

Abstract
Extensive research in recent years suggests that exposure to xenobiotic stimuli plays a critical role in autoimmunity induction and severity and that the resulting response would be exacerbated in individuals with an infection-aroused immune system. In this context, heavy metals constitute a prominent category of xenobiotic substances, known to alter divergent immune cell responses in accidentally and occupationally exposed individuals, thereby increasing the susceptibility to autoimmunity and cancer, especially when accompanied by inflammation-triggered persistent sensitization. This perception is learned from experimental models of infection and epidemiologic studies and clearly underscores the interplay of exposure to such immunomodulatory elements with pre- or postexposure infectious events. Further, the TH17 cell subset, known to be associated with a growing list of autoimmune manifestations, may be the “superstar” at the interface of xenobiotic exposure and autoimmunity. In this review, the most recently established links to this nomination are short-listed to create a framework to better understand new insights into TH17's contributions to autoimmunity.
1. Introduction
Long-term exposure to xenobiotic substances induces hyperactivity of the immune system, thereby increasing the incidence of autoimmune diseases (AD), especially in infection-aroused systems. Circumstances dating back to earlier exposure, as in case of heavy-metal industry workers or current exposure as in individuals harboring amalgam teeth filling, favor incidence of inflammatory processes and most likely AD [1–4]. Exposure to infectious agents leads to the induction of various cellular pathways essential to the microbe's infectivity, survival, and virulence, thus making it difficult for the microbe to go undetected by the host's immune system [5]. Upon pathogen recognition, production of a proinflammatory response, primarily by macrophages, NK and NKT cells, is the subsequent event in the early phase of the infection [5, 6]. Further, the coordination between innate and adaptive immune defense systems ensures a successful eradication of pathogens, and such developed cytokine milieu determines the induction of a specific T-cell-mediated response that is critical for an effective and complete pathogen clearance. However, whether the induction of a strong host inflammation constitutes an adaptive advantage to the host or pathogen remains debated. Indeed, many disorders, including AD [7] and cancer [8], are associated with and maintained by chronic inflammation; for review, see [9, 10]. The association of cancer incidence with exposure to heavy metals, such as cadmium [11], or following attainment of chronic inflammation, as in case of colitis-associated cancer, has been widely anticipated.
Research on TH17 cells has suggested a crucial role in autoimmunity. Despite developing autoimmune signs in the absence of detectable IL-17 levels, as in case of choriomeningitis-virus-induced model of type 1 diabetes [12], a key role of TH17 cells and their related molecules was underscored in many previously assigned “TH1-mediated” AD including rheumatoid arthritis (RA), psoriasis, systemic lupus erythematosus (SLE), and multiple sclerosis (MS), as well as, the experimental autoimmune encephalomyelitis—EAE [7, 9, 13–15]. Variations in disease susceptibility or outcome may be a result of co-exposure to one or multiple xenobiotic substances or infectious pathogens, so that a xenobiotic-induced polarized immune response triggers the development of AD in genetically predisposed individuals [1, 2, 4, 16–19]. The IL-17 response, while constituting a protective arm defending the body against various infections, also functions as a double-edged sword constituting a risk factor that mediates the development and/or induction of AD, mostly manifested following pathogenic and xenobiotic-induced chronic inflammation; it then acts as a double-edged sword, constituting a risk factor that mediates the development and/or induction of AD, mostly manifested following pathogenic and xenobiotic-induced chronic inflammation. In the next sections, we revisit our view on the TH17 cells' role in autoimmunity [9] and provide a brief description of the double-sided role of TH17 cells and their related molecules IL-17, IL-21, and IL-22 and their participation at the initiation/induction of autoimmunity as a consequence of xenobiotic exposure.
2. TH17 Cells and Their Associated Molecules Link Infection to Autoimmunity
T cells differentiate and expand into distinct lineages including TH1, TH2, iTReg, and TH17 cells [9], whereas iTReg cells differentiate under subimmunogenic antigen presentation both during chronic inflammation and under normal homeostatic conditions of the gut and function to control severe chronic allergic inflammation and as a barrier to the eradication of tumors [20, 21]. TH17 cells derive from CD161+ precursors in umbilical cord blood and newborn thymus [22] and likely constitute the most prominent T cell subset at the crossroads of infection and autoimmunity. The contributions of TH17 cells have prompted and were the results of intensive scientific research, which is reflected by a growing list of publications in this field (Figure 1), and have in turn led to identification of TH17 cells' markers, as well as, their differentiation and commitment program [23]. Figure 2 demonstrates the major T cell subsets, their interaction with TH17 cells and the main contributions of the latter.
Figure 1.
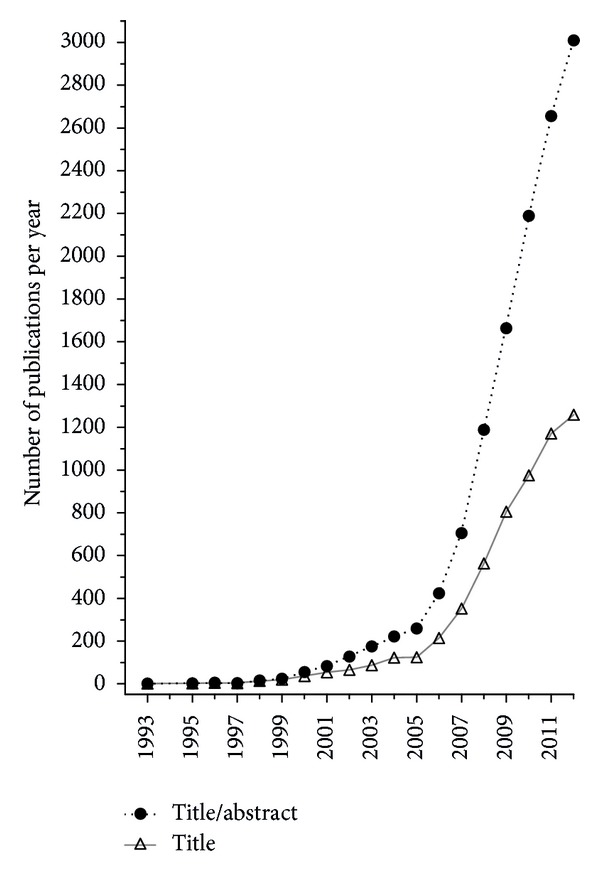
Frequencies of IL-17-producing T helper (TH)17 cells and their related molecules as appeared in title or title/abstract of PubMed publications. The “keywords” {TH17} OR {IL-17∖*} OR {IL-21} OR {IL-22} OR {IL-23∖*} OR {CTLA-8} OR {CCL20} OR {ROR*} and their synonyms combined with “publication date” {1993} through {2011} were given as search parameters.
Figure 2.
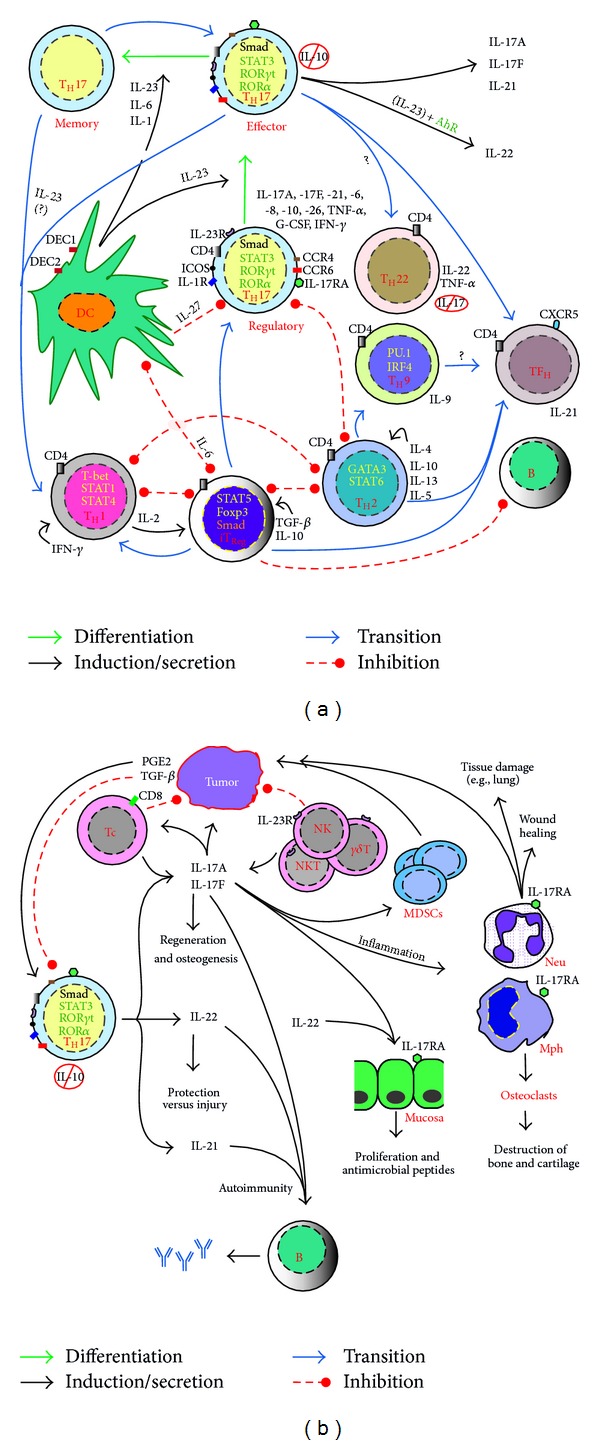
Differentiation and commitment of IL-17-producing T helper (TH)17 cells in the midpoint of other coacting cells in favor of and against the harboring individual. Upon antigen recognition, presentation and cosignaling, naïve (TH0) cells differentiate in the presence of distinct cytokine milieu into effecter (TH1, TH2, TH9, TH17, TH22, and TFH) and T regulatory (TReg) cells. Signaling cytokine and other molecules activate lineage-unique transcription factors that ultimately mediate cell differentiation and maturation. Whereas activation of STAT1 induces T-bet expression, STAT6 signaling upregulates GATA3 expression; both cell lineages reciprocally regulate each other and regulate the generation of TH17 cells through their hallmark effecter cytokines, IFN-γ and IL-4, respectively, though IFN-γ is produced by TH17 cells in some disease settings and in response to certain infections. Differentiation of TH17 necessitates costimulatory signals of CD28 and ICOS (the last is not mandatory) and the absence of TH1 and TH2 cytokines (IL-12/IFN- and IL-4) and their master transcription factors; a task taken over by TGF-β is to constrain TH1 and TH2 during TH17 differentiation program. TH17 and TReg are likely descendants of the same ancestor cell lineage, which differentiates in presence of low levels of TGF-β and IL-6/IL-21 or other proinflammatory cytokines (IL-1, TNF-α, and IL-18) into TH17 cells; TGF-β signals through Smad2 protein pathway and is indispensable for induction of expression RORγt. High levels of TGF-β alone induce Foxp3 expression and hence TReg cell differentiation. Following their final commitment and upregulation of IL-23R, TH17 cells require IL-23 signaling that is crucial for their survival and effecter functions including production of IL-22, as well as, cell plasticity including later production of IFN-γ. TH22 may differentiate from TH0 cells or through a local commitment of TH17 cells homed in the epidermis. Contributions of TH17 cells entail activations of aryl hydrocarbon receptor (AhR) signaling and production of IL-22 upon exposure to xenobiotic substances. IL-17 and IL-17F increase production of IL-6, IL-8, prostaglandin E2 (PGE2), monocyte chemotactic protein-(MCP-) 1, and the granulocyte colony-stimulating factor (G-CSF) by various cells including macrophages, fibroblasts, keratinocytes, and epithelial and endothelial cells and ultimately promote inflammatory diseases, AD, and/or cancer. These cytokines together with IL-21 and IL-22 are also implicated in mediating protective as well as pathogenic processes in various disease settings.
