

Abstract
Autonomous retrotransposons lacking long terminal repeats (LTR) account for much of the variation in genome size and structure among vertebrates. Mammalian genomes contain hundreds of thousands of non-LTR retrotransposon copies, mostly resulting from the amplification of a single clade known as L1. The genomes of teleost fish and squamate reptiles contain a much more diverse array of non-LTR retrotransposon families, whereas copy number is relatively low. The majority of non-LTR retrotransposon insertions in nonmammalian vertebrates also appear to be very recent, suggesting strong purifying selection limits the accumulation of non-LTR retrotransposon copies. It is however unclear whether this turnover model, originally proposed in Drosophila, applies to nonmammalian vertebrates. Here, we studied the population dynamics of L1 in the green anole lizard (Anolis carolinensis). We found that although most L1 elements are recent in this genome, truncated insertions accumulate readily, and many are fixed at both the population and species level. In contrast, full-length L1 insertions are found at lower population frequencies, suggesting that the turnover model only applies to longer L1 elements in Anolis. We also found that full-length L1 inserts are more likely to be fixed in populations of small effective size, suggesting that the strength of purifying selection against deleterious alleles is highly dependent on host demographic history. Similar mechanisms seem to be controlling the fate of non-LTR retrotransposons in both Anolis and teleostean fish, which suggests that mammals have considerably diverged from the ancestral vertebrate in terms of how they interact with their intragenomic parasites.
Keywords: non-LTR retrotransposon, Anolis carolinensis, green anole, population genomics
Introduction
Autonomous non-long terminal repeat retrotransposons (nLTR-RTs) are transposable elements (TEs) that can “copy and paste” themselves by an RNA intermediate in a process mediated by their own reverse transcriptase domain. nLTR-RTs have proliferated with great success in eukaryote genomes and they are the main drivers of genome size and structural variation among vertebrate lineages (Lander et al. 2001; Furano et al. 2004; Tollis and Boissinot 2011). A single type of nLTR-RT known as LINE-1 (Long Interspersed Nuclear Element, L1 hereafter) dominates the human genome (Lander et al. 2001), and ancient L1 fossils and their nonautonomous counterparts, including the Alu interspersed repeats, may account for over two-thirds of the human genome (de Koning et al. 2011). Most L1 DNA in human is the result of past amplifications from millions of years of placental mammalian evolution (Boissinot et al. 2000; Khan et al. 2006), which is typical of eutherians (Waterston et al. 2002; Gibbs et al. 2004, 2007; Pontius et al. 2007; Wade et al. 2009). In contrast to mammals, compact teleost fish genomes contain several active and highly diverse types of nLTR-RT (sometimes including L1), many of which have produced recent copies; however, they do not accumulate as they do in mammals (Volff et al. 2003; Duvernell et al. 2004; Furano et al. 2004). Meanwhile, recent analyses of squamate reptile genomes (Novick et al. 2009; Alfoldi et al. 2011; Castoe et al. 2011) have revealed several highly divergent repetitive landscapes that are more fish-like than mammal-like.
Differences between the respective nLTR-RT landscapes of the vertebrate lineages may be due to differences in the strength of purifying selection against deleterious element-containing loci in populations (Charlesworth B and Charlesworth D 1983; Charlesworth et al. 1994; Le Rouzic and Deceliere 2005). It was suggested that purifying selection is the mechanism causing a high turnover of elements in Drosophila, thus preventing TE accumulation and contributing to the low copy number in the genome (Charlesworth 1989; Biemont et al. 1994, 1997). In the human genome, the majority of L1 copies seem to be selectively neutral and accumulate readily (Boissinot et al. 2000, 2001). However, as in Drosophila (Petrov et al. 2003), human full-length (FL) nLTR-RT insertions may behave as deleterious alleles due to their ability to mediate ectopic recombination (Langley et al. 1988; Boissinot et al. 2001). As parasitic DNA sequences, TEs are dependent on the demographic history, and especially the effective population size (Ne), of the host. Therefore, changes in Ne can alter the strength of purifying selection against deleterious alleles (Charlesworth 2009), making TE fixation in a genome more likely (Le Rouzic and Deceliere 2005). The differential fixation of TE copies in colonized versus native populations of Drosophila subobscura (Garcia Guerreiro et al. 2008) and Arabidopsis lyrata (Lockton et al. 2008) strongly suggests that random processes in the host demographic history such as founder effect can also influence the fate of TEs in the genome. The mutagenic ability of TEs to disrupt or change genetic pathways has provided an important source of evolutionary novelties for host genomes, and it is clear that TEs interact with their hosts in numerous ways (Oliver and Greene 2009).
The differences in TE copy number and abundance between mammals and nonmammals suggest that these lineages differ greatly in terms of how they deal with their intragenomic parasites, and that the TE profiles of mammalian genomes have significantly diverged from the first land vertebrate. However, the divergence history of the modern amniote lineages spans approximately 310 Myr (Donoghue and Benton 2007) and studies of TE population dynamics in nonmammalian vertebrates have so far been limited to teleost fish (Neafsey et al. 2004; Blass et al. 2012). An evolutionarily less distant comparison with mammals than offered by teleost fish is sorely needed. As the sister group to mammals, reptiles make a more ideal system for resolving ancestral states in vertebrate genome evolution (Janes et al. 2010), yet there have been few reptilian genomic models available until very recently (Alfoldi et al. 2011; Castoe et al. 2011; Shaffer et al. 2013; Wang et al. 2013). The first fully sequenced reptile genome was that of the green anole lizard (Anolis carolinensis), which is a model organism that has long been studied in the fields of neuroscience and behavior (Lovern et al. 2004; Wade 2012). The lizard genome contains five divergent clades of nLTR-RT including CR1, R2, L2, RTE, and L1. Within L1 alone, there are 20 distinct families (fig. 1). Copy number within these families is relatively low, and the low divergence within each Anolis L1 family suggests most inserts in the genome are recent (Novick et al. 2009). The L1 profile in the Anolis genome suggests strong purifying selection, as in Drosophila. However, there has never been a test of the turnover model in reptiles, and as a result there is a large gap in our knowledge of how nonmammalian vertebrates interact with their intragenomic parasites.
Fig. 1.—

Neighbor-joining phylogenetic tree depicting the evolutionary relationships between the ORF2 consensus sequences of the 20 L1 families found in the Anolis genome. Node support was assessed with 1,000 bootstrap replicates (greater than 95% is shown). Tips are labeled with the L1 family name and within each parenthesis are the copy number and percent pair wise divergence from consensus sequence as reported in Novick et al. (2009).
Our goal here is to provide the first study of TE population dynamics in a reptile, A. carolinensis. The green anole is a very suitable model because the species is widespread and abundant in the southeastern United States, making population genetic analysis feasible, and its recent evolutionary history is well characterized (Campbell-Staton et al. 2012; Tollis et al. 2012). In this study, we consider five distinct evolutionary lineages, the geographic distributions of which are depicted in figure 2: 1) the Everglades population, which is geographically limited to the southern part of the Florida peninsula; 2) the Suwannee population, which inhabits the Gulf Coast of the Florida peninsula; 3) the Central Florida population, which primarily is restricted to the Atlantic Coast of peninsular Florida; 4) the North Carolina population, which exists in that state at the northern limits of the species range along the Atlantic Coast; and 5) the Gulf–Atlantic population, which extends from South Carolina and Georgia, along the Gulf Coastal Plain and across the Mississippi River into Texas. In terms of the demographic history, the oldest and most stable populations exist in peninsular Florida (Campbell-Staton et al. 2012; Tollis et al. 2012). On the continental mainland, North Carolina was estimated to have the smallest Ne (Tollis et al. 2012) and the Gulf–Atlantic experienced a recent and rapid westward expansion (Campbell-Staton et al. 2012; Tollis et al. 2012). Both of these populations are likely candidates for scenarios where genetic drift has been relatively strong.
Fig. 2.—

The geographic distribution of localities from which the green anole samples used in this study were collected is indicated by solid black circles. The geographic distribution of the five major evolutionary lineages of green anoles, summarized from Tollis et al. (2012) and Campbell-Staton et al. (2012) is indicated by colored polygons: Everglades (magenta), Suwannee (blue), Central Florida (brown), Gulf–Atlantic (green), and North Carolina (yellow).
