3-MO show marked reduction of vps45 when normalized with β-actin, which was used as a protein-loading control. In Panel C, in situ hybridization of embryos 5 days after fertilization supports functional knockdown of vps45. Shown are representative images of uninjected zebrafish embryos (left), embryos injected with ATG-MO (middle), and embryos injected with I2E3-MO (right). Results of whole-mount in situ hybridization with the use of a digoxigenin-labeled RNA probe against zebrafish myeloperoxidase are shown. The myeloperoxidase detects neutrophils in the caudal hematopoietic tissue (rectangle). In uninjected embryos, the myeloperoxidase signals are readily seen (arrowheads). However, in ATG-MO–injected and I2E3-MO–injected embryos, there is a marked reduction in myeloperoxidase staining, suggestive of a decreased number of mature neutrophils.
Discussion
We identified five children in four families who had the same homozygous mutation (p.Thr224Asn) in VPS45 and two children in one family who had a different homozygous mutation (p.Glu238Lys) in VPS45. These seven children had a new disease characterized primarily by neutropenia and neutrophil dysfunction, a lack of response to G-CSF, life-threatening infections, bone marrow fibrosis, and renal extramedullary hematopoiesis. The absence of dysmorphic features such as albinism or short stature distinguishes this syndrome from other known syndromes associated with neutropenia, such as the Chédiak–Higashi syndrome, Griscelli's syndrome type 2, the Hermansky–Pudlak syndrome type 2, and p14 deficiency.5,25,26 Neurologic defects, including developmental delays, in the two affected children from the Moroccan family may reflect differences in the VPS45 mutation or the presence of another genetic defect in this consanguineous pedigree.
The VPS45 mutations were shown to cause this disease on the basis of the following findings: they fully segregated with the disease, were absent from 250 alleles of normal controls with the same genetic background and from published databases, caused structural alterations in VPS45, and were located in residues highly conserved among species. Furthermore, in both neutrophils and fibroblasts, the Thr224Asn mutation led to decreased VPS45 levels, affected proteins that interact with VPS45, impaired cell migration, and increased apoptosis. In addition, a zebrafish model with reduced vps45 protein had severe neutropenia, resembling that in the patients.
VPS45, a highly conserved protein,27 is a member of the Sec1p/Munc18-like (SM) family that binds soluble N-ethylmaleimide-sensitive factor attachment protein receptors (known as SNARE).28 Rab5 GTPase, a protein that regulates trafficking of membrane and proteins through endocytic and secretory pathways,29 specifically recruits VPS45 and rabenosyn-5 as cytosolic effector proteins30; in neutrophils, these proteins function in de-granulation and intracellular trafficking.31 Vps45p is associated with cellular membranes, including those of the Golgi apparatus, endosomes, and other vesicles.32 Mutations in yeast vps45 result in accumulation of membrane vesicles, and human VPS45 functions in protein trafficking and the release of inflammatory mediators, particularly in leukocytes.33 In addition, vps45 interacts with rabenosyn-5 in yeast, Caenorhabditis elegans, and drosophila and directs localization of syntaxin-16 and syntaxin-6.34-36 In human cells, the direct interaction between endogenous human VPS45 and rabenosyn-5 stabilizes both proteins and functions in β1-integrin recycling between the endocytic compartment and the plasma membrane, a process required for cell motility.37
Mutations in VPS45 affected all these functions in the cells of the children we describe. In particular, the Thr224Asn mutation resulted in significantly reduced fibroblast and neutrophil levels of VPS45 protein and its three known interacting proteins — rabenosyn-5, β1 integrin, and syntaxin-16. Moreover, the neutrophils and fibroblasts in these children were characterized by abnormal migration, presumably caused by impaired recycling of β1 integrin as a result of VPS45 deficiency37; indeed, these children had reduced numbers of β1-integrin–positive cells in circulation (Fig. 2B, and Fig. S7 in the Supplementary Appendix). In a similar fashion, the lack of response to G-CSF might be due to impaired delivery to the plasma membrane of proteins (i.e., integrins) required for neutrophil function.38 The abnormal fibroblast migration, as well as reduced levels of rabenosyn-5 and syntaxin-16, was corrected by transfection of VPS45-mutant cells with VPS45-nonmutated plasmid.
