

Abstract
The study of genetic and phenotypic variation is fundamental for understanding the dynamics of bacterial genome evolution and untangling the evolution and epidemiology of bacterial pathogens. Neisseria meningitidis (Nm) is among the most intriguing bacterial pathogens in genomic studies due to its dynamic population structure and complex forms of pathogenicity. Extensive genomic variation within identical clonal complexes (CCs) in Nm has been recently reported and suggested to be the result of homologous recombination, but the extent to which recombination contributes to genomic variation within identical CCs has remained unclear. In this study, we sequenced two Nm strains of identical serogroup (C) and multi-locus sequence type (ST60), and conducted a systematic analysis with an additional 34 Nm genomes. Our results revealed that all gene content variation between the two ST60 genomes was introduced by homologous recombination at the conserved flanking genes, and 94.25% or more of sequence divergence was caused by homologous recombination. Recombination was found in genes associated with virulence factors, antigenic outer membrane proteins, and vaccine targets, suggesting an important role of homologous recombination in rapidly altering the pathogenicity and antigenicity of Nm. Recombination was also evident in genes of the restriction and modification systems, which may undermine barriers to DNA exchange. In conclusion, homologous recombination can drive both gene content variation and sequence divergence in Nm. These findings shed new light on the understanding of the rapid pathoadaptive evolution of Nm and other recombinogenic bacterial pathogens.
Keywords: horizontal gene transfer, homologous recombination, clonal complex, MLST, nonvertical gene, restriction modification systems
Introduction
Neisseria meningitidis (Nm), a leading cause of bacterial meningitis (Stephens et al. 2007), has long been recognized to have a dynamic population structure due to its substantial genomic plasticity (Caugant et al. 1986; Smith et al. 1993; Holmes et al. 1999). A multi-locus sequence typing (MLST) scheme, based on the sequence similarity of DNA fragments from seven housekeeping genes, is widely used for classification of Nm. Isolates can be identified as sequence types (STs) and then grouped into clonal complexes (CCs) by their similarity to a central allelic profile (Maiden et al. 1998). Fine-scale relationships can be further obtained by analyzing more loci or a large amount of shared genomic sequences (Touchon et al. 2009; Bennett et al. 2012; Jolley and Maiden 2013).
The genomic diversity of Nm has been shown to be extensive at the levels of both gene content variation and sequence diversity (Schoen et al. 2008, 2009; Budroni et al. 2011). This is attributed to the natural competence of Nm cells for transformation and homologous recombination (Chen and Dubnau 2004; Findlay and Redfield 2009). In fact, a high frequency of homologous recombination was consistently observed in a variety of Nm studies (Feil et al. 1999; Holmes et al. 1999; Vos and Didelot 2009; Hao et al. 2011; Joseph et al. 2011). Homologous recombination can bring mutations arising in different genomes together and have a strong impact on pathogenic adaptation (Marri et al. 2010; Moradigaravand and Engelstadter 2012). For instance, recombination was found in the penicillin-binding-protein-2 gene penA (Bowler et al. 1994), in capsule gene cluster and vaccine-target genes (Hao et al. 2011), and other virulence-associated genes (Hao et al. 2011; Joseph et al. 2011). Extensive genomic variation has been evident between strains of identical STs in Nm (Hao et al. 2011) and other bacterial pathogens (Hao et al. 2012; Paul et al. 2013). The unexpectedly high level of dissimilarity but short divergence time between strains of identical STs provide an excellent system to address how homologous recombination drives genomic variation during bacterial genome evolution.
The restriction and modification system (RMS) in bacteria generally consists of a sequence-specific restriction enzyme (restriction endonuclease) and a modification enzyme (methyltransferase) encoded by adjacent genes on the chromosome (Wilson and Murray 1991). It is well appreciated that RMSs play an important role in protecting bacteria from the invasion of foreign DNA, such as bacteriophage (Wilson and Murray 1991; Bickle and Kruger 1993). RMSs have been suggested to serve as a barrier to genetic exchange between Nm strains (Jeltsch 2003; Budroni et al. 2011), but the role of RMSs in limiting recombination between subpopulations within a species remains subject to debate (Fraser et al. 2007) and many homologous recombination events between different CCs of Nm have previously been documented (Feil et al. 1999; Holmes et al. 1999; Hao et al. 2011). It is therefore important to systematically examine the significance of RMS in limiting DNA exchange in Nm.
In this study, we sequenced two invasive Nm strains, Nm10259 and Nm9418, both of which belong to the same ST (ST60) and the same serogroup (C). Previous Nm sequencing efforts have primarily focused on the serogroup B strains (Budroni et al. 2011) and the serogroup C strains are significantly underrepresented in the published genomes. Furthermore, no ST60 strains have been sequenced. The genomic information of this pair of ST60 genomes would be an excellent source for understanding Nm genomic diversity and could also provide insights on the rapid genomic changes between very closely related pathogens. We identified 35 genes, clustered in nine genomic regions, which were not shared between the two ST60 strains. All of these nine genomic regions exhibited hallmarks of homologous recombination at their conserved flanking genes. Our analysis also identified 152 genes that were present in both ST60 strains but differed significantly at the sequence level due to homologous recombination. These 152 horizontally acquired homologous genes, described as nonvertical genes in this study, contributed to approximately 94.25% of sequence divergence between the two ST60 genomes. Furthermore, many of the genes that have undergone homologous recombination have functions associated with pathogenicity. This suggests a significant role of homologous recombination in driving genomic variation at both the sequence level and gene content level. Homologous recombination has affected many RMS genes, which may undermine their function as a barrier to DNA exchange. Our findings suggest a transient role of RMSs in preventing homologous recombination between Nm clades.
Materials and Methods
Genomes Sequenced and Analyzed
The two invasive Nm strains, Nm10259 and Nm9418, were isolated in Ontario, Canada. Nm10259 was isolated from the blood of a 21-year-old woman in 2005, and Nm9418 was a cerebrospinal fluid isolate obtained from a 33-year-old man in 2006. Genome sequences were generated using GS-FLX pyrosequencing (Roche, 454 Life Sciences, Brantford, CT). Draft genomes were assembled using gsAssembler (Roche) and CAP3 (Huang and Madan 1999) with the help of the Phred/Phrap/Consed package (Gordon et al. 2001) as in Hao et al. (2011). The two draft genomes have been submitted to GenBank and the accession numbers are ARYF00000000 (Nm10259) and ARYG00000000 (Nm9418). Publically available genome sequences for additional 34 Nm, 1 N. lactamica (Nl), and 3 N. gonorrhoeae (Ng) strains were obtained from GenBank (supplementary table S1, Supplementary Material online).
Homolog Identification and Genome Comparisons
Annotated genes from the Nm genomes NEM8013, Z2491, MC58, FAM18, 053442, and alpha14 were downloaded from the NeMeSys database (Rusniok et al. 2009), as these genomes were manually (re)annotated and believed to be more accurate than the corresponding initial annotation. Annotated ORFs in other Nm genomes, when not present in the NeMeSys genomes, were also extracted. These annotated genes were used as query sequences to determine homologs in Nm10259 and Nm9418. Genes were categorized based on the Clusters of Orthologous Groups of proteins (COG) classification (Tatusov et al. 2000). Homologs were identified by a BlastN search (Camacho et al. 2009) with a cut off of E value <10−20 and at least 85% length match. DNA sequences in Nm10259 and Nm9418 without any match in other Nm genomes, when larger than 200 nucleotides, were used to Blast against the UniProtKB/Swiss-Prot database in a BlastX search and the Reference Genomic Sequences database in a BlastN search at NCBI to identify putative function and likely origin.