Materials and Methods
We studied 158 green anoles collected across the US states of North Carolina, South Carolina, Georgia, Alabama, Florida, Tennessee, Arkansas, and Louisiana between 2009 and 2012. A. Pires da Silva provided specimens from Texas. Collecting localities for all of these specimens are shown in figure 2 and GPS coordinates are available in the supplementary files of this article (Supplementary Material online) and of Tollis et al. (2012). Specimens were caught by hand or noose and tissue samples were taken in the form of tail clippings or, if dissected, muscle or liver, which were preserved in ethanol. Protocols were established in accordance with and approved by the Queens College Institutional Animal Care and Use Committee (Animal Welfare Assurance Number: A32721-01; protocol number: 135). DNA was extracted from all tissues with the Promega Wizard Genomic DNA Purification kit.
To minimize bias in collecting L1-containing loci, we obtained L1 inserts that were missing from the February 2007 and May 2010 releases of the Anolis genome with the following cloning strategy. The 3′-ends and genomic flanking regions of Anolis L1 inserts were cloned from each of the five green anole populations: Everglades, Suwannee, Central Florida, Gulf–Atlantic, and North Carolina. For each population, the genomic DNA from five individuals was pooled in equal proportion to obtain approximately 2 µg, the concentration and purity of which was verified using a NanoDrop 2000 spectrophotometer. We digested the pooled DNA samples with NEBNext dsDNA Fragmentase to obtain randomized genomic fragments of 1–2.5 kb, which was verified by electrophoresis on a 1% agarose gel. The DNA fragments contain overhangs, which were polished by incubation at 12 °C for 30 min with T4 DNA polymerase followed by heat-inactivation (20 min at 75 °C) of the polymerase to produce blunt ends. The 3′ hydroxyl groups were phosphorylated by incubation at 37 °C for 30 min with T4 polynucleotide kinase (with 5% polyethylene glycol) followed by heat inactivation of the kinase at 75 °C for 20 min. The DNA fragments were then ligated to 10 µM of double-stranded anchor (5′-TAGCTACAGCTGTAGCTGACAT-3′) with T4 DNA ligase at room temperature for 3 h. To ensure that the anchors ligated sufficiently, we performed a PCR using the putatively ligated DNA with the double-stranded anchor as a primer, and checked for DNA smears of appropriate size (1–2.5 kb) on a 1% agarose gel.
We then took a series of enrichment steps to ensure the capture of L1-containing loci from different Anolis L1 families. The L1 families we focused on were L1AC18 and L1AC20 as described in Novick et al. (2009); we chose these families because they represent a wide range of copy numbers found within Anolis L1 families: L1AC18 contains 144 copies including 24 full length (FL) and 120 truncated (TR), and L1AC20 contains 75 copies including 22 FL and 53 TR. For each L1 family, we used the consensus sequence from Novick et al. (2009) in a BLAT search (Kent 2002) of the May 2010 release (Broad Institute version AnoCar2.0) of the Anolis genome on the UCSC Genome Browser (Kent et al. 2002) (www.genome.ucsc.edu, last accessed May 21, 2013). PCR primers were designed using BioEdit (Hall 1999). Primers have been provided as supplementary files, Supplementary Material online. We performed an asymmetrical PCR on the anchor-ligated DNA with a 5 to 1 volumetric ratio of a 10 µM family-specific L1 biotinylated primer and the 10 µM single strand anchor. These PCR products were then captured using streptavidin-coated magnetic beads (M-280 Dynabeads) following the procedure recommended by the manufacturer, after which a second enrichment PCR was performed using bead-captured DNA, the single strand anchor as a primer and a second nested L1 family-specific primer. Purified PCR products were ligated into plasmids using a pGEM-T Easy Vector kit (Promega), and the ligated vectors were transformed into JM109 Escherichia coli competent cells. Bacterial colonies were grown overnight on plates with LB agar + ampicillin + IPTG + X-gal and were blue-white screened. Positive clones were picked and incubated overnight in 300 µL of LB media with ampicillin in 96-well plates. We amplified the cloned products by PCR using primers located in the plasmids (Sp6 and T7). As the goal was to determine whether our captured L1 insertions were unique to populations and individuals, we needed enough flanking region that could be mapped to the database. Therefore, our biotinylated and nested primers were designed to be less than 150 bases from element 3′-ends, and we selected PCR products that were at least approximately 500 bp in length. The vector primers were used for Sanger sequencing by the HT-Seq facility at the University of Washington, Seattle, WA. Forward and reverse reads for each sequenced clone were assembled into contigs using Geneious v5.5 (Drummond et al. 2010), and their consensus sequences were extracted and used for further analysis. After removing vector sequence, each L1-containing clone with enough flanking region (∼50 bp) was used in another BLAT search of the Anolis genome. If the entire query, consisting of an L1 3′-end and flanking sequence, could be matched unambiguously to a specific location in the genome then the insertion site was deemed occupied. A novel insertion was recorded when the BLAT returned a match of only the flank with no upstream L1 3′-end, indicating an empty insertion site in the database.
We determined the polymorphism of any novel cloned inserts by using a presence/absence ascertainment with a series of flanking and internal primers. PCRs were performed on a panel comprised of the individuals whose genomic DNA was originally pooled for the enrichment method. The primers were designed in Primer3 (Rozen and Skaletsky 2000) after a BLAT search to locate the insertion site in the Anolis genome database and collection of 300 bp upstream and downstream of the insertion site. Primers for presence detection were performed with reverse flanking primer and one of the L1 family-specific internal (forward) primers. The specificity of each reaction was verified with the in silico PCR tool on the UCSC Genome Browser. We then used touchdown PCR to optimize reaction specificity (Korbie and Mattick 2008). PCRs for presence/absence detection included a 1:00 hold at 94 °C followed by 30 cycles of 0:30 denaturing at 94 °C, 0:30 annealing at 55–62 °C (depending on the melting temperatures of the primer pairs given in the supplementary files, Supplementary Material online), and 0:30 extension at 72 °C. Upstream or downstream alternative primers were designed and tested in cases where gel bands were ambiguous. Where we could not avoid ambiguities, those loci were removed from the analysis. To determine the size of these novel elements, we conducted PCRs using genomic DNA from an individual that successfully amplified for element presence with the forward flanking primers and three reverse primers located at various distances from the 5′-end of the consensus sequence for each family: 500 bp, 1 kb, and 2 kb (table 1). These PCRs included a 1:00 hold at 94 °C followed by 30 cycles of 0:30 denaturing at 94 °C, 0:30 annealing at 55 °C, and 1:30 extension at 72 °C. Successful amplification with these primers allowed us to determine to what extent these novel insertions extended toward their 5′-ends.
Table 1.
