

Abstract
Aim
We examined the association between birth weight and methylation in the imprinted IGF/H19 loci, the nonimprinted gene NR3C1 and repetitive element DNA (LINE-1 and Alu).
Materials & methods
We collected umbilical cord venous blood from 219 infants born in Mexico City (Mexico) as part of a prospective birth cohort study and analyzed DNA methylation using pyrosequencing.
Results
Birth weight was not associated with DNA methylation of the regions studied. One of the CpG dinucleotides in the IGF2 imprinting control region (ICR)1 includes a potential C–T SNP. Among individuals with an absence of methylation at this site, probably due to a paternally inherited T allele, birth weight was associated with mean methylation status of both IGF2 ICR1 and ICR2. However, this association would not have survived adjustment for multiple testing.
Conclusion
While we did not detect an association between DNA methylation and birth weight, our study suggests a potential gene–epigene interaction between a T allele in the IGF2 ICR1 and methylation of ICRs of IGF2, and fetal growth.
Keywords: Alu, birth weight, DNA methylation, fetal growth, glucocorticoid receptor, IGF2, imprinting, LINE-1, NR3C1, SNP
Inadequate fetal growth is associated with multiple neonatal morbidities requiring neonatal intensive care, including preterm birth, hypoglycemia and hypothermia [1]. It is also associated with increased risk of later life adverse health outcomes, including metabolic disorders, such as Type 2 diabetes and hypertension [2–4]. Fetal growth is dependent on maternal health, nutrition, placental integrity, exposure to toxicants and genetics [1]. Epigenetic mechanisms are also critical as fetal growth factors are common among imprinted genes. In addition, nonimprinted genes, such as the glucocorticoid receptor (known as NR3C1), may play a part given their role in nutrient metabolism and responsiveness to the environment.
Chromosome 11p15 contains an imprinting cluster that includes the IGF2 gene, which is imprinted in tandem with H19, a long ncRNA that is associated with growth suppression (Figure 1). IGF2/H19 expression is regulated by at least two imprinting control regions (ICRs; ICR1 and ICR2). ICR1 is also known as the H19 differentially methylated region (DMR). ICR2 is also known as KCNQ1OT1. Loss of imprinting (LOI) within either ICR has been associated with both fetal undergrowth (Russell–Silver syndrome – ICR1) [5,6] and overgrowth (Beckwith–Wiedemann syndrome [BWS] – ICR2) [7] depending on whether maternally or paternally expressed genes are affected. Investigators have also recently reported an association between DNA methylation of this region in cord blood and birth weight in nonsyndromic infants [8,9].
Figure 1. The IGF2 cluster and parent-of-origin imprinting patterns.
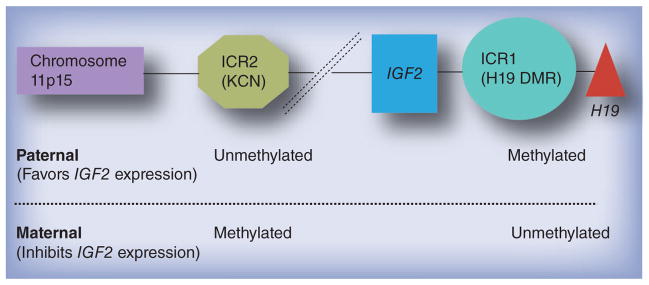
DMR: Differential methylation region; ICR: Imprinting control region.
In addition to imprinted genes, nonimprinted genes that regulate nutrient metabolism may also be involved in fetal growth, and methylation of their CpG islands, as well as less CpG dense areas, may correlate with fetal growth. A potential candidate gene for such effects is NR3C1. Corticosteroid-regulated nutrient metabolism [10] and exogenous corticosteroid exposure during pregnancy [11–13] have both been associated with restricted fetal growth in previous studies. Risk factors that affect fetal growth, such as maternal depression [14], also predict offspring DNA methylation patterns within the NR3C1 promoter [15,16]. Taken together, cortisol-related genes such as NR3C1 are potential candidate genes for the epigenetic regulation of fetal growth.
In addition to studying the DNA methylation of particular genes and their promoters, investigating the DNA methylation of repetitive elements has revealed numerous associations with human health. Transposons and retrotransposons are repetitive elements of viral origin. These DNA sequences comprise approximately half of all DNA [17]. Owing to their frequency in the human genome, measures of their methylation are often used as proxies of global DNA methylation [18]. Two common repetitive elements include a long interspersed nuclear element known as LINE-1 and a short interspersed nuclear element known as Alu [19]. Hypomethylation of repetitive elements (LINE-1 and Alu) has been associated, albeit inconsistently, with inflammatory markers [20], vascular disease [21] and toxic exposures [22,23], which might be associated with altered fetal growth [24–27]. Furthermore, one recent study demonstrated a positive association between DNA methylation of LINE-1 and Alu in placental tissue and birth weight [28]. Another found that normal birth weight infants had higher cord blood LINE-1 DNA methylation than low and high birth weight infants [29].
To better understand whether DNA methylation may affect fetal growth, we examined the cross-sectional relationship between birth weight and DNA methylation of the IGF2 gene, the glucocorticoid receptor gene and LINE-1/Alu in umbilical cord blood leukocytes from a cohort of Mexican newborns.
Materials & methods
Study sample
Between 2007 and 2011 we recruited healthy pregnant women at 12–24 weeks’ gestation through the Mexican social security system (Instituo Mexicano del Seguro Social). Instituo Mexicano del Seguro Social is funded by the federal government, employers and employees to provide healthcare to private sector, middle class workers and their families. It provides medical coverage to over 34.3% of Mexico City residents (numbers according to the 2010 National Census). Inclusion criteria for the study included maternal age of 18 years or older, access to a telephone and a plan to reside within Mexico City for the following 3 years. Exclusion criteria included diagnosis of heart or kidney disease, use of steroids or antiepilepsy drugs, daily alcohol consumption or pregnancy of over 20 weeks’ gestation. We invited 3898 women to participate in the cohort. Among these, 624 (16%) were ineligible. A total of 1054 (27%) agreed to participate, signed a letter of informed consent and were formally enrolled. Institutional review boards of the participating institutions approved the study. We studied a convenience sample of 226 of 1054 (21%) mother–infant pairs participating in an ongoing prospective birth cohort in Mexico City. As this was a pilot study, the selection of samples was predicated on the availability of funding and the timing of an infant’s delivery. We excluded two infants for gestational age <36 weeks, two for having a missing gestational age and three for having missing covariate data, giving a final number of 219 infants. Compared with the larger cohort of women, the subsample we analyzed had similar sociodemographic, perinatal and anthropometric characteristics, except that the subsample had a slightly higher proportion of nulliparous women (37 vs 32%).