Phylogenetic Analyses
Homologous sequences were aligned using the MUSCLE program (Edgar 2004). Pairwise DNA distance was measured using Dnadist implemented in the PHYLIP package v3.6 (Felsenstein 1989). Phylogenetic trees were constructed using the RAxML program (Stamatakis 2006) under a GTR + Γ + I substitution model. In each phylogenetic reconstruction, 100 bootstrap iterations were performed. The phylogenetic relationship of the 36 Nm strains was first constructed using the concatenated sequences of 1,069 single-copy genes present in all 36 Nm genomes and 3 Ng genomes, with the Ng genomes as the outgroup. Then, all single-copy genes present in all 36 Nm genomes (1,090 genes in total) were examined, regardless of the presence of any homologs in the 3 Ng genomes. An unrooted tree was constructed using the concatenated sequences of the 1,090 genes. A phylogenetic tree was also constructed based on the allelic profile of all the 1,090 genes as described in Jolley and Maiden (2010). In brief, the number of different alleles between any two Nm genomes was treated as pairwise distance and computed using the neighbor-joining algorithm (Saitou and Nei 1987) implemented in the PHYLIP package (Felsenstein 1989). A network relationship of the 36 Nm genomes based on the concatenated sequences of the 1,090 genes was constructed using SplitsTree4.12.6 (Huson and Bryant 2006).
Analysis of the MLST Loci
The sequences of the seven MLST loci, namely abcZ, adk, aroE, fumC, gdh, pdhC, and pgm, were downloaded from the PubMLST website (Jolley and Maiden 2010), and used as query sequences to Blast and extract the corresponding loci from all studied genomes. Maximum likelihood trees were constructed for each individual MLST locus and also for the concatenated sequence of the seven MLST loci. The concatenated sequence was further analyzed using the ClonalFrame package (Didelot and Falush 2007) to infer relationships of the 36 Nm genomes and detect homologous recombination along the seven MLST loci.
Identification of Nonvertical Homologous Genes in ST60
Nonvertical gene transfer refers to the horizontal acquisition of genes by each of the ST60 genomes via homologous recombination. The term nonvertical is preferred because it avoids any assumption of gene origin, whereas horizontal gene transfer is often used to designate DNA exchange between species. Polymorphic single-copy genes between Nm10259 and Nm9418 were extracted and examined for nonvertical evolution, which was based on the probability of nucleotide changes in each examined gene at a maximally allowed genome-wide divergence given the observed identical seven MLST loci (Hao et al. 2011; Hao 2013). In brief, if all nucleotide differences between two genomes of identical STs were derived from independent random point mutations, there would exist an upper bound of genome-wide nucleotide divergence at which it was still possible to observe identical nucleotides in all seven MLST loci. Homologous genes that had greater than expected nucleotide changes at a significance level of P < 0.001 were deemed as nonvertical genes. In this study, all nonvertical genes were determined by unexpected nucleotide changes between the homologs in Nm10259 and Nm9418 as described here.
Identification of Recombination Partners of the ST60 Genomes
To infer the possible recombination partners of the ST60 strains, we first employed a stringent criterion that sequences in other Nm genomes were required to be identical to the ST60 genes. The number of identical sequences with the ST60 genes was counted for each strain and an average was obtained from all strains of the same CC. The analysis was conducted on the nonvertical ST60 genes as well as the genes identical between Nm10259 and Nm9418. As the nonvertical homologs are different between Nm10259 and Nm9418, genes from both genomes were examined. To make the results comparable, only single-copy genes present in all 36 Nm genomes were examined. Identical sequences were determined by zero DNA distance to encounter possible homopolymer sequencing errors (Hao et al. 2012). Furthermore, a less stringent criterion was employed for recombination partner identification. Instead of requiring identical sequences between the ST60 and non-ST60 strains, the recombination partners were determined by the non-ST60 strains with the nearest DNA distance to the ST60 homologs.
Identification and Analysis of RMSs
The sequences of all documented DNA endonuclease (restriction) and DNA methyltransferase (modification) genes in Nm were downloaded from the REBASE database (Roberts et al. 2010) and used as query sequences in a BlastN search to identify homologs. EcoPI-ModA (or ModA), EcoPI-ModB (or ModB), and EcoPI-ModD (or ModD) contain phase-variable repeats, which can confound the identification of these RMSs (Srikhanta et al. 2009; Seib et al. 2011; Gawthorne et al. 2012). Sequences of all allele types from ModA, ModB, and ModD (Srikhanta et al. 2009; Seib et al. 2011; Gawthorne et al. 2012) have been downloaded to search for homologs. An RMS was identified as present when both the restriction and modification genes were present and adjacent on the chromosome. If either the restriction or modification gene was truncated or disrupted by large internal insertion or deletion, the RMS was classified as disrupted. Possible frame-shift mutations and phase variation in the restriction and modification genes, which could also be explained by sequencing errors, were not classified as disrupted (Budroni et al. 2011). The presence/absence pattern of each RMS was determined in all the studied Nm genomes. Phylogenetic incongruence between the restriction and modification genes of each RMS was examined by the approximately unbiased (AU) test (Shimodaira 2002) implemented in the CONSEL program (Shimodaira and Hasegawa 2001).
To examine the role of RMSs in homologous recombination, we first examined the NmeSI system in the CC41/44 clade. We chose NmeSI, because NmeSI is only present in CC41/44 and CC22/CC167, and likely to be functional, as newly acquired RMSs are often initially active but gradually inactivated during evolution (Bart et al. 2001; Lin et al. 2001). Genes containing NmeSI recognition sites (5′-AGTACT-3′) were detected using a custom PERL script. Homologous recombination events between the NmeSI-containing clades and the NmeSI-lacking clades were identified. We further identified two genes only present in ST60 (ST60-specific). Together with the modification gene of EcoPI-ModD1 only present in three of the five CC41/44 strains, we determined whether these clade-specific genes contained the recognition sites of any RMSs present in the clade.
Results
Well-Supported CCs but Poorly Resolved Deeper Relationships
On the phylogenetic tree of the concatenated sequences of 1,069 single-copy genes present in all 36 Nm genomes and 3 Ng genomes (fig. 1A), clades of individual CCs were all supported by a 100% bootstrap value, whereas deeper relationships among different CCs were weakly supported. Similarly, a poor deep phylogenetic relationship was observed when using all single-copy genes (1,090 genes in total) shared by the 36 Nm genomes (fig. 1B). The 21 additional genes included in figure 1B were genes absent from at least one Ng genome. Three clades containing more than one CC were supported by a 100% bootstrap value. Among these clades, CC4/CC5 (Z2491/WUE2594), CC8/CC11, and CC32/CC269, the latter two clades have been previously reported in Budroni et al. (2011). Nm10259 and Nm9418 were always clustered together, but they were clustered together with a different CC on each phylogeny, for example, with N1568 in figure 1A and with alpha14 in figure 1B.
Fig. 1.—

Phylogenetic relationships of all Nm strains analyzed in this study. (A) Maximum likelihood tree constructed using the concatenated sequences of 1,069 single-copy genes present in all studied Nm and Ng genomes. The phylogeny was rooted using three Ng genomes (Ng TCDC-NG08107, Ng NCCP11945, and Ng FA1090) as outgroups (not shown). (B) Unrooted maximum likelihood tree constructed using the concatenated sequences of 1,090 single-copy genes present in all studied Nm genomes. One hundred bootstrap iterations were performed in each phylogenetic construction and any bootstrap values <100 are shown. (C) Majority rule consensus tree of the concatenated sequenced of the seven MLST loci constructed by the ClonalFrame program. The branches leading to individual CCs (labeled from A to I) were chosen to show evidence for recombination in supplementary figure S1, Supplementary Material online. (D) Neighbor-joining tree based on the number of gene alleles that differ between any two Nm genomes.
The seven MLST loci in the 36 Nm strains were analyzed using the ClonalFrame program. The majority rule consensus tree clustered the two ST60 strains with the CC41/44 clade (fig. 1C). Homologous recombination was evident in at least one MLST locus on all branches leading to individual CCs (supplementary fig. S1, Supplementary Material online). Phylogenetic analysis was performed on each individual locus and the concatenated sequence of the seven MLST loci, and significant phylogenetic incongruence was found between each individual-gene tree against the concatenated-sequence tree (supplementary table S2, Supplementary Material online). Furthermore, the phylogeny based on the allelic profile of the 1,090 single-copy genes showed different deeper relationships and the two ST60 strains were clustered with NEM8013 (fig. 1D). Similarly, the network analysis revealed an overall star-like topological structure with a web-like structure at the deep relationship level (supplementary fig. S2, Supplementary Material online). The poorly resolved deep relationships could be explained by selective sweeps, rapid population growth, and high levels of recombination in Nm populations, all of which can remove phylogenetic signals (Smith et al. 1993).