Summary of L1 Inserts Collected during the Cloning Experiment for Each Green Anole Population and Information about Polymorphism
| Cloning |
||||||
|---|---|---|---|---|---|---|
| Everglades | Suwannee | Central Florida | North Carolina | Gulf–Atlantic | Total | |
| Clones collected and sequenced | 480 | |||||
| Clones containing an L1 element | 380 | |||||
| Total sequences mapped to database | 265 | |||||
| Number of different L1AC18 inserts | ||||||
| Flanking sequences located in database | 55 | 23 | 43 | 22 | 29 | 172 |
| Insertion sites occupied in database | 42 | 20 | 37 | 17 | 21 | 137 |
| Insertion sites empty in database | 13 | 3 | 6 | 5 | 8 | 35 |
| Tested by PCR | 10 | 3 | 5 | 4 | 6 | 28 |
| Proportion <50% polymorphism | 80% | 63% | 83% | 100% | 0 | |
| No. FL inserts | 0 | 2 | 2 | 1 | 4 | 9 |
| Proportion FL >50% | – | 0 | 0 | 0 | 100% | |
| Number of different L1AC20 inserts | ||||||
| Flanking sequences located in database | 18 | 12 | 34 | 14 | 15 | 93 |
| Insertion sites occupied in database | 12 | 9 | 26 | 14 | 12 | 73 |
| Insertion sites empty in database | 6 | 3 | 6 | 0 | 3 | 18 |
| Tested by PCR | 6 | 2 | 5 | – | 2 | 15 |
| Proportion <50% polymorphism | 100% | 50% | 100% | – | 50% | |
| No. FL inserts | 0 | 2 | 0 | 0 | 0 | 2 |
| Proportion FL >50% (%) | – | 50 | – | – | – | |
We added to this data set a collection of L1-containing loci from the February 2007 and May 2010 releases of the Anolis genome, both of which are available on the UCSC Genome Browser. We used a consensus sequence query for each Anolis L1 family described in Novick et al. (2009) in a BLAT search to retrieve elements from the Anolis genome. We then aligned the collected elements to their family consensus sequences and calculated their divergence from family consensus using the Kimura 2-Parameter corrected distance method in MEGA5 (Tamura et al. 2011) as a proxy for their age. For each insert in the output, we collected 2,500 bp of upstream and downstream genomic flank. Flanking regions were submitted to Repeat Masker (Smit et al. 1996–2010), which screened for single sequence repeats, short tandem repeats, or TEs, which would interfere with PCR primer design. Primers were designed in flanking regions either manually or using Primer3. For inserts longer than 2 kb, we designed family-specific internal primers near the element 3′-ends from sequence alignments using ClustalW (Larkin et al. 2007). All primer pairs were tested for specificity using the in silico PCR tool available on the UCSC Genome Browser. We measured the population frequencies of L1 loci retrieved from the database using the presence/absence PCR ascertainment method described earlier. Individuals from each population were genotyped according to amplified fragment size after electrophoresis on a 1% agarose gel with ethidium bromide and a Promega BenchTop 1 kb DNA ladder.
Within each population for each locus, we recorded the total number of present and absent insertions, and population frequencies were calculated as the number of present alleles divided by the number of total chromosomes. We also examined the population frequencies of elements that differ by length categories. To determine whether purifying selection is acting against full-length insertions in green anole populations, we compared the frequency distribution of full-length and truncated L1 elements. For this purpose, elements extending all the way from their 3′- to 5′-ends were counted as full-length, while those missing more than 10% of their 5′-ends were counted as truncated. Using the Wilcoxon rank-sum test (Mann–Whitney U test), we aimed to detect statistically significant differences in allele frequencies between truncated and full-length loci both within and between populations. We used the Kolmogorov–Smirnov test to determine whether the shape of the frequency distributions between the two insertion types is significantly different.
Results
We collected L1-containing loci from two sources: the Anolis genome database and through the direct cloning of inserts from the genomic DNA of individuals. The reasoning behind this two-pronged approach was to minimize ascertainment bias. As the database was constructed from the sequencing of a single individual, it may be less likely to contain low-frequency polymorphisms, which are integral to any study of purifying selection. Therefore, the cloning afforded us the opportunity to more closely approximate the amount of genetic variation in natural populations. The five green anole populations we studied here are treated as distinct entities, and we measured the allelic frequencies of L1 loci within each population separately. This is because it has been shown that these five populations constitute independently evolving lineages with minimal gene flow between them (Campbell-Staton et al. 2012; Tollis et al. 2012).
The genomic coordinates of the L1 inserts that we were able to map to the database are provided in the supplementary files, Supplementary Material online, and the results from the cloning experiments are summarized in table 1. We sequenced 480 clones and identified 380 L1 insertions. Using BLAT, we were able to unambiguously identify 265 flanking regions that could be mapped onto the Anolis genome database. Forty-seven of these represented insertion sites we sequenced more than once because they were captured multiple times, and thus we captured 218 unique L1 insertion sites. Of these, we identified 148 elements from the L1AC18 family and 70 from the L1AC20 family, representing, respectively, 100.2% and 93% of the copy number estimates of these families from Novick et al. (2009). The remaining cloned L1 either did not contain enough flanking region to allow the determination of the insertion site or contained repetitive DNA in the flank and thus their insertion sites were ambiguous. Of the 218 unique L1 insertion sites found in the database, 51 (23%) were not occupied by an L1 element. These elements were probably not present in the individual who was sequenced for the Anole Genome Project and are most likely polymorphic in green anole populations. The polymorphism data and the status of novel full-length insertions in green anole populations are also given in table 1. We were able to successfully measure the polymorphism for 28 of 35 (80%) novel insertion loci from the L1AC18 family and 15 of 18 (83%) novel insertion loci from the L1AC20 family (these primers are given in the supplementary files, Supplementary Material online). We were able to successfully ascertain the size of 18 of 28 (64%) novel L1AC18 inserts, of which 9 were full-length and 9 truncated, and 6 of 15 (40%) novel L1AC20 inserts, of which 2 were full-length and 4 were truncated.
The results from the survey of L1 polymorphism using insertion loci from the database are summarized in table 2. Three of the truncated insertion loci designed from the database were also captured by our cloning method, which was not an unexpected result since with that method we were able to retrieve a high proportion of the total copy numbers of the studied L1 families. These loci were L1AC18_128 and L1AC18_223 from L1 family L1AC18, which were fixed across all populations, and L1AC20_150 from L1 family L1AC20, which ranged in population frequency from 88% to total fixation. The high population frequency of these elements is not surprising because they were retrieved from multiple populations during the cloning. Overall, we were able to collect population frequency data on 52 insertion loci from 16 of the 20 Anolis L1 families described in Novick et al. (2009), including 22 full-length and 30 truncated insertions.
Table 2.
Locus-Specific Information for 52 L1 Loci Retrieved from the Anolis Genome and Their Frequencies in Five Green Anole Populations
| Locus | Coordinates | Length (bp) | FL or TR | % Divergence from Consensus | North Carolina | Suwannee | Central Florida | Everglades | Gulf–Atlantic |
|---|---|---|---|---|---|---|---|---|---|
| L1AC20_684 | chr4:68403974–68404330 | 357 | TR | 3.80 | 0.50 | 0.57 | 0.84 | 0.44 | 0.90 |
| L1AC12s_4:12 | chr4:126979289–126979702 | 414 | TR | 2.50 | 1.00 | 0.96 | 1.00 | 1.00 | 1.00 |
| L1AC12s_GL3 | chrUn_GL343596:105315–105733 | 419 | TR | 3.70 | 1.00 | 0.75 | 0.59 | 0.00 | 1.00 |
