

Abstract
Thanks to the microarray technology, our understanding of transcriptome evolution at the genome level has been considerably advanced in the past decade. Yet, further investigation was challenged by several technical limitations of this technology. Recent innovation of next-generation sequencing, particularly the invention of RNA-seq technology, has shed insightful lights on resolving this problem. Though a number of statistical and computational methods have been developed to analyze RNA-seq data, the analytical framework specifically designed for evolutionary genomics remains an open question. In this article we develop a new method for estimating the genome expression distance from the RNA-seq data, which has explicit interpretations under the model of gene expression evolution. Moreover, this distance measure takes the data overdispersion, gene length variation, and sequencing depth variation into account so that it can be applied to multiple genomes from different species. Using mammalian RNA-seq data as example, we demonstrated that this expression distance is useful in phylogenomic analysis.
Keywords: transcriptome evolution, RNA-seq, genome expression distance
Introduction
Despite exciting achievements in transcriptome changes in genome evolution, mainly based on microarrays (Enard et al. 2002; Caceres et al. 2003; Gu and Gu 2003; Makova and Li 2003; Rifkin et al. 2003; Huminiecki and Wolfe 2004; Khaitovich et al. 2004; Gu et al. 2005; Gu and Su 2007), further investigation has been challenged by the availability of robust gene expression data across a broad range of species and tissues (Wang et al. 2009). Nevertheless, recent technological innovations called next-generation sequencing, particularly the development of RNA-seq technology, have shed some light to this problem, which can generate tens of millions of short sequence reads. These reads can be mapped to each gene through the reference genome or de novo assembling, enabling researchers to quantify the transcription level in ultra-high resolution (Cloonan et al. 2009; Morozova et al. 2009; Wang et al. 2009). Indeed, RNA-seq technology has already made unprecedented advances for revealing the complexity of transcriptional phenomena, ranging from the expression profiling, dissection of isoform, and allelic expression, to the extension of 3′-UTR regions, novel splice junctions, modes of antisense regulation, and intragenic expression (Carninci et al. 2005; Eveland et al. 2008; Mortazavi et al. 2008; Nagalakshmi et al. 2008; Sultan et al. 2008; Graveley et al. 2010; Trapnell et al. 2010).
The power of RNA-seq in the study of transcriptome evolution was well demonstrated by the recent work of Brawand et al. (2011). They reported a large-scale RNA-seq analysis of six mammalian tissues and showed the dynamics of transcriptome evolution that may underlie many phenotypic differences between species. However, despite many studies in RNA-seq data analysis (Lu et al. 2005; Robinson and Smyth 2007, 2008; Anders and Huber 2010; Di et al. 2011; Zhou et al. 2011; McCarthy et al. 2012), statistical methods designed specifically for evolutionary genomics have not been well developed. In this article we report a new method for estimating the genome expression distance based on RNA-seq data, which has explicit interpretations under the model of gene expression evolution. Using mammalian RNA-seq data as example, we show that this expression distance can be used in phylogenomic reconstruction and related phylogeny-based expression analysis.
Materials and Methods
New Methods
Statistical Framework of Transcriptome Evolution
Because RNA-seq technology provides readout counts, the sampling property is similar to some earlier data types such as SAGE (Velculescu et al. 1995) or EST (Audic and Claverie 1997; Ewing and Claverie 2000). A variety of statistical methods were proposed; see Zhou et al. (2011), McCarthy et al. (2012), and Di et al. (2011) for recent advances and references therein. Simply to say, these methods considered RNA-seq overdispersion as well as data normalization to remove nonbiological effects in the data processing, which will be also addressed in our method.