Variation of Genomic Content between the ST60 Genomes
Nm10259 and Nm9418 shared almost identical gene content, except 15 Nm10259 genes absent from Nm9418 and 20 Nm9418 genes absent from Nm10259 (fig. 2; supplementary table S3, Supplementary Material online). These 35 variable genes were located between conserved flanking genes in nine regions, and each region contained up to eight genes (figs. 2A–I ). Among the nine gene regions, five harbored distinct genes between Nm10259 and Nm9418, while four harbored genes in one genome that were missing from the other. It is striking that none of these regions were truly unique to Nm10259 or Nm9418. For each, a region with identical gene organization was found in at least one other Nm genome (fig. 2J ). Homologous recombination at the conserved flanking genes would account for this phenomenon whereby gene organization was identical between distantly related genomes but different between the closely related ST60 genomes. These regions displayed characteristics of minimal mobile elements, which were defined as variable regions among the closest relatives in sequence composition and gene content between specific conserved flanking genes (Saunders and Snyder 2002; Snyder et al. 2007). Among the nine regions, only the glnD-guaB region (fig. 2H ) has been previously identified (Snyder et al. 2007).
Fig. 2.—

Regions of variable gene content between Nm10259 and Nm9418. (A–I) Gene organization in regions of variable gene content between Nm10259 (at the top of each panel) and Nm9418 (at the bottom of each panel). Genes are shown as arrows (white for variable genes, gray for conserved genes). Empty space between genes indicates an intergenic region. In the hisS-NMB0867 region of Nm10259, there are two copies of NMA1079 (B). (J) Distribution of each gene organization in the studied Nm genomes. Each gene organization, though distinct between Nm10259 and Nm9418, was found in other Nm genomes.
Although a majority of the variable genes were poorly characterized (2 genes in the COG category R, 4 genes in the COG category S, and 24 genes not in COG), some genes are most likely of functional significance (supplementary table S3, Supplementary Material online). Two clustered-regularly interspaced short palindromic repeats (CRISPR) coupled with CRISPR-associated protein genes cas1 and cas2 were found in the gph-dnaE region in Nm10259 (fig. 2A). The CRISPR/Cas system has been described as a bacterial immune system for protection against potentially dangerous foreign DNA molecules such as bacteriophages (Makarova et al. 2006; Barrangou et al. 2007). The presence/absence polymorphism of the CRISPR/Cas system between Nm10259 and Nm9418 suggested a rapid alteration of immune ability between very closely related genomes facilitated by homologous recombination. The nadA gene, encoding an adhesin/invasin auto-transporter, was found in the glnB-NMB1993 region in Nm10259 but absent from Nm9418 (fig. 2F). The nadA-encoded protein is one antigen-target of the newly developed four-component 4CMenB vaccine (Toneatto et al. 2011). Here, both the glnB and NMB1993 genes are among the identified nonvertical ST60 homologs (discussed later) that underwent homologous recombination, and the nadA gene sequences varied substantially in Nm (fig. 3). The fast turnover of both the nadA sequences and sequence types in Nm can rapidly alter human host immune response and affect vaccine efficacy. Taken together, gene content variation via homologous recombination at the conserved flanking genes could lead to antigenic shift and modify the resistance to foreign DNA and virulence of the pathogenic strains.
Fig. 3.—

Sequence alignments of the glnB-NMB1993 gene region (A) and dotplot of the nadA homologs (B). (A) Only informative sites are shown with the coordinates at the top. The topology from figure 1A is shown on the left of the alignment. As glnB and NMB1993, in most of the genomes, were found adjacent to each other but on the opposite strand, the coordinates for glnB and NMB1993 are shown following the direction of the genomic sequence (same with the direction of glnB). The glnB gene was not found in the 961–5,945 genome (labeled as *). (B) The nadA sequences varied substantially among the Nm genomes. The nadA sequences labeled in black were found in five genomes; the ones in blue were disrupted as they were found on two different contigs; the one in green was disrupted by IS1301; the one in gold was truncated in 961–5,945, and its adjacent gene glnB was also not found in the draft genome; the ones in cyan were involved in an internal deletion; the ones in magenta and in red contained a region which is nonhomologous with the nadA in black, and the region is tandem-repeated in the magenta ones.
Nonvertical Homologs in ST60 Acquired from Non-ST60 Strains
The Nm10259 and Nm9418 genomes shared 1,745 single-copy genes, and two of those genes were not found in any of the other 34 Nm genomes but shared homologs in Neisseria species other than Nm (table 1). Out of the 1,745 genes, 1,424 genes (81.6%) showed 100% nucleotide identity and 321 genes were polymorphic between Nm10259 and Nm9418. Among the 321 polymorphic genes, a subset of 152 genes was determined to be nonvertical genes. These nonvertical ST60 genes are predicted to have been acquired via homologous recombination and thus may have originated from, or exhibit homology to, genes in non-ST60 strains. To validate our method of detecting nonvertical genes, phylogenetic positions of Nm10259 and Nm9418 were examined for the genes present in all 36 Nm genomes (86 nonvertical genes, in total). Separated phylogenetic positions of Nm10259 and Nm9418 were evident in 84 out of the 86 nonvertical genes (supplementary fig. S3A, Supplementary Material online, for one example). The concatenated sequences of the 86 nonvertical genes showed strong phylogenetic incongruence (P < 0.001, AU test) with either figure 1A or B, as the two ST60 strains were well separated (supplementary fig. S3B, Supplementary Material online). Two genes, murF and kpsF, were identified as nonvertical genes, but the Nm10259 and Nm9418 loci were clustered together on each phylogeny (supplementary fig. S4, Supplementary Material online). Further analysis revealed that the murF sequence at positions 281–978 in Nm10259 is identical with M13399, whereas the remaining murF sequence in Nm10259 is identical with Nm9418 (supplementary fig. S5A, Supplementary Material online). The kpsF sequences between Nm10259 and Nm9418 differed by 14 nucleotides. Although there is no single genome that could be identified as the donor, all nucleotide differences, with only one exception, the nucleotide position 492, can be found in other Nm genomes (supplementary fig. S5B, Supplementary Material online). Similarly, we have previously shown that fine-scale recombination does not always lead to phylogenetic incongruence, but sometimes causes significant branch elongation (Hao and Palmer 2011), which was indeed observed in both murF and kpsF (supplementary fig. S4, Supplementary Material online). In other words, the detection of nonvertical genes was very reliable, and the nonvertical homologs in ST60 have undergone homologous recombination at both the gene and subgenic levels.
Table 1.
Genes Shared by Nm10259 and Nm9418, but Not Present in Any Other Nm Genomes in This Study
| Genes | Source | Function |
|---|---|---|
| NgonD_04508 | Neisseria gonorrhoeae DGI18 | Hypothetical protein |
| NLA_20160a | Neisseria lactamica 020-06 | Cold-shock DNA-binding domain protein |
aThe NLA_20160 gene contains the recognition sites of NmeAI, which is present in both Nm10259 and Nm9418.
The 152 nonvertical genes predicted to have undergone direct homologous recombination were distributed in 20 different functional COG categories (supplementary fig. S6, Supplementary Material online), and 43 nonvertical genes were classified in COG categories R (general function prediction only) and S (function unknown), or not classified in COG (the full list is available at http://haolab.wayne.edu). In contrast, among the 35 variable genes between Nm10259 and Nm9418 (supplementary table S3, Supplementary Material online) that arose from homologous recombination at the conserved flanking genes, 29 variable genes were classified in COG categories R and S, or not classified in COG. The higher proportion of nonvertical genes of well-characterized function than that of variable genes is statistically significant (P = 0.0006, Fisher’s exact test). In other words, though both associated with homologous recombination with non-ST60 strains, the nonvertical ST60 genes were more likely involving genes of characterized function than the variable genes that distinctly present in only one of the ST60 strains.