| L1AC11s_6:33 | chr6:33204599–33205038 | 440 | TR | 0.00 | 0.25 | 0.07 | 0.03 | 0.00 | 0.28 |
| L1AC20_150 | chr1:150575039–150575547 | 509 | TR | 7.30 | 1.00 | 0.97 | 1.00 | 0.94 | 0.88 |
| L1AC16s_GL3 | chrUn_GL343395:465703–466212 | 510 | TR | 4.30 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| L1AC16s_GL4 | chrUn_GL343471:34099–34667 | 569 | TR | 1.80 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| L1AC16s_GL5 | chrUn_GL344110:24189–24796 | 608 | TR | 1.50 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| L1AC20_257 | chr1:257099845–257100469 | 625 | TR | 12.20 | 1.00 | 1.00 | 1.00 | 0.63 | 1.00 |
| L1AC20_227 | chr5:22764139–22764850 | 712 | TR | 11.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| L1AC18_223 | chr1:223912127–223912841 | 715 | TR | 3.90 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| TE_3 | chr1:214783982–214784696 | 715 | TR | 1.40 | 0.04 | 0.00 | 0.00 | 0.00 | 0.21 |
| L1AC13s_4:27 | chr4:27512892–27513664 | 773 | TR | 3.00 | 0.40 | 0.00 | 0.00 | 0.00 | 0.91 |
| L1AC18_128 | chr1:128475510–128476320 | 811 | TR | 3.60 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| L1AC18_543 | chr5:54332386–54333253 | 868 | TR | 5.20 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| L1AC20_660 | chr5:66011824–66012703 | 880 | TR | 4.70 | 0.38 | 0.27 | 0.33 | 0.13 | 0.71 |
| L1AC17s_1:544 | chr1:54502268–54503155 | 888 | TR | 2.70 | 0.82 | 0.67 | 0.71 | 1.00 | 1.00 |
| L1AC17s_Gly | chrUn_GL343200:1968310–1969771 | 916 | TR | 1.40 | 0.27 | 0.00 | 0.03 | 0.00 | 0.00 |
| L1AC19_139 | chr3:139851678–139852602 | 925 | TR | 1.90 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| L1AC19_2:144 | chr2:144963722–144964741 | 1,020 | TR | 1.70 | 0.88 | 0.45 | 0.45 | 0.00 | 0.25 |
| L1AC18_107 | chr1:107831209–107832288 | 1,080 | TR | 1.20 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| L1AC2.26 | chrUn_GL343239:906018–907246 | 1,229 | TR | 1.10 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| L1AC15s_1:87 | chr1:87962249–87963655 | 1,407 | TR | 0.42 | 0.10 | 0.00 | 0.00 | 0.00 | 0.03 |
| L1AC3.25 | chr2:172762715–172767090 | 4,376 | TR | 0.45 | 1.00 | 0.94 | 1.00 | 1.00 | 0.97 |
| L1AC3.24 | scaffold_24:516031–520,687a | 4,657 | TR | 0.53 | 1.00 | 0.71 | 1.00 | 0.83 | 0.81 |
| L1AC3.21 | chrUn_GL343497:464966–469686 | 4,721 | TR | 0.45 | 0.50 | 0.03 | 0.00 | 0.00 | 0.07 |
| L1AC4.17 | scaffold_125:1567058–1571933a | 4,876 | TR | 0.54 | 1.00 | 1.00 | 1.00 | 0.93 | 1.00 |
| L1AC4.18 | scaffold_43:3503254–3508180a | 4,927 | TR | 0.54 | 1.00 | 0.00 | 0.05 | 0.07 | 0.25 |
| L1AC4.15 | chr3:96424624–96429616 | 4,993 | TR | 0.50 | n/a | 0.70 | 0.77 | 1.00 | 0.96 |
| L1AC3.18 | chrUn_GL343280:1636141–1641248 | 5,108 | TR | 0.57 | 1.00 | n/a | n/a | 1.00 | 1.00 |
| L1AC4.8 | chr5:19962314–19967530 | 5,217 | FL | 0.39 | n/a | 0.00 | n/a | 0.00 | n/a |
| L1AC4.19 | chrUn_GL343243:1081190–1086410 | 5,221 | FL | 0.52 | 1.00 | 0.50 | 0.41 | 1.00 | 0.93 |
| L1AC4.22 | chr2:90589288–90594512 | 5,225 | FL | 0.46 | n/a | n/a | n/a | 0.00 | 0.31 |
| L1AC4.20 | scaffold_527:549438–554665a | 5,228 | FL | 0.85 | 0.90 | 0.13 | 0.69 | 0.13 | 0.88 |
| L1AC4.11 | chr3:178322659–178327899 | 5,241 | FL | 0.91 | n/a | 0.56 | n/a | 0.00 | 0.83 |
| L1AC4.2 | scaffold_85:3499711–3504951a | 5,241 | FL | 0.35 | 0.31 | 0.03 | 0.00 | 0.00 | 0.54 |
| L1AC4.25 | chr3:170477780–170483021 | 5,242 | FL | 0.46 | 1.00 | 0.00 | 0.00 | 0.00 | 0.24 |
| L1AC4.26 | chr3:159015596–159020837 | 5,242 | FL | 0.50 | 0.80 | 0.17 | 0.18 | 0.40 | 0.65 |
| L1AC20_3:170 | chr3:170477780–170483022 | 5,243 | FL | 0.31 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| L1AC4.21 | chr3:32972264–32977686 | 5,243 | FL | 0.66 | 0.63 | 0.00 | 0.00 | 0.25 | 0.68 |
| L1AC4.4 | scaffold_30:3968578–3973822a | 5,245 | FL | 0.52 | 0.00 | 0.18 | 0.10 | 0.50 | 0.23 |
| L1AC4.1 | chr2:172917348–172922593 | 5,246 | FL | 0.35 | 0.00 | 0.00 | 0.03 | 0.00 | 0.00 |
| L1AC8_1:108 | chr1:108322088–108328343 | 5,334 | FL | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| L1AC3.4 | chr6:54998113–55004259 | 6,147 | FL | 0.47 | 0.00 | 0.13 | 0.03 | 0.00 | 0.38 |
| L1AC3.10 | chrUn_GL343295:80571–86721 | 6,151 | FL | 0.36 | 1.00 | 0.06 | 0.23 | 0.00 | 0.78 |
| L1AC15_5:14 | chr5:142569373–142575532 | 6,160 | FL | 0.10 | 0.80 | 0.22 | 0.56 | 0.00 | 0.69 |
| L1AC3.8 | Chr3:168587085–168593244 | 6,160 | FL | 0.31 | 1.00 | 0.18 | 0.10 | 0.00 | 0.61 |
| L1AC15_2:15 | chr2:153639275–153645435 | 6,161 | FL | 0.31 | 0.70 | 0.56 | 0.87 | 0.00 | 1.00 |
| L1AC3.3 | scaffold_57:1761641–1767805a | 6,165 | FL | 0.39 | 0.27 | 0.28 | 0.03 | 0.20 | 0.23 |
| L1AC11_2:10 | chr2:107077315–107083913 | 6,599 | FL | 0.06 | 0.42 | 0.11 | 0.28 | 0.00 | 0.05 |
| L1AC14_GL | chrUn_GL343255:672694–679409 | 6,716 | FL | 0.70 | 0.92 | 0.00 | 0.23 | n/a | 0.85 |
| L1AC10.2 | chr3:172235202–172242019 | 6,818 | FL | 0.73 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
Note.—n/a, not applicable.
aAn insert collected from the February 2007 version that we were not able to map onto the May 2010 version.
Widespread Fixation of L1 in Anolis
Many L1 inserts were fixed in green anole populations. For instance, 22 out of the 30 truncated insertions (73%) collected from the database have reached fixation in at least one of the five green anole populations, as well as 5 out of the 22 full-length insertions (23%). This widespread presence of fixed insertions was surprising, because L1 copy number is very low in the Anolis genome and ancient insertions are extremely rare. Therefore, we decided to estimate the number of fixed L1 insertions in the genome. To do this, we first looked at the frequencies of L1 inserts with varying levels of divergence in each population. In an attempt to remove the potentially confounding effects of demographic history, we focused at first on only the Gulf–Atlantic and North Carolina populations (fig. 3). This is because the individual that was sequenced for the Anole Genome Project was collected in Aiken, SC, and is an admixed individual whose genome is derived from both of these populations (Tollis M and Boissinot S, unpublished data). In the Gulf–Atlantic and North Carolina populations, 70% and 66% of L1 inserts that diverge from their consensus by more than 1% are fixed, respectively. This does not mean that only old elements become fixed; the fraction of elements that are both fixed and younger than 1% divergent is somewhat smaller—10% in the Gulf–Atlantic and 34% in North Carolina—which suggests that at least some elements can reach fixation rather quickly. From the divergence curve, we multiplied the proportion of total elements that are fixed by the 1,006 total L1 copies in the Anolis genome reported by Novick et al. (2009) and estimated the total number of fixed inserts to be 342 in the Gulf–Atlantic population and 482 in North Carolina. Although these numbers do not comprise a majority of the L1 repertoire in the Anolis genome, they do amount to a significant proportion of fixed elements.
Fig. 3.—

Number of fixed and polymorphic L1 elements extrapolated from population data according to their divergence from consensus.
It is possible that the unique demographic histories of these populations may be affecting the number of L1 inserts that become fixed. The Gulf–Atlantic and North Carolina populations are relatively young when compared with their conspecifics living on the Florida peninsula (Campbell-Staton et al. 2012; Tollis et al. 2012), and they may have smaller effective population sizes (Ne). When Ne is small, it can affect the efficiency of purifying selection, causing otherwise harmful alleles to drift toward fixation, as well as contribute to an overall higher rate of allele fixation (Charlesworth 2009). Therefore, we decided to look at the number of fixed elements in the Central Florida population (fig. 3), which has considerably more genetic diversity, suggesting a larger Ne (Campbell-Staton et al. 2012). We found that 62% of elements diverging from their consensus by more than 1% are fixed in this population, and 11% of elements younger than 1% divergent are fixed as well. This translates into an estimated 335 total fixed L1 inserts in the Central Florida population, which is still an appreciable amount of fixed elements, but is lower than what was estimated for the Gulf–Atlantic population, and even more so than the North Carolina population.
