

Abstract
Expression quantitative trait loci (eQTLs) have been found to be enriched in trait-associated single-nucleotide polymorphisms (SNPs). However, whether eQTLs are adaptive to different environmental factors and its relative evolutionary significance compared with nonsynonymous SNPs (NS SNPs) are still elusive. Compiling environmental correlation data from three studies for more than 500,000 SNPs and 42 environmental factors, including climate, subsistence, pathogens, and dietary patterns, we performed a systematic examination of the adaptive patterns of eQTLs to local environment. Compared with intergenic SNPs, eQTLs are significantly enriched in the lower tail of a transformed rank statistic in the environmental correlation analysis, indicating possible adaptation of eQTLs to the majority of 42 environmental factors. The mean enrichment of eQTLs across 42 environmental factors is as great as, if not greater than, that of NS SNPs. The enrichment of eQTLs, although significant across all levels of recombination rate, is inversely correlated with recombination rate, suggesting the presence of selective sweep or background selection. Further pathway enrichment analysis identified a number of pathways with possible environmental adaption from eQTLs. These pathways are mostly related with immune function and metabolism. Our results indicate that eQTLs might have played an important role in recent and ongoing human adaptation and are of special importance for some environmental factors and biological pathways.
Keywords: eQTLs, environmental adaptation, population genetics
Introduction
There has been a long-standing debate about the evolutionary significance of regulatory mutations and their prevalence in adaptation to local environments (King and Wilson 1975; Carroll 2005; Wray 2007; Zheng et al. 2011; Wittkopp and Kalay 2012). Most of our current knowledge on the evolutionary patterns of mutations and their phenotypic consequences have been mainly gained from mutations in coding regions, whose identification and functional assessment are much easier than regulatory variation (Carroll 2005; Wray 2007; Dermitzakis 2008). However, evidence of regulatory adaptation during human evolution, including recent and ongoing adaptation to local environment, has been mounting (Wray 2007). One classic example is the parallel adaptations of multiple regulatory mutations in LCT for milk consumption in North Europeans (Enattah et al. 2002; Bersaglieri et al. 2004), East Africans (Tishkoff et al. 2007), and Saudi Arabians (Enattah et al. 2008). Another case in point is the near fixation of a regulatory mutation in the promoter of DARC in the sub-Saharan African populations, which abolishes the expression of DARC in erythrocytes and benefits its homozygous carriers with complete resistance to malarial parasites, Plasmodium vivax (Tournamille et al. 1995; Hamblin and Di Rienzo 2000). Such studies have highlighted the importance of regulatory mutations in shaping human physiology, behavior, and cognition (Prabhakar et al. 2006; Haygood et al. 2007; Wray 2007).
The difficulty of accurate and comprehensive identification of regulatory mutations has hindered evolutionary studies on their functional importance. The recent expansion of expression quantitative trait loci (eQTLs) that are identified by the genome-wide association studies (GWAS) between genotypes and gene expression levels provides a unique opportunity for genome-wide evolutionary assessments of the underlying regulatory variants (Myers et al. 2007; Stranger et al. 2007; Schadt et al. 2008; Veyrieras et al. 2008; Dimas et al. 2009; Montgomery et al. 2010; Pickrell et al. 2010; Zeller et al. 2010). As proxies of regulatory variants, eQTLs have been shown to be enriched in single-nucleotide polymorphisms (SNPs) associated with complex traits and diseases (Dermitzakis 2008; Nica et al. 2010; Nicolae et al. 2010). However, the significance of genome-wide eQTLs in the evolutionary context has remained nearly unexplored. Preliminary analysis found that eQTLs tend to overlap with signals of incomplete selective sweeps as detected by integrated haplotype score (iHS) (Kudaravalli et al. 2009), warranting further verifications and systemic examinations with varying selection–detecting approaches, which capture different aspects or types of selection signals (Hancock, Alkorta-Aranburu et al. 2010; Hancock, Witonsky et al. 2010).
Environmental correlation is a way of detecting adaptation by testing whether the spatial distribution of the frequency of an allele could be explained by an environmental factor. This selection–detection method has a special advantage of providing an ecological context that exerts selection pressure and is capable of detecting different types of selection, including hard and soft selective sweeps, and adaptation by subtle allele frequency shift (Hancock, Alkorta-Aranburu et al. 2010; Pritchard et al. 2010; Ye and Gu 2011). A specific method of environmental correlation based on a Bayesian linear model was recently developed (Coop et al. 2010) and applied to more than 500,000 SNPs and more than 35 environmental factors (Hancock, Witonsky et al. 2010; Hancock et al. 2011; Fumagalli et al. 2011). This method effectively quantifies the correlation between allele frequency and environmental factor while controlling for population structure. For each SNP and each environmental factor, this method yields a Bayes Factor (BF), which indicates the strength of evidence that the environmental factor influences the frequency of the SNP in local populations. With this method, genic and nonsynonymous (NS) SNPs have been shown to be enriched in signals of adaptation to a wide spectrum of environmental factors, including climate, subsistence, dietary patterns, and pathogens (Hancock, Witonsky et al. 2010; Hancock et al. 2011; Fumagalli et al. 2011). This method, with the currently available eQTL data set, provides a great opportunity to investigate the importance of regulatory variants in recent and ongoing human environmental adaptation and to compare qualitatively and quantitatively their relative importance to NS SNPs. In this study, we conducted such analyses and present strong evidence that regulatory mutations played important roles in recent and ongoing human adaptation to local environment.
Material and Methods
Environmental Correlation Data
We collected the environmental correlation data from three large-scale studies, which span more than 550,000 SNPs and 42 environmental factors (36 individual environmental factors and 6 environmental categories, fig. 1A and B). We referred to these three studies as Hancock et al. PNAS (Hancock, Witonsky et al. 2010), Hancock et al. Plos (Hancock et al. 2011), and Fumagalli et al. Plos (Fumagalli et al. 2011), and labeled environmental factors from each study with prefixes 1-, 2-, and 3-, respectively. Hancock et al. PNAS and Hancock et al. Plos used the same sample of 61 worldwide human populations, including 52 Human Genome Diversity Project panel populations, 4 HapMap phase III populations, and 5 additionally genotyped populations. Hancock et al. PNAS includes 595,891 SNPs and 11 environmental factors (1-Polar domain, 1-Humid temperate domain, 1-Dry domain, 1-Humid tropical domain, 1-Foraging, 1-Horticulture, 1-Pastoralism, 1-Agriculture, 1-Cereals, 1-Roots and tubers, 1-Fats, meat, and milk). Hancock et al. Plos contains 623,318 SNPs and 11 environmental factors (2-Latitude, 2-Minimum temperature [Winter], 2-Maximum temperature [Summer], 2-Precipitation rate [Summer], 2-Precipitation rate [Winter], 2-Short wave radiation flux [Summer], 2-Short wave radiation flux [Winter], 2-Relative humidity [Summer], 2-Relative humidity [Winter], 2-Absolute latitude, 2-Longitude). Fumagalli et al. Plos used 55 human populations by joining data from the Human Genome Diversity Project and HapMap Phase III. It has 552,134 SNPs and 14 environmental factors (3-Distance from the sea, 3-Virus diversity, 3-Bacteria diversity, 3-Protozoa diversity, 3-Helminths diversity, 3-Relative humidity, 3-Temperature [annual mean], 3-Precipitation rate [annual mean]; 3-Net short wave radiation flux; 3-Gathering; 3-Hunting; 3-Fishing; 3-Animal husbandry; 3-Agriculture). The climate-related environmental factors in Fumagalli et al. Plos are annual means whereas those in Hancock et al. Plos are separated into Summer and Winter components. The subsistence-related environmental factors in Fumagalli et al. Plos are continuous, representing the percentage of time spent on a specific activity whereas those in Hancock et al. PNAS are binary.