Putative virulence genes have been identified in previous studies (Parkhill et al. 2000; Tettelin et al. 2000; Marri et al. 2010). In this study, 12 nonvertical genes (supplementary table S4, Supplementary Material online) were among the previously identified putative virulence genes. These genes include the antigenic outer membrane gene porB, pilin glycosylation genes pglC and pglD, iron transporter substrate-binding protein gene fetB, transferrin binding protein gene tbpB, and DNA-binding/iron metalloprotein/AP endonuclease gene gcp. Homologous recombination events in these virulence genes would create antigenic variation, produce novel virulence adaption in disease epidemiology, and contribute to pathoadaptive diversification. Furthermore, 11 genes of the 14-gene nuo gene cluster, which encodes subunits of the NADH dehydrogenase complex I, were identified as nonvertical in ST60. The ClonalFrame analysis revealed that homologous recombination has taken place in different sets of genes in the nuo gene cluster on different lineages (supplementary fig. S7, Supplementary Material online). Given the important function of NADH dehydrogenase, the highly dynamic nature of the nuo gene cluster is likely of significant functional consequence in Nm.
Nonvertical Genes and Sequence Diversity
Single-nucleotide polymorphisms (SNPs) were counted in the single-copy genes shared between Nm10259 and Nm9418. In total, 6,174 SNPs were found in the 321 polymorphic genes. Among the 6,174 SNPs between Nm10259 and Nm9418, 5,819 SNPs (94.25%) were in the subset of 152 nonvertical genes. In other words, 94.25% of SNPs observed between Nm10259 and Nm9418 were associated with homologous recombination but not point mutations. Thus, homologous recombination plays a major role in driving sequence diversity during Nm genome evolution.
Different Recombination Partners between the Nonvertical and Identical Genes in ST60
To identify the possible recombination partners of the ST60 genes, sequences identical with the ST60 genes were identified in other Nm genomes. When a non-ST60 Nm genome had sequences identical with a ST60 gene, we considered the non-ST60 genome as a putative recombination partner of the ST60 genome for the analyzed gene. Recombination partners were successfully identified for 462 identical ST60 genes (i.e., genes identical between both strains), 37 nonvertical genes in Nm10259, and 29 nonvertical genes in Nm9418 (fig. 4). The recombination events involved in the nonvertical ST60 genes most likely have occurred after the split between Nm10259 and Nm9418, while the recombination events involved in the identical ST60 genes most likely have occurred before the two genomes split. It is possible that there is more than one recombination partner of a single ST60 gene due to multiple recombination events involving the same sequence. The recombination partners of the identical ST60 genes were distributed in all analyzed CCs, ranging from 59 to 122 identical genes in each CC with the highest number in CC8. The nonvertical ST60 genes, however, showed a significantly different distribution of recombination partners from the identical ST60 genes (P = 0.0008, χ2 test). The highest number of recombination partners of the nonvertical ST60 genes was found in CC11 (fig. 4 and supplementary fig. S8, Supplementary Material online). As the data were averages of all strains in each CC, the results were not an artifact of the high number of sequenced genomes in CC11. In this study, no attempt was made to infer the directionality of individual recombination events, as recombination is very frequent and also there is no well-supported closely related clade to the ST60 strains. It is noteworthy that separate analyses of the recombination partners on Nm10259 and Nm9418 both revealed the highest numbers of recombination partners of the nonvertical genes consistently found in CC11 (data not shown).
Fig. 4.—

Distribution of recombination partners in CCs of the identical ST60 genes (A) and nonvertical ST60 genes (B). The recombination partners were determined by identical sequences. The total number of recombination partners in each CC was presented.
Highly Dynamic RMSs
The 28 RMSs identified in this study were distributed sporadically among the Nm genomes, and none of the RMSs were shared by all 36 genomes (fig. 5). The most abundant RMSs are Nme53442ORF787P and NlaIV, which were found in 34 of the 36 Nm genomes. Seven RMSs were only found in a single genome. Different RMS combinations were evident among different CCs, and even within the same CC. For instance, among the five strains in the CC41/44 clade, M0579 and alpha710 carried the EcoPI-ModB1 system, M01-240149 and NZ-05/33 contained the EcoPI-ModB2 system, while OX99.30304 carried the EcoPI-ModB3 system. Interestingly, there was no evidence of M0579 and alpha710 being clustered together within the CC41/44 clade (fig. 1). This suggests that RMSs are highly dynamic during Nm genome evolution.
Fig. 5.—

Presence/absence matrix of 28 RMSs. Each row of the matrix represents one Nm genome, and is color coded as per figure 1. Each column represents one RMS with the RMS type in parenthesis. The names of RMSs corresponded to the common names used in the literature, and to the reference names used in the REBASE database when common names could not be found in the literature. A black square indicates the presence of an RMS in the strain, while white indicates absence. A gray square indicates an RMS containing a disrupted restriction or modification gene. The topology on the left was the same as figure 1A.
RMSs were disrupted in the forms of partial gene deletion, internal sequence duplication, and insertion (IS) elements (fig. 5). For example, a 1,025-nucleotide deletion in the modification gene of the Nme53442ORF1140 system was found in both Nm3127 and Nm8187. The NmeAI restriction gene in N1568 was interrupted by an IS element ISNme3. Tandem repeats of a 411-nucleotide region were found in the NlaIV restriction gene (nucleotide positions 173–583 and 584–994) in M04-240196. As disrupted genes, truncated genes, and IS elements are often rapidly removed from the genome (Wagner 2006; Hao and Golding 2009, 2010), the high number of disrupted restriction and modification genes would support a fast turnover of the RMSs.
Among the 28 RMSs, 15 RMSs were present in more than four genomes. Phylogenetic incongruence was examined between the modification and restriction genes of each RMS. It is striking that phylogenetic incongruence was found between the restriction and modification genes of the same RMS (see fig. 6 for examples), which suggests that the restriction and modification genes of the same RMS in the same strain could be of different origins. The AU test revealed significant phylogenetic incongruence between the modification and restriction genes in 10 RMSs (table 2). For instance, the modification gene of the Nme18ORF295P system in N1568 is identical with M01-240355, whereas the restriction gene of Nme18ORF295P in N1568 is identical with genomes in CC11 (from K1207 to FAM18 in fig. 6). The CC11 genomes and N1568 shared an IS1655 insertion (also identical in sequence) between the restriction and modification genes of Nme18ORF295P (fig. 6A). The NmeAI system contains three genes, the modification gene, restriction gene, and a patch repair gene (fig. 6B). The three genes showed different evolutionary origins from one another. ATCC13091 and alpha14 shared identical modification genes of NmeAI, the restriction gene in ATCC13091 differed from alpha14 by 47 nucleotides, and the short patch repair genes only differed by one nucleotide between ATCC13091 and alpha14. The nonsignificant P value = 0.091 in the AU test (table 2) between the restriction gene and short patch repair gene is likely due to the lack of statistical power of the short patch repair gene.
Fig. 6.—

Sequence alignments of two RMSs, Nme18ORF295P (A) and NmeAI (B). Only informative sites are shown with the coordinates at the top. The topology from figure 1A is shown on the left of each alignment. The letters in parenthesis indicate the gene organization in each strain (supplementary figs. S10A and D, Supplementary Material online). The restriction and modification genes of Nme18ORF295P were separated by IS1655 in six genomes, and the coordinates for the restriction and modification genes were counted separately for each gene. IS element was observed in only one strain for the NmeAI system, the coordinates were counted by the NmeAI system as a whole.
Table 2.
Phylogenetic Incongruence between the Modification and Restriction Genes of Each RMS in Nm
| RMS Names | Number of Genomes |
P Valuesa |
|
|---|---|---|---|
| Modification– Restriction | Other | ||
| Nme53442ORF1140 | 5 | NSb | |
| EcoR124II | 25 | 2.0E-06 | |
| Nme53442ORF787P | 34 | 3.5E−05 | |
| NlaIV | 33 | 0.008 | |
| NmeBI | 12 | NS | 0.031c |
| EcoRII | 22 | 3.1E−06 | |
| NmeAI | 16 | 1.5E−18 | NSd |
| NmeDI | 7 | NS | NSd |
| Nme18ORF295P | 13 | 1.3E−05 | |
| NmeSI | 7 | NS | NSe |
| BsePI | 17 | 0.031 | |
| EcoPI-ModA11 | 9 | 0.023 | |
| EcoPI-ModA12 | 24 | 1.0E−09 | |
| EcoPI-ModB1 | 10 | NS | |
| EcoPI-ModB2 | 13 | 0.006 | |
aTo examine the incongruence between the restriction and modification gene-phylogenies of each RMS using both the restriction sequences and the modification sequences, 10 iterations of the AU test were performed. The median P value of the 10 iterations of each test was selected, and the larger (or less significant). P value is shown for each sequence-pair. If an RMS contained a third gene linked to the restriction or modification genes, the AU test was also performed between this third gene and the adjacent restriction or modification gene (shown as other).
bNS, not significant.
cThere were two modification genes in the NmeBI system. The P value was between the second modification gene and the restriction gene.
dThe system contained a short patch repair gene encoding a type II nicking enzyme in addition to the restriction and modification genes. The P value was between the patch repair gene and the restriction gene.
eThe system contained a gene encoding a control protein in addition to the restriction and modification genes. The P value was between the control protein gene and the restriction gene.