Selection against Full-Length L1 Elements
Our estimates of the amount of fixed L1 elements in green anole populations suggest that nLTR-RTs accumulate in the Anolis genome, which is in contrast to previous suggestions (Novick et al. 2009; Tollis and Boissinot 2011). However, this does not necessarily mean that all L1 insertions are selectively neutral. Figure 4 shows the proportion of inserts that are either fixed or polymorphic according to whether they are FL or TR. The figure shows that in all populations, TR elements are much more likely to be fixed than FL elements, and, conversely, FL elements are much more likely to be polymorphic than TR ones. That a much larger proportion of TR inserts are fixed suggests that FL elements are prevented from reaching fixation, and perhaps this is because they are subjected to stronger purifying selection. Sixteen FL insertions (73%) were completely absent in at least one green anole population, compared with 8 TR (27%), including 16 out of the 22 full-length inserts we screened in the Everglades population. It is difficult to say whether purifying selection keeps these inserts at such low population frequencies that we failed to detect them in our sample. Another explanation is that they may have recently inserted into the host genome, sometime after the split in the population histories. The Everglades lineage likely split off from the rest of the species relatively early in its history (Campbell-Staton et al. 2012; Tollis et al. 2012), so this latter situation is plausible. Therefore, to detect purifying selection within each population, we excluded all loci for which we failed to detect presence on a single chromosome. This should not prevent us from detecting selection, because full-length and truncated inserts are generated by the same biological mechanism, and any bias against low frequency alleles will similarly shift the frequency distribution for inserts that are both full-length and truncated.
Fig. 4.—
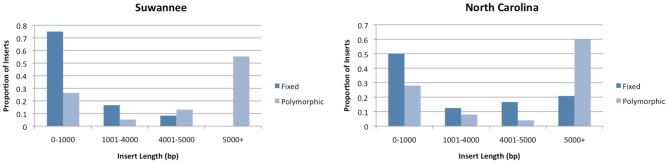
Fraction of polymorphic and fixed L1 elements according to their length in green anole populations. The distribution is based on 52 L1-containing loci retrieved from the Anolis genome database.
Table 3 gives the total number of TR and FL insertions compared within each population, their average population frequencies, and the statistical significances of the differences in the population frequency means and of the shapes of their distributions. The average frequency of TR elements was higher than the average frequency of FL elements in all populations; however, this difference was not statistically significant in the Everglades and North Carolina populations. The statistical significance of the difference in population frequency between FL and TR elements was significant in the Gulf–Atlantic population (P < 0.01), and highly significant in the Suwannee and Central Florida populations (P < 0.001). The shapes of the frequency distributions between TR and FL elements were significantly different in all populations that were tested except for North Carolina. The shape of the distribution could not be estimated for FL elements in the Everglades, because the number of FL elements in this population was too small to draw any conclusions.
Table 3.
Comparison of TR and FL Allele Frequencies in Five Green Anole Populations
| Population | TR | FL | Wilcoxon Rank-Sum Test | Kolmogorov–Smirnov Test |
|
|---|---|---|---|---|---|
| P Value | D | ||||
| Gulf–Atlantic | N = 30 | N = 21 | <0.01 | 0.002 | 0.510 |
| 0.74 | 0.58 | ||||
| North Carolina | N = 30 | N = 14 | ns | ns | 0.348 |
| 0.78 | 0.71 | ||||
| Everglades | N = 27 | N = 6 | ns | – | – |
| 0.81 | 0.41 | ||||
| Suwannee | N = 25 | N = 17 | <0.001 | 0 | 0.800 |
| 0.78 | 0.24 | ||||
| Central Florida | N = 27 | N = 15 | <0.001 | 0 | 0.674 |
| 0.84 | 0.29 | ||||
Note.—The number of inserts measured (N) for each category is indicated above the average population frequency. The P value for the Wilcoxon rank-sum test is given. D indicates the largest difference between the cumulative distributions of each sample. ns, not significant.
Host Demography Affects the Fixation of Full-length L1 Elements
It is possible that if purifying selection is acting against FL elements, its efficiency may be different across populations if the effective population sizes of those populations are different. We used the full data set to compare the frequencies of TR and FL insertions within and between each population and found that while the frequencies of TR elements are not significantly different between any of the populations, the frequencies of FL elements are highly significantly different between populations of starkly different demographic histories (table 4). Figure 5 shows the frequency distributions of FL and TR L1 elements in each green anole population. From this, it is evident that FL inserts segregate very differently in the Florida populations versus the mainland populations. For instance, the proportion of FL inserts below 50% population frequency is 83% in the Everglades, 76% in Suwannee, and 80% in Central Florida. FL L1 elements are much more common in the mainland populations, with only 29% below 50% population frequency in both North Carolina the Gulf–Atlantic. As these two populations have either a small estimated Ne or have recently experienced a dramatic range expansion (Tollis et al. 2012), it is likely that relaxed purifying selection due to stronger genetic drift is generating a higher rate of fixation for FL L1 insertions in these populations.
Table 4.
Pair Wise Population Comparisons of FL and TR L1 Insertions
| Populations Compared | Wilcoxon Rank-Sum Test |
|
|---|---|---|
| TR | FL | |
| North Carolina–Suwannee | ns | <0.001 |
| North Carolina–Central Florida | ns | <0.001 |
| North Carolina–Everglades | ns | ns |
| North Carolina–Gulf/Atlantic | ns | ns |
| Gulf/Atlantic–Suwannee | ns | <0.001 |
| Gulf/Atlantic–Central Florida | ns | <0.001 |
| Gulf/Atlantic–Everglades | ns | ns |
| Suwannee–Everglades | ns | ns |
| Suwannee–Central Florida | ns | ns |
| Everglades–Central Florida | ns | ns |
Note.—ns, not significant.
Fig. 5.—

The frequency distributions of FL and TR L1 elements in five green anole populations.
Discussion
We present here the first study of retrotransposon population dynamics in a reptile, based on a double-sided approach: We collected L1 inserts directly from the genomic DNA of individuals via cloning, and we developed population genetic markers from the Anolis genome database. We have three main conclusions: 1) L1 elements are able to reach fixation in Anolis more readily than previously thought; 2) TR elements are more likely to accumulate in the Anolis genome while FL elements are subjected to purifying selection and thus do not accumulate; and 3) the efficiency of purifying selection to remove FL elements is highly dependent on the demographic history of the population, such that FL elements are more likely to be fixed in populations of small Ne. Thus, the selective turnover model as it applies to TEs in Drosophila cannot fully explain the L1 profile of Anolis. In fact, the L1 profile in Anolis is remarkably similar to the nLTR-RT landscape in stickleback fish (Gasterosteus aculeatus), in which TR insertions accumulate while FL elements are subjected to purifying selection (Blass et al. 2012).
Because of the fact that our database-generated markers result from the sequencing a single individual, there could be an ascertainment bias that could skew our estimation of the number of fixed elements in the Anolis genome as well as the certainty with which we could detect purifying selection (Clark et al. 2005). Indeed, within the Florida populations, novel L1 insertions were found at low population frequencies (table 1), which might suggest that using the database caused us to miss rare alleles. Yet, the frequency distribution of all elements, including those retrieved from the database, shows an overabundance of rare inserts in Florida. In addition, all of the novel cloned L1 inserts we collected from the Gulf–Atlantic population were either at very high population frequency (>50%) or were fixed, suggesting that the genetic variation we captured with this method closely mirrors what is in the database. The database was not more likely to yield fixed inserts than the cloning, as our PCR presence/absence study of cloned novel insertions was able to retrieve some elements that were fixed (10%). Therefore, our conclusion that a significant number of L1 has reached fixation in Anolis is accurate and supported by a more unbiased assessment. Even if we were able to completely remove all bias and sample more rare alleles, the frequency distribution of FL elements, which are rare as suggested by our data, would still be shifted toward zero and would not change the fact that many TR insertions are fixed; this would actually strengthen our conclusion that purifying selection is acting against FL elements.