Fig. 1.—

eQTLs are enriched in signals of environmental adaptation. The ERs of eQTLs to intergenic SNPs in the 5% tail of the transformed rank statistics for (A) 36 individual environmental factors and (B) 6 environmental categories. Mean and standard error for each ER are estimated from 1,000 whole-genome block bootstraps. Six environmental categories are defined by grouping individual environmental factors as indicated at the left. Colors blue, orange, magenta, and green represent ecoregion, subsistence, climate, and pathogen, respectively. (C) The box plot of empirical ERs of eQTLs to intergenic SNPs in the tail of the transformed rank statistics for 42 environmental factors under three tail cutoffs. The ERs for each environmental factor under three cutoffs are connected with dashed lines. Environmental categories are represented by dash lines with the same colors as figure 1B. The red squares, connected by a red line, are the means of all 42 empirical ERs under three tail cutoffs. The P values are for generalized paired Z tests of whether eQTLs with more stringent tail cutoffs have higher mean ERs. (D) Progressive enrichment of eQTL SNPs in comparison to intergenic SNPs for increasing values of prediction accuracy (Q2) of 14 environmental factors. Red line indicates the median value of enrichment of eQTLs from 1,000 block bootstraps whereas blue line is that for intergenic SNPs. Pink region denotes the 90th confidence interval for eQTL enrichment.
All three studies applied the same Bayesian linear model method (Coop et al. 2010) to test the association between a SNP and an environmental factor. For each SNP and each environmental factor, this method yields a BF, which indicates the strength of evidence that the environmental factor influences the frequency of the SNP in local populations. To further examine the statistical significant of a BF, a transformed rank statistic (also known as empirical p value) was calculated based on its rank in BFs for a group of SNPs in the same ascertainment panel and within the same allele frequency bin. We obtained the data of transformed rank statistics for each SNP and environmental factors from dbCLINE (http://genapps2.uchicago.edu:8081/dbcline/main.jsp, last accessed January 20, 2012) or directly from the authors, Fumagalli et al. To summarize the evidence of association for each SNP with the six categories of variables (1-Ecoregion, 1-Subsistence, 2-Climate, 3-Subsistence, 3-Climate, and 3-Pathogen), we followed the method used in the original studies by assigning the minimum of transformed rank statistics across all individual variables in the category (Hancock, Witonsky et al. 2010; Hancock et al. 2011).
eQTLs Data
eQTLs data were downloaded from http://eqtl.uchicago.edu/cgi-bin/gbrowse/eqtl/ (last accessed February 10, 2012) which is a compilation of eQTLs identified in eight large-scale studies in different human tissues (Zeller et al. 2010). All eQTLs were used when the analyses were restricted to eQTLs itself. But when we did comparison between eQTLs and genic (or NS) SNPs, only eQTLs associated with RefSeq-supported protein-coding genes (see definition below) were used. We denoted these two groups of eQTLs as eQTLs-all and eQTLs-for comparison, respectively.
Definitions of Genomic Regions
Gene annotations based on hg18 were downloaded from UCSC Genome Browser database. Only autosomal genes were retrieved because environmental correlation data were only available for autosomal SNPs. In total, there were 24,814 autosomal genes, 18,396 of which were coding genes whereas the rest were noncoding genes. For genes with multiple isoforms, the longest transcript was used. If there were multiple transcripts of the same length, one was arbitrarily chosen. Based on these 24,814 autosomal genes, we defined intergenic SNPs as those at least 50 kb away from known genes, either coding or noncoding. There were 16,914 autosomal coding genes with support from RefSeq and thus they were used in the definition of genic SNPs and NS SNPs. Genic SNPs were defined as those within 5 kb of a gene. Annotation to SNPs is also downloaded from UCSC Genome Browser database. A SNP is called NS based on the longest transcript used in the analysis. To compare the enrichment ratio (ER) of eQTLs with that of genic SNPs (or NS SNPs), only eQTLs for these 16,914 genes were used. Further, to control for the potential difference between genes with and without eQTLs, we restricted this comparison for only genes with eQTLs. We denoted genic SNPs for this group of gene as e-genic SNPs and NS SNPs as e-NS SNPs. The numbers of SNPs in each group and each data set are available in supplementary table S11, Supplementary Material online. All data are available upon request.
Enrichment of eQTLs, Genic, and NS SNPs in the Lower Tail of the Transformed Rank Statistic Distribution
To examine whether there is an excess of tested SNPs in the lower tail of the transformed rank statistic distribution, which is the empirical distribution of the transformed rank statistics for genome-wide SNPs, we calculated an ER, which is the ratio of the percentage of tested SNPs in the lower tail to the corresponding percentage of intergenic SNPs. The tested SNPs could be genic SNPs, NS SNPs, or eQTLs, whereas the intergenic SNPs were treated as the neutral control. Although not all intergenic SNPs are neutral, the presence of adaptive SNPs in the control could only lead to a smaller ER, which will make our results more conservative. An ER >1 indicates that there is an excess of tested SNPs in the lower tail compared with intergenic SNPs. To avoid the arbitrary choice of cutoffs in the definition of the lower tail, we used three cutoffs, 5%, 1%, and 0.5%. To assess the significance of an observed excess, we applied a block bootstrap resampling procedure which corrected for the nonindependence of nearby loci because of linkage disequilibrium (LD). To this end, we broke the genome into 500-kb nonoverlapping segments and bootstrap resampled a number of segments to make a pseudo-genome of the same length as the real genome. For the pseudo-genome, we calculated the ER just as we did for the empirical genome. We performed 1,000 bootstrap replicates to obtain a confidence interval for the observed excess. We considered an observed excess significant if at least 95% of the bootstrap replicates produced ERs >1.
One specific feature for eQTLs, different from genic SNPs and NS SNPs, is that they tend to cluster together because they are identified with association analysis. If the clustering of eQTL overlaps with the clustering of significant correlative signals indicated by the BF, the block bootstrap may not be powerful to control for this effect because each time a block of SNPs are drawn rather than a single SNP. To totally break down the association among eQTLs for a gene, we performed the following random simulation. For genes with multiple eQTLs, only one eQTL was randomly chosen and used for the calculation of the ER. We repeated this procedure 1000 times and we considered an observed excess significant if 95% of the simulations reveal ER >1. This random simulation procedure gave essentially the same pattern of significance (supplementary table S2, Supplementary Material online) and thus for most analyses we only used the block bootstrap procedure.
Enrichment of eQTLs, Genic, and NS SNPs in the Higher Tail of Prediction Accuracy (Q2) in Relative to Intergenic SNPs
Prediction accuracy data were obtained from Fumagalli et al. (2011) and data were available for 552,136 SNPs for each of four categories of environmental factors (all environmental factors, climate, subsistence, and pathogen). Following the method used in the original study, we divided the distribution of Q2 into bins. The number of bins was chosen to ensure that each bin has a large enough number for block bootstrap and the calculation of enrichment. Enrichment here is defined as the proportion of tested SNPs (genic, NS, eQTLs, and intergenic) in the bin divided by the total proportion of tested SNPs. Intergenic SNPs are used as control for other tested SNPs (genic, NS, and eQTLs). For example, when calculating the enrichment for eQTLs in comparison to intergenic SNPs, the total proportion of eQTLs is the number of eQTLs divided by the total number of eQTLs and intergenic SNPs, whereas the proportion of eQTLs in a specific bin is the number of eQTLs in the bin divided by the total number of eQTLs and intergenic SNPs in the bin. We applied the same block bootstrap procedure as described in the previous section to estimate the confidence interval of each enrichment value.