Inefficient or Transient Function of RMSs
Clade-specific genes, which are mostly recently acquired genes, were examined for the recognition sites of the RMSs present in the clade. The ST60-specific gene NLA_20160 was found to have the recognition sites of NmeAI, which was present in both ST60 genomes. The NMBM01-240149_1214 gene, which encodes the EcoPI-ModD1 modification gene, is only present in three of the five CC41/44 genomes, suggesting recent acquisition of EcoPI-ModD1. Both ends of this gene share homology with the EcoPI-ModD2 or EcoPI-ModD3 (in N. lactamica) modification gene, while no homology was found at the middle of the gene among the three ModD types (fig. 7). The 3′ 633-nucleotide region in the M0579 strain differs from the other two C44/44 strains by 53 nucleotides, and all these 53 changed nucleotides can be found in the EcoPI-ModD2 gene in CC22 or the EcoPI-ModD3 gene in N. lactamica (fig. 7). A feasible explanation is that the 3′ region into the M0579 strain results from a more recent acquisition after the acquisition of EcoPI-ModD1 into the ancestor of the three CC41/44 strains. As both acquisition events in the EcoPI-ModD1 sequence occurred after the split of CC41/44, the recognition sites the sequences carry might provide useful insight into the functionality of the RMSs present in CC41/44. We observed that the 3′ 633-nucleotide region in M0579 and the nonhomologous region in the three CC41/44 strains contain the recognition sites of NmeBI, which is present in CC41/44 but absent from CC22 and N. lactamica (fig. 7). This suggests that genes containing the recognition sites of a RMS cannot be completely prevented from transferring into the genome by the present RMS.
Fig. 7.—
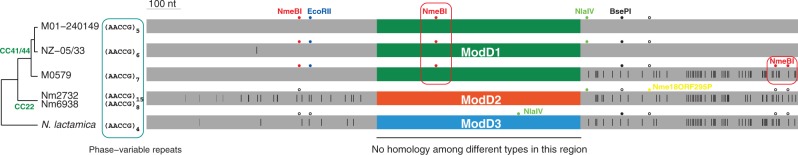
Sequence variation of the EcoPI-ModD modification genes. Genes in CC41/44, CC22, and Neisseria lactamica were distinct, and no homology was found in the middle part of the gene among different types (colored in different colors). The nonhomologous region varies in length (592 nt in CC41/44, 583 nt in CC22, and 577 nt in N. lactamica), but for illustration purposes, they are shown in the same length. Phase-variable repeats are present at the beginning of the gene. The two CC22 strains have identical sequences except different number of phase-variable repeat units. Nucleotide sites different from the NMBM01240149_1214 gene (in M01-240149) are shown as vertical bars. Colored filled circles represent recognition sites of other RMSs present in the corresponding genome. Conserved recognition sites in the genome lacking the RMS are shown in open circles. The NmeBI recognition sites in sequence regions that have recently been introduced into CC41/44 are boxed.
A previous study has shown that recently acquired RMSs would be active, whereas more ancient ones would be inactivated during bacterial genome evolution (Lin et al. 2001). We chose the NmeSI system for detailed analysis, as the NmeSI system was only found in eight strains from two clades, CC22/CC167 and CC41/CC44 (fig. 5). All the NmeSI genes were identical at the sequence level except a structural disruption in the restriction gene in Nm6938 (supplementary fig. S9, Supplementary Material online), suggesting a relatively recent acquisition of this system and therefore likely active function (Lin et al. 2001). In fact, the functionality of the NmeSI system has been experimentally verified in an Nm strain (Bart et al. 2001). Among the single-copy genes present in all 36 Nm genome, 21 genes contained the recognition sites (5′-AGTACT-3′) of NmeSI. In 12 of these 21 genes, identical sequences were found between at least one NmeSI-containing strain and one NmeSI-lacking strain, suggesting recombination between the NmeSI-containing and NmeSI-lacking strains. For instance, M0579 contained the NmeSI system and shared two genes (NMB0868 and frr; fig. 8) identical with Nm10259, which is lack of NmeSI. As both genes in M0579 contain the NmeSI recognition sites and are relatively distinct from the homologs in other CC41/44 strains and the NmeSI recognition sites are not located within the region of possible recombination breakpoints, it is likely that the NmeSI system in M0579 has not been completely efficient in preventing the introduction of recognition site-containing sequences. The evolutionary role of RMSs as a barrier to DNA exchange, if any, might be inefficient and/or transient.
Fig. 8.—

Maximum likelihood trees of the NMB0868 (A) and frr (B) genes. The strains that contained the NmeSI system are labeled by a filled square. The NMB0868 sequences containing the recognition sites of NmeSI were boxed, and all frr sequences contained the NmeSI recognition sites. One hundred bootstrap iterations were performed for each phylogeny, and bootstrap values when >85 are shown. Strains are color coded as per figure 1.
Discussion
Frequent Recombination Resulted in Poorly Resolved Deep Phylogenetic Relationships
Maximum likelihood analysis based on the concatenated sequences (figs. 1A and B) supported the grouping of each CC with a 100% bootstrap value. However, deeper relationships were weakly supported, and different deeper relationships were evident when a slightly different number of genes were used in the phylogenetic construction (e.g., 1,069 vs. 1,090). These findings are an interesting exception to the expectation in phylogenomic analysis that a large number of concatenated genes generally lead to robust phylogenies with high bootstrap supports (Rokas et al. 2003). Our explanation is that the high frequency of recombination in Nm resulted in conflicting phylogenetic signals and the addition of recombinant genes into the concatenated sequences could have weakened the overall phylogenetic signal. In previous studies, unreliable phylogenetic reconstruction of concatenated genes has been demonstrated in the presence of horizontal gene transfer (Jeffroy et al. 2006; Lapierre et al. 2013).
The primary focus of this study was on the genes with very strong phylogenetic signals for homologous recombination. We did not attempt to identify the genes that have never been horizontally recombined, because 1) the deep relationship in Nm is unknown such that it would be challenging to accurately identify the bona fide vertically inherited genes in all Nm genomes; 2) the number of vertically inherited genes conserved among all 36 Nm genomes is likely to be very small. In this study, among the 1,090 genes present in all 36 Nm genomes, 86 genes, on the conservative side, have been identified as nonvertical (7.89%) between the two ST60 genomes. Assuming the same fraction (x = 7.89%) of genes under homologous recombination in each genome and the recombinant genes are randomly distributed then, as shown in Hao (2012), among all 36 Nm genomes, the proportion of genes that have never been affected by homologous recombination among the 36 genomes will be (1 − x)36, which is only 5.19% with x = 0.0789.
In this study, frequent homologous recombination was evident on multiple branches of the phylogeny in many genes, including the seven MLST loci (supplementary fig. S1, Supplementary Material online). In contrast to the highly frequent recombination observed in the MLST loci among the Nm strains, Hanage et al. (2005) have reported that, by concatenating the seven MLST loci, most Neisseria species can be delineated. Although this may seem contradictory to the findings presented here, it is possible that the recombination frequency has been greatly reduced between Neisseria species and some phylogenetic signal is preserved at the species level, given the fact that the frequency of homologous recombination correlates positively with nucleotide sequence similarity and decreases sharply with the level of relatedness between the two parental sequences (Majewski and Cohan 1999).