Compared with the human genome, L1 in Anolis is relatively low in copy number, and the few elements that are found in the genome are of very recent age (Novick et al. 2009). These features of the nLTR-RT landscape in Anolis are reminiscent of what is found in the teleost fish genomes that have been studied so far (Volff et al. 2003; Duvernell et al. 2004; Furano et al. 2004; Neafsey et al. 2004; Blass et al. 2012). To explain these observations in teleost fish, it was originally proposed that TE accumulation was prevented by a high rate of turnover (Furano et al. 2004) in which the insertion of new elements is offset by the selective loss of insertions, and it was hypothesized that many TEs would exist in populations at low frequencies. This model was initially proposed for and supported by studies of TE dynamics in Drosophila (Charlesworth 1989; Biemont et al. 1994). However, the turnover hypothesis was rejected when it was tested in two teleost fish models: stickleback (Blass et al. 2012) and pufferfish (Neafsey et al. 2004). In pufferfish, the majority of the nLTR-RT insertions that were studied were found at middle or high population frequencies, which suggests that these elements are not subjected to strong purifying selection. This was a surprising finding as the pufferfish genome is so devoid of nLTR-RTs. However, this analysis only looked at short elements and therefore it may have been biased towards neutral or nearly neutral alleles. In stickleback, all FL insertions were polymorphic, which suggests that purifying selection acts preferentially against FL elements in this genome, whereas a large number of TR insertions were fixed in populations.
We found that 38 out of 43 novel cloned inserts and 44 out of 52 database-recovered L1-containing loci were polymorphic in at least one green anole population, which is a significant amount of polymorphism that is greater than, for instance, what was observed in the Ta-1 family of L1 inserts of the human genome (86% vs. 69%, respectively) (Boissinot et al. 2000). However, five of the novel cloned loci (12%) were fixed in their population of origin, and eight (15%) of the loci from the database were fixed in every population—and therefore in the entire species—which suggests that L1 is quite capable of reaching fixation in the Anolis genome. This widespread fixation of L1 elements suggests that, as in stickleback, the turnover model cannot explain the scarcity and young age of L1 elements found in Anolis. An alternative explanation would be that L1 has no effect on host fitness, which would be consistent with the conclusion of selective neutrality suggested by Neafsey et al. (2004). We find here that the vast majority of L1 elements that are fixed in Anolis are TR, and that TR insertions make up the vast majority of older elements. This suggests that at least short L1 insertions may behave as neutral alleles, which would be consistent with the fact that in both Drosophila (Petrov et al. 2003) and human (Boissinot et al. 2006) TR elements seem to be neutral or at least are under much weaker selection than FL insertions.
Universal neutrality of L1 in Anolis is an unlikely scenario, however, because the data suggest that some elements are subjected to purifying selection. FL elements are rare within all Anolis L1 families, comprising about 18% of all L1 in the genome (Novick et al. 2009), and within all natural populations they are found at lower population frequencies relative to TR elements. The scarcity of FL elements in Anolis is similar to what was found in a study of teleost fish genomes that included zebrafish, Medaka, stickleback, and pufferfish (Basta et al. 2007), and their low frequencies in green anole is reminiscent of stickleback as well (Blass et al. 2012). This suggests not only that the Anolis genome is similar to fish in its autonomous nLTR-RT repertoire, but also that a similar mechanism is preventing the fixation of FL elements in nonmammalian genomes. As similar patterns of element decay was reported in stickleback and Anolis (Novick et al. 2009; Blass et al. 2012), it is possible that a high rate of DNA loss could account for the scarcity of fixed FL elements found in both fish and reptiles. However, large DNA deletions would also remove TR insertions at the same rate, and we now have evidence that TR elements do become fixed, therefore it is more likely that the turnover model actually does apply to Anolis—but only to FL elements.
Element length was reported to be the main driver of purifying selection against nLTR-RTs in both Drosophila, (Petrov et al. 2003, 2011) human (Boissinot et al. 2006), and stickleback (Blass et al. 2012) and the patterns we are reporting for Anolis are consistent with that. In both Drosophila and human, longer elements are probably more likely than TR ones to be involved in ectopic recombination, which can cause extremely deleterious chromosomal breaks (Langley et al. 1988). Another line of evidence used to support the ectopic recombination model in Drosophila and human was that FL elements accumulate only in genomic regions that are nonrecombining (Boissinot et al. 2001; Petrov et al. 2003, 2011; Song and Boissinot 2007). In fact, it has been proposed that an overall low rate of ectopic recombination rate may be a factor that has allowed mammalian genomes to be more tolerant of significant L1 accumulation (Eickbush and Furano 2002). However, recombination rates are not yet known in the Anolis genome or for reptiles in general, so we cannot rule out a mechanism of purifying selection against L1 other than ectopic recombination.
As FL elements contain the open reading frames and promoter necessary for autonomous retrotransposition, another possibility could be that purifying selection acts against the deleterious effect of this process itself (Nuzhdin et al. 1996; Brookfield and Badge 1997). It is clear from our study that in Anolis FL elements are limited not only in genomic copy number but also population frequency; these factors would undoubtedly act to reduce the number of active copies capable of retrotransposition. The mouse genome contains 2,000–3,000 potentially active L1 copies (Akagi et al. 2008), which is in stark contrast to the approximately 90 Anolis L1 copies that contain both ORFs and are therefore potentially active (Novick et al. 2009). The human genome contains 80–100 potentially active L1 copies (Brouha et al. 2003), yet it seems that purifying selection against FL elements in the human genome is not strong enough to prevent fixation and accumulation of some active copies (Boissinot et al. 2000, 2001). If potentially active FL elements were at very low frequencies in populations, then the transposition rate would be lower than in genomes with more common active elements. The overall result of this would be a relatively low copy number of elements, which is the case in reptiles and fish. Regardless of the mechanism, the low population frequencies of FL L1 inserts, especially in conjunction with the fact that the only old and fixed inserts are TR, strongly suggest that purifying selection is limiting the ability of FL elements to become fixed in the Anolis genome.
Whether FL or TR L1 elements are being subjected to varying degrees of purifying selection, all TEs are parasites that proliferate within a host genome, and they are therefore dependent on the evolutionary history of their host. Theoretical and empirical studies of TE dynamics in eukaryotes have shown that any change in the effective population size (Ne) of the host can affect the efficiency of purifying selection (Charlesworth B and Charlesworth D 1983; Le Rouzic and Deceliere 2005), and in populations of small Ne, otherwise deleterious alleles are able to reach higher population frequencies due to stronger genetic drift (Charlesworth 2009). The Everglades, Suwannee, and Central Florida populations are the oldest green anole populations, they are the most demographically stable, and by every measure contain high neutral genetic diversity (Campbell-Staton et al. 2012; Tollis et al. 2012); all of these aspects are associated with a large Ne. In contrast, the North Carolina population was estimated to have the smallest Ne of the green anole lineages (Tollis et al. 2012); and the largest number of fixed L1 insertions was estimated in this population. The Gulf–Atlantic population experienced a recent expansion in Ne that may be the result of a westward dispersal of anoles across the Gulf Coastal Plain (Tollis et al. 2012), and we observed a high number of fixed TE insertions in this population. It is thought that strong genetic drift at the wave front of an expansion causes higher fixation rates, leading to the spread of fixed alleles across the territory of a population (Lohmueller et al. 2008; Slatkin and Excoffier 2012). The extensive fixation of L1 insertions in the Gulf–Atlantic green anole population adds to recent empirical evidence of this kind of allele surfing in reptiles (Gracia et al. 2013).
The different fixation rates of full-length L1 insertions in green anole populations with different demographic histories show us how important genetic drift can be for genomic evolution. For instance, if a FL element is purged from a population via purifying selection, it will be unable to produce new copies. This may result in the removal of harmful alleles, but it might also be the case that the species will potentially lose a source of genetic variation that throughout the history of life, particularly in reptiles (Di-Poi et al. 2009, 2010) has been co-opted in adaptive ways (Bowen and Jordan 2007; Oliver and Greene 2009). In a landmark paper, Lynch and Conery (2003) suggested that the origins of eukaryote genome complexity might be a direct result of the shift in the selection-drift balance that occurred during the evolution of smaller effective population sizes. Indeed, variation in GC content has been correlated with certain life history traits including Ne across mammals (Romiguier et al. 2010), and our results suggest this may also apply other genomic features such as TEs. It leaves the intriguing possibility that the waxing and waning of Ne during lineage diversification can have far-reaching consequences and may account for the divergent patterns of TE evolution observed across amniotes.