Though a simple Poisson distribution model p(x; λ), characterized by the variance equal to the mean (λ), can effectively handle substantial zero counts, many studies have shown that RNA-seq counts exhibit a greater variance across biological replicates than expected (Di et al. 2011; McCarthy et al. 2012). This phenomenon is called overdispersion in statistics. Among a number of statistical models proposed to remedy this problem (Lu et al. 2005; Robinson and Smyth 2007, 2008; Anders and Huber 2010; Di et al. 2011; Zhou et al. 2011; McCarthy et al. 2012), our study adopts the widely used negative binomial distribution (NBD). We choose a special form denoted by p(x; λ, ω), characterized by the mean parameter (λ) and the overdispersion parameter (ω) (see eq. 4 in Data Processing section). A large value of ω indicates a strong over-dispersion, and vice versa. When ω = 0, p(x; λ, ω) is reduced to the Poisson model.
Next we model the mean parameter λ as a random variable to describe the expression variability among genes. A typical RNA-seq sample may include many thousands of genes, showing a highly skewed distribution of read counts. For instance, in mammalian tissues (Brawand et al. 2011), the top 5% highly expressed genes received roughly 102–105 RNA-seq counts, whereas the bottom 40% lowly expressed genes received roughly 0–10 counts. We therefore implement a lognormal distribution, analogous to the log-transformation in the microarray data analysis (Kerr and Churchill 2001; Irizarry et al. 2003); the log of λ follows a normal distribution with the mean µ and variance η2. Together, the RNA-seq counts in a sample follow a negative binomial-lognormal distribution denoted by f(x). Though the analytical form of f(x) is not available, the mean and variance of f(x) can be derived straightforwardly; see equation (5) in Data Processing section.
In the case of two RNA-seq samples of the same tissue from two species (genomes) X and Y, the mean parameter λX (or λY) of genome X (or Y) follows a lognormal distribution accounting for the among-gene expression variability. Because λX and λY are correlated by the evolutionary relatedness of genomes X and Y, without loss of generality the joint model of λX and λY can be written as follows (Gu 2004):
| (1) |
where αXY is the ancestral genetic component shared by X and Y, βX and βY are the independent genetic effects, and μX and μY are the ground means. Together, αXY, βX, and βY describe the evolutionally correlated structure of the underlying regulatory machinery. To implement this model, we further assume that αXY, βX, and βY are mutually independent, each of which follows a normal distribution with the mean 0, and the variance ρ2, v2X, or v2Y, respectively. As shown in figure 1, the variance component ρ2 measures the expression variability at the common ancestor of species X and Y. Meanwhile, the variance component v2X (or v2Y) measures the expression variability generated during the evolution from the common ancestor to the current species X (or Y). For the current genome X, the marginal expression variability is given by γX = αXY + βX so that the variance of among-gene variability is given by η2X = ρ2 + v2X. Similarly, for genome Y, we have γY = αXY + βY and η2Y = ρ2 + v2Y.
Fig. 1.—

Model of transcriptome evolution between two species. (A) A schematic illustration for a rooted two-gene tree: ρ2 refers to among-gene expression variability at the common ancestor of species X and Y; v2X and v2Y measure the among-gene expression variability in lineage X and Y since the split of common ancestor, respectively. (B) The variance–covariance matrix of genome expression between for current genomes X and Y. (C) The expression distance UXY plotted against the evolutionary time t. Expression divergence is an accelerated process under the adaptive model, a constant-rate process under the neutral model, and a decelerated process under the stabilizing model. In particular, when W→0, we have UXY →2σ2t, i.e., the stabilizing selection model is reduced to the neutral model; and when t→∞, UXY →1/W, i.e., the expression divergence approaches a saturated level.
Definition of Expression Distance
For a given tissue, the expression distance should measure the expression divergence between two species that had diverged t time units ago, reflecting the underlying regulatory divergence. Because two variance components v2X and v2Y characterize the expression divergence along the lineages from the common ancestor to species X and Y, respectively, following our previous work (Gu 2004; Gu and Su 2007) we define the expression distance between species X and Y as
| (2) |
The biological interpretation of equation (2) can be briefly summarized as follows; also see figure 1C for numerical illustrations.