Given the poorly resolved deeper relationships in Nm, the phylogenetic position of the two ST60 strains varied among the four phylogenies (fig. 1). The strains that clustered most closely with the ST60 strains were N1568 (fig. 1A), alpha14 (fig. 1B), the CC41/44 clade (fig. 1C), and NEM8013 (fig. 1D), respectively. In other words, the clade most closely related to the two ST60 strains could not be reliably identified. The well-supported grouping of individual CCs was likely due to the high genome-wide similarity within each CC and should not be interpreted as contradictory to the observed extensive homologous recombination. In fact, at least 152 nonvertical genes have been acquired into ST60 from other CCs via homologous recombination.
Homologous Recombination Solely Drives Gene Content Variation in ST60
The 35 genes variable between Nm10259 and Nm9418 were clustered in nine regions and flanked by conserved genes. These regions were not unique such that the gene organizations observed in each of the two ST60 strains were also found in at least one other Nm genome. All the genes flanking both sides of each of the nine variable regions between Nm10259 and Nm9418 have undergone homologous recombination, as they were all among the 152 identified nonvertical genes. Homologous recombination of conserved flanking regions has been previously proposed to contribute gene content variation (Lawrence and Retchless 2009; Polz et al. 2013). In our study, the gene content variation between Nm10259 and Nm9418 was solely due to homologous recombination at the conserved neighbor genes. Therefore, homologous recombination could have played a much more significant role in driving gene content variation than previously appreciated.
Although a majority (29 of 35, or 82.86%) of the variable genes were poorly characterized, some genes are of clear physiological significance, including the nadA gene. This is consistent with the hypothesis that many recently acquired genes are under rapid and relaxed evolution, and a few genes are of adaptive value (Hao and Golding 2006). The NadA protein is one antigen target of the newly developed four-component 4CMenB vaccine (Toneatto et al. 2011). It has been previously shown that the nadA gene is not present in all Nm strains (Lucidarme et al. 2010). Our current findings suggest that homologous recombination facilitates the rapid turnover of both the nadA sequences and sequence types, and thus can quickly alter human host immune response and affect vaccine efficacy.
Homologous Recombination as a Major Driving Force in Sequence Diversity
The 152 nonvertical homologous genes contained 94.25% of the SNPs observed between Nm10259 and Nm9418. The association of SNPs with nonvertical genes is a trend that holds true in other strains of identical STs (data not shown). Homologous recombination should therefore be considered as a predominant contributor to sequence divergence in Nm. With 94.25% of the SNPs in the nonvertical genes, the ratio of SNPs caused by recombination relative to mutation (r/m) can be calculated. If all remaining SNPs (5.75%) were caused by mutation, then the r/m ratio would be calculated as 94.25/5.75 = 16.39. As suggested in Hao et al. (2011) that the identification of nonvertical genes was likely conservative, the r/m ratio 16.39 would probably have been underestimated. Nevertheless, the r/m ratio calculated based on the nonvertical genes is much higher than the ones estimated in previous Nm studies, such as 4.75 (Feil et al. 2001), 5.37 (Hao et al. 2011), and 6.71 (Vos and Didelot 2009). This is, at least in part, due to our nonvertical-gene detection method developed specifically for clonally related strains. Similarly, Guttman and Dykhuizen (1994) have examined the clonal divergence of Escherichia coli strains in the ECOR group A and found that recombination has occurred at a rate 50-fold higher than the mutation rate (r/m ≈ 50) in four loci, which is much greater than the estimated r/m ratio (0.7) in E. coli based on the MLST sequences (Vos and Didelot 2009).
RMSs as the Victims of Homologous Recombination
RMSs in Nm are sporadically distributed and many are involved in horizontal gene transfer (Budroni et al. 2011). This study observed that a number of RMSs have been disrupted by partial gene duplication, deletion, repeat-associated phase variation, and IS elements, suggesting a fast rate of RMS turnover during the Nm genome evolution. There is no significant association between the average sequence divergence of the RMS genes and the number of genomes present (P = 0.14, Spearman’s rank correlation test, supplementary fig. S9, Supplementary Material online), supporting that the spread of RMSs was not straightly vertical. In fact, the adjacent restriction and modification genes in many RMSs were found to have remarkably different evolutionary histories. In other words, the RMSs in Nm, though once capable of limiting DNA exchange, are often involved in DNA exchange.
Transient Role of RMSs in Limiting DNA Exchange
RMSs are well recognized to protect bacteria against foreign DNA such as phage (Bickle and Kruger 1993). Although RMSs might be capable to serve as a barrier to genetic exchange between Nm strains (Jeltsch 2003; Budroni et al. 2011), it still remains unclear whether RMS plays an absolute role in limiting recombination between subpopulations or phylogenetic clades within a species (Fraser et al. 2007). In this study, extensive homologous recombination was observed among different CCs in genes of all functional categories. In another highly recombinogenic bacterium, Helicobacter pylori, 48% of recombination endpoints were found within regions that overlapped with the recognition sites of active RMSs (Lin et al. 2009). In the M0579 EcoPI-ModD1 gene (fig. 7), the homologous recombination endpoint is likely located close to the end of the nonhomologous region, but there is no recognition site of any RMSs present in M0579 near this region. Moreover, our study observed that genes containing the NmeSI recognition sites have been exchanged between the NmeSI-containing and NmeSI-lacking clades (fig. 8) and the NmeSI recognition site is not located within regions of recombination endpoints. These findings are consistent with the hypothesis RMS plays a transient or inefficient role in limiting DNA exchange among the Nm strains (Bart et al. 2001). A similar conclusion has been drawn in another bacterial defense system against foreign DNA, E. coli CRISPRs. That is, CRISPRs do not seem to be efficient barriers to the spread of plasmids carrying an antibiotic resistance gene (Touchon et al. 2012).
It has been previously shown that RMS genes are under rapid degradation and lack of selective pressure for maintenance (Seshasayee et al. 2012), consequently, active function can most often be found in recently acquired RMSs (Lin et al. 2001). In Nm, identical RMS sequences have been found between distantly related strains and a number of RMSs have been disrupted. This is in agreement with a transient functional role of RMSs in Nm. Furthermore, RMSs, even when functional, might not always be efficient in limiting DNA exchange in Nm. This role is unlike that in H. pylori, in which transformation is mediated by double-strand DNA (dsDNA) and active RMSs play an important role in limiting recombination length by cleaving dsDNA (Lin et al. 2009), homologous recombination in Nm generally occurs via transformation in competent strains, mediated by fragments of single-stranded DNA (ssDNA) (Dubnau 1999). As ssDNA has been evident to be resistant to restriction endonucleases (Bickle and Kruger 1993), it is possible that the uptake and integration of linear ssDNA fragments in Nm may be largely unaffected by different RMSs. Among the identified RMSs, some of them, especially the recently acquired ones, are believed to be functional. Additional studies and functional analysis of these RMSs could help define their role in Nm and answer the question whether active RMSs limit the recombination length in Nm as in H. pylori.
Is the Recombination Network in Nm Dynamic?
Different recombination partners were observed in recent recombination events compared with relatively ancient recombination events. The recombination partners in figure 4 were determined based on identical sequences, and a similar trend was observed when recombination partners were determined based on the nearest phylogenetic neighbor by DNA distance (supplementary fig. S8, Supplementary Material online). Consistently, CC11 was found as the top recent recombination-partner clade with each of the two ST60 strains. The most straightforward explanation would be the ST60 strains shared similar ecological niches with strains in CC11, because DNA exchange tends to take place among strains in similar environments or ecological niches (Jain et al. 2003; Didelot et al. 2012). The two ST60 strains belong to serogroup C and three out of the four ST11 strains belong to serogroup C. There is a possibility that the ST60 strains shared ecological niches with some CC11 serogroup C strains and recurrent homologous recombination of each ST60 strain with the CC11 strains has taken place after the split of the two ST60 strains. Interestingly, CC11 was not among the major CCs (ST41/44, ST269, ST32, ST35, and ST162) recently identified for group B Nm isolated from invasive meningococcal disease (IMD) cases in Ontario (Jamieson et al. 2013). The overrepresentation of CC11 as the ST60 recombination partner but a low proportion of CC11 isolates in recent IMD cases might be due to the very dynamic epidemiology of meningococcal disease (Baccarini et al. 2013) and many ST60 recombination events involving largely commensal Nm strains not causing IMD cases.