Recently, several groups delivered compelling evidence of the effects of TH17-associated cytokines, namely IL-17, IL-21, IL-22, and IL-23, on inflammatory responses elicited by extracellular, as well as, facultative and obligate intracellular pathogens including bacteria and fungi. Exemplified contributions of IL-17 response to some infectious diseases are summarized in Table 1.
Table 1.
Mechanistic investigations on the role of TH17 cells and their related molecules in various infectious diseases.
| Diagnosis | Role | Observations on TH17-associated molecules | Citations |
|---|---|---|---|
| (1) Bacterial infections | |||
|
| |||
| Bacillus subtilis | Pathogenic/protective | Increased lung inflammation and collagen deposition; delay in bacterial clearance in IL-17R−/− compared with WT counterparts | [118] |
| Francisella tularensis | Pathogenic | Intranasal inoculation induces TH17 response and PGE2 production in the lung; inhibition of PGE2 production increased IFN-γ and decreased bacteremia | [119] |
| Saccharopolyspora rectivirgula | Pathogenic | Induction of IL-17-mediated hypersensitivity pneumonitis in mice; reduced lung inflammation and fibrosis in IL-17R−/− mice | [120, 121] |
| Klebsiella pneumoniae, Bordetella pertussis, & S. pneumoniae | Protective | Mounting of an IL-17 and IL-22 response; defects in TH17 response increased susceptibility | [12, 122] |
| Staphylococcus aureus | Protective | High infection incidence correlated with defect in TH17 response | [59] |
| Listeria monocytogenes | Protective | IL-17-mediated cross-protection following immunization with M. pulmonis; blockade of bacterial growth following transfer of IL-17-producing γδ and double negative αβ T cells into RAG2−/− mice | [30, 60] |
| Shigella flexneri | Protective | Restriction of bacterial growth mediated by TH17 response | [28] |
| Citrobacter and Salmonella sp. | Protective | Innate TH17 response-dependent protection; protective effect of IL-17 and IL-22; decrease in phagocytic activity and increase in bacterial burden upon IL-17 neutralization and its correlation with TH17 response in Hg-exposed mice | [29, 35, 38] and Hemdan and Abul El-Saad, unpublished |
| Mycobacterium tuberculosis and M. bovis | Protective | IL-17−/− mice reveal a reduced IFN-γ production by CD4+ T cells, impaired granuloma formation, and chemokine expression | [61] |
| Mycobacterium tuberculosis | Protective | Correlation of reduced TH17 responses in patients with active tuberculosis with decreased expression of IL-6R on CD4+ T cells | [123] |
| Chlamydia sp. | Protective | Enhanced bacterial growth and decreased mouse survival upon applying anti-IL-17 mAb | [34] |
| Pathogenic | Applying IL-17RA antagonist reversed the susceptible phenotype of C3H/HeN mice | [58] | |
| Inflammatory bowel disease—IBD (Crohn's disease and ulcerative colitis) | Pathogenic | Enhanced differentiation TH17 and IL-17 expression levels and NK activities in IBD | [124, 125] |
| Protective | IL-22 mediated protection against IBD | [54] | |
|
| |||
| (2) Protozoal infection | |||
|
| |||
| Toxoplasma gondii | Pathogenic | IL-23-mediated IL-22 and MMP-2 upregulation in the ileum of infected mice; MMP-2 deficiency offered protection | [47] |
| Protective | Increased mortality in IL-17−/− mice | [126] | |
|
| |||
| (3) Fungal infections | |||
|
| |||
| Candida sp. | Protective | Involvement of IL-17, IL-17F, IL-22, and IL-23 in mediating natural defense against candidiasis | [45] |
| Aspergillus fumigatus | Induced IL-17 response mediates pathogen clearance | [127] | |
|
| |||
| (4) Viral infection | |||
|
| |||
| Theiler's murine encephalomyelitis virus infection | Pathogenic | Induction of antiapoptotic molecules by IL-17 and thereby promoting persistent infection; boosting lytic function of CTLs and ameliorating disease upon neutralizing IL-17; association of lower TH17 with higher virus-specific CD8+ T cell responses in resistant mouse than in susceptible strain | [128] |
| Respiratory syncytial virus (RSV) | Pathogenic | Elevated IL-6 and IL-17 levels in tracheal aspirate samples from severely ill infants and in infected mice; IL-17 blockade decreased the exacerbated disease via increasing RSV-specific CD8+ T cells, T-bet, IFN-γ, eomesodermin, and granzyme B | [129] |
| HBV | Pathogenic/protective | Distinct effects associated with heterogeneous TH17 populations: IL-17 with inflammation and ALT levels, IL-22 with protection of hepatocytes, and IL-21 with virus clearance | [130] |
| HCV | Pathogenic | Hepatitis-C-virus-infected patient revealed upregulated TH17 cell cytokines that became downregulated by combined treatment with pegylated IFN and ribavirin | [131] |
| Simian immunodeficiency virus (SIV)/HIV | Pathogenic | Induction of TGF-β and IL-18 during the acute phase in SIV-infected rhesus macaques proposed to be associated with induction of IL-17-producing NKT cells | [132] |
| Protective | Association of disease progression with loss of TH17 and induction of TReg cells; TH17 cell frequency correlated negatively with viral load | [63, 133, 134] | |
| Herpes simplex virus (HSV-1) | Infiltration of TH1 preceded TH17 cells, the latter showed lower responsiveness ability to HSV-1; diminished stromal keratitis severity in IL-17R−/−-infected mice and upon IL-17 neutralization in WT mice | [135] | |
|
| |||
| (5) Nematode infection | |||
|
| |||
| Trichinella spiralis | Pathogenic | Correlation of TH17 response with increase of smooth muscle contraction probably causing gut dysfunction; association of IL-17/IL-23 axis induction with increased mortality in mice coinfected with malaria and nematode | [136] |
2.1. TH17 Cells-Associated Molecules and Their Contributions to Anti-Infectious Responses
In comparison to the frequent appearance of TH1, the relative rarity of TH17 in inflamed tissues was attributed to their RorC-dependent expression of the oxidase IL4I1, which impairs CD3 signaling and hence constrains IL-2 production and cell proliferation [24]. As we recently reviewed, the recruitment of TH17 cells to inflammatory tissues accompanies the expression of the chemokine receptor CCR6, in addition to CCR4, IL-23R (involved in the survival/maturation program of TH17 cells) [9], and CD161 [22]. TH17 cells are considered as potent inflammation inducers that, in addition to production of IL-17, differentially produce IL-6, IL-2, IL-8, IL-9, TNF-α, IL-17F, IL-21, IL-22, IL-26, IFN-γ, and the chemokine CCL20 and induce activation and recruitment of other cells including neutrophils that are pivotal in inflammation and AD [9, 25]. Through their cytokine/chemokine production, TH17 cells act on a broad range of cell types initiating the expression of antibodies, metalloproteinases, prostaglandin E2 (PGE2), and antimicrobial peptides and inducing cyclooxygenase 2 activity [9, 26], constituting, thereby, a link between innate and adaptive immune responses. In addition to its role as an arm of adaptive immunity, the current perception categorizes IL-17 also as an innate cytokine, produced mainly by NK cells [27] as well as by γδ T cells [28–31], CD8+ T cells, and mast cells [9, 32]. Indeed, detecting functional TH17 cells and production of protective IL-17 during the early phase of the immune response [33–35] and the activation of TH17 that even precedes the differentiation of TH1 cells [33], together with the later contribution of TH17 [36], highlight the crucial importance of IL-17 and other TH17-related cytokines in the early, as well as, the late phase of infection. The upregulation of TLR1 and TLR2 and dectin 1 by IL-17-producing γδ T cells [37] supports this belief. Moreover, studies on nucleotide oligomerization domain knockout mice (Nod1−/− and Nod2−/−) demonstrated that this “early” TH17 response was Nod1- and Nod2-dependent, and hence they have been given the name innate (i)TH17 cells [35]. Therefore, the new look of IL-17-producing cells comprises their contribution in building the first line of host defense, besides mediating and shaping adaptive responses required for ultimate clearance.