(i) Under the simple Brownian model that represents a selectively neutral expression evolution (Gu 2004), we have UXY = 2σ2t, where σ2 is the rate of mutational variance. Hence, under the neutral expression model, the expression distance UXY increases proportionally with the evolutionary time t, and the rate (r) of expression divergence equals to the rate of mutational variance, i.e., r = σ2.
(ii) Under the Ornstein-Uhlenback (OU) model (Gu and Su 2007), gene expression has been maintained around its optimum by the stabilizing selection and any deviation of expression profile may reduce the organismal fitness. It has been shown (Gu and Su 2007) that the expression distance is expected to be UXY = (1 − e−2βt)/W, where W describes the selection strength and the decay rate β = Wσ2. Importantly, when t→∞, UXY→1/W, which means that the expression distance approaches a saturated level determined by the strength of stabilizing selection. In a special case of W→0, i.e., very weak stabilizing selection, one can show UXY→2σ2t, i.e., the neutral Brownian model. Intuitively, expression divergence under the stabilizing model evolves more slowly than the neutral expectation. Indeed, the rate of expression divergence (r) under the stabilizing model can be symbolically written by r = σ2f, where the expression constraint f < 1 measures the effect of purifying selection. In short, stabilizing selection model of expression divergence is consistent with the nearly neutral model.
(iii) Despite many forms of adaptive expression divergence, the general pattern is that the rate of expression divergence can be accelerated by the adaptive evolution, i.e., r > σ2. For instance, gradual directive selection (Gu 2004) predicts that the expression distance is proportional to t2.
Estimation of Expression Distance from RNA-seq Data
Suppose that we have RNA-seq data of a tissue from genomes X and Y, both of which contain n orthologous genes with RNA-seq counts denoted by x1, … , xn and y1, … , yn. When RNA-seq data contain multiple biological replicates, we use a simple mean. It is thus straightforward to obtain the estimates of first, second, and cross moments E[x], E[x2], E[y], E[y2], and E[xy], respectively; for instance, the estimate of E[xy] is given by Σi xi yi/n. On the other hand, the expectations of these moments under the NBD-lognormal model can be found in equations (5) and (6) in Data Processing section, allowing us to develop a simple method to estimate the expression distance UXY = v2X + v2Y. To this end, we first define three basic quantities: JXX = E[x2] − E[x], JYY = E[y2] − E[y], and JXY = E[xy]. The (mean-corrected) second moments JXX and JYY represent the expression variability in genomes X and Y, respectively, and the cross-product JXY measures the co-expression pattern. Putting together with equation (2) and equations (5) and (6), one can derive the relationships of JXX, JYY, and JXY with the underlying model parameters (presented in the second column of table 1). It follows that the expression distance defined by equation (2) can be rewritten as follows:
| (3) |
where ΩX = ln(1 + ωX /LX) and ΩY = ln(1 + ωY /LY) are the effects of overdispersion; LX and LY are the numbers of biological replicates of genomes X and Y, respectively.
Table 1.
Definitions, Theoretical Expectations, and Formulas of Statistical Estimation for Three Quantities JXX, JYY, and JXY
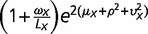
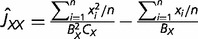
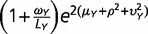
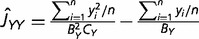
aE[.] is short form for expectation.
bDerivation of each expectation can be found in Materials and Methods. See figure 1 and the text for the description of model parameters.
cxi. (or yi.) is the mean RNA-seq count of gene i over its biological replicates in genome X (or Y); and n is the number of genes under study.
The flow chart in figure 2 shows the statistical procedure for the estimation of UXY; see Data Processing section for technical details. In the first step of data normalization, we introduced two correction constants of each genome (X) to remove the overestimation of expression distance: constant CX accounts for the effect caused by the sequence length variation among genes and BX for the sequencing depth variation among genomes. After data normalization, one can compute JXX, JYY, and JXY, respectively, by the formulas in the third column of table 1. When genes have the same sequence length and genomes have the same sequencing depth, we have CX = 1 and BX = 1; in this case, JXX, JYY, and JXY are simply calculated by the method of moments. The next issue is to estimate overdispersion. We implemented a simple method to estimate ΩX and ΩY even for only two biological replicates available. One may see Results and Discussion for a special treatment in the case of single biological replicate. Finally, the sampling variance of the estimated UXY can be empirically determined by the bootstrapping approach or a simple approximate method.