Although purifying selection seems to be limiting the number of FL elements, TR insertions do accumulate readily in the Anolis genome. However, as there is a complete absence of L1 insertions that are anywhere near the order of divergence that is typical of some L1 families in the human genome, which can be up to 30% (Khan et al. 2006), it appears that L1 elements are removed from the Anolis genome soon after they become fixed. Novick et al. (2009) analyzed the decay of Anolis nLTR-RTs in the RTE clade and reported that large-scale deletions account for the heavily fragmented copies of this group of insert. A similar pattern was found in the Expander nLTR-RT clade in the stickleback genome (Blass et al. 2012). In both of these cases, these elements were much more fragmented than human L1 insertions of similar age, suggesting that DNA loss in the form of large deletions is counteracting the accumulation of retrotransposon copies in fish and reptiles, thus limiting the expansion of the sizes of these genomes. In contrast, large deletions are rare in mammals, which may account for the large size of mammalian genomes. It is therefore possible that DNA loss is a major factor controlling genome size and structure more than previously thought. However, if this were true then some of the TR elements studied here may have once been FL inserts that became fixed but subsequently accumulated deletions over time. To confirm that TR and FL elements are indeed separate classes and not simply at varying stages of the drift-deletion process due to differences in age (Blumenstiel et al. 2012), we compared the population frequencies of TR and FL insertions of similar age (<1% divergent from their consensus sequence) in the Central Florida green anole population. We determined the population frequency distributions of these age-matched sets of insertions to be significantly different (P < 0.05, Mann–Whitney U test), thus strengthening our conclusion that TR elements can reach fixation relatively quickly and are subsequently removed by large deletions. The role of large deletions is still a controversial subject, as Petrov (2002) found that small deletions are actually more common in small insect genomes, and suggested that large deletions are probably too deleterious to be common. However, this may apply only to the compact genomes of insects, as the larger genomes of most vertebrates contain vast intergenic regions that could possibly experience large deletions without consequence.
Conclusion
We have provided here the first study of TE population dynamics in reptiles. Contrary to earlier suggestions in which strong purifying selection limits the accumulation of nLTR-RTs in the Anolis genome, we find that the L1 retrotransposon actually accumulates readily in this genome. By studying the population frequencies of L1 inserts collected by direct cloning from genomic DNA and by marker design from the genomic database, we found that TR L1 insertions are very often fixed in green anole populations, and some appear to be fixed across the entire species. This suggests that short elements behave neutrally in populations and may have little to no effect on host fitness. In contrast, FL inserts are rare in green anole populations, and none are fixed at the species level, suggesting that purifying selection is at least acting on long L1 elements. The deleteriousness of FL L1 elements may stem from their ability to mediate ectopic recombination or their potential for retrotransposition activity. We also found that the demographic history of populations is an important factor that affects the strength of selection against FL elements. By comparing the frequency spectrum of L1 elements by length in different populations, we found that FL elements are found at significantly higher frequencies in populations where genetic drift is likely to be very strong. Meanwhile, FL elements are found at significantly lower frequencies in populations of large Ne and demographic stability, suggesting purifying selection is much more efficient at removing harmful alleles in these populations. The deleterious effect of FL elements does not appear to completely prevent fixation of L1 elements, yet there are very few ancient elements in the Anolis genome. Therefore, we suggest that DNA loss plays a major role in removing L1 insertions after they become fixed. This interplay of selection, demography, and large-scale deletions may account for the differences between the high-copy number L1 profile of mammalian genomes and the low-copy number profile of the genomes of nonmammalian vertebrates.
Supplementary Material
Supplementary files are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by PSC-CUNY (City University of New York) grant 63799-00-41, National Institutes of Health grant R15GM096267-01 (to S.B.)., and a CUNY Doctoral Research Grant and an American Museum of Natural History Theodore Roosevelt Memorial grant (to M.T.). The work was conducted in part with equipment from the Core Facility for Imaging, Cellular and Molecular Biology at Queens College.
Literature Cited
- Akagi K, Li J, Stephens RM, Volfovsky N, Symer DE. Extensive variation between inbred mouse strains due to endogenous L1 retrotransposition. Genome Res. 2008;18:869–880. doi: 10.1101/gr.075770.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfoldi J, et al. The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature. 2011;477:587–591. doi: 10.1038/nature10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basta HA, Buzak AJ, McClure MA. Identification of novel retroid agents in Danio rerio, Oryzias latipes, Gasterosteus aculeatus and Tetraodon nigroviridis. Evol Bioinform Online. 2007;3:179–195. [PMC free article] [PubMed] [Google Scholar]
- Biemont C, et al. Population dynamics of the copia, mdg1, mdg3, gypsy, and P transposable elements in a natural population of Drosophila melanogaster. Genet Res. 1994;63:197–212. doi: 10.1017/s0016672300032353. [DOI] [PubMed] [Google Scholar]
- Biemont C, et al. Maintenance of transposable element copy number in natural populations of Drosophila melanogaster and D. simulans. Genetica. 1997;100:161–166. [PubMed] [Google Scholar]
- Blass E, Bell M, Boissinot S. Accumulation and rapid decay of non-LTR retrotransposons in the genome of the three-spine stickleback. Genome Biol Evol. 2012;4:687–702. doi: 10.1093/gbe/evs044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenstiel JP, He M, Bergman CM. An age-of-allele test of neutrality for transposable element insertions not at equilibrium. 2012 doi: 10.1534/genetics.113.158147. arXiv arXiv:1209.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissinot S, Chevret P, Furano AV. L1 (LINE-1) retrotransposon evolution and amplification in recent human history. Mol Biol Evol. 2000;17:915–928. doi: 10.1093/oxfordjournals.molbev.a026372. [DOI] [PubMed] [Google Scholar]
- Boissinot S, Davis J, Entezam A, Petrov D, Furano AV. Fitness cost of LINE-1 (L1) activity in humans. Proc Natl Acad Sci U S A. 2006;103:9590–9594. doi: 10.1073/pnas.0603334103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissinot S, Entezam A, Furano AV. Selection against deleterious LINE-1-containing loci in the human lineage. Mol Biol Evol. 2001;18:926–935. doi: 10.1093/oxfordjournals.molbev.a003893. [DOI] [PubMed] [Google Scholar]
- Bowen NJ, Jordan IK. Exaptation of protein coding sequences from transposable elements. Genome Dyn. 2007;3:147–162. doi: 10.1159/000107609. [DOI] [PubMed] [Google Scholar]
- Brookfield JF, Badge RM. Population genetics models of transposable elements. Genetica. 1997;100:281–294. [PubMed] [Google Scholar]
- Brouha B, et al. Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci U S A. 2003;100:5280–5285. doi: 10.1073/pnas.0831042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell-Staton SC, et al. Out of Florida: mtDNA reveals patterns of migration and Pleistocene range expansion of the Green Anole lizard (Anolis carolinensis) Ecol Evol. 2012;2:2274–2284. doi: 10.1002/ece3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoe TA, et al. Discovery of highly divergent repeat landscapes in snake genomes using high throughput sequencing. Genome Biol Evol. 2011;3:641–653. doi: 10.1093/gbe/evr043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. Transposable elements in natural populations of Drosophila. Prog Nucleic Acid Res Mol Biol. 1989;36:25–36. doi: 10.1016/s0079-6603(08)60158-0. [DOI] [PubMed] [Google Scholar]
- Charlesworth B. Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nat Rev Genet. 2009;10:195–205. doi: 10.1038/nrg2526. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D. The population dynamics of transposable elements. Genet Res. 1983;42:1–27. [Google Scholar]
- Charlesworth B, Jarne P, Assimacopoulos S. The distribution of transposable elements within and between chromosomes in a population of Drosophila melanogaster. III. Element abundances in heterochromatin. Genet Res. 1994;64:183–197. doi: 10.1017/s0016672300032845. [DOI] [PubMed] [Google Scholar]
- Clark AG, Hubisz MJ, Bustamante CD, Williamson SH, Nielsen R. Ascertainment bias in studies of human genome-wide polymorphism. Genome Res. 2005;15:1496–1502. doi: 10.1101/gr.4107905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011;7:e1002384. doi: 10.1371/journal.pgen.1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di-Poi N, Montoya-Burgos JI, Duboule D. Atypical relaxation of structural constraints in Hox gene clusters of the green anole lizard. Genome Res. 2009;19:602–610. doi: 10.1101/gr.087932.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di-Poi N, et al. Changes in Hox genes' structure and function during the evolution of the squamate body plan. Nature. 2010;464:99–103. doi: 10.1038/nature08789. [DOI] [PubMed] [Google Scholar]
- Donoghue PC, Benton MJ. Rocks and clocks: calibrating the tree of life using fossils and molecules. Trends Ecol Evol. 2007;22:424–431. doi: 10.1016/j.tree.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, et al. Geneious v5.5. 2010. [cited 2013 May 21]. Available from: http://www.geneious.com.