Based on a wealth of experimental data, the contribution of TH17 cells to infection is manifold. As in the case of oral infection, the importance of IL-17 in protection against infection seems to be crucial to attain a mucosal barrier in the intestine as in case of Salmonella [30, 38], in mediating protection against oral brucellosis [39], promoting granulopoiesis through induction of granulocyte colony-stimulating factor G-CSF [40], and neutrophil influx through inducing neutrophil chemotactic CXCL8 (IL-8), macrophage chemotactic protein (MCP)-1, and macrophage inflammatory proteins- (MIP-) 1 and MIP-2 [9, 40, 41]. Additionally, we and others found that IL-17 activates phagocytosis and neutrophil cytotoxic activity [42]. Therefore, IL-17R−/− mice revealed increased systemic dissemination of S. typhimurium from the gut [29]. The same strategy seems to be attained to combat extracellular pathogens such as Klebsiella pneumoniae [12] and fungal infections (e.g., Candida sp.), mainly due to defective IL-17 immunity [43, 44], mediated by eliciting production of autoantibodies (AAs) against IL-17, IL-17F, and IL-22 that contribute to chronic mucocutaneous candidiasis [45, 46]. A third arm of TH17 cells is built through IL-22, which is also produced by other cell types including NK22 and lymphoid tissue inducer cells [47], as well as by skin homing TH22 cells [48, 49]. Besides its accepted role against infection, it induces tissue repair offering protection against injury [49]. Although both IL-22 and IL-17 or IL-17F synergize to stimulate expression of human beta-defensin- (HBD-) 2, S100 calcium binding protein A9 (S100A9) and enhanced the expression of S100A7 and S100A8 [9], IL-22, rather than IL-17, seems to contribute more to the epidermal and mucosal immunity [47, 49]. It synergizes with TNF-α to induce secretion of initial complement factors C1r and C1s, antimicrobial peptides S100A7 and HBD-2, and antimicrobial chemokines CXCL-9/-10/-11 in primary human keratinocytes [50]. In a three-dimensional skin infection model, stimulation of keratinocytes with TH22 supernatants or by adding IL-22 plus TNF-α effectively inhibited C. albicans growth and maintained epithelial survival, and the combinatorial stimulation of keratinocytes with IL-22 plus TNF-α most effectively conserved the integrity of the epidermal barrier as compared with IFN-γ, IL-17, IL-22, or TNF-α alone [50]. IL-22 also functions to induce an acute phase systemic response that extends beyond IL-22R-expressing cells and revealed diverse significant impact on coagulation and cellular constituents of blood, in addition to induction of thymic atrophy, body weight loss, and renal proximal tubule metabolic activity and biochemical changes in the liver, including induction of fibrinogen, CXCL1, and serum amyloid A [51]. Besides its contribution to protection against bacterial infection [50, 52], IL-22 plays an important role in protection against viral infection, for example, hepatitis B virus [53]. On the other side, IL-22 is implicated in the induction of IBD [54] and AD such as experimental autoimmune myocarditis [9] and psoriatic disease through the induction of keratinocyte proliferation and cytokine and chemokine release [7]. This reflects the dark side of the TH17 story; that is, a promoted TH17 response may reflect a current or predict incidence of AD.
2.2. TH1-TH17 Cells Interaction during Infection
Although a protective role against intracellular bacteria such as Listeria monocytogenes [55] or S. typhimurium (N. Y. A. Hemdan and A. M. Abu El-Saad, unpublished data) may be attributed to TH17 response, this may be rather compensatory to a defective IL-12/IFN-γ axis as previously demonstrated by N. N. Orgun et al. [56], or a complementary function to indirectly induce type 1 response mediated by APCs endowing thereby a protection against infection, as in case of the obligate intracellular bacteria Chlamydia muridarum [57]. In case of infection with S. typhimurium and C. muridarum, neutralizing IL-17 significantly reduced pathogen-specific TH1 but promoted higher TH2 responses. DCs isolated from IL-17-neutralized mice demonstrated lower expression of CD40, MHC II, and IL-12 production, but higher level of IL-10 compared with control mice [57]. Furthermore, neutralizing IL-17 in case of S. typhimurium significantly reduced phagocytosis as well as TH1 cytokine production (N. Y. A. Hemdan and A. M. Abu El-Saad, unpublished data). Moreover, delivery of an IL-17R antagonist that resulted in a 50% reduction in the neutrophilic infiltration in lungs following Chlamydia infection reversed the susceptible phenotype of C3H/HeN mice [58], indicating a key role of IL-17 in induction of neutrophil infiltration. The compromised IL-17 response in HIS (Job's syndrome) patients that contributed to higher susceptibility to Staphylococcus aureus infection [59] is evidenced by a recent finding that coinfection with influenza A abrogated host defense, which was rescued by overexpression of IL-23 and markedly improved bacterial clearance [52]. Influenza A was found to inhibit TH17 differentiation and substantially decreased IL-17, IL-22, and IL-23 production after S. aureus infection. Interestingly, IL-17-mediated cross-protection against secondary L. monocytogenes infection has been demonstrated following immunization with Mycoplasma pulmonis [60].
In addition to the function of TH17 cells as a substitute for a defective TH1 response, synergism between TH17 and TH1 cells is proposed following infection or postvaccination challenge with Mycobacterium sp., based on the observation that IL-17−/− mice revealed a reduced IFN-γ production by CD4+ T cells and impaired granuloma formation and expression of chemokines CXCL9, CXCL10, and CXCL11 [61]. Also, an enhanced TH1 memory response in the lungs of vaccinated mice infected with M. tuberculosis was dependent upon IL-23/IL-17 axis [61]. In a model of TCR αβ −/− mouse [62], where adoptive transfer of either TH1 or TH17 cells restored bacterial burdens and innate immune cell infiltrates to wild-type animals level, TH17 transferred cells revealed plasticity within the CNS compartment with an ultimate TH1-like cytokine profile, and this might be the reason for restoration of a strong innate immune response against infection with pyogenic bacteria; for review on various forms of TH17 cell plasticity, refer to Hemdan [10]. Furthermore, the importance of TH17 cells in combating HIV-associated bacterial infections has been recently elucidated [63].
2.3. Interaction of TH17 Cells with Commensal Bacteria
Of a crucial importance is the recent clue linking the induction of TH17 response with gut commensal bacteria. Colonization of the small intestine of mice with a single commensal microbe, segmented filamentous bacterium (SFB), was sufficient to induce IL-17- and IL-22-producing TH17 cell responses in the lamina propria, and this was correlated with enhanced expression of inflammation- and antimicrobial-associated genes and increased resistance to the intestinal pathogens Citrobacter and Salmonella [35, 64]. Induction of TH17 cells mediated autoimmune arthritis in K/BxN mice [65], whereas when the same mice were held under germ-free conditions, autoimmune arthritis was strongly attenuated and mice revealed reductions in serum AAs titers, splenic AAs-secreting cells, germinal centers, and splenic TH17 cells as well as the lack of TH17 cells in the small intestinal lamina propria [65]. These findings suggest the role of TH17 cells not only in defending the gastrointestinal tract against pathogens, but also in mediating AD (Table 1). How does the immune system monitor the resident intestinal microbes and coordinate between host defense and tolerance and how do dysregulated host-microbe interactions lead to intestinal inflammation were recently discussed [66]. The increased production of IL-17 and IL-23 by PBMCs derived from patients of primary Sjogren's syndrome upon TLR2, TLR4, and TLR6 stimulation [67] highlights the link between TLR ligation and autoimmune induction in such disease settings, where the participation of TH17 cytokines in their pathogenesis is evident [68, 69].
2.4. TH17 Cells in the Bone Disease
One of the most important contributions of TH17 cells involves bone metabolism and bone disease. The coincidence of chronic inflammation and osteoporosis (OP) or osteoarthritis (OA) is quite anticipated and raised a debate about IL-17's contribution. Several hallmark inflammatory mediators including TNF-α, IL-1, IL-6, IFN-γ, receptor activator of NF-κB (RANK), and RANK ligand (RANKL) are of crucial importance not only at the primary inflammation site, but also in bone metabolism [70, 71]. Although a protective role of IL-17 against bone loss has been described [72], induction of osteoclastogenesis by TH17 cells has been suggested in various inflammatory models [73]. Proinflammatory cytokines correlated with osteoclastogenic or antiosteoclastogenic manifestations in human OP and OA, for example, negative correlations of hip bone mineral density (BMD) with TNF-α in OA and with RANKL/RANK in OP [70]. In a mouse model of type II diabetes, whereas osteocalcin and osteoprotegerin (osteoblast-specific bone forming markers) were decreased, osteoclast-driven bone resorption markers such as IL-6 and RANK were elevated and coincided with enhanced RANKL and IL-17 expression by CD4+ cells; IL-17 induction was directly promoted upon leptin treatment [74]. The authors proposed that leptin and IL-6 stimulate IL-17 production and, thereby, induce RANKL-mediated osteoclastogenesis. A direct link of IL-17 to osteoclast induction was proved in cultures of PBMCs drained from patients with Crohn's disease [71]. Altogether, IL-17 may be a valuable target for controlling bone diseases, at least those accompanying chronic inflammations as in Crohn's disease or inflammatory arthritis.