Fig. 2.—

Flow chart for illustrating the statistical procedure of expression distance estimation.
Data Sets
We downloaded the mammalian RNA-seq data in five tissues (brain, cerebellum, liver, heart, and kidney) from Brawand et al. (2011). For simplicity, we used the total reads of all 5,636 1:1 orthologous genes, suggested by the original authors. Nevertheless, we obtain the RNA-seq counts independently from the raw reads and found virtually the same results.
Data Processing
Calculation of Moments
The specific form of NBD we used in our study is as follows:
| (4) |
where α = 1/ω. Let φ(λ) be a lognormal distribution φ(λ) such that the log of λ follows a normal distribution with the mean µ and variance η2. Then, the negative binomial-lognormal distribution for RNA-seq counts (x) of genes is given by f (x) = ∫p(x; λ, ω) φ(λ) d λ. Next we derive first and second moments. From the conditional expectation E[x| λ] = λ according to equation (4), we have E[x] = E[E[x| λ]] = E[λ]. Similarly, we have E[x2] = E[E[x2| λ]] = E[λ + (ω + 1)λ2]. With respective the lognormal distribution φ(λ), we obtain
 |
(5) |
In the case of two genomes X and Y, the first and second moments of x or y are given by equation (5). For the cross-moment of x and y, from equation (4) we have E[xy] = E[E[xy| λXλY]] = E[λXλY]. Together with the independent assumption of three components in equation (1) and the lognormal distribution φ(λ), we derive E[λXλY] = E[exp(μX + αXY + βX) exp(μY + αXY + βY)] = exp(μX + μY) E[exp(2αXY)] E[exp(βX)] E[exp (βY)], resulting in
| (6) |
When the mean RNA-seq counts over L number of biological replicates is used to estimate the expression distance, the first, second, and cross-moments can be derived with a similar approach, except for the overdispersion parameters ω. = ω/L (omitting the subscripts X or Y).
RNA-seq Data Normalizations
Two main nonbiological effects inherited in the RNA-seq data processing need to be removed to avoid potential biases in the estimation of expression distance: sequence length variation and sequencing depth variation. To this end, we assume that the RNA-seq count of any gene (xi) can be written as
| (7) |
where ci and BX are the normalization constants and variable zi is the normalized count when all genes have the same length (equal to the genome mean) and the same sequencing depth (equal to the mean over the genomes under study). Similar to RPKM (reads per kilobase per million mapped reads), we set ci = li/l, where li is the sequence length of gene i and l is the genome mean of sequence lengths. To correct sequencing depth variation, one has to consider the factor that the number (N) of genes may vary among genomes. Here we used a relative measure for any genome X by defining RX = Total counts/N. That is, we actually normalize the data such that the mean count per gene is roughly the same among the genomes under study. Moreover, we choose BX = RX/R0, where R0 is the mean over all genomes under study.
Next we derive the formulas in the third column of table 1 to estimate the expression distance after the data normalization. From equation (7) we claim that the expectations E[x] is given by E[x] = BX E[z] Σi ci/n = BXE[z] because Σi ci/n = 1. Similarly, we have E[x2] = BX2E[z2] Σi ci2/n = BX2CXE[z2], where CX = Σi ci2/n. Therefore, after data normalization, we have JXX = E[z2] − E[z] = E[x2]/(BX2 CX) − E[x]/BX. In the same manner, we have JYY = E[y2]/(BY2 CY) − E[y]/BY, and JXY = E[xy]/(BX BY CXY), where CXY = Σi ci,X ci,Y/n. After replacing these moments by their corresponding sampling moments, we obtain the results as shown in table 1.