Statistical Tests Comparing ERs
To compare the ERs of different groups of SNPs, we did the analysis at two levels: for each individual environmental factor/category and for all environmental factors. When doing analysis for a specific environmental factor, we used ERs from 1,000 bootstraps. For example, when we compared eQTLs with NS SNPs for the climate category, we had 1,000 pairs of ERs from 1,000 bootstraps and we performed a paired one-tailed two-sample t-test.
When doing the analysis for all environmental factors as a whole to detect the general trend, we cannot directly apply t-test because observations for different environmental factors are not independent of each other. Therefore to test whether there is difference between the means of ERs for two groups of SNPs, we developed a generalized paired Z-test by incorporating the correlations among different environmental factors. The test was developed as follows. Let 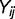 denote the response for environmental factor
denote the response for environmental factor 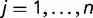 in group
in group 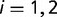 . In our case,
. In our case, 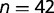 . We can apply a mixed effect model:
. We can apply a mixed effect model:
where 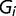 is the fixed effect of group
is the fixed effect of group 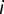 ,
, 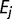 is the random effect of environmental factor
is the random effect of environmental factor 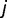 , and
, and 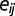 is the random error. The random errors for environmental factors within the same group are correlated. With the assumption of multivariate normal distribution, we have
is the random error. The random errors for environmental factors within the same group are correlated. With the assumption of multivariate normal distribution, we have 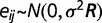 . The correlation matrix (
. The correlation matrix (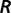 ) among different environmental factors was estimated from all SNPs with the transformed rank statistics for all 42 environmental factors. By pairing
) among different environmental factors was estimated from all SNPs with the transformed rank statistics for all 42 environmental factors. By pairing 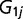 with
with 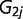 to take into account the dependence between the two observations for each environmental factor, we have a simpler model:
to take into account the dependence between the two observations for each environmental factor, we have a simpler model:
where 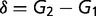 and
and 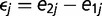 with
with 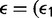
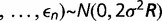 . The observed differences for each environmental factor are
. The observed differences for each environmental factor are 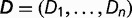 . The mean difference
. The mean difference 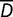 has variance:
has variance:
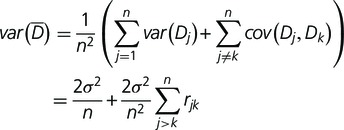 |
 and
and 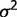 can be estimated using maximum likelihood estimation from observed differences
can be estimated using maximum likelihood estimation from observed differences 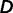 while taking into account correlations among environmental factors. Assuming multivariate normal distribution for
while taking into account correlations among environmental factors. Assuming multivariate normal distribution for 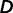 , we have the following probability density function:
, we have the following probability density function:
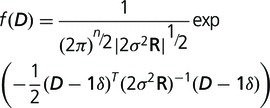 |
where 1 is a vector of n 1 s. By taking derivatives of the log-likelihood function with respect to  and
and 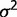 and setting the resulted derivatives to be zero, we solved the equations and got:
and setting the resulted derivatives to be zero, we solved the equations and got:
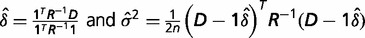
A test for difference between two groups can be based on a Z statistic,
where 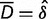 and
and 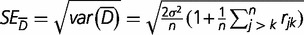 . The P value of an observed Z statistic was found from a standard normal distribution.
. The P value of an observed Z statistic was found from a standard normal distribution.
Controlling for Recombination Rate
Recombination rate data were downloaded from Hapmap (hapmap.ncbi.nlm.nih.gov/downloads/recombination/2008-03_rel22_B36/rates/, last accessed September 17, 2013). These data were calculated from four continental populations and thus were only approximation of recombination rates in global populations. Combining three data sets (Hancock et al. PNAS, Hancock et al. Plos, and Fumagalli et al. Plos), there were 639,663 SNPs, out of which 633,340 had point estimate of recombination rate. These SNPs were grouped into five equal-sized bins, corresponding to median recombination rate of 0.0354, 0.187, 0.445, 1.016, and 4.236 cM/Mb. Five bins were chosen to ensure that for each type of variation, there was enough number of SNPs in each bin for the calculation of ER. For different types of variations, the numbers of SNPs for each bin were different. Each bin of recombination rate was denoted as the log(median recombination rate). The proportion of significant SNPs in the lower tail (defined with three cutoffs, 5%, 1%, and 0.5%) was calculated for each bin and each type of variation, including intergenic SNPs. The ER of a specific type of variation (genic, NS, or eQTLs) for each environmental factor in each bin of recombination rate was calculated using corresponding intergenic SNPs in the bin as control. To demonstrate the general trend of the effect of recombination rate on the proportion of significant SNPs (or ER), simple linear regressions were performed between the median of recombination rate and the mean of the proportion of significant SNPs (or the mean of ER) in each bin.
In addition to using population-based estimates of recombination rate, we further confirmed our analysis with a pedigree-derived sex-averaged recombination map from deCODE (Kong et al. 2010), from which recombination rate for each SNP was calculated with a 500 kb window centered on the SNP.
Canonical Pathway Enrichment
To determine whether there is an enrichment of eQTLs in a canonical pathway, for each environmental factor (or category), we did the following analyses. For each environmental factor, we calculated ERs separately for both eQTLs and genic SNPs. However, it is of note that ERs calculated here is different from those for genome-wide pattern. The control we used here was not intergenic SNPs. Rather, for eQTLs we used all other eQTLs not in the pathway as control, and for genic SNPs we used all other genic SNPs not in the pathway. We did the analyses for three different cutoffs (5%, 1%, and 0.5%). The significance of enrichment was assessed with 1,000 block bootstraps as described before. Pathways of interest are those that have significant enrichment signals for eQTLs across three cutoffs but have no consistent significant enrichment signals for genic SNPs.
Results
We extracted environmental correlation data from three large-scale studies, which span more than 550,000 SNPs and 42 environmental factors (36 individual environmental factors and 6 environmental categories, fig. 1A and B). We referred to these three studies as Hancock et al. PNAS (Hancock, Witonsky et al. 2010), Hancock et al. Plos (Hancock et al. 2011), and Fumagalli et al. Plos (Fumagalli et al. 2011), and labeled environmental factors from each study with prefixes 1-, 2 -, and 3-, respectively. Climate- and subsistence-related environmental factors are included in two of the studies but they represent different aspects of the same environmental variables. For instance, climate-related factors in Study 2 are separated into winter and summer components whereas those in Study 3 are annual means. Subsistence-related factors in Study 1 are treated as binary variables whereas those in Study 3 are continuous and representing the percentage of time a population spent on a specific activity. For each SNP, we retrieved its transformed rank statistic (also known as empirical P value), for each of the 42 environmental factors. Although the transformed rank statistics of a SNP are correlated among environmental factors (supplementary fig. S1, Supplementary Material online), to fully exploit the information available, we included all 42 environmental factors in our analysis while accounting for their correlations. A statistic called ER, the ratio of the proportion of tested SNPs to that of intergenic neutral SNPs in the lower tail of the transformed statistic, was used to test whether a group of SNPs is enriched in signals of adaptation to a specific environmental factor (Hancock, Witonsky et al. 2010; Hancock et al. 2011). Groups of SNPs important for adaptation will be enriched in the lower tail and have ERs >1. The more important a group of SNPs is in adaptation, the larger the ER.