Overall, in addition to expecting counterprotective impacts of TH17-associated cytokines, the induction of TH17 response seems to be an intrinsic feature originally evolved to fight bacterial, viral, and fungal infections. However, what drives such an immune arm to react against the body's own elements, that is, the loss of tolerance, remains elusive. Upon infection, it seems to be a failure to eliminate the invader, whereby an inflammation-potent cell response is amplified, whose army calls for other inflammation competent cells that might have lost the ability to recognize the body's own MHC molecules and therefore attack the self and/or induce production of AAs. Such a modified response attained through a persistent infection constitutes an additional load against the system's strategy of pathogen clearance and the culmination of the immune response to its steady state thereafter. In other words, boosting such a potent inflammatory cell type as TH17 through an initial inflammation, for example, through inflammatory cytokine-mediated induction of NF-κB, see next, should normally be accompanied by induction of a regulation program; otherwise autoimmunity occurs. On the basis of current understanding, we propose that the TH17-driven autoimmune response is manifold, attaining its incidence through (i) activating TH1 responses and the later conversion of TH17 themselves into TH1-like cells or double TH1/TH17 cytokine producers having the inflammatory potency of both subsets; (ii) activating B cells and their production of AAs, especially through IL-21-dominated responses; (iii) inducing inflammatory cells like macrophages and neutrophils and their recruitment through induction of chemokines, facilitating thereby tissue destruction and release of intrinsic cellular factors, which, in turn, leads to local or systemic induction of the autoimmune traits; and (iv) promoting cytotoxicity of NK and CD8+ cells and the conversion of the latter cells into IL-17 producers that further magnify the whole response. On the basis of current knowledge, introducing the TH17 efficacy as a potent inflammatory lineage, targeting IL-23/TH17 axis, may be a promising approach that paves the way for additive and alternative treatment of chronic inflammation and AD [75].
3. TH17 Cells Are Key Players in Heavy-Metal-Elicited Autoimmunity
Whereas some heavy metals (e.g., copper, selenium, iron, and zinc) are essential to maintain our metabolism, the majority of heavy metals are non-essential, for example, arsenic (As3+, As4+), cadmium (Cd2+), chromium (Cr3+, Cr4+), mercury (Hg2+), and lead (Pb2+), and are ranked among the most highly toxic substances. Great evidence exists that various heavy metals elicit immunomodulation increasing thereby the incidence of human AD and cancer [3, 18, 76]. It has been recently found that patients with autoimmune thyroiditis (AT) and other AD, including MS, psoriasis, SLE, and atopic eczema, showed increased lymphocyte reactivity to inorganic Hg2+, Ni3+, and other metals and that replacement of amalgam in Hg2+-allergic subjects resulted in improvement of health in about 70% of AT patients [3]. Furthermore, recent data implied that exposure of mice to low micromolar concentrations of Cd2+ and Hg2+ induces a robust TH17 response, that was also inferred by mild but significant increase of IL-17 profile in serum of individuals occupationally exposed to the same metals, as well as a robust ex vivo TH17 response (N. Y. A. Hemdan et al., unpublished data).
3.1. Ligation of Metal Ion with the Aryl Hydrocarbon Receptor
Like other xenobiotic stimuli [77], one mechanism so far delineated is the ligation of metal ion with the aryl hydrocarbon receptor (AhR) as in the case of Cd2+, As3+, Cr6+ [78, 79], and Pb2+ [80]. Such ligand-specific activation of the AhR was found to regulate the balance between TH17 and TReg cell responses [81]. Whereas AhR activation by TCDD (dioxin) induces functional TReg cells that suppress EAE, activation by 6-formylindolo[3,2-b]carbazole induces TH17 cells and ultimately the disease severity. AhR is expressed by TH17 cells, γδ T cells, and DCs [82, 83] and is indispensible for IL-22 production as evidenced by AhR−/− mouse studies [82] and by downregulation of the AhR on RNA-mediated interference, as well as, by applying AhR agonists [48], where it substantially altered the balance of IL-22- versus IL-17-producing cells. In DCs, activation of AhR induces expression of IDO1 and IDO2 that mediate induction of TReg cells [83]. Therefore, we hypothesized that exposure to heavy metals may mediate autoimmune initiation/induction through metal ligation of AhR. Recent works of other groups and our unpublished data indicate the association of IL-22 with the appearance of autoimmune signs, as inferred in the pathogenesis of psoriasis [7]. These data raise the AhR as a sequential segment linking such potent inflammatory TH17 cells with the heavy-metal-induced autoimmune induction and reveal a mechanism for further differentiation of TH17 into TH22 under organ- or pathogen-specific conditions, including AhR ligation pathways. Therefore, targeting AhR may offer a possibility for differential regulation of TH17 cytokines and thereby reduction of autoimmune susceptibility in heavy-metal occupationally exposed individuals; however, the paradoxical effect of various AhR ligands should be considered.
AhR-mediated immunomodulation by heavy metals may involve several mechanisms; one of which is the regulation of CYP1A1 expression, for example, in the case of Pb2+ [80] that coincided with increase of heme-oxygenase- (HO-) 1 mRNA level and production of reactive oxygen species (ROS). Induction of CYP1A1 expression by AhR ligation is well documented; its downstream signal mediates cellular responses probably through modifying cytokine secretion, including IL-6 [84], IL-17, and IL-22 [37, 82, 85]. Induction of oxidative stress has been reported in case of Pb2+ [86], Cd2+ [87–89], and Hg2+ exposure [90], inferred by reduced activity of antioxidant enzymes catalase (CAT), superoxide dismutase (SOD), and glutathione peroxidase (GPx) and reduction of glutathione (GSH). Smoking, a major source of Cd [91], also initiates ROS production accompanied by augmented cell signaling pathways implicated in the pathogenesis of AD such as psoriasis that implicates mitogen-activated protein kinase (MAPK), NF-κB and Janus kinase JAK/STAT [92], and reduced antioxidants malondialdehyde and SOD [93]. Oxidative stress comprising production of free radicals such as reactive oxygen and nitrogen species is closely related to inflammation and discussed as an underlying mechanism of inflammatory diseases, accompanying activation of the leucocytes and generation of peroxynitrite, at the early stages of induction, or appearing before the incidence of various AD such as IBD [94, 95], systemic sclerosis [96], SLE [97], psoriasis [93, 98], and in cardiovascular inflammation [99]. Therefore, depletion of endogenous antioxidants such as GSH, Cu, and ZnSOD was manifested in experimental models of IBD [100], accompanied, however, by induction of HO-1. The latter catalyzes CO production, and, although it is considered as a prooxidant due to iron released from HO activity [101], it may be a compensatory response to oxidative stress and chronic inflammation [102], whose protective function has been elucidated in various AD including MS [103].
The protective role of antioxidants in combating inflammation has been clarified in various models by applying exogenous antioxidants or by manipulating expression of endogenous antioxidants. Depletion of NF-E2-related factor 2 (Nrf2), for instance, markedly enhanced susceptibility of experimental IBD [104]. Moreover, by disrupting GSH metabolism through targeting GSH peroxidase (GPx) 1 and GPx2 derived development of colitis [105], or by depleting GSH though curcumin-elicited glyoxalase 1 activity inhibition that enhanced the anti-inflammatory response as well as anticarcinogenetic potency [106], it became clear how close the metabolic stress is related to cell response and cell survival, an assumption that is confirmed by many studies [107]. Unfortunately, although the role of TH17 cells-related products such as IL-17 in the previous experimental settings has not been addressed, TH17 together with TH1 cells are drawn as being major players in the IBD as well as in other inflammatory concerts [108]. Therefore, the elevation of ROS levels links heavy metal exposure to induction of inflammation and cancer. Indeed, induction of TH17 cell response by heavy metals is likely a downstream event of hydrogen-peroxide- (H2O2-) mediated IL-6 induction, which is found to protect resident lung cells from ROS-induced injury [109]. Ye et al. [110] found that fumarates induced type II DCs as a result of initial GSH depletion followed by induction of HO-1, which interacts with AP-1 and NF-κB sites of Il23p19 promoter and inactivates STAT1 and thereby improves TH1- and TH17-mediated AD including MS and psoriasis.
3.2. Modification of NF-κB Signaling
A key pathway through which heavy metals, amongst other xenobiotic substances, exert impacts on the immune response occurs via modifying NF-κB signaling. Several distinct NF-κB activation pathways are identified, including responses to various cell stresses and stimuli such as proinflammatory cytokines TNF-α and IL-1, bacterial products, genotoxic stimuli such as ionizing radiation and some chemotherapeutic drugs [88], in addition to exposure of various cell types to heavy metals such as Cd2+ [88, 111]. This may highlight NF-κB activation as a trait of carcinogenicity assigned to heavy metals including Cd2+ [112]. The increased production of cytokines, for example, IL-6 and IL-17, in murine models and in heavy-metal-exposed individuals (Hemdan & Abul El-Saad, unpublished data), might ultimately lead to excessive induction of NF-κB-mediated chronic inflammation [113]. This is consistent with the involvement of IL-17 in the differentiation of plasma cells mediated by NF-κB-regulated TF Twist-1 [114]; we recall the correlation of higher IL-17 levels with the severity of various AD and the appearance of autoimmune signs accompanying exposure to heavy metals. Therefore, delicate intervention to regulate NF-κB activation may help prevent chronic inflammation, simultaneous tissue cell damage, and reduce incidence of AD in individuals occupationally exposed to heavy metals or those with an accidental exposure history.