Outlier Control
There are always a few outlier, i.e., extremely highly expressed genes. Their expression variations are very sensitive to the physiological or developmental condition when the sample was obtained. Because the distribution of RNA-seq is highly skewed, estimation of expression distance could be distorted by these outliers. As the first attempt, we implemented a simple cutoff to alleviate this problem: for the top 2.5% of highly expressed genes, we reset their RNA-seq counts to the value of the 97.5% quantile. Our preliminary analysis indicates that this approach is efficient and not sensitive to the selected cutoff (not shown).
Estimation of Overdispersion
If the number of biological replicates in RNA-seq data set is small, estimation of gene-specific overdispersion remains a difficult task. To deal with this problem, a number of statistical methods were proposed by sharing a certain amount of information between genes. For the practical reason, we implemented a fast but robust method to estimate the genome-wide overdispersion parameters ω (for ωX or ωY) by maximizing the joint likelihood function of NBDs.
We use genome X for illustration. Suppose that xik is the RNA-seq count of the k-th biological replicate of gene i. The log-likelihood function of gene i, denoted by liki(λi, ωX|xik), is formulated according to the NBD, whereas the mean (λi) is gene-specific and ωX is the common parameter. Thus, the overall likelihood function Lik over all genes is the sum of all liki(λi, ωX|xik). A standard numerical procedure can be applied to obtain the maximum likelihood estimate of ωX, which is converged rapidly when the moment estimate is used as an initial value: Let xi. and Vi,X be the sampling mean and variance of gene i. The initial estimate of ωX can be calculated as Σi (Vi,X − xi.)/Σi xi.2.
A Simple Method for Estimating Sampling Variance of UXY
The sampling variance of the estimated expression distance can be numerically calculated by the bootstrapping method. Nevertheless, by computer simulations we found that the following simple formula is close to the bootstrapping result: Var(UXY) = q/[(1 − q)n], where q = J2XY/JXX JYY, and n is the number of genes.
Results and Discussion
Mammalian Tissue Expression Evolution
We used mammalian RNA-seq data (Brawand et al. 2011) in brain, cerebellum, kidney, heart, and lung to demonstrate the application of our newly developed method. For simplicity, we used the RNA-seq counts of 5,636 1:1 orthologous genes used by the original authors. We estimated CX for the effect of sequence length variation in each genome. Since we observed that it has only a small-scale variation among genomes, we used the averaged correction constant C = 1.324 in the following analysis. By contrast, each tissue we have studied reveals a great deal of BX variation, suggesting that the sequencing depth variation among genomes should be corrected appropriately (see table 2 for examples). After estimating the effects of overdispersion, we calculated the pairwise expression distances between mammalian genomes (the up diagonal in table 3 for brain and the down diagonal for cerebellum); the sampling variances of expression distance are presented in the form of standard error. Apparently, the expression distance is small between phylogenetically closely related genomes and large between distantly related genomes. Based on the expression distance matrices, we reconstructed the genome expression phylogeny by the neighbor-joining method. For illustration, figure 3A shows the expression phylogeny for the mammalian brain. With some minor exceptions, the inferred tree is consistent with the known mammalian phylogeny, which correctly resolved the lineage of placentals, or eutherians from marsupials and monotremes, and separated two major eutherian lineages (primates and rodents). On the other hand, we mapped the expression distances onto the known mammalian phylogeny, as shown in figure 3B. We have performed all analyses in the other four tissues. All inferred expression phylogenies are roughly consistent with the known mammalian phylogeny. Similar to Brawand et al. (2011), we found that different tissues and lineages may show different expression distances. For instance, the expression rate of testes is more rapid than the rest of tissues. Because of the space limit, we will show these results in detail elsewhere.
Table 2.