Quantification of DNA methylation
We collected umbilical cord venous blood at the time of delivery and stored whole blood in PAX-gene™ (PreAnalytiX GmbH, Hombrechtikon Switzerland) tubes and extracted DNA using PAXgene kits. The DNA subsequently underwent bisulfite treatment. We treated 500 ng DNA (concentration: 25 ng/μl) using the EZ DNA Methylation-Gold Kit (Zymo Research, CA, USA) according to the manufacturer’s protocol. Final elution was performed with 30 μl M-Elution Buffer. We used PCR and pyrosequencing to quantify the percentage methylation at each gene within the population of leukocytes obtained from each individual. We used built-in controls to verify bisulfite conversion efficiency. Every sample was tested twice for each marker. Results from the two technical replicates were averaged to reduce the variability of our results. Pearson’s correlation coefficients across the two runs ranged from 0.26 (Alu) to 0.99 (ICR1). Twelve of the individual sites had correlations >0.8 across the two runs with two of the Alu sites being the lowest. We expressed the measured degree of methylation as the percentage of methylated cytosines divided by the sum of methylated and unmethylated cytosines (%5mC).
We analyzed the average methylation across four sites of the ICR1 locus, four sites of the ICR2 locus, two sites of the H19 promoter, four sites of LINE-1, three sites of Alu, five sites of NR3C1 and one site of another region of the glucocorticoid receptor. We list the details of the promoters, amplicons, primers and sequences analyzed in Supplementary Table 1 (see www.futuremedicine.com/doi/suppl/10.2217/EPI.13.24). The genome used was GRCh37/h19 (2009). The coordinates were obtained using Genomatix software (Genomatix Software GmbH, Munich, Germany). To obtain gene-specific methylation of the IGF2 regulatory loci, we designed primers to analyze sequences in the same region described by Byun et al. [30]. The region analyzed within ICR1 is one of the seven core binding sites in this ICR that bind to CTCF protein when unmethylated on the maternally inherited allele [31]. For the glucocorticoid receptor, we analyzed two promoter regions on chromosome 5, a region we called ‘region 1’ or ‘GCR’, which is a CpG shore adjacent to a CpG island [32], and NR3C1 as described by Oberlander et al. [16]. We chose to use only the first sequencing primer described by Oberlander et al., and to analyze five out of 13 CpGs, as the authors defined CpG 3 and 4 as potential NGFI-A consensus binding sites. To quantify methylation in the repetitive elements, LINE-1 and Alu, we used previously described methods [17,33].
Birth weight
We obtained birth weight from the hospital medical records and estimated gestational age based on a maternal report of last menstrual period (LMP). We performed an additional ana lysis comparing pregnancy dating estimates among a subset of the parent cohort who had ultrasound data in addition to LMP dating. We calculated the birth weight for gestational age Z-scores based on a Mexican reference population to adjust for the nonlinearity of fetal growth [34]. However, when our results did not change based on this approach versus adjusting for gestational age, for ease of interpretability we used gestational age-adjusted birth weight in grams as the primary outcome variable.
Covariates
At the time of enrollment, study staff interviewed mothers to obtain demographic data, including age, education, smoking, parity and height. Staff weighed and measured women at enrollment. At the time of birth, study staff recorded infant sex. We also obtained a complete blood count, including a white blood cell differential analysis to account for the variety of leukocyte cell types.
Statistical analysis
We first conducted univariate descriptive analyses (means, standard deviations [SDs] and proportions) of the cohort, as well as the distribution and variation of percentage methylation at each of the genes. We used multivariable linear regression to estimate the association between methylation and gestational age-adjusted birth weight, with further adjustment for maternal age, weight, parity, education and infant sex. We also performed additional analyses to stratify participants by lack of methylation as a result of a SNP that occurs at one of the CpG sites within ICR1 or lack of methylation of the cytosine (Figure 2). The SNP will eliminate methylation at this CpG and the distribution of methylation predictably appears similar to bimodal patterns seen with LOI. However, this is not true LOI as only this one CpG is affected rather than the whole ICR. We also excluded some women from our H19 promoter (n = 1), GCR (n = 2), LINE-1 (n = 1) and NR3C1 (n = 1) analyses. These women had extraordinarily high leverage in our analyses because of very low or very high methylation values. We conducted two sensitivity analyses to verify the robustness of our results to confounding factors. First, among a subset of 186 participants with leukocyte differential counts we added the percentage of neutrophils, monocytes, eosinophils, and basophils to the model. Second, we adjusted for maternal BMI instead of weight in models with 196 mother–child pairs.
Figure 2. DNA methylation of the IGF2 imprinting control region 1 (H19 differential methylation region), CpG site 2.
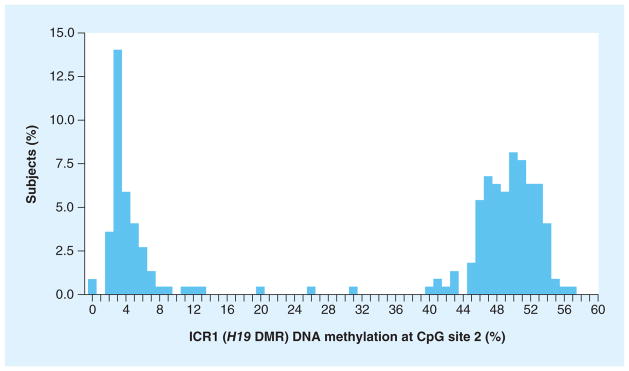
DMR: Differential methylation region; ICR: Imprinting control region.