Any or all of these functional cellular defects could contribute to the increased apoptosis of VPS45-deficient neutrophils, which could be the proximate cause of neutropenia in this disorder; apoptosis also occurs in cells from patients with other variants of congenital neutropenia, such as HAX1, adenylate kinase 2 (AK2), and serine–threonine kinase 4 (STK4)39 deficiencies. In contrast to mutant ELANE and G6PC3 cells, the apoptosis did not appear to be associated with increased stress on the endoplasmic reticulum.40-42 In all studied cases, significant bone marrow fibrosis was evident and the cellular distribution of VPS45 was abnormal, suggesting a role for VPS45 in the specialized hematopoietic niche.43
VPS45 deficiency should be considered in patients with neutropenia, bone marrow fibrosis, nephromegaly, migration defects, and severe bacterial and fungal infections. The cellular defects in this new disease suggest that other immuno-deficiency disorders may also result from impaired vesicle trafficking.
Supplementary Material
Acknowledgments
Supported by the Intramural Research Program of the National Human Genome Research Institute, an advanced grant (EXPLORE) from the European Research Council, the Gottfried-Wilhelm-Leibniz Program of the Deutsche Forschungsgemeinschaft, the Care-for-Rare Foundation, and the German Network on Primary Immunodeficiency Diseases. Dr. Somech is the recipient of grants from the Jeffrey Modell Foundation, the Israeli Science Foundation, and the Chief Scientist Office of the Israeli Ministry of Health.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
We thank all the study participants and their families for their cooperation, Patra Yeetong and Aleixa Triot for Sanger validations, the NIH Intramural Sequencing Center for performing whole-exome sequencing, and Pedro Cruz for analyzing the sequence data.
References
- 1.Malech HL, Gallin JI. Immunology: neutrophils in human diseases. N Engl J Med. 1987;317:687–94. doi: 10.1056/NEJM198709103171107. [DOI] [PubMed] [Google Scholar]
- 2.Klein C. Congenital neutropenia. Hematol Oncol Clin North Am. 2013;27(3):xi–xii. doi: 10.1016/j.hoc.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Klein C. Genetic defects in severe congenital neutropenia: emerging insights into life and death of human neutrophil granulocytes. Annu Rev Immunol. 2011;29:399–413. doi: 10.1146/annurev-immunol-030409-101259. [DOI] [PubMed] [Google Scholar]
- 4.Dell'Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell. 1999;3:11–21. doi: 10.1016/s1097-2765(00)80170-7. [DOI] [PubMed] [Google Scholar]
- 5.Bohn G, Allroth A, Brandes G, et al. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14. Nat Med. 2007;13:38–45. doi: 10.1038/nm1528. [DOI] [PubMed] [Google Scholar]
- 6.Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol. 2008;9:781–95. doi: 10.1038/nrm2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gissen P, Tee L, Johnson CA, et al. Clinical and molecular genetic features of ARC syndrome. Hum Genet. 2006;120:396–409. doi: 10.1007/s00439-006-0232-z. [DOI] [PubMed] [Google Scholar]
- 8.Horn D, Krebsova A, Kunze J, Reis A. Homozygosity mapping in a family with microcephaly, mental retardation, and short stature to a Cohen syndrome region on 8q21.3-8q22.1: redefining a clinical entity. Am J Med Genet. 2000;92:285–92. [PubMed] [Google Scholar]
- 9.Rubio JP, Danek A, Stone C, et al. Chorea-acanthocytosis: genetic linkage to chromosome 9q21. Am J Hum Genet. 1997;61:899–908. doi: 10.1086/514876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wider C, Skipper L, Solida A, et al. Autosomal dominant dopa-responsive parkinsonism in a multigenerational Swiss family. Parkinsonism Relat Disord. 2008;14:465–70. doi: 10.1016/j.parkreldis.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 11.Peiffer DA, Le JM, Steemers FJ, et al. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136–48. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steemers FJ, Gunderson KL. Whole genome genotyping technologies on the BeadArray platform. Biotechnol J. 2007;2:41–9. doi: 10.1002/biot.200600213. [DOI] [PubMed] [Google Scholar]