Summary for the Estimates of Deep-Sequencing Parameters and Overdispersed Parameters in Mammalian Brains and Cerebellums
|
Bx |
ΩX |
|||
|---|---|---|---|---|
| Brain | Cerebellum | Brain | Cerebellum | |
| Human | 0.619 | 1.183 | 0.165 | 0.034 |
| Chimpanzee | 0.660 | 0.831 | 0.102 | 0.049 |
| Gorilla | 1.215 | 1.063 | 0.051 | 0.034 |
| Orangutan | 1.462 | 0.970 | 0.039 | 0.033 |
| Macaque | 0.846 | 0.598 | 0.046 | 0.009 |
| Mouse | 1.439 | 0.876 | 0.162 | 0.054 |
| Opossum | 1.030 | 0.746 | 0.153 | 0.003 |
| Platypus | 1.093 | 0.999 | 0.034 | 0.013 |
Table 3.
Pairwise Tissue Expression Distance (UXY) Matrix of Brain and Cerebellum in Mammals
| Human | Chimpanzee | Gorilla | Orangutan | Macaque | Mouse | Opossum | Platypus | |
|---|---|---|---|---|---|---|---|---|
| Human | 0 | 0.116 ± 0.038 | 0.174 ± 0.031 | 0.338 ± 0.021 | 0.247 ± 0.025 | 0.248 ± 0.025 | 0.473 ± 0.017 | 0.797 ± 0.012 |
| Chimpanzee | 0.304 ± 0.023 | 0 | 0.191 ± 0.029 | 0.300 ± 0.023 | 0.258 ± 0.025 | 0.333 ± 0.021 | 0.494 ± 0.017 | 0.799 ± 0.012 |
| Gorilla | 0.357 ± 0.021 | 0.329 ± 0.022 | 0 | 0.348 ± 0.021 | 0.299 ± 0.023 | 0.379 ± 0.020 | 0.512 ± 0.016 | 0.890 ± 0.011 |
| Orangutan | 0.523 ± 0.016 | 0.393 ± 0.019 | 0.511 ± 0.016 | 0 | 0.302 ± 0.023 | 0.426 ± 0.018 | 0.535 ± 0.016 | 0.912 ± 0.011 |
| Macaque | 0.468 ± 0.017 | 0.343 ± 0.021 | 0.456 ± 0.018 | 0.459 ± 0.018 | 0 | 0.306 ± 0.023 | 0.464 ± 0.018 | 0.852 ± 0.012 |
| Mouse | 0.493 ± 0.017 | 0.467 ± 0.017 | 0.549 ± 0.016 | 0.680 ± 0.014 | 0.518 ± 0.016 | 0 | 0.361 ± 0.020 | 0.704 ± 0.013 |
| Opossum | 0.810 ± 0.012 | 0.699 ± 0.013 | 0.785 ± 0.012 | 0.821 ± 0.012 | 0.672 ± 0.014 | 0.676 ± 0.014 | 0 | 0.512 ± 0.016 |
| Platypus | 1.010 ± 0.010 | 0.842 ± 0.012 | 0.976 ± 0.010 | 0.992 ± 0.010 | 0.823 ± 0.012 | 0.786 ± 0.012 | 0.777 ± 0.012 | 0 |
Note.—Up diagonal for brain and down diagonal for cerebellum; the sampling variances of expression distance are presented in the form of standard error.
Fig. 3.—

Mammalian brain expression phylogeny. (A) Expression phylogeny inferred by the neighbor-joining method based on expression distance matrix of brains. Nodes with * means bootstrapping values >0.95 and with ** values >0.99. (B) The result of mapping the expression distance to a given species tree, which is extracted from the tree of life (http://tolweb.org/, last accessed September 18, 2013).
Some Technical Comments
There are several ad hoc distance measures that have been used to analyze the divergence in expression. For instance, Brawand et al. (2011) used 1−R, where R is the Spearman’s correlation coefficient, and the Euclidean distance in their analyses. Although these measures are useful, our model-based expression distance has a unique strength for the study of transcriptome evolution because it provides a basis to generate testable hypotheses under the phylogenetic framework. In addition, our method has considered the effects of sampling and data processing so that the user can justify whether a conclusion is sensitive to the high throughput-dependent noise.