We combined eQTLs that are identified in eight large-scale studies in different human tissues (Myers et al. 2007; Stranger et al. 2007; Schadt et al. 2008; Veyrieras et al. 2008; Dimas et al. 2009; Montgomery et al. 2010; Pickrell et al. 2010; Zeller et al. 2010). By integrating the eQTLs data and the environmental correlation data, we examined whether eQTLs are enriched in signals of environmental correlation at the genome-wide level and compared their enrichment pattern with that of NS SNPs.
Genome-Wide Enrichment of eQTLs in Environmental Correlation
The ERs of eQTLs to intergenic SNPs were calculated for each of the 36 individual environmental factors and 6 environmental categories. The significance of each ER is assessed by a whole-genome block bootstrap procedure. To avoid the arbitrary use of tail cutoff, we used 5%, 1%, and 0.5% to define the lower tails of the statistic distribution. For the 36 individual environmental factors (fig. 1A and supplementary table S1, Supplementary Material online), 32 (89%) have ER significantly larger than 1 under at least one tail cutoff and 18 (50%) have significant ERs across all three tail cutoffs. Under the tail cutoff of 5%, short-wave radiation flux in winter has the largest ER of 1.44, suggesting that the enrichment of eQTLs in the 5% lower tail is 44% higher than neutral expectation. Consistently, the annual mean of net short wave radiation flux from Study 3 has the second largest ER of 1.31, 31% higher than neutral expectation. Individual factors in categories other than climate also exhibit significant ERs, such as polar domain (1.29), foraging (1.24), helminths diversity (1.18), roots and tubers (1.22), and absolute latitude (1.30). Taken together, eQTLs are observed to be enriched in the lower tail of the transformed rank statistics for a majority of environmental factors examined here, including ecoregion, climate, subsistence, pathogen, and dietary patterns.
For all six environmental categories examined (fig. 1B and supplementary table S1, Supplementary Material online), the ERs of eQTLs are consistently and significantly larger than 1. Under the tail cutoff of 5%, when summarizing over the summer and winter components of all related environmental factors from Study 2, the climate category has an empirical ER of 1.13. Consistently, when summarizing over the annual means of all related factors from Study 3, the empirical ER for the climate category is 1.14. For the subsistence category, when all related factors are treated as binary, the summarized ER is 1.10 and it is 1.11 when all related factors are treated as continuous. Moreover, the ERs for the categories of ecoregion and pathogens are 1.11 and 1.08, respectively. Over all 42 environmental factors, the mean empirical ERs of eQTLs are 1.15, 1.27, and 1.31, respectively, for the three tail cutoffs (fig. 1C). They are significantly larger than 1 (P values are 1.02 × 10−13, 3.36 × 10−14, and 3.09 × 10−10, respectively, with generalized one-tailed paired Z tests). As previously observed for genic and NS SNPs (Hancock, Witonsky et al. 2010; Hancock et al. 2011), ERs of eQTL SNPs increase with more stringent tail cutoffs, suggesting that in general eQTLs are enriched in signals of environmental adaptation.
One observation worth pointing out is that for the same type of climate-related environmental factors, such as temperature, humidity, and precipitation rate, separating them into summer and winter components provides stronger evidence of enrichment for eQTLs. For instance, while the annual mean of temperature has significant eQTLs enrichment under two tail cutoffs, the minimum temperature in winter and the maximum temperature in summer have significant signals over all three tail cutoffs (supplementary table S1, Supplementary Material online). Another interesting case is precipitation. Although the annual mean of precipitation rate has significant signal only in the 1% tail, the precipitation rate in winter has significant enrichment under all three cutoffs and the precipitation rate in summer has no significant results (supplementary table S1, Supplementary Material online). This pattern suggests the presence of season-specific selection pressures from different environment factors during human evolution.
Another statistic, prediction accuracy (Q2), from a different implementation of environmental correlation provides us an opportunity to verify our observation that eQTLs are enriched in signals of environmental adaptation. Prediction accuracy, developed by Fumagalli et al. (2011), is a measure of how well a group of environmental variables predict the global frequency distribution of an allele. The prediction accuracy for SNPs that are adaptive to the tested environmental factor is expected to be higher than neutral SNPs. We retrieved prediction accuracy data for more than 550,000 SNPs for 14 climate, subsistence, and pathogen-related environmental factors that were used in Fumagalli et al. (2011). When all environmental factors were combined as a single predictor, we observed a significant enrichment of eQTLs compared with intergenic SNPs in the highest bin of prediction accuracy (fig. 1D). The median value for the resampled distribution of the enrichment statistic is 1.08 (P < 0.05). These observations verified that environmental adaptation has shaped the differential allele frequency distribution of eQTLs among human populations. If these 14 environmental factors are separated into three categories (climate, subsistence, and pathogen), we observed a similar trend of progressive enrichment of eQTLs for higher values of prediction accuracy (supplementary figs. S2–S5, Supplementary Material online).
eQTLs and NS SNPs in Environmental Adaptation
Our data allowed us to investigate whether regulatory variations are qualitatively and quantitatively distinct from coding variations in environmental adaptations. To compare the relative prevalence of eQTLs and NS SNPs in environmental adaptation, we tested whether the ER of eQTLs is significantly larger than that of NS SNPs. As shown in figure 2, under the tail cutoff of 5%, the ER of eQTLs is 1.16, which is significantly higher than that of genic SNPs, 1.04 (p = 1.500e−09). The mean ER of eQTLs is also significantly higher than that of genome-wide NS SNPs, 1.11 (P = 0.0102). Because not all genes have eQTLs, it is possible that genes with eQTLs may have different ERs from genome-wide patterns, thus leading to a biased comparison. To correct for this, we further calculated ERs for genic and NS SNPs only for the genes with eQTLs (denoted as e-genic and e-NS SNPs). Under the same cutoff, the comparison between eQTLs and e-NS SNPs supports the above trends, but the ER difference between these two categories of SNPs is not statistically significant. Similar patterns were observed under the other two tail cutoffs (1% and 0.5%) (supplementary fig. S6 and table S3, Supplementary Material online). Taken together, these results indicate that in general eQTLs are as prevalent as, if not more prevalent than, NS SNPs in environmental adaptation.
Fig. 2.—
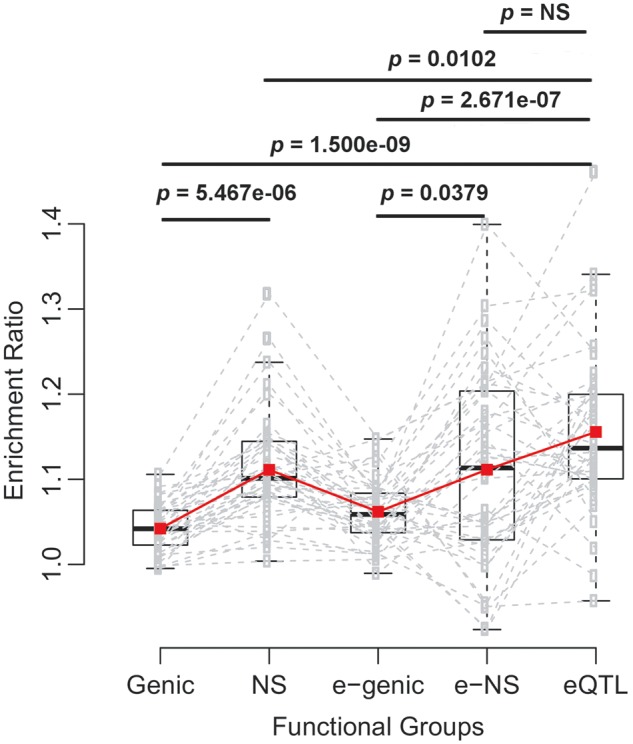
ERs of different types of SNPs in the tail of the transformed rank statistic. Five types of SNPs were analyzed, including genome-wide genic and NS SNPs (Genic and NS), genic and NS SNPs for genes with eQTLs (e-genic and e-NS), and eQTLs whose associated genes were included in our analyses. For each type of SNPs, ERs were calculated for all 42 environmental factors. Each dashed line connects ERs calculated from one environmental factor. The red line connects mean ERs of each SNP group. The P values are for generalized paired Z tests of whether the mean ERs of one group is larger than that of the other group. The tail cutoff is 5%. The patterns are similar for cutoffs of 1% and 0.5% (see supplementary fig. S6, Supplementary Material online).