An alternative explanation to the significantly different distributions of the recombination partners would be that the genes observed identical between Nm10259 and Nm9418 have evolved at lower evolutionary rates than the nonvertical ST60 genes. To address such a concern, we measured the DNA distance between the Nl (N. lactamica) genome and the three Ng genomes, and compared that with the pairwise distance among the Nm genomes. It is clear that although the average DNA distance among the homologs of the nonvertical ST60 genes is significantly larger within Nm, the homologs of the nonvertical ST60 genes did not show accelerated divergence between the Nl and Ng genomes (supplementary fig. S11, Supplementary Material online). It is therefore possible that the high similarity among the genes observed to be identical between Nm10259 and Nm9418 is largely due to frequent recombination within Nm.
In this study, we observed extensive genomic variation between strains with identical ST. On the other hand, there has been a general trend based on the epidemiological data that hyperinvasive lineages are stable over time with life spans of many years (Bertrand et al. 2011). The discrepancy could be explained by the substantial difference in time scale of the comparisons, namely, between evolutionary time (in comparative genomics) and real time (in epidemiology). Another possible explanation is that strains in the same circulating hyperinvasive lineage might be of significant genomic variation, but overlooked by the current typing methods. As many more epidemiologically related sequencing data are being generated, it is becoming more feasible to test these hypotheses.
Supplementary Material
Supplementary figures S1–S11 and tables S1–S4 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Dr McCutcheon and two anonymous reviewers for their thoughtful suggestions. The authors are grateful for the grid computing service from Computing & Information Technology of Wayne State University. This work was supported by funds from Sanofi-Pasteur to D.E.L. and funds from Wayne State University to W.H.
Literature Cited
- Baccarini C, Ternouth A, Wieffer H, Vyse A. The changing epidemiology of meningococcal disease in North America 1945-2010. Hum Vaccin Immunother. 2013;9:162–171. doi: 10.4161/hv.22302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- Bart A, Pannekoek Y, Dankert J, van der Ende A. NmeSI restriction-modification system identified by representational difference analysis of a hypervirulent Neisseria meningitidis strain. Infect Immun. 2001;69:1816–1820. doi: 10.1128/IAI.69.3.1816-1820.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett JS, et al. A genomic approach to bacterial taxonomy: an examination and proposed reclassification of species within the genus Neisseria. Microbiology. 2012;158:1570–1580. doi: 10.1099/mic.0.056077-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand S, et al. Detection of a geographical and endemic cluster of hyper-invasive meningococcal strains. Microbes Infect. 2011;13:684–690. doi: 10.1016/j.micinf.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Bickle TA, Kruger DH. Biology of DNA restriction. Microbiol Rev. 1993;57:434–450. doi: 10.1128/mr.57.2.434-450.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowler LD, Zhang QY, Riou JY, Spratt BG. Interspecies recombination between the penA genes of Neisseria meningitidis and commensal Neisseria species during the emergence of penicillin resistance in N. meningitidis: natural events and laboratory simulation. J Bacteriol. 1994;176:333–337. doi: 10.1128/jb.176.2.333-337.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budroni S, et al. Neisseria meningitidis is structured in clades associated with restriction modification systems that modulate homologous recombination. Proc Natl Acad Sci U S A. 2011;108:4494–4499. doi: 10.1073/pnas.1019751108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caugant DA, et al. Intercontinental spread of a genetically distinctive complex of clones of Neisseria meningitidis causing epidemic disease. Proc Natl Acad Sci U S A. 1986;83:4927–4931. doi: 10.1073/pnas.83.13.4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I, Dubnau D. DNA uptake during bacterial transformation. Nat Rev Microbiol. 2004;2:241–249. doi: 10.1038/nrmicro844. [DOI] [PubMed] [Google Scholar]
- Didelot X, Falush D. Inference of bacterial microevolution using multilocus sequence data. Genetics. 2007;175:1251–1266. doi: 10.1534/genetics.106.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Meric G, Falush D, Darling AE. Impact of homologous and non-homologous recombination in the genomic evolution of Escherichia coli. BMC Genomics. 2012;13:256. doi: 10.1186/1471-2164-13-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubnau D. DNA uptake in bacteria. Annu Rev Microbiol. 1999;53:217–244. doi: 10.1146/annurev.micro.53.1.217. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil EJ, Maiden MC, Achtman M, Spratt BG. The relative contributions of recombination and mutation to the divergence of clones of Neisseria meningitidis. Mol Biol Evol. 1999;16:1496–1502. doi: 10.1093/oxfordjournals.molbev.a026061. [DOI] [PubMed] [Google Scholar]
- Feil EJ, et al. Recombination within natural populations of pathogenic bacteria: short-term empirical estimates and long-term phylogenetic consequences. Proc Natl Acad Sci U S A. 2001;98:182–187. doi: 10.1073/pnas.98.1.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP—phylogeny inference package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- Findlay WA, Redfield RJ. Coevolution of DNA uptake sequences and bacterial proteomes. Genome Biol Evol. 2009;1:45–55. doi: 10.1093/gbe/evp005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser C, Hanage WP, Spratt BG. Recombination and the nature of bacterial speciation. Science. 2007;315:476–480. doi: 10.1126/science.1127573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawthorne JA, Beatson SA, Srikhanta YN, Fox KL, Jennings MP. Origin of the diversity in DNA recognition domains in phasevarion associated modA genes of pathogenic Neisseria and Haemophilus influenzae. PLoS One. 2012;7:e32337. doi: 10.1371/journal.pone.0032337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Desmarais C, Green P. Automated finishing with autofinish. Genome Res. 2001;11:614–625. doi: 10.1101/gr.171401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman DS, Dykhuizen DE. Clonal divergence in Escherichia coli as a result of recombination, not mutation. Science. 1994;266:1380–1383. doi: 10.1126/science.7973728. [DOI] [PubMed] [Google Scholar]
- Hanage WP, Fraser C, Spratt BG. Fuzzy species among recombinogenic bacteria. BMC Biol. 2005;3:6. doi: 10.1186/1741-7007-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W. Fast rates of evolution in bacteria due to horizontal gene transfer. In: Singh RS, Xu J, Kulathinal RJ, editors. Rapidly evolving genes and genetic systems. Oxford: Oxford University Press; 2012. pp. 64–72. [Google Scholar]
- Hao W. Extensive genomic variation within clonal bacterial groups resulted from homologous recombination. Mob Genet Elements. 2013;3:1–5. doi: 10.4161/mge.23463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, Allen VG, Jamieson FB, Low DE, Alexander DC. Phylogenetic incongruence in E. coli O104: understanding the evolutionary relationships of emerging pathogens in the face of homologous recombination. PLoS One. 2012;7:e33971. doi: 10.1371/journal.pone.0033971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, Golding GB. The fate of laterally transferred genes: life in the fast lane to adaptation or death. Genome Res. 2006;16:636–643. doi: 10.1101/gr.4746406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, Golding GB. Does gene translocation accelerate the evolution of laterally transferred genes? Genetics. 2009;182:1365–1375. doi: 10.1534/genetics.109.104216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, Golding GB. Inferring bacterial genome flux while considering truncated genes. Genetics. 2010;186:411–426. doi: 10.1534/genetics.110.118448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, Palmer JD. HGT turbulence: confounding phylogenetic influence of duplicative horizontal transfer and differential gene conversion. Mob Genet Elements. 2011;1:256–261. doi: 10.4161/mge.19030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, et al. Extensive genomic variation within clonal complexes of Neisseria meningitidis. Genome Biol Evol. 2011;3:1406–1418. doi: 10.1093/gbe/evr119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes EC, Urwin R, Maiden MC. The influence of recombination on the population structure and evolution of the human pathogen Neisseria meningitidis. Mol Biol Evol. 1999;16:741–749. doi: 10.1093/oxfordjournals.molbev.a026159. [DOI] [PubMed] [Google Scholar]