Our model implements a NBD to account for data over-dispersion. Though it is a common practice in statistics, some studies suggested that it may not be sufficient in RNA-seq analysis. Meanwhile, we use the lognormal-normal distribution to account for highly skewed RAN-seq variability. It remains our further work to evaluate whether the current model is the most appropriate for RNA-seq data, and how to improve the robustness of our method in the estimation of expression distance.
In real data analysis, application of new expression distance is difficult in the case of no biological replicate, because ΩX and ΩY cannot be estimated. To resolve this problem, we suggest a modified expression distance by omitting the overdispersion effects, that is,
| (8) |
Though U*XY tends to overestimate the expression distance, one can show that U*XY satisfies the “four-point condition” (Gu and Li 1996). In other words, U*XY is a paralinear distance to UXY, which has the following properties: 1) Under the strict additivity, the phylogenetic topology inferred from U*XY is the same as that from UXY. 2) External branch lengths tend to be overestimated, whereas internal branch lengths are expected to be unbiased. Our software has the option of paralinear expression distance estimation.
Software Availability
We have developed a software system, called PhyExp, short for phylogenomic analysis of expression profiles, to help the evolutionary analysis of RNA-seq data. There are several commercially available platforms such as Illumina, SOLiD, or 454 Genome Sequencer, but the RNA-seq data processing and analysis is about the same. Two distribution R packages, compatible with Windows and Linux operating systems, respectively, are available at http://www.xungulab.com (last accessed September 23, 2013). The first version, PhyExp1.0, has implemented the following options: 1) After the input file (RNA-Seq counts of genes) has been loaded, the expression distance matrix, including the paralinear distances, as well as their sampling variances are calculated.2) Infer the expression phylogeny by the neighbor-joining method; the statistical reliability can be examined via the bootstrapping. 3) PhyExp1.0 has the option to input the amino acid sequence alignment, which allows the user to map the expression distances onto the inferred molecular phylogeny or to a user-provided phylogeny.
There are several directions in further improvements: 1) Implement a suite of phylogeny-based analysis tools, including testing asymmetry of expression divergence, ancestral expression inference, and phylogeny-dependent detection of differentially expressed genes (unpublished results). 2) Develop and implement advanced methods for dealing with data normalization and data overdispersion. And 3) for the practical purpose, implement the option of expression divergence analysis based on microarray data. Moreover, we are particularly interested how expression divergence is correlated with sequence divergence as well as related phenotypes along the phylogeny (Lartillot and Poujol 2011).
Acknowledgments
This work was supported by the National Science Foundation of China (31272299) and the China State Key Basic Research Program (2012CB910101), and grants from Fudan University and Iowa State University. Y.Z. was supported by Specialized Research Fund for the Doctoral Program of Higher Education of China (20120071120009).