Results
Mean maternal age was 27.6 years (SD: 5.3) (Table 1). Women reported a mean of 11.6 years of education (SD: 2.8). In general, this was a lean cohort, with a mean maternal second trimester weight 64.5 kg (SD: 11.0) and a BMI of 26.9 kg/m2 (SD: 4.2). Multiparous women comprised 68% of the cohort. Mean birth weight was 3123 g (SD: 403) and mean gestational age was 39.1 weeks (SD: 1.1).
Table 1.
Characteristics of 219 mother–infant pairs in a birth cohort in Mexico City, Mexico.
| Characteristic | Mean (SD) |
|---|---|
| Maternal age (years) | 27.6 (5.3) |
| Maternal education (years) | 11.6 (2.9) |
| Maternal weight (kg) | 64.6 (11.0) |
| Maternal BMI (kg/m2) | 26.9 (4.2) |
| Birth weight (g) | 3129 (401) |
| Gestational age (weeks) | 39.1 (1.1) |
| Birth weight for gestational age (Z-score) | −0.5 (1.0) |
| Male infant; n (%) | 115 (52.5) |
| Multiparous mother; n (%) | 151 (68.9) |
SD: Standard deviation.
The mean methylation of each of the CpG sites with a gene/ICR/repetitive element was averaged to give a mean methylation value across each region/gene. The mean percentage of methylated cytosines varied widely across regions analyzed from 1.8 %5mC (SD: 0.8) for GCR gene locus NR3C1 to 64.1 %5mC (SD: 6.7) for ICR2 and 85.0 %5mC (SD: 1.3) for LINE-1 (Table 2). In addition, the range of methylation within regions analyzed varied from within 5 and 7% for Alu and NR3C1 to >30% for the GCR and ICR2.
Table 2.
Gene-specific and global DNA methylation in cord blood leukocytes of 219 infant participants in a birth cohort in Mexico City, Mexico.
| Gene/locus | Participants (n†) | DNA methylation (%5mC) | |||||
|---|---|---|---|---|---|---|---|
| Mean (SD) | Min | 25th percentile | 50th percentile | 75th percentile | Max | ||
| IGF2 regulatory complex | |||||||
| ICR1 | 215 | 42.6 (6.3) | 28.5 | 37.0 | 43.6 | 48.0 | 54.0 |
| ICR2 | 219 | 64.0 (6.7) | 49.4 | 58.3 | 64.3 | 69.5 | 80.3 |
| H19 promoter | 218 | 46.8 (2.5) | 39.3 | 45.2 | 46.6 | 48.7 | 54.9 |
| Glucocorticoid receptor | |||||||
| GCR | 214 | 61.3 (4.1) | 44.0 | 59.1 | 61.7 | 63.5 | 74.7 |
| NR3C1 (region of the glucocorticoid receptor promoter) | 207 | 1.8 (0.8) | 0.0 | 1.4 | 1.6 | 2.0 | 6.8 |
| Repetitive elements | |||||||
| LINE-1 | 218 | 85.0 (1.3) | 81.9 | 84.1 | 85.1 | 86.0 | 88.5 |
| Alu | 219 | 24.7 (1.1) | 21.8 | 24.4 | 25.0 | 25.5 | 26.3 |
n varies based on the success of the pyrosequencing assay.
%5mC: Percentage of methylated cytosines divided by the sum of methylated and unmethylated cytosines; ICR: Imprinting control region; Max: Maximum; Min: Minimum; SD: Standard deviation.
With respect to the average methylation across each gene/region, we did not detect statistically significant (p < 0.05) associations between gestational age-adjusted birth weight and DNA methylation of any of the regions we studied (Table 3). However, we noted a suggestive trend between NR3C1 methylation and birth weight in our unadjusted models, where each 1 SD increase in methylation was associated with a 49 g (95% CI: −103, 5) decrease in birth weight. Adjusting for maternal age, parity, second trimester weight, education and infant sex did not substantially alter the magnitude of our estimates. Stratified ana lysis based on infant sex revealed that, while the methylation generally differed by sex, we did not detect an association between methylation and birth weight in either males or females (data not shown).
Table 3.
Birth weight differences associated with 1 standard deviation increase in gene-specific and global DNA methylation in cord blood leukocytes of 219 infant participants in a prospective birth cohort in Mexico City, Mexico.
| Gene/locus | Mean weight for model 1†; g (95% CI) | Mean weight for model 2‡; g (95% CI) |
|---|---|---|
| IGF2 regulatory complex | ||
| ICR1 | 6 (−47, 58) | 3 (−50, 56) |
| ICR2 | −24 (−77, 28) | −20 (−72, 32) |
| H19 promoter | 10 (−42, 63) | 4 (−49, 57) |
| Glucocorticoid receptor | ||
| GCR | −4 (−58, 49) | −1 (−53, 52) |
| NR3C1 | −49 (−103, 5) | −39 (−94, 15) |
| Repetitive elements | ||
| LINE-1 | 25 (−27, 78) | 16 (−38, 70) |
| Alu | 2 (−50, 55) | −8 (−61, 44) |
Adjusted for gestational age.
Model 1 + adjustment for parity, child sex, maternal age, education and second trimester maternal weight. ICR: Imprinting control region.
We next examined the effects at specific loci within the ICR regions. At one site we noted a bimodal distribution of methylation at the second position analyzed within the ICR1 of IGF2 (Figure 2). A C–T SNP exists at this potential CpG site (rs10732516) (Figure 3). In our study sample we found that 36% of subjects had almost a complete absence of methylation at this site, which could be due to an unmethylated cytosine, but is probably due to a paternally inherited SNP that codes for the presence of a minor T allele, as thymine is not methylated. Paternally inherited cytosines at this site are typically methylated.
Figure 3.
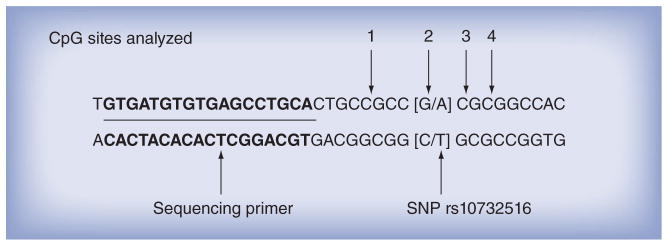
Location of the rs10732516 SNP within the imprinting control region 1 assay of the IGF2 gene.