- Huang X, Madan A. CAP3: a DNA sequence assembly program. Genome Res. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- Jain R, Rivera MC, Moore JE, Lake JA. Horizontal gene transfer accelerates genome innovation and evolution. Mol Biol Evol. 2003;20:1598–1602. doi: 10.1093/molbev/msg154. [DOI] [PubMed] [Google Scholar]
- Jamieson FB, et al. Genetic and antigenic characterization of invasive endemic serogroup B Neisseria meningitidis from Ontario, Canada, in 2001-2010. J Med Microbiol. 2013;62:46–55. doi: 10.1099/jmm.0.050369-0. [DOI] [PubMed] [Google Scholar]
- Jeffroy O, Brinkmann H, Delsuc F, Philippe H. Phylogenomics: the beginning of incongruence? Trends Genet. 2006;22:225–231. doi: 10.1016/j.tig.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Jeltsch A. Maintenance of species identity and controlling speciation of bacteria: a new function for restriction/modification systems? Gene. 2003;317:13–16. doi: 10.1016/s0378-1119(03)00652-8. [DOI] [PubMed] [Google Scholar]
- Jolley KA, Maiden MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley KA, Maiden MC. Automated extraction of typing information for bacterial pathogens from whole genome sequence data: Neisseria meningitidis as an exemplar. Euro Surveill. 2013;18:20379. doi: 10.2807/ese.18.04.20379-en. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph B, et al. Virulence evolution of the human pathogen Neisseria meningitidis by recombination in the core and accessory genome. PLoS One. 2011;6:e18441. doi: 10.1371/journal.pone.0018441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapierre P, Lasek-Nesselquist E, Gogarten JP. The impact of HGT on phylogenomic reconstruction methods. Brief Bioinform. 2013 doi: 10.1093/bib/bbs050. doi:10.1093/bib/bbs050. Advance Access publication August 20, 2012. [DOI] [PubMed] [Google Scholar]
- Lawrence JG, Retchless AC. The interplay of homologous recombination and horizontal gene transfer in bacterial speciation. Methods Mol Biol. 2009;532:29–53. doi: 10.1007/978-1-60327-853-9_3. [DOI] [PubMed] [Google Scholar]
- Lin LF, Posfai J, Roberts RJ, Kong H. Comparative genomics of the restriction-modification systems in Helicobacter pylori. Proc Natl Acad Sci U S A. 2001;98:2740–2745. doi: 10.1073/pnas.051612298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin EA, et al. Natural transformation of Helicobacter pylori involves the integration of short DNA fragments interrupted by gaps of variable size. PLoS Pathog. 2009;5:e1000337. doi: 10.1371/journal.ppat.1000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucidarme J, et al. Characterization of fHbp, nhba (gna2132), nadA, porA, and sequence type in group B meningococcal case isolates collected in England and Wales during January 2008 and potential coverage of an investigational group B meningococcal vaccine. Clin Vaccine Immunol. 2010;17:919–929. doi: 10.1128/CVI.00027-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden MC, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998;95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski J, Cohan FM. DNA sequence similarity requirements for interspecific recombination in Bacillus. Genetics. 1999;153:1525–1533. doi: 10.1093/genetics/153.4.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct. 2006;1:7. doi: 10.1186/1745-6150-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marri PR, et al. Genome sequencing reveals widespread virulence gene exchange among human Neisseria species. PLoS One. 2010;5:e11835. doi: 10.1371/journal.pone.0011835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradigaravand D, Engelstadter J. The effect of bacterial recombination on adaptation on fitness landscapes with limited peak accessibility. PLoS Comput Biol. 2012;8:e1002735. doi: 10.1371/journal.pcbi.1002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhill J, et al. Complete DNA sequence of a serogroup A strain of Neisseria meningitidis Z2491. Nature. 2000;404:502–506. doi: 10.1038/35006655. [DOI] [PubMed] [Google Scholar]
- Paul S, et al. Role of homologous recombination in adaptive diversification of extraintestinal Escherichia coli. J Bacteriol. 2013;195:231–242. doi: 10.1128/JB.01524-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polz MF, Alm EJ, Hanage WP. Horizontal gene transfer and the evolution of bacterial and archaeal population structure. Trends Genet. 2013;29:170–175. doi: 10.1016/j.tig.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2010;38:D234–D236. doi: 10.1093/nar/gkp874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokas A, Williams BL, King N, Carroll SB. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature. 2003;425:798–804. doi: 10.1038/nature02053. [DOI] [PubMed] [Google Scholar]
- Rusniok C, et al. NeMeSys: a biological resource for narrowing the gap between sequence and function in the human pathogen Neisseria meningitidis. Genome Biol. 2009;10:R110. doi: 10.1186/gb-2009-10-10-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Saunders NJ, Snyder LA. The minimal mobile element. Microbiology. 2002;148:3756–3760. doi: 10.1099/00221287-148-12-3756. [DOI] [PubMed] [Google Scholar]
- Schoen C, Tettelin H, Parkhill J, Frosch M. Genome flexibility in Neisseria meningitidis. Vaccine. 2009;27(2 Suppl):B103–B111. doi: 10.1016/j.vaccine.2009.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoen C, et al. Whole-genome comparison of disease and carriage strains provides insights into virulence evolution in Neisseria meningitidis. Proc Natl Acad Sci U S A. 2008;105:3473–3478. doi: 10.1073/pnas.0800151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seib KL, et al. A novel epigenetic regulator associated with the hypervirulent Neisseria meningitidis clonal complex 41/44. FASEB J. 2011;25:3622–3633. doi: 10.1096/fj.11-183590. [DOI] [PubMed] [Google Scholar]
- Seshasayee AS, Singh P, Krishna S. Context-dependent conservation of DNA methyltransferases in bacteria. Nucleic Acids Res. 2012;40:7066–7073. doi: 10.1093/nar/gks390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimodaira H. An approximately unbiased test of phylogenetic tree selection. Syst Biol. 2002;51:492–508. doi: 10.1080/10635150290069913. [DOI] [PubMed] [Google Scholar]
- Shimodaira H, Hasegawa M. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 2001;17:1246–1247. doi: 10.1093/bioinformatics/17.12.1246. [DOI] [PubMed] [Google Scholar]
- Smith JM, Smith NH, O′Rourke M, Spratt BG. How clonal are bacteria? Proc Natl Acad Sci U S A. 1993;90:4384–4388. doi: 10.1073/pnas.90.10.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder LA, et al. The repertoire of minimal mobile elements in the Neisseria species and evidence that these are involved in horizontal gene transfer in other bacteria. Mol Biol Evol. 2007;24:2802–2815. doi: 10.1093/molbev/msm215. [DOI] [PubMed] [Google Scholar]
- Srikhanta YN, et al. Phasevarions mediate random switching of gene expression in pathogenic Neisseria. PLoS Pathog. 2009;5:e1000400. doi: 10.1371/journal.ppat.1000400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Stephens DS, Greenwood B, Brandtzaeg P. Epidemic meningitis, meningococcaemia, and Neisseria meningitidis. Lancet. 2007;369:2196–2210. doi: 10.1016/S0140-6736(07)61016-2. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettelin H, et al. Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science. 2000;287:1809–1815. doi: 10.1126/science.287.5459.1809. [DOI] [PubMed] [Google Scholar]
- Toneatto D, et al. The first use of an investigational multicomponent meningococcal serogroup B vaccine (4CMenB) in humans. Hum Vaccin. 2011;7:646–653. doi: 10.4161/hv.7.6.15482. [DOI] [PubMed] [Google Scholar]
- Touchon M, et al. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 2009;5:e1000344. doi: 10.1371/journal.pgen.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchon M, et al. Antibiotic resistance plasmids spread among natural isolates of Escherichia coli in spite of CRISPR elements. Microbiology. 2012;158:2997–3004. doi: 10.1099/mic.0.060814-0. [DOI] [PubMed] [Google Scholar]
- Vos M, Didelot X. A comparison of homologous recombination rates in bacteria and archaea. ISME J. 2009;3:199–208. doi: 10.1038/ismej.2008.93. [DOI] [PubMed] [Google Scholar]
- Wagner A. Periodic extinctions of transposable elements in bacterial lineages: evidence from intragenomic variation in multiple genomes. Mol Biol Evol. 2006;23:723–733. doi: 10.1093/molbev/msj085. [DOI] [PubMed] [Google Scholar]
- Wilson GG, Murray NE. Restriction and modification systems. Annu Rev Genet. 1991;25:585–627. doi: 10.1146/annurev.ge.25.120191.003101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.