- 13.Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–9. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cullinane AR, Vilboux T, O'Brien K, et al. Homozygosity mapping and whole-exome sequencing to detect SLC45A2 and G6PC3 mutations in a single patient with oculocutaneous albinism and neutropenia. J Invest Dermatol. 2011;131:2017–25. doi: 10.1038/jid.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westerfield M. The zebrafish book: a guide for the laboratory use of zebrafish Danio (Brachydanio) rerio. Eugene: University of Oregon Press; 2000. [Google Scholar]
- 16.Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- 17.Sood R, Liu P. Novel insights into the genetic controls of primitive and definitive hematopoiesis from zebrafish models. Adv Hematol. 2012;2012:830703. doi: 10.1155/2012/830703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Guyader D, Redd MJ, Colucci-Guyon E, et al. Origins and unconventional behavior of neutrophils in developing zebrafish. Blood. 2008;111:132–41. doi: 10.1182/blood-2007-06-095398. [DOI] [PubMed] [Google Scholar]
- 19.Zucker-Franklin D, L'Esperance P, Good RA. Congenital neutropenia: an intrinsic cell defect demonstrated by electron microscopy of soft agar colonies. Blood. 1977;49:425–36. [PubMed] [Google Scholar]
- 20.Bishop JB, Tani Y, Witt K, et al. Mitochondrial damage revealed by morphometric and semiquantitative analysis of mouse pup cardiomyocytes following in utero and postnatal exposure to zidovudine and lamivudine. Toxicol Sci. 2004;81:512–7. doi: 10.1093/toxsci/kfh208. [DOI] [PubMed] [Google Scholar]
- 21.Amitai G, Shemesh A, Sitbon E, et al. Network analysis of protein structures identifies functional residues. J Mol Biol. 2004;344:1135–46. doi: 10.1016/j.jmb.2004.10.055. [DOI] [PubMed] [Google Scholar]
- 22.Jovic M, Naslavsky N, Rapaport D, Horowitz M, Caplan S. EHD1 regulates beta1 integrin endosomal transport: effects on focal adhesions, cell spreading and migration. J Cell Sci. 2007;120(Pt 5):802–14. doi: 10.1242/jcs.03383. [DOI] [PubMed] [Google Scholar]
- 23.Schäffer AA, Klein C. Genetic heterogeneity in severe congenital neutropenia: how many aberrant pathways can kill a neutrophil? Curr Opin Allergy Clin Immunol. 2007;7:481–94. doi: 10.1097/ACI.0b013e3282f1d690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bennett CM, Kanki JP, Rhodes J, et al. Myelopoiesis in the zebrafish, Danio rerio. Blood. 2001;98:643–51. doi: 10.1182/blood.v98.3.643. [DOI] [PubMed] [Google Scholar]
- 25.Gahl WA, Brantly M, Kaiser-Kupfer MI, et al. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky–Pudlak syndrome) N Engl J Med. 1998;338:1258–64. doi: 10.1056/NEJM199804303381803. [DOI] [PubMed] [Google Scholar]
- 26.Tomita Y, Suzuki T. Genetics of pigmentary disorders. Am J Med Genet C Semin Med Genet. 2004;131C:75–81. doi: 10.1002/ajmg.c.30036. [DOI] [PubMed] [Google Scholar]
- 27.Bock JB, Matern HT, Peden AA, Scheller RH. A genomic perspective on membrane compartment organization. Nature. 2001;409:839–41. doi: 10.1038/35057024. [DOI] [PubMed] [Google Scholar]