We re-analyzed our results after stratifying based on the bimodal methylation distribution at this locus. We excluded two infants with methylation values between 20 and 40% who did not clearly fit into one of the two distributions. While mean birth weight was not significantly different among infants with or without methylation at this site (3109 g, SD: 383 g; vs 3133 g, SD: 408 g), the presence or absence of methylation at this locus appeared to modify the associations of mean methylation of ICR1 (p = 0.04), ICR2 (p = 0.04) and LINE-1 (p = 0.07) with birth weight. In fact, while not always statistically significant, the effect estimates of nearly all of the genes/ICRs and repetitive elements differed in direction in the stratified analysis (Table 4), and the overall pattern appeared consistent. Among infants with the absence of methylation at this position, increased mean methylation of ICR1 was associated with a lower birth weight (−214 g; 95% CI: −422, −6). When we excluded ICR1 position 2 from the mean methylation, this finding was slightly diminished but persisted with borderline statistical significance (−77 g; 95% CI: −165, 10). Similarly, among these infants with an absence of methylation at ICR1 position 2, there was a trend between increased methylation of ICR2 and lower birth weight (−92 g; 95% CI: −177, 15).
Table 4.
Associations of birth weight with DNA methylation stratified by IGF2 CpG site methylation at a SNP.
| Gene/locus | Allele | Mean (SD) | Birth weight difference (β); g (95% CI) | Interaction p-value |
|---|---|---|---|---|
|
IGF2 regulatory complex
| ||||
| ICR1 | SNP† (T allele) | 35.2 (2.7) | −187 (−389, 15) | 0.04 |
| Major C allele | 46.7 (3.3) | 65 (−61, 190) | ||
|
| ||||
| ICR2 | SNP (T allele) | 64.4 (6.9) | −92 (−177, −7) | 0.04 |
| Major C allele | 63.7 (6.6) | 21 (−44, 87) | ||
|
| ||||
| H19 promoter | SNP (T allele) | 46.4 (2.7) | 62 (−20, 143) | 0.12 |
| Major C allele | 47.1 (2.3) | −25 (−96, 46) | ||
|
| ||||
|
Glucocorticoid receptor
| ||||
| GCR | SNP (T allele) | 61.3 (4.2) | 0 (−86, 85) | 0.94 |
| Major C allele | 61.3 (4.0) | −4 (−72, 63) | ||
|
| ||||
| NR3C1 | SNP (T allele) | 1.8 (0.9) | −64 (−149, 22) | 0.50 |
| Major C allele | 1.8 (0.8) | −25 (−98, 49) | ||
|
| ||||
|
Repetitive elements
| ||||
| LINE-1 | SNP (T allele) | 84.9 (1.4) | −43 (−125, 40) | 0.07 |
| Major C allele | 85.1 (1.2) | 55 (−14, 125) | ||
|
| ||||
| Alu | SNP (T allele) | 24.6 (1.2) | 9 (−70, 89) | 0.60 |
| Major C allele | 24.8 (1.0) | −19 (−89, 51) | ||
Birth weight differences per SD increment of DNA methylation. Data are stratified by DNA methylation of IGF2, ICR1 position 2 (<20 vs >40% 5-methylcytosines of SNP C–T; rs10732516) and adjusted for infant gestational age, sex, maternal age, education, parity and second trimester weight.
SNP inferred from a lack of methylation probably caused by the paternally inherited T allele, but does not exclude the possibility of a lack of methylation of a paternally inherited C allele (usually methylated).
ICR: Imprinting control region; SD: Standard deviation.
In sensitivity analyses, adjusting for proportion of lymphocytes, monocytes, eosinophils and basophils did not substantively change the estimates from our models. Using maternal BMI instead of weight did not appreciably change our results. Recognizing the uncertainty of gestational age based on LMP alone, we performed an analysis on the subset of the parent cohort who had ultrasounds in addition to dating by LMP on a subset of 98 women. The Pearson’s correlation between the gestational age calculated by either method on these women was 0.89 (p < 0.0001).
Discussion
In this study, we did not detect an association between overall mean methylation within cord blood DNA for the two regulatory loci of the IGF2 gene (ICR1 and ICR2), the promoter regions of the glucocorticoid receptor gene or repetitive elements with gestational age-adjusted birth weight. However, we did find a trend towards decreased birth weight among infants with higher methylation of NR3C1. Perhaps our most intriguing finding was that a SNP coding for a C-T transfer within the IGF2 ICR1 (or less likely, absent methylation of a typically methylated, paternally inherited cytosine) appeared to modify the association between DNA methylation at multiple loci and birth weight. This is notable because this SNP prevents DNA methylation at this imprinted CpG site, although not within the entire ICR locus. At this one potential CpG site, the presence of this SNP may have similar conformational effects as a lack of methylation at this position. Functional deficits from such a SNP may be expected to be subtle, as opposed to true LOI, which would affect multiple CpG sites, and could explain the observed differences in associations of birth weight with DNA methylation of many of the other genes when we stratified by methylation status of IGF2 ICR1 position 2.
While animal [5–7,35–37] and human studies of syndromes involved in extreme fetal growth dys-regulation [5–7,35] have demonstrated relationships between LOI and birth weight, few human studies have examined the role of DNA methylation among nonsyndromic newborns, among either imprinted or nonimprinted genes. In a study of multiple fetal tissues, Tabano et al. compared methylation patterns between ten infants with birth weights appropriate for gestational age (AGA) and five infants with intrauterine growth restriction [38]. In umbilical cord blood, they found that DNA methylation within IGF2 regulatory loci and LINE-1 was similar in intrauterine growth restriction and AGA infants (ICR1: 46.2 vs 46.1%; DMR2 [ICR2]: 48.2 vs 49.0%; and H19 promoter: 48.4 vs 48.2%; LINE-1 values not reported). Our mean methylation at both ICR1 and the H19 promoter revealed a similar range of values, but our mean ICR2 methylation (64%) differed substantially from theirs. This difference may be due to different assays, which focused on different CpG sites within ICR2, but could also be due to differences between source populations. While this study also reports mean methylation at each locus, the authors did not comment on whether they analyzed the SNPs within the ICRs of IGF2. In another small study, Guo et al. found no difference in IGF2 methylation status in cord blood in ten small-for-gestational age (SGA) infants compared with nine AGA infants and reported that neonatal cord blood methylation was ‘normal’ in both SGA and AGA infants, but their placental analyses revealed differences [39]. The investigators used Southern blotting to analyze methylation in the umbilical cord blood to achieve a methylation index, which may not be comparable to our values obtained by pyrosequencing. Neither of these two studies examined continuous birth weight. In a prospective study of very preterm (<32 weeks’ gestation) and/or very low birth weight babies (<1500 g), Tobi et al. compared DNA methylation of IGF2 from blood drawn at 19 years of age among 75 born AGA and 38 born SGA [40]. They report no difference in DNA methylation of IGF2 between SGA and AGA individuals. This same group previously found that individuals who were exposed to the Dutch famine in utero maintained persistent epigenetic differences compared with unexposed siblings in late adulthood, specifically in the DNA methylation of IGF2 [41]. These prior studies did not address the potential presence of SNPs. We did not directly analyze IGF2 itself, only the two ICRs.