We further identified environmental factors to which eQTLs are more likely to be adaptive than NS SNPs. To this end, we required that the ER of eQTLs be significantly larger than those of genic, NS, e-genic, and e-NS SNPs across all three tail cutoffs. Among 42 environmental factors examined, 8t have consistent and significant signals (fig. 3 and supplementary fig. S7, Supplementary Material online). They include cereals, latitude, short-wave radiation flux in summer, short-wave radiation flux in winter, relative humidity in winter, virus diversity, fishing, and the climate category summarizing over seasonal components. Although these eight factors encompass categories of climate, subsistence, pathogens, and dietary patterns, four of them are climate-related and climate is the only environmental category identified. It is noteworthy that these climate-related factors are continuous, as are the other three environmental factors except cereals. The prevalence of eQTLs over NS SNPs in adaptation to continuous environmental factors underlies the possibility that gene expression, because of its continuous nature, is more suitable than protein function to be fine-tuned to meet the dynamic range of continuous environmental factors (Wray 2007).
Fig. 3.—
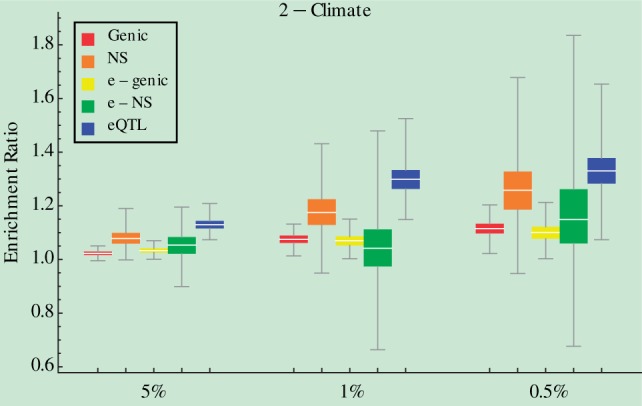
Box plots of ERs from 1,000 whole-genome block bootstraps for five groups of SNPs under three tail cutoffs. Enrichment was examined at the level of each environmental factor/category. “2-Climate” is presented here. Another seven cases could be found in supplementary figure S7, Supplementary Material online. P values for paired t-tests of whether ERs of eQTLs are larger than other types of SNPs for each individual environmental factor could be found in supplementary table S4, Supplementary Material online.
Greater Enrichment of Adaptive eQTLs in Regions of Low Recombination
Signals of background selection and selective sweep tend to extend longer in genomic regions of low recombination. Therefore, in those regions, the selective signal on a causal locus is more likely to be captured by nearby linked loci (Cai et al. 2009; Keinan and Reich 2010). If the underlying regulatory variations tagged by eQTLs are adaptive, eQTLs in region of low recombination are expected to show stronger footprint of adaptation. To explore the effect of recombination rate on the ERs of eQTLs, we divided all SNPs into five bins of equal number of SNPs based on their recombination rates and calculated ERs for each of 42 environmental factors in each bin. Linear regression analyses were performed between the natural logarithm of median recombination rate and the mean of ERs over 42 environmental factors. Significant inverse correlations were observed between local recombination rate and ERs of eQTLs SNPs (fig. 4 and supplementary fig. S8, Supplementary Material online). Similar results are also observed for the genic, NS, and synonymous SNPs.
Fig. 4.—
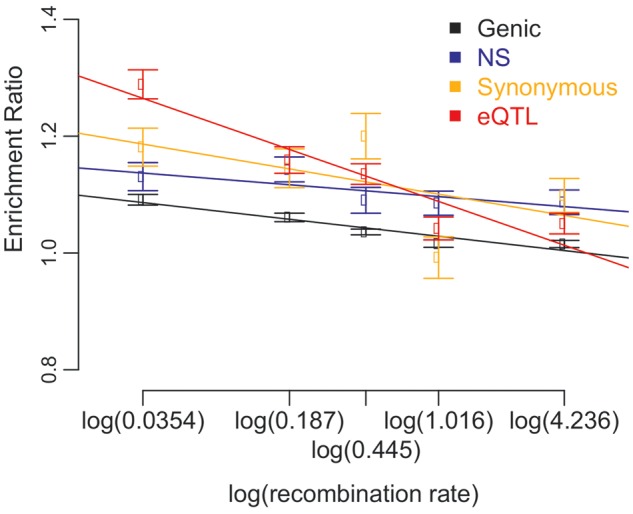
The effect of recombination rate on ER of eQTLs, genic, NS, and synonymous SNPs. The tail cutoff is 5%. The patterns are similar for cutoffs of 1% and 0.5% (supplementary fig. S8, Supplementary Material online). Only a subset of eQTLs associated with RefSeq-supported protein-coding genes (eQTLs-for comparison) were used in these analyses. The results for all eQTLs (eQTLs-all) have similar patterns (supplementary fig. S9, Supplementary Material online). The P values for generalized paired Z tests of whether the mean ERs are larger than 1 in each bin of recombination rate could be found in supplementary table S5, Supplementary Material online.
Over all 42 environmental factors, the mean ERs of eQTLs in the 5% tail are significantly larger than 1 across all five bins of recombination rate (fig. 4 and supplementary table S5, Supplementary Material online). Similar patterns are observed under the other two cutoffs (supplementary fig. S8 and table S5, Supplementary Material online). Furthermore, we compared the ERs of different types of SNPs in the five recombination rate bins. Considering the significance over all three tail cutoffs, ERs of eQTLs are larger than that of genic SNPs for the first three recombination rate bins whereas ERs of NS SNPs are larger than that of genic SNPs for the first two bins, indicating the genome-wide patterns of higher ERs of eQTLs and NS SNPs than genic SNPs are mainly driven by SNPs from the regions of low recombination.
Enrichment of eQTLs in Environmental Adaptation for Specific Biological Functions
To identify biological pathways undergoing regulatory environmental adaptation, for each environmental factor, we identified biological pathways with consistent and significant enrichment of eQTLs, but not genic SNPs, in the lower tail of the transformed rank statistic. As indicated in table 1 and supplementary table S6, Supplementary Material online, most of the significant pathways we identified are mainly related to immune system, cellular signaling, and metabolism. For instance, eQTLs associated with genes involved in biological oxidations are significantly enriched in the lower tail of the transformed rank statistic for the environmental factor of dry domain with significant ERs of 2.11, 3.88, and 6.02, respectively for three tail cutoffs. In comparison, genic SNPs in the same pathway are not significantly enriched with ERs of 0.93, 0.57, and 0.94, respectively. For the pathway of peroxisomal lipid metabolism, eQTLs are enriched for short-wave radiation flux in summer with significant ERs of 2.86, 9.36, and 17.30, much higher than those of the genic SNPs. For genes involved in signaling in immune system, eQTLs are significantly enriched in signals of environmental correlation for the subsistence category with binary individual factors, whose ERs are 1.44, 1.85, and 2.27.