- Duvernell DD, Pryor SR, Adams SM. Teleost fish genomes contain a diverse array of L1 retrotransposon lineages that exhibit a low copy number and high rate of turnover. J Mol Evol. 2004;59:298–308. doi: 10.1007/s00239-004-2625-8. [DOI] [PubMed] [Google Scholar]
- Eickbush TH, Furano AV. Fruit flies and humans respond differently to retrotransposons. Curr Opin Genet Dev. 2002;12:669–674. doi: 10.1016/s0959-437x(02)00359-3. [DOI] [PubMed] [Google Scholar]
- Furano AV, Duvernell DD, Boissinot S. L1 (LINE-1) retrotransposon diversity differs dramatically between mammals and fish. Trends Genet. 2004;20:9–14. doi: 10.1016/j.tig.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Garcia Guerreiro MP, Chavez-Sandoval BE, Balanya J, Serra L, Fontdevila A. Distribution of the transposable elements bilbo and gypsy in original and colonizing populations of Drosophila subobscura. BMC Evol Biol. 2008;8:234. doi: 10.1186/1471-2148-8-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RA, et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Gibbs RA, et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–234. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- Gracia E, et al. Surfing in tortoises? Empirical signs of genetic structuring owing to range expansion. Biol Lett. 2013;9:20121091. doi: 10.1098/rsbl.2012.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall T. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Janes DE, Organ CL, Fujita MK, Shedlock AM, Edwards SV. Genome evolution in Reptilia, the sister group of mammals. Annu Rev Genomics Hum Genet. 2010;11:239–264. doi: 10.1146/annurev-genom-082509-141646. [DOI] [PubMed] [Google Scholar]
- Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan H, Smit A, Boissinot S. Molecular evolution and tempo of amplification of human LINE-1 retrotransposons since the origin of primates. Genome Res. 2006;16:78–87. doi: 10.1101/gr.4001406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbie DJ, Mattick JS. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nat Protoc. 2008;3:1452–1456. doi: 10.1038/nprot.2008.133. [DOI] [PubMed] [Google Scholar]
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Langley CH, Montgomery E, Hudson R, Kaplan N, Charlesworth B. On the role of unequal exchange in the containment of transposable element copy number. Genet Res. 1988;52:223–235. doi: 10.1017/s0016672300027695. [DOI] [PubMed] [Google Scholar]
- Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Le Rouzic A, Deceliere G. Models of the population genetics of transposable elements. Genet Res. 2005;85:171–181. doi: 10.1017/S0016672305007585. [DOI] [PubMed] [Google Scholar]
- Lockton S, Ross-Ibarra J, Gaut BS. Demography and weak selection drive patterns of transposable element diversity in natural populations of Arabidopsis lyrata. Proc Natl Acad Sci U S A. 2008;105:13965–13970. doi: 10.1073/pnas.0804671105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller KE, et al. Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451:994–997. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovern MB, Holmes MM, Wade J. The green anole (Anolis carolinensis): a reptilian model for laboratory studies of reproductive morphology and behavior. ILAR J. 2004;45:54–64. doi: 10.1093/ilar.45.1.54. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The origins of genome complexity. Science. 2003;302:1401–1404. doi: 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]
- Neafsey DE, Blumenstiel JP, Hartl DL. Different regulatory mechanisms underlie similar transposable element profiles in pufferfish and fruitflies. Mol Biol Evol. 2004;21:2310–2318. doi: 10.1093/molbev/msh243. [DOI] [PubMed] [Google Scholar]
- Novick PA, Basta H, Floumanhaft M, McClure MA, Boissinot S. The evolutionary dynamics of autonomous non-LTR retrotransposons in the lizard Anolis carolinensis shows more similarity to fish than mammals. Mol Biol Evol. 2009;26:1811–1822. doi: 10.1093/molbev/msp090. [DOI] [PubMed] [Google Scholar]
- Nuzhdin SV, Pasyukova EG, Mackay TF. Positive association between copia transposition rate and copy number in Drosophila melanogaster. Proc Biol Sci. 1996;263:823–831. doi: 10.1098/rspb.1996.0122. [DOI] [PubMed] [Google Scholar]
- Oliver KR, Greene WK. Transposable elements: powerful facilitators of evolution. Bioessays. 2009;31:703–714. doi: 10.1002/bies.200800219. [DOI] [PubMed] [Google Scholar]
- Petrov DA. Mutational equilibrium model of genome size evolution. Theor Popul Biol. 2002;61:531–544. doi: 10.1006/tpbi.2002.1605. [DOI] [PubMed] [Google Scholar]
- Petrov DA, Aminetzach YT, Davis JC, Bensasson D, Hirsh AE. Size matters: non-LTR retrotransposable elements and ectopic recombination in Drosophila. Mol Biol Evol. 2003;20:880–892. doi: 10.1093/molbev/msg102. [DOI] [PubMed] [Google Scholar]
- Petrov DA, Fiston-Lavier AS, Lipatov M, Lenkov K, Gonzalez J. Population genomics of transposable elements in Drosophila melanogaster. Mol Biol Evol. 2011;28:1633–1644. doi: 10.1093/molbev/msq337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontius JU, et al. Initial sequence and comparative analysis of the cat genome. Genome Res. 2007;17:1675–1689. doi: 10.1101/gr.6380007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romiguier J, Ranwez V, Douzery EJ, Galtier N. Contrasting GC-content dynamics across 33 mammalian genomes: relationship with life-history traits and chromosome sizes. Genome Res. 2010;20:1001–1009. doi: 10.1101/gr.104372.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Shaffer HB, et al. The western painted turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome Biol. 2013;14:R28. doi: 10.1186/gb-2013-14-3-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M, Excoffier L. Serial founder effects during range expansion: a spatial analog of genetic drift. Genetics. 2012;191:171–181. doi: 10.1534/genetics.112.139022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A, Hublley R, Green P. RepeatMasker Open-3.0. 1996–2010 [cited 2013 May 21]. Available from: http://www.repeatmasker.org/ [Google Scholar]
- Song M, Boissinot S. Selection against LINE-1 retrotransposons results principally from their ability to mediate ectopic recombination. Gene. 2007;390:206–213. doi: 10.1016/j.gene.2006.09.033. [DOI] [PubMed] [Google Scholar]
- Tamura K, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollis M, Ausubel G, Ghimire D, Boissinot S. Multi-locus phylogeographic and population genetic analysis of Anolis carolinensis: historical demography of a genomic model species. PLoS One. 2012;7:e38474. doi: 10.1371/journal.pone.0038474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollis M, Boissinot S. The transposable element profile of the anolis genome: how a lizard can provide insights into the evolution of vertebrate genome size and structure. Mob Genet Elements. 2011;1:107–111. doi: 10.4161/mge.1.2.17733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volff JN, Bouneau L, Ozouf-Costaz C, Fischer C. Diversity of retrotransposable elements in compact pufferfish genomes. Trends Genet. 2003;19:674–678. doi: 10.1016/j.tig.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Wade J. Sculpting reproductive circuits: relationships among hormones, morphology and behavior in anole lizards. Gen Comp Endocrinol. 2012;176:456–460. doi: 10.1016/j.ygcen.2011.12.011. [DOI] [PubMed] [Google Scholar]
- Wade CM, et al. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science. 2009;326:865–867. doi: 10.1126/science.1178158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, et al. The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan. Nat Genet. 2013;45:701–706. doi: 10.1038/ng.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.