Literature Cited
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7:986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- Brawand D, et al. The evolution of gene expression levels in mammalian organs. Nature. 2011;478:343–348. doi: 10.1038/nature10532. [DOI] [PubMed] [Google Scholar]
- Caceres M, et al. Elevated gene expression levels distinguish human from non-human primate brains. Proc Natl Acad Sci U S A. 2003;100:13030–13035. doi: 10.1073/pnas.2135499100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- Cloonan N, et al. RNA-MATE: a recursive mapping strategy for high-throughput RNA-sequencing data. Bioinformatics. 2009;25:2615–2616. doi: 10.1093/bioinformatics/btp459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Y, Schafer DW, Cumbie JS, Chang JH. The NBP negative binomial model for assessing differential gene expression from RNA-Seq. Stat Appl Genet Mol Biol. 2011;10:1–28. [Google Scholar]
- Enard W, et al. Intra- and interspecific variation in primate gene expression patterns. Science. 2002;296:340–343. doi: 10.1126/science.1068996. [DOI] [PubMed] [Google Scholar]
- Eveland AL, McCarty DR, Koch KE. Transcript profiling by 3′-untranslated region sequencing resolves expression of gene families. Plant Physiol. 2008;146:32–44. doi: 10.1104/pp.107.108597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing RM, Claverie JM. EST databases as multi-conditional gene expression datasets. Pac Symp Biocomput. 2000;5:427–439. doi: 10.1142/9789814447331_0041. [DOI] [PubMed] [Google Scholar]
- Graveley BR, et al. The developmental transcriptome of Drosophila melanogaster. Nature. 2010;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X. Statistical framework for phylogenomic analysis of gene family expression profiles. Genetics. 2004;167:531–542. doi: 10.1534/genetics.167.1.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Gu X. Induced gene expression in human brain after the split from chimpanzee. Trends Genet. 2003;19:63–65. doi: 10.1016/s0168-9525(02)00040-9. [DOI] [PubMed] [Google Scholar]
- Gu X, Li WH. A general additive distance with time-reversibility and rate variation among nucleotide sites. Proc Natl Acad Sci U S A. 1996;93:4671–4676. doi: 10.1073/pnas.93.10.4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Su Z. Tissue-driven hypothesis of genomic evolution and sequence-expression correlations. Proc Natl Acad Sci U S A. 2007;104:2779–2784. doi: 10.1073/pnas.0610797104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Zhang Z, Huang W. Rapid evolution of expression and regulatory divergences after yeast gene duplication. Proc Natl Acad Sci U S A. 2005;102:707–712. doi: 10.1073/pnas.0409186102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huminiecki L, Wolfe KH. Divergence of spatial gene expression profiles following species-specific gene duplications in human and mouse. Genome Res. 2004;14:1870–1879. doi: 10.1101/gr.2705204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Kerr MK, Churchill GA. Bootstrapping cluster analysis: assessing the reliability of conclusions from microarray experiments. Proc Natl Acad Sci U S A. 2001;98:8961–8965. doi: 10.1073/pnas.161273698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitovich P, et al. A neutral model of transcriptome evolution. PLoS Biol. 2004;2: E132. doi: 10.1371/journal.pbio.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartillot N, Poujol R. A phylogenetic model for investigating correlated evolution of substitution rates and continuous phenotypic characters. Mol Biol Evol. 2011;28:729–744. doi: 10.1093/molbev/msq244. [DOI] [PubMed] [Google Scholar]
- Lu J, Tomfohr JK, Kepler TB. Identifying differential expression in multiple SAGE libraries: an overdispersed log-linear model approach. BMC Bioinformatics. 2005;6:165. doi: 10.1186/1471-2105-6-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makova KD, Li WH. Divergence in the spatial pattern of gene expression between human duplicate genes. Genome Res. 2003;13:1638–1645. doi: 10.1101/gr.1133803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozova O, Hirst M, Marra MA. Applications of new sequencing technologies for transcriptome analysis. Annu Rev Genomics Hum Genet. 2009;10:135–151. doi: 10.1146/annurev-genom-082908-145957. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifkin SA, Kim J, White KP. Evolution of gene expression in the Drosophila melanogaster subgroup. Nat Genet. 2003;33:138–144. doi: 10.1038/ng1086. [DOI] [PubMed] [Google Scholar]
- Robinson MD, Smyth GK. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics. 2007;23:2881–2887. doi: 10.1093/bioinformatics/btm453. [DOI] [PubMed] [Google Scholar]
- Robinson MD, Smyth GK. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics. 2008;9:321–332. doi: 10.1093/biostatistics/kxm030. [DOI] [PubMed] [Google Scholar]
- Sultan M, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YH, Xia K, Wright FA. A powerful and flexible approach to the analysis of RNA sequence count data. Bioinformatics. 2011;27:2672–2678. doi: 10.1093/bioinformatics/btr449. [DOI] [PMC free article] [PubMed] [Google Scholar]