Table 1.
A Subset of Biological Pathways with Consistently Significant ERs for eQTLs but Not Genic SNPs
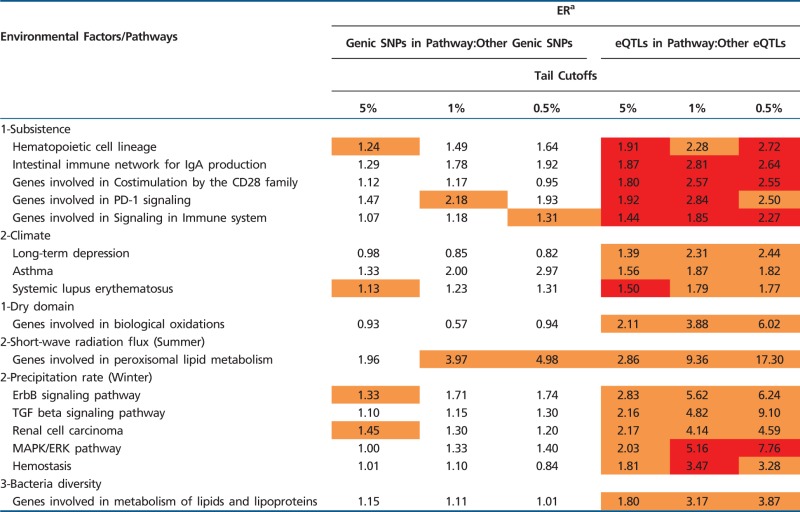 |
Note.—Red and orange indicate, respectively, >99% and >95% of bootstrap replicates having ERs > 1. A complete list of identified pathways is available in supplementary table S6, Supplementary Material online.
aThe level of significance was estimated by whole-genome block bootstrap.
The prevalence of immune-related genes with significant regulatory adaptation is consistent with previous findings that active binding sites for immune-related TFs are among the most highly eQTL-enriched regions in the genome (Gaffney et al. 2012). It has been found that eQTLs of genes interacting with HIV proteins tend to overlap with signals of incomplete selective sweep as measured by iHS (Kudaravalli et al. 2009). One of the cases is HLA-C, which has a cluster of eQTLs overlapped with signals of selection in both Europeans and Asians (Kudaravalli et al. 2009). Interestingly, we found the same cluster of eQTLs have significant signals of association with multiple environmental factors. One eQTL (rs6931332) has strong evidence of environmental association signals (transformed rank statistic = 0.0025 for the climate category with seasonal components, 0.03 for the climate category with annual means, 0.0016 for the subsistence category with continuous individual factors, and 0.034 for pathogen). Another eQTL (rs9264942), which has been found to be associated HIV-1 viral load (Fellay et al. 2007), has significant association with the climate category with seasonal components (transformed rank statistic = 0.026) and suggestive evidence of association with pathogen (0.068). These findings suggest that HLA-C has regulatory variants that might be adaptive to certain climate- or pathogen-related environmental factor(s), which further provided protective effect against HIV infection in the near past.
Lipid metabolism is the most prominent metabolic pathway to exhibit consistently significant ERs for eQTLs but not genic SNPs. In addition to the above-mentioned example of peroxisomal lipid metabolism with enrichment signal to short-wave radiation flux in summer, the pathway of metabolism of lipids and lipoproteins also shows significant eQTLs enrichment to the environmental factor of bacteria diversity with ERs of 1.80, 3.17, and 3.87. These observations are consistent with the fact that lipid metabolism plays a wide variety of roles in numerous signaling and regulatory process and host–pathogen interactions (Wenk 2006; van der Meer-Janssen et al. 2010). Our analysis also highlights top candidate genes in these two pathways for future detailed examination. For peroxisomal lipid metabolism, the top three SNPs with most significant correlative signal with short wave radiation flux in summer are found in ACOX1 (rs8065144, transformed rank statistic = 8.11e−4), SCP2 (rs11206043, 0.0018), and AMACR (rs35414, 0.0021). And for metabolism of lipids and lipoproteins, the top three correlative signals with bacteria diversity are located in NCOR2 (rs701078, 0.00047), LSS (rs2280957, 0.0016), and SGPP2 (rs4673024, 0.0021).
Discussions
The Observed Enrichment Patterns of eQTLs Are Biologically Meaningful
In this study we are able to show that compared with intergenic neutral SNPs, eQTLs are significantly more likely to show association with 42 environmental factors. Our results are unlikely to be statistical artifacts because of the following reasons. First, these enrichments are consistent across environmental variables, and also across different methods for assessing environmental correlations. Second, our results of eQTLs enrichment are also consolidated by the observation that similar environmental factors from different studies (Hancock, Witonsky et al. 2010; Hancock et al. 2011; Fumagalli et al. 2011) exhibit consistently significant patterns. For instance, the environmental category of climate, either summarized over the summer and winter components or the annual means, exhibits similar level of enrichment across three tail cutoffs. So does the category of subsistence, either summarized over binary individual factors or continuous ones. Similarly, consistent patterns are observed at the level of individual environmental factors from different studies (fig. 1A and supplementary table S1, Supplementary Material online). Third, we observed a greater enrichment of adaptive eQTLs in regions of lower recombination, which is consistent with theoretical expectations. And fourth, we observed enrichment of eQTLs in environmental adaptation for specific functions, indicating that the patterns we observed are of biological significance.
Regulatory Variants Is Underlying the Significant Enrichment of eQTLs
The statistical association between eQTLs and environmental variables appears to be strong. However, NS SNPs, rather than regulatory variants, could be the underlying explanation if a large proportion of eQTLs are themselves NS SNPs or they are in strong LD with NS SNPs. First, we ruled out the possibility that the enrichment of eQTLs is a direct effect of NS SNPs because only ∼3% of eQTLs are NS SNPs and excluding them has no influence on the observed patterns. Second, to exclude the indirect effect of NS SNPs as the underlying driver, we examined the LD patterns between eQTLs and NS SNPs. Under the environmental correlation framework, an eQTL could only capture the adaptive effect of a NS SNP if a strong LD between the two is present in most human populations tested. However, LD structure is different across populations and it has only been well-studied for a subset of populations included in the environmental correlation analysis. Therefore, we performed a preliminary and conservative analysis using LD data from Hapmap 3 (Altshuler et al. 2010). We excluded eQTLs that are in strong LD (r2 > 0.8) with any NS SNP in any of the 11 populations, which account for ∼20% of all eQTLs, much higher than the number of eQTLs (∼2%) that are in strong LD with any NS SNPs in all 11 populations (supplementary table S7, Supplementary Material online). By further excluding eQTLs that are also NS SNPs, we obtained a group of eQTLs that are less dependent on NS SNPs than all eQTLs. However, as observed for all eQTLs, this group of eQTLs is also significantly enriched in signal of environmental correlation (supplementary fig. S11, Supplementary Material online, P = 1.76e − 09, 2.21e − 08, 2.84e − 07, respectively, for three tail cutoffs).