A recent study examined the association between IGF2 methylation and birth weight. Hoyo et al. found associations between IGF2 and H19 DNA methylation and IGF2 gene expression [9]. The investigators studied 300 cord blood samples for DNA methylation at these two regions using pyrosequencing of different CpG sites within these regions to those used in our study. Similar to our study, they did not find an association between birth weight and IGF2 or H19 DNA methylation; infants ≤2500 g versus >2500 g had similar percentage methylation of the IGF2 DMR (p = 0.9) and H19 DMR (p = 0.2). Their study did not report associations with continuous birth weight and did not adjust for gestational age at the time of delivery. Positive associations between IGF2 concentrations and birth weight may be due to confounding by gestational age. It is interesting that DNA methylation was inversely correlated with IGF2 expression and expression was positively associated with birth weight, but that methylation was not associated with dichotomized birth weight. This finding supports the notion that DNA methylation could have more subtle effects on birth weight.
Like most imprinted loci, 11p15 codes for two genes with opposite imprinting patterns, and presumably opposing metabolic functions that affect the expression of IGF2. IGF2 is a growth factor gene and its biallelic expression has been associated with BWS and several cancers [42], while H19 is a long ncRNA believed to function as a tumor suppressor [43]. Regulatory loci controlled by methylation include two ICRs, ICR1 and ICR2, as well as the H19 promoter, which is proximal to ICR1. Typically, ICR1 is unmethylated on the maternal allele and, thus, binds to CTCF proteins, which in turn regulate the access of enhancers to the IGF2 promoters in cis [44]. When ICR1 methylation is lost on the paternal allele, this could lead to biallelic expression of H19, placing the fetus at increased risk of Russell–Silver syndrome, characterized by severe fetal and childhood growth restriction [5,6, 35,45,46]. By contrast, LOI in the maternal ICR2 region leads to biallelic expression of IGF2 and increased risk of BWS, characterized by fetal overgrowth and other anomalies [7].
We speculate that with normal IGF2 imprinting, small differences in mean methylation within the ICRs of IGF2 may not directly affect fetal growth. However, the presence of a functional SNP within a CpG site could lead to subtle changes in conformation or facilitate the ability of CTCF proteins to bind, affecting IGF2 expression to a small degree. Whether a SNP at one site could make such a difference is unknown, but remains possible. These small changes might not be sufficient to directly affect growth but could alter the effects of methylation at other methylation sites that control growth. One twin study showed that this T allele, when maternally inherited (usually unmethylated allele), is associated with greater methylation over the entire H19 ICR region [47].
A study by St-Pierre et al. examined IGF2 DNA methylation of placental tissue and infant genotypes of the same SNP that we studied (rs10732516) [8]. Similarly to our study, they saw no difference in birth weight among the 50 infants based on C/C, C/T or T/T genotypes (p = 0.12). They found an association between genotype at this SNP, and head and thorax circumference, which we did not measure. They did not evaluate whether the genotype modified the association between DNA methylation and birth weight. We observed a stronger inverse relationship between ICR2 methylation and birth weight among infants without methylation at this site within ICR1 (probably due to a paternally inherited T allele). This may be due to the combined effect of increased methylation at ICR2, which inhibits IGF2 expression (Figure 1), and the lack of methylation at this SNP in ICR1, which would allow for more H19 transcription and, thus, inhibition of IGF2 expression. Owing to the lack of mRNA collection, we could not directly test this hypothesis in our study. In addition, our relatively modest sample size, which was further reduced when we stratified by the ICR1 position 2 methylation status, limited our ability to precisely examine these associations.
We did not formally adjust for multiple comparisons and examined numerous associations in the process of this stratification, which may have led to chance findings and, thus, assert that our finding should be considered hypothesis-generating as opposed to conclusive. However, we found compelling evidence for a functional nature of the ICR1 SNP, in that this C–T SNP (or lack of methylation at this CpG site) modified the effect of methylation of several methylation sites we analyzed, not just ICR1. SNPs within imprinted loci could impart a loss of methylation in a subset of cytosines in imprinted loci. In fact, there is evidence that methylated cytosines are mutagenic and prone to C–T conversions [48]. Furthermore, genetic sequence and DNA methylation can jointly affect phenotype [49,50]. If DNA methylation is lost within a subset of cytosine sites within ICRs, it is possible that subclinical effects on fetal growth may be seen due to slight transformational changes in DNA packaging produced by the inability to methylate these loci, even with otherwise normal imprinting. Alternatively, the lack of methylation could facilitate binding factors and transcription.