3.3. Disruption of Ca2+ Homeostasis
A third event by which heavy metals like Cd2+ modify cell survival and function is modifying Ca2+ displacement and ultimately adherence/tight junctions, mediated by disrupting expression and translocation of E-cadherin/β-catenin, in a way that mimics Wnt-signaling [88, 115], providing a clue for the carcinogenicity of heavy metals. Previous studies [116] as well as our unpublished data identified some protein kinases including ROCK-II as a target of Cd2+- and Hg2+-mediated induction of IL-17, which activates NF-κB through CIKS/Act1 adaptor proteins, inducing thereby chronic inflammation and cancer.
3.4. Modification of Zinc Metabolism
A further important element in the interplay of heavy metals such as Cd2+ with immunomodulation is represented by its interaction with zinc. Zinc was found to inhibit cancer through regulating various oncogenic pathways including NF-κB, AP-1, Notch-1, and PI3K/Akt, apoptosis, cytotoxicity, regulating tumor suppressors such as p53 and macrophage phagocytic activities and increased production of ROS and inflammatory cytokines TNF-α, IL-1β, IL-8, VCAM, and MCP-1 [112]. Similarly, Zn probably inhibits STAT3 activation and thereby TH17-mediated collagen-induced arthritis [117]. Via targeting such oncogenic pathways, Zn supplements may participate in attaining promising antitumor approaches, at least in cases where Cd2+ is considered to have a carcinogenic potential.
4. Concluding Remarks
Ongoing research provides a preponderance of evidence that TH17 cells and related molecules act as double agents both in favor of but also against the harboring individual. They elicit various antimicrobial mechanisms on one hand, but, on the other hand, when dysregulated, likely triggered by xenobiotic agents including pollutants and infectious agents, initiate/promote chronic inflammatory/autoimmune manifestations. A delineation of the underlying mechanisms that culminate into hyperactivation of TH17 cells and the resultant production of related mediators would facilitate the development of potential therapeutic approaches to combat their deteriorating effects but simultaneously allow their benefits to act. Therefore, various research directions gave more attention in the last decade to TH17-cells-related molecules to help attain and evaluate valuable therapeutic strategies. Manipulating TH17 differentiation and function by targeting differentiation/promoting cytokines, transcription factors, or commensal-bacteria-elicited immune induction may be valuable for treating AD; however, the risk of increasing the vulnerability of attacking infections should remain in focus. Therefore, it may be of worth to apply prophylactic antibacterial and antifungal therapy in case of treating patients of AD with IL-17/-22/-23 inhibitors.
Take-Home Messages
TH17 cells and their related products act as double agents both to mediate various antimicrobial mechanisms and to initiate/promote chronic inflammatory/autoimmune manifestations.
Simultaneous or successive exposure to xenobiotic substances and infectious agents renders the genetically susceptible individual vulnerable to autoimmune incidence via induction of inflammatory mediators.
A compromise should be met to facilitate development of potential therapeutics aiming at targeting AD via inhibiting differentiation of TH17 cells and/or commitment factors for the benefit of attaining the antimicrobial response intact, for example, by applying prophylactic therapy combined with IL-17/-22/-23 inhibitors.
Acknowledgments
The work presented in this paper was funded in part by the German Federal Ministry of Education and Research (BMBF 1315883) and by the Faculty of Science, University of Alexandria, Egypt. The authors thank Andreas Thuy (Jena University Hospital, Clinic for Anesthesiology and Intensive Medicine, SG Sepsis Research) for reading through the paper.
References
- 1.Schiraldi M, Monestier M. How can a chemical element elicit complex immunopathology? Lessons from mercury-induced autoimmunity. Trends in Immunology. 2009;30(10):502–509. doi: 10.1016/j.it.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 2.Pigatto PD, Guzzi G. Linking mercury amalgam to autoimmunity. Trends in Immunology. 2010;31(2):48–49. doi: 10.1016/j.it.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 3.Hybenova M, Hrda P, Prochazkova J, Stejskal V, Sterzl I. The role of environmental factors in autoimmune thyroiditis. Neuroendocrinology Letters. 2010;31(3):283–289. [PubMed] [Google Scholar]
- 4.Al-Mogairen SM. Induction of autoimmunity in brown Norway rats by oral and parenteral administration of nickel chloride. Lupus. 2010;19(3):262–267. doi: 10.1177/0961203309351728. [DOI] [PubMed] [Google Scholar]
- 5.Layton AN, Galyov EE. Salmonella-induced enteritis: molecular pathogenesis and therapeutic implications. Expert Reviews in Molecular Medicine. 2007;9(18):1–17. doi: 10.1017/S1462399407000373. [DOI] [PubMed] [Google Scholar]
- 6.Haraga A, Ohlson MB, Miller SI. Salmonellae interplay with host cells. Nature Reviews Microbiology. 2008;6(1):53–66. doi: 10.1038/nrmicro1788. [DOI] [PubMed] [Google Scholar]
- 7.Raychaudhuri SP. Role of IL-17 in psoriasis and psoriatic arthritis. Clinical Reviews in Allergy and Immunology. 2013;44(2):183–193. doi: 10.1007/s12016-012-8307-1. [DOI] [PubMed] [Google Scholar]
- 8.Kryczek I, Wu K, Zhao E, et al. IL-17+ regulatory T Cells in the microenvironments of chronic inflammation and cancer. Journal of Immunology. 2011;186(7):4388–4395. doi: 10.4049/jimmunol.1003251. [DOI] [PubMed] [Google Scholar]
- 9.Hemdan NY, Birkenmeier G, Wichmann G, et al. Interleukin-17-producing T helper cells in autoimmunity. Autoimmunity Reviews. 2010;9(11):785–792. doi: 10.1016/j.autrev.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Hemdan NY. Anti-cancer versus cancer-promoting effects of the interleukin-17-producing T helper cells. Immunology Letters. 2013;149:123–133. doi: 10.1016/j.imlet.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 11.Sawada N, Iwasaki M, Inoue M, et al. Long-term dietary cadmium intake and cancer incidence. Epidemiology. 2012;23(3):368–376. doi: 10.1097/EDE.0b013e31824d063c. [DOI] [PubMed] [Google Scholar]
- 12.Van Belle TL, Esplugues E, Liao J, Juntti T, Flavell RA, von Herrath MG. Development of autoimmune diabetes in the absence of detectable IL-17A in a CD8-driven virally induced model. Journal of Immunology. 2011;187(6):2915–2922. doi: 10.4049/jimmunol.1000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delgoffe GM, Pollizzi KN, Waickman AT, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nature Immunology. 2011;12(4):295–304. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma J, Wang R, Fang X, Ding Y, Sun Z. Critical role of TCF-1 in repression of the IL-17 gene. PLoS ONE. 2011;6(9) doi: 10.1371/journal.pone.0024768.e24768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirota K, Duarte JH, Veldhoen M, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nature Immunology. 2011;12(3):255–263. doi: 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hemdan NYA, Emmrich F, Adham K, et al. Dose-dependent modulation of the in vitro cytokine production of human immune competent cells by lead salts. Toxicological Sciences. 2005;86(1):75–83. doi: 10.1093/toxsci/kfi177. [DOI] [PubMed] [Google Scholar]
- 17.Hemdan NYA, Emmrich F, Sack U, et al. The in vitro immune modulation by cadmium depends on the way of cell activation. Toxicology. 2006;222(1-2):37–45. doi: 10.1016/j.tox.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 18.Hemdan NYA, Emmrich F, Faber S, Lehmann J, Sack U. Alterations of Th1/Th2 reactivity by heavy metals: possible consequences include induction of autoimmune diseases. Annals of the New York Academy of Sciences. 2007;1109:129–137. doi: 10.1196/annals.1398.015. [DOI] [PubMed] [Google Scholar]
- 19.Hemdan NY. The role of interleukin-12 in the heavy metal-elicited immunomodulation: relevance of various evaluation methods. Journal of Occupational Medicine and Toxicology. 2008;3(1, article 25) doi: 10.1186/1745-6673-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive Foxp3+ regulatory T cells: more of the same or a division of labor? Immunity. 2009;30(5):626–635. doi: 10.1016/j.immuni.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 21.Issazadeh-Navikas S, Teimer R, Bockermann R. Influence of dietary components on regulatory T cells. Molecular Medicine. 2012;18(1):95–110. doi: 10.2119/molmed.2011.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maggi L, Santarlasci V, Capone M, et al. CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. European Journal of Immunology. 2010;40(8):2174–2181. doi: 10.1002/eji.200940257. [DOI] [PubMed] [Google Scholar]