- 28.Shanks SG, Carpp LN, Struthers MS, McCann RK, Bryant NJ. The Sec1/Munc18 protein Vps45 regulates cellular levels of its SNARE binding partners Tlg2 and Snc2 in Saccharomyces cerevisiae. PLoS One. 2012;7(11):e49628. doi: 10.1371/journal.pone.0049628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mellman I. Endocytosis and molecular sorting. Annu Rev Cell Dev Biol. 1996;12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- 30.Nielsen E, Christoforidis S, Uttenweiler-Joseph S, et al. Rabenosyn-5, a novel Rab5 effector, is complexed with hVPS45 and recruited to endosomes through a FYVE finger domain. J Cell Biol. 2000;151:601–12. doi: 10.1083/jcb.151.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perskvist N, Roberg K, Kulyte A, Stendahl O. Rab5a GTPase regulates fusion between pathogen-containing phagosomes and cytoplasmic organelles in human neutrophils. J Cell Sci. 2002;115:1321–30. doi: 10.1242/jcs.115.6.1321. [DOI] [PubMed] [Google Scholar]
- 32.Cowles CR, Emr SD, Horazdovsky BF. Mutations in the VPS45 gene, a SEC1 homologue, result in vacuolar protein sorting defects and accumulation of membrane vesicles. J Cell Sci. 1994;107:3449–59. doi: 10.1242/jcs.107.12.3449. [DOI] [PubMed] [Google Scholar]
- 33.Rajasekariah P, Eyre HJ, Stanley KK, Walls RS, Sutherland GR. Molecular cloning and characterization of a cDNA encoding the human leucocyte vacuolar protein sorting (h1Vps45) Int J Biochem Cell Biol. 1999;31:683–94. doi: 10.1016/s1357-2725(99)00017-5. [DOI] [PubMed] [Google Scholar]
- 34.Tall GG, Hama H, DeWald DB, Horazdovsky BF. The phosphatidylinositol 3-phosphate binding protein Vac1p interacts with a Rab GTPase and a Sec1p homologue to facilitate vesicle-mediated vacuolar protein sorting. Mol Biol Cell. 1999;10:1873–89. doi: 10.1091/mbc.10.6.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gengyo-Ando K, Kuroyanagi H, Kobayashi T, et al. The SM protein VPS-45 is required for RAB-5-dependent endocytic transport in Caenorhabditis elegans. EMBO Rep. 2007;8:152–7. doi: 10.1038/sj.embor.7400882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morrison HA, Dionne H, Rusten TE, et al. Regulation of early endosomal entry by the Drosophila tumor suppressors Rabenosyn and Vps45. Mol Biol Cell. 2008;19:4167–76. doi: 10.1091/mbc.E08-07-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rahajeng J, Caplan S, Naslavsky N. Common and distinct roles for the binding partners Rabenosyn-5 and Vps45 in the regulation of endocytic trafficking in mammalian cells. Exp Cell Res. 2010;316:859–74. doi: 10.1016/j.yexcr.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen C, Huang X, Atakilit A, Zhu QS, Corey SJ, Sheppard D. The Integrin alpha-9beta1 contributes to granulopoiesis by enhancing granulocyte colony-stimulating factor receptor signaling. Immunity. 2006;25:895–906. doi: 10.1016/j.immuni.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 39.Abdollahpour H, Appaswamy G, Kotlarz D, et al. The phenotype of human STK4 deficiency. Blood. 2012;119:3450–7. doi: 10.1182/blood-2011-09-378158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boztug K, Appaswamy G, Ashikov A, et al. A syndrome with congenital neutropenia and mutations in G6PC3. N Engl J Med. 2009;360:32–43. doi: 10.1056/NEJMoa0805051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grenda DS, Murakami M, Ghatak J, et al. Mutations of the ELA2 gene found in patients with severe congenital neutropenia induce the unfolded protein response and cellular apoptosis. Blood. 2007;110:4179–87. doi: 10.1182/blood-2006-11-057299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nanua S, Murakami M, Xia J, et al. Activation of the unfolded protein response is associated with impaired granulopoiesis in transgenic mice expressing mutant Elane. Blood. 2011;117:3539–47. doi: 10.1182/blood-2010-10-311704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang LD, Wagers AJ. Dynamic niches in the origination and differentiation of haematopoietic stem cells. Nat Rev Mol Cell Biol. 2011;12:643–55. doi: 10.1038/nrm3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.