Adkins et al. analyzed multiple SNPs within IGF2/H19 and the IGF2 receptor gene, and offspring birth weight [51]. To assess for parent-of-origin associations, they only considered homozygous mothers who transmitted a C allele to their offspring within an H19 SNP (rs4929984) instead of the minor A allele. Infants who received the A (vs C) allele from their mothers had significantly lower birth weight Z-scores regardless of which allele they received from their fathers. The authors did not measure methylation in this study. However, in a study analyzing gene expression related to brain activity, Ursini et al. demonstrated that DNA methylation that differed by genotype at C–T SNP loci can affect gene expression [52]. We did not perform allele-specific DNA methylation or genotyping but surmise that the absence of methylation at ICR1 position 2 is due to a paternally inherited SNP, since typically the CpG site at this locus would be methylated (in a normal imprinting pattern), but a thymine would not. While we are not aware of the Mexican frequency of the minor T allele, if it is similar to the Brazilian population (44%) [35] or a French–Canadian study population (46%) [8], the predicted paternal transmission to the offspring would be approximately 45%, slightly higher than our observed frequency of lack of methylation of 36% at this site in our study. Using only methylation analyses we cannot distinguish between methylated cytosine and a maternally inherited SNP at this locus, since both the SNP and the CpG site would be expected to be unmethylated. Further work using allele-specific methylation in combination with genotyping would be necessary to confirm our assertion that this is a paternally inherited SNP and determine whether a maternally inherited SNP could also be functional.
Our study has several strengths, including the recognition of SNPs within CpG islands and a relatively large sample size for epigenetic analyses. Nonetheless, we used a convenience subset of a larger birth cohort and inadequate power may be responsible for the lack of association between methylation and birth weight at the various loci studied, specifically the glucocorticoid receptor gene. Host factors, such as depression, regulate glucocorticoid metabolism and methylation of the glucocorticoid receptor gene [16]. Given that depression [14] and glucocorticoids [11] are known to affect fetal growth, this lends biologic plausibility to a relationship with birth weight. Furthermore, Filiberto et al. demonstrated in a study of placental tissue, that DNA methylation of the glucocorticoid receptor promoter region was inversely associated with large-for-gestational-age status in newborns [53].
We studied cord blood leukocyte DNA methylation and not other target tissues that might be more directly responsible for fetal growth, such as the placenta, fetal liver or skeletal muscle. Investigators often use cord or peripheral blood DNA methylation analyses in epidemiologic studies because such samples are convenient to collect. White blood cells represent a potential target tissue for epigenetic research into fetal growth since they mediate inflammatory processes that may be relevant to fetal growth [24]. The IGF2 regulatory complex is an imprinted region in which methylation is maintained in somatic cells throughout cell differentiation [54]. In the case of imprinting disorders, peripheral blood leukocytes are thought to be representative of all tissues, and, thus, are routinely collected for diagnostic purposes [101]. However, subtle differences in methylation patterns within the imprinted loci or other regions we analyzed may not be correlated across tissues or time.
Conclusion
In this study, we did not detect an association between birth weight and DNA methylation within two known regulatory loci of IGF2, glucocorticoid receptor or repetitive elements (LINE-1 and Alu). However, our results suggest that some associations between DNA methylation and birth weight may be modified by the presence of a SNP at a potential CpG site in the ICR1 region. Future studies should determine whether SNPs associated with CpG sites within imprinted genes modify the relationship between DNA methylation and fetal growth. Further investigation into the role of gene–epigene interactions and perinatal health is warranted.
Future perspective
The future of gene–epigene interactions in perinatal epidemiology is wide open. How environmental and social factors affect birth outcomes remains largely unknown and epigenomic data may fill some of these knowledge gaps. For example, mechanisms underlying disparities in fetal growth and preterm birth associated with poverty, African–American race/ethnicity, young maternal age and environmental toxin exposures may be due to epigenetic variation. Elucidating such pathophysiology is the challenge in the next 5–10 years, with the goal of discovering interventions to improve perinatal outcomes.
Supplementary Material
Executive summary.
DNA methylation of IGF2 & birth weight
We found no association between the mean DNA methylation of the imprinting control regions (ICR) 1 and 2 of IGF2 and birth weight in a cohort of healthy Mexican infants.
In a subset of infants with a probably paternally inherited T allele or unmethylated cytosine at one potential CpG site within ICR1 of the IGF2 gene, DNA methylation of both ICR1 and ICR2 were associated with a decreased birth weight, suggesting a potential gene–epigene interaction.
DNA methylation of the glucocorticoid receptor gene & birth weight
We did not detect a statistically significant association between DNA methylation of the glucocorticoid receptor and birth weight in a cohort of healthy Mexican infants.
However, our results do not exclude the possibility of such an association because models that adjust for gestational age revealed a 49 g (95% CI: 103, 5) reduction in birth weight for each standard deviation increase in NR3C1 DNA methylation.
Repetitive element LINE-1 & Alu DNA methylation & birth weight
We did not detect an association between DNA methylation of repetitive elements (LINE-1 and Alu) and birth weight in a cohort of healthy Mexican infants.
Conclusion
Further work elucidating the roles of the genome and epigenome in fetal growth is warranted.
Future studies in larger cohorts should evaluate the association between DNA methylation of the glucocorticoid receptor gene as it could be important in understanding how social and environmental stressors affect fetal growth.
Acknowledgments
The authors would like to thank the cohort participants and staff in Mexico City (Mexico).