Moreover, although ∼66% of eQTLs locate within 5 kb of a gene and ∼49% are within introns (supplementary table S8, Supplementary Material online), the difference between the ER for eQTLs located in genic region and that for eQTLs in non-genic regions is not consistently significant (supplementary fig. S10, Supplementary Material online). In addition, if the enrichment of eQTLs is only caused by their LD with NS SNPs, the enrichment of eQTLs should be smaller than that of NS SNPs, as the trend observed for synonymous SNPs (supplementary fig. S6, Supplementary Material online). However, under all three tail cutoffs, the mean/median ER of eQTLs across 42 environmental factors is slightly higher than that of NS SNPs, although the difference is not significant. Taken together, we rule out the possibility that the enrichment of eQTLs is an indirect effect of NS SNPs through their LD with eQTLs. Therefore, the most parsimonious explanation for the enrichment of eQTLs in signals of environmental correlation is the adaptive effect of regulatory variants, which could be eQTLs themselves or the underlying causal variants in strong LD with eQTLs.
eQTLs Are As Prevalent As, If Not More Prevalent Than, NS SNPs in Local Adaptation
We also observed a general trend that the degree of enrichment over all environmental factors is higher for eQTLs than NS SNPs, although this trend is only significant under the tail cutoff of 5%. However, the interpretation of this observation is not straightforward. First, these two groups of SNPs are not independent of each other and strong LD between these two may be the underlying cause of similar enrichment. To relieve the complication of LD, we obtained a group of eQTLs that are less dependent on NS SNPs as defined above and similarly a group of NS SNPs that are less dependent on eQTLs (supplementary table S7, Supplementary Material online). The same trend is observed that the less dependent eQTLs have higher ER than the less dependent NS SNPs. And this difference is also significant under the tail cutoff of 5% (P = 0.0035, supplementary fig. S11, Supplementary Material online). These additional observations suggest that the similar degree of enrichment in signals of environmental correlation is unlikely to be explained by their LD with each other.
Second, recombination rate may confound the comparison of ERs between two SNPs groups. As we demonstrated previously, SNPs in regions of low recombination rate tend to have higher ER. The recombination rate of genome-wide NS SNPs (median 0.29) is not significantly different than that of eQTLs (median 0.30) (Wilcoxon test, P = 0.62). However, the recombination rate of NS SNPs for genes with eQTLs (median 0.27) is significantly lower than that of eQTLs (P = 0.0022), indicating that our identification of environmental factors, to which eQTLs adaptation is more prevalent than NS SNPs, is conservative because recombination rate make the comparison bias toward NS SNPs.
Therefore, the observed slightly higher ER for eQTLs than NS SNPs suggests that eQTLs, at least the current collection of eQTLs, are as prevalent as, if not more prevalent than, NS SNPs in local environmental adaptation. Consistent with this conclusion, a new study recently published while our manuscript was under review utilized part of the data sets used in our study (Hancock et al. Plos) and found that loci under local adaptation are 10-fold more likely to overlap with eQTLs than NS SNPs, supporting the important role of regulatory variants in human adaptation (Fraser 2013). Despite our effort to compile eQTLs identified from different tissues and populations, they may represent only a small subset of all regulatory variants that are functional in human regulatory networks. It is unclear how many eQTLs remain to be identified and how these newly identified eQTLs will affect the patterns observed in our study.
Different Functional Aspects of eQTLs May Impact Enrichment Patterns
Tissue-Specific eQTLs
eQTLs for specific tissues may show unique enrichment patterns for different environmental factors. The eQTLs data we used were mainly identified in lymphoblastoid cell lines (LCL) and monocytes (supplementary table S9, Supplementary Material online). So it is possible that the enrichment patterns we observed only reflect the properties of these two cell types. For instance, enrichment analysis for tissue-specific eQTLs of LCL, monocytes, and liver revealed that LCL-specific eQTLs tend to exhibit enrichment to a larger number of environmental factors than the other two (supplementary table S10, Supplementary Material online). And several environmental factors only show significant enrichment of LCL-specific eQTLs. Moreover, although eQTLs identified in multiple tissues tend to have higher ER than eQTLs identified in a single tissue (P = 0.032, supplementary fig. S12, Supplementary Material online), interpreting the role of tissue specificity is not straightforward because eQTLs identified in multiple tissues also tend to locate at regions of low recombination rate (P < 2.2e−16). Therefore, the presence of specific evolutionary pattern for tissue-specific eQTLs needs future investigation when more tissue-specific eQTLs become available.
Cis- and Trans eQTLs
Our analyses here included all eQTLs, without separating them into cis- and trans-eQTLs. It is a very important question to examine whether cis- and trans-eQTLs exhibit different evolutionary properties. However, most eQTLs identified by far are cis-eQTLs. Trans-eQTLs are much less identified, probably due to the fact that they are much less frequent or they exert much smaller effects than cis-eQTLs. And a much larger burden of multiple testing also prevents the identification of trans-eQTLs (Gilad et al. 2008). In the data set of eQTLs used in our analysis, only a small fraction (<5%) are trans-eQTLs, precluding our analyses with trans-eQTLs. Due to the same reason, our observations may only reflect the evolutionary patterns of cis-eQTLs. A larger collection of trans-eQTLs is needed to characterize their evolutionary significance using the method applied in our study.
The Number of Genes Regulated by an eQTL
Master regulators (eQTLs regulating multiple genes) (Gilad et al. 2008) may be functionally more important and exhibit different evolutionary properties from those regulating only one gene. It is observed that the ERs across 44 environmental factors are higher for master regulator than those for eQTLs associated with only one gene (P = 7.96e − 5, supplementary fig. S12, Supplementary Material online). However, master regulators also tend to locate in regions of lower recombination (P < 2.2e − 16), confounding the interpretation of the higher ERs.
The Effect Size of an eQTL
The effect size of an eQTL refers to the degree to which the eQTL could influence the expression level of the associated gene. If an eQTL has higher effect size, it may be more likely to play a role in adaptation. To explore whether eQTLs with larger effect size exhibit higher ERs, we took advantage of an eQTLs data set (Zeller et al. 2010) that provides the effect size and P value of association for each eQTL. Since the effect size of an eQTL is inversely correlated with the P value of association (ρ = −0.31, P = 2.2e − 16), a statistic called expression score (Nicolae et al. 2010) was developed to measure how likely an eQTL is a true positive and how strong its effect is. The more likely an eQTL is a true positive and the larger its effect size, the higher its expression score. Similar to the cases of tissue-specific eQTLs and master regulators, although eQTLs with higher expression score show stronger enrichment, they are also associated with lower recombination rate (supplementary fig. S12, Supplementary Material online).
Environmental Correlation Study Assists the Elucidation of Regulatory Adaptation
Our analyses highlight a number of environmental factors and biological pathways for which eQTLs are of special evolutionary importance. The underlying biological mechanisms making eQTLs important for environmental adaptation need further investigation. After identifying the adaptation signals and ecological context, the natural next step is to elucidate the underlying mechanistic processes of how a regulatory change can result in phenotypic differences (Gilad et al. 2008). There are a growing number of cases of regulatory adaptation whose molecular and evolutionary mechanism have been elucidated. For example, a causal eQTL (rs9493857) regulating the expression level of SGK1, a key gene in response to environmental stress, was found to be associated with multiple environmental factors, including latitude (Luca et al. 2009). This SNP (rs9493857) is not present in the data set used in our study, but an available proxy SNP (rs4896028, r2 with rs9493857 is 0.736 in Europeans) is significantly associated with short-wave radiation flux in summer (transformed rank statistic = 0.016). Combining the functional context of eQTLs and the ecological context of environmental correlation, many more cases of regulatory adaptation could be illustrated in the near future.