- 23.Hemdan NY, Birkenmeier G, Wichmann G. Key molecules in the differentiation and commitment programof T helper 17 (Th17) cells up-to-date. Immunology Letters. 2012;148:97–109. doi: 10.1016/j.imlet.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 24.Santarlasci V, Maggi L, Capone M, et al. Rarity of human T helper 17 cells is due to retinoic acid orphan receptor-dependent mechanisms that limit their expansion. Immunity. 2012;36(2):201–214. doi: 10.1016/j.immuni.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 25.Ankathatti Munegowda M, Deng Y, Mulligan SJ, Xiang J. Th17 and Th17-stimulated CD8+ T cells play a distinct role in Th17-induced preventive and therapeutic antitumor immunity. Cancer Immunology, Immunotherapy. 2011;60(10):1473–1484. doi: 10.1007/s00262-011-1054-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinto LG, Cunha TM, Vieira SM, et al. IL-17 mediates articular hypernociception in antigen-induced arthritis in mice. Pain. 2010;148(2):247–256. doi: 10.1016/j.pain.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 27.Passos ST, Silver JS, O’Hara AC, Sehy D, Stumhofer JS, Hunter CA. IL-6 promotes NK cell production of IL-17 during toxoplasmosis. Journal of Immunology. 2010;184(4):1776–1783. doi: 10.4049/jimmunol.0901843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sellge G, Magalhaes JG, Konradt C, et al. Th17 cells are the dominant T cell subtype primed by Shigella flexneri mediating protective immunity. Journal of Immunology. 2010;184(4):2076–2085. doi: 10.4049/jimmunol.0900978. [DOI] [PubMed] [Google Scholar]
- 29.Raffatellu M, Santos RL, Verhoeven DE, et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nature Medicine. 2008;14(4):421–428. doi: 10.1038/nm1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riol-Blanco L, Lazarevic V, Awasthi A, et al. IL-23 receptor regulates unconventional IL-17-producing T cells that control bacterial infections. Journal of Immunology. 2010;184(4):1710–1720. doi: 10.4049/jimmunol.0902796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dejima T, Shibata K, Yamada H, et al. Protective role of naturally occurring interleukin-17A-producing γδ cells in the Lung at the early stage of systemic candidiasis in mice. Infection and Immunity. 2011;79(11):4503–4510. doi: 10.1128/IAI.05799-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hueber AJ, Asquith DL, Miller AM, et al. Cutting edge: mast cells express IL-17A in rheumatoid arthritis synovium. Journal of Immunology. 2010;184(7):3336–3340. doi: 10.4049/jimmunol.0903566. [DOI] [PubMed] [Google Scholar]
- 33.Pötzl J, Botteron C, Tausch E, et al. Tracing functional antigen-specific CCR6+ Th17 cells after vaccination. PLoS ONE. 2008;3(8) doi: 10.1371/journal.pone.0002951.e2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang X, Gao L, Lei L, et al. A MyD88-dependent early IL-17 production protects mice against airway infection with the obligate intracellular pathogen Chlamydia muridarum. Journal of Immunology. 2009;183(2):1291–1300. doi: 10.4049/jimmunol.0803075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geddes K, Rubino SJ, Magalhaes JG, et al. Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nature Medicine. 2011;17(7):837–844. doi: 10.1038/nm.2391. [DOI] [PubMed] [Google Scholar]
- 36.Torchinsky MB, Garaude J, Martin AP, Blander JM. Innate immune recognition of infected apoptotic cells directs T H17 cell differentiation. Nature. 2009;458(7234):78–82. doi: 10.1038/nature07781. [DOI] [PubMed] [Google Scholar]
- 37.Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing γδ T cells selectively expand in response to pathogen products and environmental signals. Immunity. 2009;31(2):321–330. doi: 10.1016/j.immuni.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 38.Godinez I, Raffatellu M, Chu H, et al. Interleukin-23 orchestrates mucosal responses to Salmonella enterica serotype typhimurium in the intestine. Infection and Immunity. 2009;77(1):387–398. doi: 10.1128/IAI.00933-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clapp B, Skyberg JA, Yang X, Thornburg T, Walters N, Pascual WP. Protective live oral brucellosis vaccines stimulate Th1 and Th17 cell responses. Infection and Immunity. 2011;79(10):4165–4174. doi: 10.1128/IAI.05080-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ye P, Rodriguez FH, Kanaly S, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. Journal of Experimental Medicine. 2001;194(4):519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miyamoto M, Prause O, Sjöstrand M, Laan M, Lötvall J, Lindén A. Endogenous IL-17 as a mediator of neutrophil recruitment caused by endotoxin exposure in mouse airways. Journal of Immunology. 2003;170(9):4665–4672. doi: 10.4049/jimmunol.170.9.4665. [DOI] [PubMed] [Google Scholar]
- 42.Hoshino H, Laan M, Sjöstrand M, Lötvall J, Skoogh BE, Linden A. Increased elastase andmyeloperoxidase activity associated with neutrophil recruitment by IL-17 in airways in vivo. Journal of Allergy and Clinical Immunology. 2000;105(1):143–149. doi: 10.1016/s0091-6749(00)90189-1. [DOI] [PubMed] [Google Scholar]
- 43.Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332(6025):65–68. doi: 10.1126/science.1200439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu L, Okada S, Kong X-F, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. Journal of Experimental Medicine. 2011;208(18):1635–1648. doi: 10.1084/jem.20110958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kisand K, Bøe Wolff AS, Podkrajsek KT, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. Journal of Experimental Medicine. 2010;207(2):299–308. doi: 10.1084/jem.20091669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puel A, Döffinger R, Natividad A, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. Journal of Experimental Medicine. 2010;207(2):291–297. doi: 10.1084/jem.20091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muñoz M, Heimesaat MM, Danker K, et al. Interleukin (IL)-23 mediates Toxoplasma gondii-induced immunopathology in the gut via matrixmetalloproteinase-2 and IL-22 but independent of IL-17. Journal of Experimental Medicine. 2009;206(13):3047–3059. doi: 10.1084/jem.20090900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from TH-17, TH1 and TH2 cells. Nature Immunology. 2009;10(8):864–871. doi: 10.1038/ni.1770. [DOI] [PubMed] [Google Scholar]
- 49.Eyerich S, Eyerich K, Pennino D, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. Journal of Clinical Investigation. 2009;119(12):3573–3585. doi: 10.1172/JCI40202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eyerich S, Wagener J, Wenzel V, et al. IL-22 and TNF-α represent a key cytokine combination for epidermal integrity during infection with Candida albicans. European Journal of Immunology. 2011;41(7):1894–1901. doi: 10.1002/eji.201041197. [DOI] [PubMed] [Google Scholar]
- 51.Liang SC, Nickerson-Nutter C, Pittman DD, et al. IL-22 induces an acute-phase response. Journal of Immunology. 2010;185(9):5531–5538. doi: 10.4049/jimmunol.0904091. [DOI] [PubMed] [Google Scholar]
- 52.Kudva A, Scheller EV, Robinson KM, et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. Journal of Immunology. 2011;186(3):1666–1674. doi: 10.4049/jimmunol.1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y, Cobleigh MA, Lian J-Q, et al. A proinflammatory role for interleukin-22 in the immune response to hepatitis B virus. Gastroenterology. 2011;141(5):1897–1906. doi: 10.1053/j.gastro.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29(6):947–957. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miyamoto M, Emoto M, Emoto Y, et al. Neutrophilia in LFA-1-deficient mice confers resistance to listeriosis: possible contribution of granulocyte-colony-stimulating factor and IL-17. Journal of Immunology. 2003;170(10):5228–5234. doi: 10.4049/jimmunol.170.10.5228. [DOI] [PubMed] [Google Scholar]
- 56.Orgun NN, Mathis MA, Wilson CB, Sing SW. Deviation from a strong Th1-dominated to a modest Th17-dominated CD4 T cell response in the absence of IL-12p40 and type I IFNs sustains protective CD8 T cells. Journal of Immunology. 2008;180(6):4109–4115. doi: 10.4049/jimmunol.180.6.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bai H, Cheng J, Gao X, et al. IL-17/Th17 promotes type 1 T cell immunity against pulmonary intracellular bacterial infection through modulating dendritic cell function. Journal of Immunology. 2009;183(9):5886–5895. doi: 10.4049/jimmunol.0901584. [DOI] [PubMed] [Google Scholar]
- 58.Zhou X, Chen Q, Moore J, Kolls JK, Halperin S, Wang J. Critical role of the interleukin-17/interleukin-17 receptor axis in regulating host susceptibility to respiratory infection with Chlamydia species. Infection and Immunity. 2009;77(11):5059–5070. doi: 10.1128/IAI.00403-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Minegishi Y, Saito M, Nagasawa M, et al. Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. Journal of Experimental Medicine. 2009;206(6):1291–1301. doi: 10.1084/jem.20082767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sieve AN, Meeks KD, Bodhankar S, et al. A novel IL-17-dependent mechanism of cross protection: respiratory infection with mycoplasma protects against a secondary listeria infection. European Journal of Immunology. 2009;39(2):426–438. doi: 10.1002/eji.200838726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khader SA, Bell GK, Pearl JE, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nature Immunology. 2007;8(4):369–377. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 62.Holley MM, Kielian T. Th1 and Th17 cells regulate innate immune responses and bacterial clearance during central nervous system infection. Journal of Immunology. 2012;188(3):1360–1370. doi: 10.4049/jimmunol.1101660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Salgado M, Rallón NI, Rodés B, López M, Soriano V, Benito JM. Long-term non-progressors display a greater number of Th17 cells than HIV-infected typical progressors. Clinical Immunology. 2011;139(2):110–114. doi: 10.1016/j.clim.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 64.Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139(3):485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu H-J, Ivanov II, Darce J, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32(6):815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abraham C, Medzhitov R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology. 2011;140(6):1729–1737. doi: 10.1053/j.gastro.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kwok S-K, Cho M-L, Her Y-M, et al. TLR2 ligation induces the production of IL-23/IL-17 via IL-6, STAT3 and NF-kB pathway in patients with primary Sjogren’s syndrome. Arthritis Research and Therapy. 2012;14(2, article R64) doi: 10.1186/ar3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ciccia F, Guggino G, Rizzo A, et al. Potential involvement of IL-22 and IL-22-producing cells in the inflamed salivary glands of patients with Sjögren’s syndrome. Annals of the Rheumatic Diseases. 2012;71(2):295–301. doi: 10.1136/ard.2011.154013. [DOI] [PubMed] [Google Scholar]