Footnotes
For reprint orders, please contact: reprints@futuremedicine.com
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
Financial & competing interests disclosure
This work was funded by NIH-NIEHS (K23ES022242, R01ES014930, R01ES013744, R01ES020268, P42ES016454, T32ES007069, R01ES000001 and K99ES020346), the Leaves of Grass Foundation and the Klarman Scholars Program at Beth Israel Deaconess Medical Center. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- 1.Brodsky D, Christou H. Current concepts in intrauterine growth restriction. J Intensive Care Med. 2004;19(6):307–319. doi: 10.1177/0885066604269663. [DOI] [PubMed] [Google Scholar]
- 2.Ross MG, Beall MH. Adult sequelae of intrauterine growth restriction. Semin Perinatol. 2008;32(3):213–218. doi: 10.1053/j.semperi.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359(1):61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gillman MW. Developmental origins of health and disease. N Engl J Med. 2005;353(17):1848–1850. doi: 10.1056/NEJMe058187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horike S, Ferreira JC, Meguro-Horike M, et al. Screening of DNA methylation at the H19 promoter or the distal region of its ICR1 ensures efficient detection of chromosome 11p15 epimutations in Russell–Silver syndrome. Am J Med Genet A. 2009;149A(11):2415–2423. doi: 10.1002/ajmg.a.33065. [DOI] [PubMed] [Google Scholar]
- 6.Netchine I, Rossignol S, Dufourg MN, et al. 11p15 imprinting center region 1 loss of methylation is a common and specific cause of typical Russell–Silver syndrome: clinical scoring system and epigenetic–phenotypic correlations. J Clin Endocrinol Metab. 2007;92(8):3148–3154. doi: 10.1210/jc.2007-0354. [DOI] [PubMed] [Google Scholar]
- 7.Gaston V, Le Bouc Y, Soupre V, et al. Analysis of the methylation status of the KCNQ1OT and H19 genes in leukocyte DNA for the diagnosis and prognosis of Beckwith–Wiedemann syndrome. Eur J Hum Genet. 2001;9(6):409–418. doi: 10.1038/sj.ejhg.5200649. [DOI] [PubMed] [Google Scholar]
- 8.St-Pierre J, Hivert MF, Perron P, et al. IGF2 DNA methylation is a modulator of newborn’s fetal growth and development. Epigenetics. 2012;7(10):1125–1132. doi: 10.4161/epi.21855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoyo C, Fortner K, Murtha AP, et al. Association of cord blood methylation fractions at imprinted insulin-like growth factor 2 (IGF2), plasma IGF2, and birth weight. Cancer Causes Control. 2012;23(4):635–645. doi: 10.1007/s10552-012-9932-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mericq V, Medina P, Kakarieka E, Marquez L, Johnson MC, Iniguez G. Differences in expression and activity of 11beta-hydroxysteroid dehydrogenase type 1 and 2 in human placentas of term pregnancies according to birth weight and gender. Eur J Endocrinol. 2009;161(3):419–425. doi: 10.1530/EJE-09-0308. [DOI] [PubMed] [Google Scholar]
- 11.Reinisch JM, Simon NG, Karow WG, Gandelman R. Prenatal exposure to prednisone in humans and animals retards intrauterine growth. Science. 1978;202(4366):436–438. doi: 10.1126/science.705336. [DOI] [PubMed] [Google Scholar]
- 12.French NP, Hagan R, Evans SF, Godfrey M, Newnham JP. Repeated antenatal corticosteroids: size at birth and subsequent development. Am J Obstet Gynecol. 1999;180(1 Pt 1):114–121. doi: 10.1016/s0002-9378(99)70160-2. [DOI] [PubMed] [Google Scholar]
- 13.Bloom SL, Sheffield JS, McIntire DD, Leveno KJ. Antenatal dexamethasone and decreased birth weight. Obstet Gynecol. 2001;97(4):485–490. doi: 10.1016/s0029-7844(00)01206-0. [DOI] [PubMed] [Google Scholar]
- 14.Diego MA, Field T, Hernandez-Reif M, Schanberg S, Kuhn C, Gonzalez-Quintero VH. Prenatal depression restricts fetal growth. Early Hum Dev. 2009;85(1):65–70. doi: 10.1016/j.earlhumdev.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weaver IC, Cervoni N, Champagne FA, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7(8):847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 16.Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3(2):97–106. doi: 10.4161/epi.3.2.6034. [DOI] [PubMed] [Google Scholar]
- 17.Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32(3):e38. doi: 10.1093/nar/gnh032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weisenberger DJ, Campan M, Long TI, et al. Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res. 2005;33(21):6823–6836. doi: 10.1093/nar/gki987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wright RO, Schwartz J, Wright RJ, et al. Biomarkers of lead exposure and DNA methylation within retrotransposons. Environ Health Perspect. 2010;118(6):790–795. doi: 10.1289/ehp.0901429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baccarelli A, Tarantini L, Wright RO, et al. Repetitive element DNA methylation and circulating endothelial and inflammation markers in the VA normative aging study. Epigenetics. 2010;5(3):222–228. doi: 10.4161/epi.5.3.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim M, Long TI, Arakawa K, Wang R, Yu MC, Laird PW. DNA methylation as a biomarker for cardiovascular disease risk. PLoS ONE. 2010;5(3):e9692. doi: 10.1371/journal.pone.0009692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pilsner JR, Hu H, Ettinger A, et al. Influence of prenatal lead exposure on genomic methylation of cord blood DNA. Environ Health Perspect. 2009;117(9):1466–1471. doi: 10.1289/ehp.0800497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baccarelli A, Wright RO, Bollati V, et al. Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med. 2009;179(7):572–578. doi: 10.1164/rccm.200807-1097OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amarilyo G, Oren A, Mimouni FB, Ochshorn Y, Deutsch V, Mandel D. Increased cord serum inflammatory markers in small-for-gestational-age neonates. J Perinatol. 2011;31(1):30–32. doi: 10.1038/jp.2010.53. [DOI] [PubMed] [Google Scholar]
- 25.Odegard RA, Vatten LJ, Nilsen ST, Salvesen KA, Austgulen R. Preeclampsia and fetal growth. Obstet Gynecol. 2000;96(6):950–955. [PubMed] [Google Scholar]
- 26.Andrews KW, Savitz DA, Hertz-Picciotto I. Prenatal lead exposure in relation to gestational age and birth weight: a review of epidemiologic studies. Am J Ind Med. 1994;26(1):13–32. doi: 10.1002/ajim.4700260103. [DOI] [PubMed] [Google Scholar]