Conclusions
Our evolutionary analyses with eQTLs reveal that regulatory variations are as prevalent as, if not more prevalent than, NS SNPs, in driving recent and ongoing human adaptation to local environment. The importance of regulatory variations is more prominent for continuous environmental factors, such as climate, possibly due to the fine-tuning property of gene expression. Moreover, regulatory changes played an important role in some biological pathways, especially those related with signaling, immune, and metabolic functions, for their local adaptation. Combining the functional context of eQTLs and the ecological implication of environmental correlation provides important insights for future elucidation of the mechanism and selection pressure of regulatory adaptation.
Supplementary Material
Supplementary figures S1–S12 and tables S1–S11 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Dr James Booth for his help in developing the generalized Z test and Drs Alon Keinan and Andrew Clark and their lab members for their valuable comments. They also thank Drs Matteo Fumagalli and Anna Di Rienzo for providing their data. This work was supported by funds from Cornell University and ILSI Future Leader in Nutrition Award to Z.G.
Literature Cited
- Altshuler DM, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersaglieri T, et al. Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet. 2004;74:1111–1120. doi: 10.1086/421051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai JJ, Macpherson JM, Sella G, Petrov DA. Pervasive hitchhiking at coding and regulatory sites in humans. PLoS Genet. 2009;5:e1000336. doi: 10.1371/journal.pgen.1000336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SB. Evolution at two levels: on genes and form. PLoS Biol. 2005;3:e245. doi: 10.1371/journal.pbio.0030245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coop G, Witonsky D, Di Rienzo A, Pritchard JK. Using environmental correlations to identify loci underlying local adaptation. Genetics. 2010;185:1411–1423. doi: 10.1534/genetics.110.114819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis ET. Regulatory variation and evolution: implications for disease. Adv Genet. 2008;61:295–306. doi: 10.1016/S0065-2660(07)00011-9. [DOI] [PubMed] [Google Scholar]
- Dimas AS, et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enattah NS, et al. Identification of a variant associated with adult-type hypolactasia. Nat Genet. 2002;30:233–237. doi: 10.1038/ng826. [DOI] [PubMed] [Google Scholar]
- Enattah NS, et al. Independent introduction of two lactase-persistence alleles into human populations reflects different history of adaptation to milk culture. Am J Hum Genet. 2008;82:57–72. doi: 10.1016/j.ajhg.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellay J, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB. Gene expression drives local adaptation in humans. Genome Res. 2013;23:1089–1096. doi: 10.1101/gr.152710.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, et al. Signatures of environmental genetic adaptation pinpoint pathogens as the main selective pressure through human evolution. PLoS Genet. 2011;7:e1002355. doi: 10.1371/journal.pgen.1002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney DJ, et al. Dissecting the regulatory architecture of gene expression QTLs. Genome Biol. 2012;13:R7. doi: 10.1186/gb-2012-13-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y, Rifkin SA, Pritchard JK. Revealing the architecture of gene regulation: the promise of eQTL studies. Trends Genet. 2008;24:408–415. doi: 10.1016/j.tig.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin MT, Di Rienzo A. Detection of the signature of natural selection in humans: evidence from the Duffy blood group locus. Am J Hum Genet. 2000;66:1669–1679. doi: 10.1086/302879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock AM, Alkorta-Aranburu G, Witonsky DB, Di Rienzo A. Adaptations to new environments in humans: the role of subtle allele frequency shifts. Philos Trans R Soc Lond B Biol Sci. 2010;365:2459–2468. doi: 10.1098/rstb.2010.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock AM, Witonsky DB, et al. Colloquium paper: human adaptations to diet, subsistence, and ecoregion are due to subtle shifts in allele frequency. Proc Natl Acad Sci U S A. 107(Suppl. 2010;2):8924–8930. doi: 10.1073/pnas.0914625107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock AM, et al. Adaptations to climate-mediated selective pressures in humans. PLoS Genet. 2011;7:e1001375. doi: 10.1371/journal.pgen.1001375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haygood R, Fedrigo O, Hanson B, Yokoyama KD, Wray GA. Promoter regions of many neural- and nutrition-related genes have experienced positive selection during human evolution. Nat Genet. 2007;39:1140–1144. doi: 10.1038/ng2104. [DOI] [PubMed] [Google Scholar]
- Keinan A, Reich D. Human population differentiation is strongly correlated with local recombination rate. PLoS Genet. 2010;6:e1000886. doi: 10.1371/journal.pgen.1000886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- Kong A, et al. Fine-scale recombination rate differences between sexes, populations and individuals. Nature. 2010;467:1099–1103. doi: 10.1038/nature09525. [DOI] [PubMed] [Google Scholar]
- Kudaravalli S, Veyrieras JB, Stranger BE, Dermitzakis ET, Pritchard JK. Gene expression levels are a target of recent natural selection in the human genome. Mol Biol Evol. 2009;26:649–658. doi: 10.1093/molbev/msn289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca F, et al. Adaptive variation regulates the expression of the human SGK1 gene in response to stress. PLoS Genet. 2009;5:e1000489. doi: 10.1371/journal.pgen.1000489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SB, et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers AJ, et al. A survey of genetic human cortical gene expression. Nat Genet. 2007;39:1494–1499. doi: 10.1038/ng.2007.16. [DOI] [PubMed] [Google Scholar]
- Nica AC, et al. Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet. 2010;6:e1000895. doi: 10.1371/journal.pgen.1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolae DL, et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, et al. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar S, Noonan JP, Paabo S, Rubin EM. Accelerated evolution of conserved noncoding sequences in humans. Science. 2006;314:786. doi: 10.1126/science.1130738. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Pickrell JK, Coop G. The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Curr Biol. 2010;20:R208–R215. doi: 10.1016/j.cub.2009.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, et al. Population genomics of human gene expression. Nat Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, et al. Convergent adaptation of human lactase persistence in Africa and Europe. Nat Genet. 2007;39:31–40. doi: 10.1038/ng1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournamille C, Colin Y, Cartron JP, Le Van Kim C. Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy-negative individuals. Nat Genet. 1995;10:224–228. doi: 10.1038/ng0695-224. [DOI] [PubMed] [Google Scholar]
- van der Meer-Janssen YP, van Galen J, Batenburg JJ, Helms JB. Lipids in host-pathogen interactions: pathogens exploit the complexity of the host cell lipidome. Prog Lipid Res. 2010;49:1–26. doi: 10.1016/j.plipres.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veyrieras JB, et al. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet. 2008;4:e1000214. doi: 10.1371/journal.pgen.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk MR. Lipidomics of host-pathogen interactions. FEBS Lett. 2006;580:5541–5551. doi: 10.1016/j.febslet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, Kalay G. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat Rev Genet. 2012;13:59–69. doi: 10.1038/nrg3095. [DOI] [PubMed] [Google Scholar]
- Wray GA. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 2007;8:206–216. doi: 10.1038/nrg2063. [DOI] [PubMed] [Google Scholar]
- Ye K, Gu Z. Recent advances in understanding the role of nutrition in human genome evolution. Adv Nutr. 2011;2:486–496. doi: 10.3945/an.111.001024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller T, et al. Genetics and beyond—the transcriptome of human monocytes and disease susceptibility. PLoS One. 2010;5:e10693. doi: 10.1371/journal.pone.0010693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Gianoulis TA, Karczewski KJ, Zhao H, Snyder M. Regulatory variation within and between species. Annu Rev Genomics Hum Genet. 2011;12:327–346. doi: 10.1146/annurev-genom-082908-150139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.