- 69.Abdulahad WH, Kroese FGM, Vissink A, Bootsma H. Immune regulation and B-cell depletion therapy in patients with primary Sjögren’s syndrome. Journal of Autoimmunity. 2012;39:103–111. doi: 10.1016/j.jaut.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 70.Zupan J, Komadina R, Marc J. The relationship between osteoclastogenic and anti-osteoclastogenic pro-inflammatory cytokines differs in human osteoporotic and osteoarthritic bone tissues. Journal of Biomedical Science. 2012;19(1, article 28) doi: 10.1186/1423-0127-19-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oostlander AE, Everts V, Schoenmaker T, et al. T cell-mediated increased osteoclast formation from peripheral blood as a mechanism for crohn’s disease-associated bone loss. Journal of Cellular Biochemistry. 2012;113(1):260–268. doi: 10.1002/jcb.23352. [DOI] [PubMed] [Google Scholar]
- 72.Goswami J, Hernández-Santos N, Zuniga LA, Gaffen SL. A bone-protective role for IL-17 receptor signaling in ovariectomy-induced bone loss. European Journal of Immunology. 2009;39(10):2831–2839. doi: 10.1002/eji.200939670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yuan F-L, Li X, Lu W-G, et al. Type 17 T-helper cells might be a promising therapeutic target for osteoporosis. Molecular Biology Reports. 2012;39(1):771–774. doi: 10.1007/s11033-011-0797-z. [DOI] [PubMed] [Google Scholar]
- 74.Won HY, Lee J-A, Park ZS, et al. Prominent bone loss mediated by RANKL and IL-17 produced by CD4+ T cells in tallyho/JngJ mice. PLoS ONE. 2011;6(3) doi: 10.1371/journal.pone.0018168.e18168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Toussirot É. The IL23/Th17 pathway as a therapeutic target in chronic inflammatory diseases. Inflammation and Allergy. 2012;11(2):159–168. doi: 10.2174/187152812800392805. [DOI] [PubMed] [Google Scholar]
- 76.Putila JJ, Guo NL. Association of arsenic exposure with lung cancer incidence rates in the United States. PLoS ONE. 2011;6(10) doi: 10.1371/journal.pone.0025886.e25886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bock KW, Köhle C. Ah receptor: dioxin-mediated toxic responses as hints to deregulated physiologic functions. Biochemical Pharmacology. 2006;72(4):393–404. doi: 10.1016/j.bcp.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 78.Elbekai RH, El-Kadi AOS. Modulation of aryl hydrocarbon receptor-regulated gene expression by arsenite, cadmium, and chromium. Toxicology. 2004;202(3):249–269. doi: 10.1016/j.tox.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 79.Kluxen FM, Höfer N, Kretzschmar G, Degen GH, Diel P. Cadmium modulates expression of aryl hydrocarbon receptor-associated genes in rat uterus by interaction with the estrogen receptor. Archives of Toxicology. 2012;86:591–601. doi: 10.1007/s00204-011-0787-x. [DOI] [PubMed] [Google Scholar]
- 80.Korashy HM, El-Kadi AOS. Transcriptional and posttranslational mechanisms modulating the expression of the cytochrome P450 1A1 gene by lead in HepG2 cells: a role of heme oxygenase. Toxicology. 2012;291(1–3):113–121. doi: 10.1016/j.tox.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 81.Quintana FJ, Basso AS, Iglesias AH, et al. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453(7191):65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 82.Veldhoen M, Hirota K, Westendorf AM, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453(7191):106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 83.Vogel CFA, Goth SR, Dong B, Pessah IN, Matsumura F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochemical and Biophysical Research Communications. 2008;375(3):331–335. doi: 10.1016/j.bbrc.2008.07.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.DiNatale BC, Murray IA, Schroeder JC, et al. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicological Sciences. 2010;115(1):89–97. doi: 10.1093/toxsci/kfq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ramirez J-M, Brembilla NC, Sorg O, et al. Activation of the aryl hydrocarbon receptor reveals distinct requirements for IL-22 and IL-17 production by human T helper cells. European Journal of Immunology. 2010;40(9):2450–2459. doi: 10.1002/eji.201040461. [DOI] [PubMed] [Google Scholar]
- 86.Baranowska-Bosiacka I, Gutowska I, Marchlewicz M, et al. Disrupted pro- and antioxidative balance as a mechanism of neurotoxicity induced by perinatal exposure to lead. Brain Research. 2012;1435:56–71. doi: 10.1016/j.brainres.2011.11.062. [DOI] [PubMed] [Google Scholar]
- 87.Oguzturk H, Ciftci O, Aydin M, Timurkaan N, Beytur A, Yilmaz F. Ameliorative effects of curcumin against acute cadmium toxicity on male reproductive system in rats. Andrologia. 2012;44:243–249. doi: 10.1111/j.1439-0272.2012.01273.x. [DOI] [PubMed] [Google Scholar]
- 88.Thévenod F. Cadmium and cellular signaling cascades: to be or not to be? Toxicology and Applied Pharmacology. 2009;238(3):221–239. doi: 10.1016/j.taap.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 89.Galazyn-Sidorczuk M, Brzóska MM, Rogalska J, Roszczenko A, Jurczuk M. Effect of zinc supplementation on glutathione peroxidase activity and selenium concentration in the serum, liver and kidney of rats chronically exposed to cadmium. Journal of Trace Elements in Medicine and Biology. 2012;26(1):46–52. doi: 10.1016/j.jtemb.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 90.Pal M, Ghosh M. Studies on comparative efficacy of α-linolenic acid and α-eleostearic acid on prevention of organic mercury-induced oxidative stress in kidney and liver of rat. Food and Chemical Toxicology. 2012;50(3-4):1066–1072. doi: 10.1016/j.fct.2011.12.042. [DOI] [PubMed] [Google Scholar]
- 91.Tellez-Plaza M, Navas-Acien A, Caldwell KL, Menke A, Muntner P, Guallar E. Reduction in cadmium exposure in the United States population, 1988–2008: the contribution of declining smoking rates. Environmental Health Perspectives. 2012;120(2):204–209. doi: 10.1289/ehp.1104020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Armstrong AW, Armstrong EJ, Fuller EN, Sockolov ME, Voyles SV. Smoking and pathogenesis of psoriasis: a review of oxidative, inflammatory and genetic mechanisms. British Journal of Dermatology. 2011;165(6):1162–1168. doi: 10.1111/j.1365-2133.2011.10526.x. [DOI] [PubMed] [Google Scholar]
- 93.Attwa E, Swelam E. Relationship between smoking-induced oxidative stress and the clinical severity of psoriasis. Journal of the European Academy of Dermatology and Venereology. 2011;25(7):782–787. doi: 10.1111/j.1468-3083.2010.03860.x. [DOI] [PubMed] [Google Scholar]
- 94.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474(7351):307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhu H, Li YR. Oxidative stress and redox signaling mechanisms of inflammatory bowel disease: updated experimental and clinical evidence. Experimental Biology and Medicine. 2012;237(5):474–480. doi: 10.1258/ebm.2011.011358. [DOI] [PubMed] [Google Scholar]
- 96.Yamamoto T. Autoimmune mechanisms of scleroderma and a role of oxidative stress. Self/Nonself. 2011;2(1):4–10. doi: 10.4161/self.2.1.14058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mathis KW, Venegas-Pont M, Masterson CW, Stewart NJ, Wasson KL, Ryan MJ. Oxidative stress promotes hypertension and albuminuria during the autoimmune disease systemic lupus erythematosus. Hypertension. 2012;59(3):673–679. doi: 10.1161/HYPERTENSIONAHA.111.190009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gabr SA, Al-Ghadir AH. Role of cellular oxidative stress and cytochrome c in the pathogenesis of psoriasis. Archives of Dermatological Research. 2012;304(6):451–457. doi: 10.1007/s00403-012-1230-8. [DOI] [PubMed] [Google Scholar]
- 99.Profumo E, Buttari B, Rigano R. Oxidative stress in cardiovascular inflammation: its involvement in autoimmune responses. International Journal of Inflammation. 2011;2011:6 pages. doi: 10.4061/2011/295705.295705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Íşman ÇA, Yeğen BÇ, Alican Í. Methimazole-induced hypothyroidism in rats ameliorates oxidative injury in exprerimental colitis. Journal of Endocrinology. 2003;177(3):471–476. doi: 10.1677/joe.0.1770471. [DOI] [PubMed] [Google Scholar]