- 27.Wilhelm M, Ghosh JK, Su J, Cockburn M, Jerrett M, Ritz B. Traffic-related air toxics and term low birth weight in Los Angeles County, California. Environ Health Perspect. 2012;120(1):132–138. doi: 10.1289/ehp.1103408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilhelm-Benartzi CS, Houseman EA, Maccani MA, et al. In utero exposures, infant growth, and DNA methylation of repetitive elements and developmentally related genes in human placenta. Environ Health Perspect. 2011;120(2):296–302. doi: 10.1289/ehp.1103927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michels KB, Harris HR, Barault L. Birth weight, maternal weight trajectories and global DNA methylation of LINE-1 repetitive elements. PLoS ONE. 2011;6(9):e25254. doi: 10.1371/journal.pone.0025254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Byun HM, Wong HL, Birnstein EA, Wolff EM, Liang G, Yang AS. Examination of IGF2 and H19 loss of imprinting in bladder cancer. Cancer Res. 2007;67(22):10753–10758. doi: 10.1158/0008-5472.CAN-07-0329. [DOI] [PubMed] [Google Scholar]
- 31.Zhang H, Niu B, Hu JF, et al. Interruption of intrachromosomal looping by CCCTC binding factor decoy proteins abrogates genomic imprinting of human insulin-like growth factor II. J Cell Biol. 2011;193(3):475–487. doi: 10.1083/jcb.201101021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Irizarry RA, Ladd-Acosta C, Carvalho B, et al. Comprehensive high-throughput arrays for relative methylation (CHARM) Genome Res. 2008;18(5):780–790. doi: 10.1101/gr.7301508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bollati V, Baccarelli A, Hou L, et al. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007;67(3):876–880. doi: 10.1158/0008-5472.CAN-06-2995. [DOI] [PubMed] [Google Scholar]
- 34.Rios JM, Tufino-Olivares E, Reza-Lopez S, Sanin LH, Levario-Carrillo M. Birth weight percentiles by gestational age and gender for children in the North of Mexico. Paediatr Perinat Epidemiol. 2008;22(2):188–194. doi: 10.1111/j.1365-3016.2007.00898.x. [DOI] [PubMed] [Google Scholar]
- 35.Gicquel C, Rossignol S, Cabrol S, et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver–Russell syndrome. Nat Genet. 2005;37(9):1003–1007. doi: 10.1038/ng1629. [DOI] [PubMed] [Google Scholar]
- 36.Angiolini E, Fowden A, Coan P, et al. Regulation of placental efficiency for nutrient transport by imprinted genes. Placenta. 2006;27(Suppl A):S98–S102. doi: 10.1016/j.placenta.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 37.Fowden AL, Sibley C, Reik W, Constancia M. Imprinted genes, placental development and fetal growth. Horm Res. 2006;65(Suppl 3):50–58. doi: 10.1159/000091506. [DOI] [PubMed] [Google Scholar]
- 38.Tabano S, Colapietro P, Cetin I, et al. Epigenetic modulation of the IGF2/H19 imprinted domain in human embryonic and extra-embryonic compartments and its possible role in fetal growth restriction. Epigenetics. 2010;5(4):313–324. doi: 10.4161/epi.5.4.11637. [DOI] [PubMed] [Google Scholar]
- 39.Guo L, Choufani S, Ferreira J, et al. Altered gene expression and methylation of the human chromosome 11 imprinted region in small for gestational age (SGA) placentae. Dev Biol. 2008;320(1):79–91. doi: 10.1016/j.ydbio.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 40.Tobi EW, Heijmans BT, Kremer D, et al. DNA methylation of IGF2, GNASAS, INSIGF and LEP and being born small for gestational age. Epigenetics. 2011;6(2):171–176. doi: 10.4161/epi.6.2.13516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heijmans BT, Tobi EW, Stein AD, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA. 2008;105(44):17046–17049. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cui H, Onyango P, Brandenburg S, Wu Y, Hsieh CL, Feinberg AP. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 2002;62(22):6442–6446. [PubMed] [Google Scholar]
- 43.Yoshimizu T, Miroglio A, Ripoche MA, et al. The H19 locus acts in vivo as a tumor suppressor. Proc Natl Acad Sci USA. 2008;105(34):12417–12422. doi: 10.1073/pnas.0801540105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szabo PE, Tang SH, Silva FJ, Tsark WM, Mann JR. Role of CTCF binding sites in the Igf2/H19 imprinting control region. Mol Cell Biol. 2004;24(11):4791–4800. doi: 10.1128/MCB.24.11.4791-4800.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Price SM, Stanhope R, Garrett C, Preece MA, Trembath RC. The spectrum of Silver–Russell syndrome: a clinical and molecular genetic study and new diagnostic criteria. J Med Genet. 1999;36(11):837–842. [PMC free article] [PubMed] [Google Scholar]
- 46.Murrell A, Heeson S, Reik W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat Genet. 2004;36(8):889–893. doi: 10.1038/ng1402. [DOI] [PubMed] [Google Scholar]
- 47.Coolen MW, Statham AL, Qu W, et al. Impact of the genome on the epigenome is manifested in DNA methylation patterns of imprinted regions in monozygotic and dizygotic twins. PLoS ONE. 2011;6(10):e25590. doi: 10.1371/journal.pone.0025590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 49.Salam MT, Byun HM, Lurmann F, et al. Genetic and epigenetic variations in inducible nitric oxide synthase promoter, particulate pollution, and exhaled nitric oxide levels in children. J Allergy Clin Immunol. 2011;129(1):232, 239.e1–7. doi: 10.1016/j.jaci.2011.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ronneberg JA, Tost J, Solvang HK, et al. GSTP1 promoter haplotypes affect DNA methylation levels and promoter activity in breast carcinomas. Cancer Res. 2008;68(14):5562–5571. doi: 10.1158/0008-5472.CAN-07-5828. [DOI] [PubMed] [Google Scholar]
- 51.Adkins RM, Somes G, Morrison JC, et al. Association of birth weight with polymorphisms in the IGF2, H19, and IGF2R genes. Pediatr Res. 2010;68(5):429–434. doi: 10.1203/PDR.0b013e3181f1ca99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ursini G, Bollati V, Fazio L, et al. Stress-related methylation of the catechol-O-methyltransferase Val 158 allele predicts human prefrontal cognition and activity. J Neurosci. 2011;31(18):6692–6698. doi: 10.1523/JNEUROSCI.6631-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Filiberto AC, Maccani MA, Koestler D, et al. Birth weight is associated with DNA promoter methylation of the glucocorticoid receptor in human placenta. Epigenetics. 2011;6(5):566–572. doi: 10.4161/epi.6.5.15236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weaver JR, Susiarjo M, Bartolomei MS. Imprinting and epigenetic changes in the early embryo. Mamm Genome. 2009;20(9–10):532–543. doi: 10.1007/s00335-009-9225-2. [DOI] [PubMed] [Google Scholar]
Website
- 101.Mayo Clinic. [Accessed 1 February 2012];Beckwith–Wiedemann syndrome (BWS)/Russell–Silver syndrome (RSS) molecular analysis. www.mayomedicallaboratories.com/test-catalog/Specimen/61010.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.