- 101.Castellani RJ, Moreira PI, Perry G, Zhu X. The role of iron as a mediator of oxidative stress in Alzheimer disease. BioFactors. 2012;38(2):133–138. doi: 10.1002/biof.1010. [DOI] [PubMed] [Google Scholar]
- 102.Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacological Reviews. 2008;60(1):79–127. doi: 10.1124/pr.107.07104. [DOI] [PubMed] [Google Scholar]
- 103.Fagone P, Mangano K, Coco M, et al. Therapeutic potential of carbon monoxide in multiple sclerosis. Clinical and Experimental Immunology. 2012;167(2):179–187. doi: 10.1111/j.1365-2249.2011.04491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Khor TO, Huang M-T, Kwon KH, Chan JY, Reddy BS, Kong A-N. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Research. 2006;66(24):11580–11584. doi: 10.1158/0008-5472.CAN-06-3562. [DOI] [PubMed] [Google Scholar]
- 105.Esworthy RS, Aranda R, Martín MG, Binder JH, Doroshow SW, Chu F-F. Mice with combined disruption of gpx1 and gpx2 genes have colitis. The American Journal of Physiology. 2001;281(3):G848–G855. doi: 10.1152/ajpgi.2001.281.3.G848. [DOI] [PubMed] [Google Scholar]
- 106.Santel T, Pflug G, Hemdan NYA, et al. Curcumin inhibits glyoxalase 1: a possible link to its anti-inflammatory and anti-tumor activity. PLoS ONE. 2008;3(10) doi: 10.1371/journal.pone.0003508.e3508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen C-C, Chen H-L, Hsieh C-W, Yang Y-L, Wung B-S. Upregulation of NF-E2-related factor-2-dependent glutathione by carnosol provokes a cytoprotective response and enhances cell survival. Acta Pharmacologica Sinica. 2011;32(1):62–69. doi: 10.1038/aps.2010.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Olsen T, Rismo R, Cui G, Goll R, Christiansen I, Florholmen J. TH1 and TH17 interactions in untreated inflamed mucosa of inflammatory bowel disease, and their potential to mediate the inflammation. Cytokine. 2011;56(3):633–640. doi: 10.1016/j.cyto.2011.08.036. [DOI] [PubMed] [Google Scholar]
- 109.Kida H, Yoshida M, Hoshino S, et al. Protective effect of IL-6 on alveolar epithelial cell death induced by hydrogen peroxide. The American Journal of Physiology. 2005;288(2):L342–L349. doi: 10.1152/ajplung.00016.2004. [DOI] [PubMed] [Google Scholar]
- 110.Ghoreschi K, Brück J, Kellerer C, et al. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. Journal of Experimental Medicine. 2011;208(11):2291–2303. doi: 10.1084/jem.20100977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Napolitano JR, Liu M-J, Bao S, et al. Cadmium-mediated toxicity of lung epithelia is enhanced through NF-κB-mediated transcriptional activation of the human zinc transporter ZIP8. The American Journal of Physiology. 2012;302(9):L909–L918. doi: 10.1152/ajplung.00351.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bao B, Thakur A, Li Y, et al. The immunological contribution of NF-κB within the tumor microenvironment: a potential protective role of zinc as an anti-tumor agent. Biochimica et Biophysica Acta. 2012;1825(2):160–172. doi: 10.1016/j.bbcan.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li L, Huang L, Vergis AL, et al. IL-17 produced by neutrophils regulates IFN-γ-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. Journal of Clinical Investigation. 2010;120(1):331–342. doi: 10.1172/JCI38702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Doreau A, Belot A, Bastid J, et al. Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nature Immunology. 2009;10(7):778–785. doi: 10.1038/ni.1741. [DOI] [PubMed] [Google Scholar]
- 115.Lindner I, Hemdan NYA, Buchold M. α2-macroglobulin inhibits the malignant properties of astrocytoma cells by impeding β-catenin signaling. Cancer Research. 2010;70(4):277–287. doi: 10.1158/0008-5472.CAN-09-1462. [DOI] [PubMed] [Google Scholar]
- 116.Sønder SU, Saret S, Tang W, Sturdevant DE, Porcella SF, Siebenlist U. IL-17-induced NF-κB activation via CIKS/Act1 physiologic significance and signaling mechanisms. Journal of Biological Chemistry. 2011;286(15):12881–12890. doi: 10.1074/jbc.M110.199547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kitabayashi C, Fukada T, Kanamoto M, et al. Zinc suppresses Th17 development via inhibition of STAT3 activation. International Immunology. 2010;22(5):375–386. doi: 10.1093/intimm/dxq017. [DOI] [PubMed] [Google Scholar]
- 118.Simonian PL, Roark CL, Wehrmann F, et al. IL-17A-expressing T cells are essential for bacterial clearance in a murine model of hypersensitivity pneumonitis. Journal of Immunology. 2009;182(10):6540–6549. doi: 10.4049/jimmunol.0900013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Woolard MD, Hensley LL, Kawula TH, Frelinger JA. Respiratory Francisella tularensis live vaccine strain infection induces Th17 cells and prostaglandin E2, which inhibits generation of gamma interferon-positive T cells. Infection and Immunity. 2008;76(6):2651–2659. doi: 10.1128/IAI.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Joshi AD, Fong DJ, Oak SR, et al. Interleukin-17-mediated immunopathogenesis in experimental hypersensitivity pneumonitis. The American Journal of Respiratory and Critical Care Medicine. 2009;179(8):705–716. doi: 10.1164/rccm.200811-1700OC. [DOI] [PubMed] [Google Scholar]
- 121.Simonian PL, Roark CL, Wehrmann F, et al. Th17-polarized immune response in a murine model of hypersensitivity pneumonitis and lung fibrosis. Journal of Immunology. 2009;182(1):657–665. [PMC free article] [PubMed] [Google Scholar]
- 122.Wang F, Xu J, Liao Y, et al. Tim-3 ligand galectin-9 reduces IL-17 level and accelerates Klebsiella pneumoniae infection. Cellular Immunology. 2011;269(1):22–28. doi: 10.1016/j.cellimm.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 123.Chen X, Zhang M, Liao M, et al. Reduced Th17 response in patients with tuberculosis correlates with IL-6R expression on CD4+ T cells. The American Journal of Respiratory and Critical Care Medicine. 2010;181(7):734–742. doi: 10.1164/rccm.200909-1463OC. [DOI] [PubMed] [Google Scholar]
- 124.Liu Z, Yang L, Cui Y, et al. IL-21 enhances NK cell activation and cytolytic activity and induces Th17 cell differentiation in inflammatory bowel disease. Inflammatory Bowel Diseases. 2009;15(8):1133–1144. doi: 10.1002/ibd.20923. [DOI] [PubMed] [Google Scholar]
- 125.Sugihara T, Kobori A, Imaeda H, et al. The increased mucosal mRNA expressions of complement C3 and interleukin-17 in inflammatory bowel disease. Clinical and Experimental Immunology. 2010;160(3):386–393. doi: 10.1111/j.1365-2249.2010.04093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kelly MN, Kolls JK, Happel K, et al. Interteukin-17/interleukin-17 receptor-mediated signaling is important for generation of an optimal polymorphonuclear response against Toxoplasma gondii infection. Infection and Immunity. 2005;73(1):617–621. doi: 10.1128/IAI.73.1.617-621.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Werner JL, Metz AE, Horn D, et al. Requisite role for the dectin-1 β-glucan receptor in pulmonary defense against aspergillus fumigatus. Journal of Immunology. 2009;182(8):4938–4946. doi: 10.4049/jimmunol.0804250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jin Y-H, Kang HS, Mohindru M, Kim BS. Preferential induction of protective T cell responses to Theiler’s virus in resistant (C57BL/6 X SJL)F1 mice. Journal of Virology. 2011;85(6):3033–3040. doi: 10.1128/JVI.02400-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mukherjee S, Lindell DM, Berlin AA, et al. IL-17Induced pulmonary pathogenesis during respiratory viral infection and exacerbation of allergic disease. The American Journal of Pathology. 2011;179(1):248–258. doi: 10.1016/j.ajpath.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Huang Z, van Velkinburgh JC, Ni B, Wu Y. Pivotal roles of the interleukin-23/T helper 17 cell axis in hepatitis B. Liver International. 2012;32:894–901. doi: 10.1111/j.1478-3231.2012.02764.x. [DOI] [PubMed] [Google Scholar]
- 131.Jimenez-Sousa MA, Almansa R, de la Fuente C, et al. Increased Th1, Th17 and pro-fibrotic responses in hepatitis C-infected patients are down-regulated after 12 weeks of treatment with pegylated interferon plus ribavirin. European Cytokine Network. 2010;21(2):84–91. doi: 10.1684/ecn.2010.0191. [DOI] [PubMed] [Google Scholar]
- 132.Campillo-Gimenez L, Cumont M-C, Fay M, et al. AIDS progression is associated with the emergence of IL-17-producing cells early after simian immunodeficiency virus infection. Journal of Immunology. 2010;184(2):984–992. doi: 10.4049/jimmunol.0902316. [DOI] [PubMed] [Google Scholar]
- 133.Elhed A, Unutmaz D. Th17 cells and HIV infection. Current Opinion in HIV and AIDS. 2010;5(2):146–150. doi: 10.1097/COH.0b013e32833647a8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chege D, Sheth PM, Kain T, et al. Sigmoid Th17 populations, the HIV latent reservoir, and microbial translocation in men on long-term antiretroviral therapy. AIDS. 2011;25(6):741–749. doi: 10.1097/QAD.0b013e328344cefb. [DOI] [PubMed] [Google Scholar]
- 135.Suryawanshi A, Veiga-Parga T, Rajasagi NK, et al. Role of il-17 and th17 cells in herpes simplex virus-induced corneal immunopathology. Journal of Immunology. 2011;187(4):1919–1930. doi: 10.4049/jimmunol.1100736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fu Y, Wang W, Tong J, et al. Th17: a new participant in gut dysfunction in mice infected with trichinella spiralis. Mediators of Inflammation. 2009;2009:7 pages. doi: 10.1155/2009/517052.517052 [DOI] [PMC free article] [PubMed] [Google Scholar]