

Abstract
Establishment of specific characteristics of each embryonic cardiac chamber is crucial for development of a fully functional adult heart. Despite the importance of defining and maintaining unique features in ventricular and atrial cardiomyocytes, the regulatory mechanisms guiding these processes are poorly understood. Here, we show that the homeodomain transcription factors Nkx2.5 and Nkx2.7 are necessary to sustain ventricular chamber attributes through repression of atrial chamber identity. Mutation of nkx2.5 in zebrafish yields embryos with diminutive ventricular and bulbous atrial chambers. These chamber deformities emerge gradually during development, with a severe collapse in the number of ventricular cardiomyocytes and an accumulation of excess atrial cardiomyocytes as the heart matures. Removal of nkx2.7 function from nkx2.5 mutants exacerbates the loss of ventricular cells and the gain of atrial cells. Moreover, in these Nkx-deficient embryos, expression of vmhc, a ventricular gene, fades, whereas expression of amhc, an atrial gene, expands. Cell-labeling experiments suggest that ventricular cardiomyocytes can transform into atrial cardiomyocytes in the absence of Nkx gene function. Through suggestion of transdifferentiation from ventricular to atrial fate, our data reveal a pivotal role for Nkx genes in maintaining ventricular identity and highlight remarkable plasticity in differentiated myocardium. Thus, our results are relevant to the etiologies of fetal and neonatal cardiac pathology and could direct future innovations in cardiac regenerative medicine.
Keywords: nkx2.5, nkx2.7, Atrium, Ventricle, Zebrafish, Chamber identity
INTRODUCTION
The appropriation of specific molecular, cellular and physiological attributes by developing cardiomyocytes yields indispensable building blocks necessary to fabricate a fully functional mature heart. The discrete features of ventricular and atrial cardiomyocytes include electrophysiological and contractile properties and chamber-specific molecular and cellular traits (Moorman and Christoffels, 2003). Distinctive molecular characteristics include differential expression of transcription factors, structural genes, ion channels and secretory proteins (Ng et al., 2010). For example, individual myosin heavy chains are uniquely expressed in zebrafish ventricular and atrial cardiomyocytes (Berdougo et al., 2003). Characteristic cell morphologies also differ in each chamber; for instance, in the zebrafish heart tube, ventricular cardiomyocytes are cuboidal whereas atrial cardiomyocytes are squamous (Rohr et al., 2008). Only a few regulators of chamber-specific developmental programs have been identified (Bao et al., 1999; Bruneau et al., 2001a; Bruneau et al., 2001b; Koibuchi and Chin, 2007; Wu et al., 2013; Xin et al., 2007); thus, the full roster of genes guiding assignment and maintenance of chamber identity remains elusive.
Irx4 and Hey2 are two examples of transcription factors known to activate and repress gene programs that control regionalization of the heart. Irx4 is expressed in a ventricle-restricted pattern during cardiac development in the chick, rat and mouse (Bao et al., 1999; Bruneau et al., 2000; He et al., 2009). In chick, expression of Irx4 activates the ventricular gene program and suppresses the atrial gene program (Bao et al., 1999). Furthermore, disruption of the murine Irx4 gene causes induction of an atrium-specific gene expression pattern in the ventricle (Bruneau et al., 2001a). Hey2, a transcriptional repressor that acts downstream of Notch signaling, seems to play a related role, as knockout of Hey2 induces atrial gene expression in the ventricular myocardium (Koibuchi and Chin, 2007; Xin et al., 2007). Although both Irx4 and Hey2 are clearly important in regulating chamber-specific gene expression, loss of these transcription factors does not yield a complete transformation of chamber identity. We have therefore sought to identify other components of the transcriptional hierarchy responsible for chamber fate decisions.
In this study, we highlight the roles of Nkx transcription factors in chamber identity maintenance. In humans, mutations in NKX2-5 are associated with particular types of congenital heart defects (Benson et al., 1999; Elliott et al., 2003; Jay et al., 2003; McElhinney et al., 2003; Schott et al., 1998). Investigations in multiple model organisms have led to insights into the conserved roles of NKX2-5 homologs in cardiac specification, morphogenesis and maturation (Azpiazu and Frasch, 1993; Bodmer, 1993; Grow and Krieg, 1998; Lyons et al., 1995; Prall et al., 2007; Tanaka et al., 1999; Targoff et al., 2008; Tu et al., 2009). In addition, studies of the murine genes Nkx2-5 and Nkx2-6 have hinted at roles of Nkx genes in regulating chamber-specific gene expression. Loss of Nkx2-5 function results in dysmorphic chambers and downregulation of the ventricle-specific myosin gene Mlc2v (Myl2 - Mouse Genome Informatics) (Biben et al., 2000; Lyons et al., 1995; Tanaka et al., 1999). When Nkx2-6 function is also depleted, this aspect of the cardiac phenotype is exacerbated; Mlc2v is expressed in the ventricular myocardium at low levels, and Mlc2a (Myl7 - Mouse Genome Informatics), an atrium-specific myosin gene, persists throughout the heart, rather than becoming restricted to the atrium (Tanaka et al., 2001). Because Mlc2a is initially expressed broadly throughout the primitive myocardium, this phenotype has been suggested to represent an arrest in a primitive state of maturation; less attention has been paid to the possibility that this phenotype could indicate a defect in the assignment of ventricular identity. The notion that Nkx2-5 influences ventricular gene expression has been reinforced by the finding that Nkx2-5-/-;Hand2-/- double mutant embryos produce a single-chambered heart expressing markers of atrial identity (Yamagishi et al., 2001). However, it has been suggested that the loss of ventricular myocardium in this context is the consequence of cell death or hypoplasia, rather than of defects in chamber identity assignment. Altogether, the precise influence of Nkx genes on the acquisition and maintenance of chamber identity remains unclear.
The zebrafish genome contains two NKX2-5 homologs that are expressed in the heart: nkx2.5 and nkx2.7 (Chen and Fishman, 1996; Lee et al., 1996). Our previous studies in zebrafish have revealed roles for nkx2.5 and nkx2.7 in limiting atrial cell number and promoting ventricular cell number (Targoff et al., 2008). Here, through characterization of a newly identified nkx2.5 mutation, we find that loss of nkx2.5 function causes formation of a voluminous atrial chamber and a miniscule ventricular chamber despite normal specification of chamber-specific precursor populations. Furthermore, loss of both nkx2.5 and nkx2.7 function leads to the gradual elimination of all ventricular cardiomyocytes. In Nkx-deficient embryos, ventricular depletion coincides with encroachment of atrial gene expression into the ventricular territory, and cell-labeling experiments suggest that Nkx-deficient ventricular cardiomyocytes transdifferentiate to become atrial cardiomyocytes. We therefore conclude that Nkx genes play a pivotal role in maintaining the distinctive identity of the ventricular myocardium. These studies expand our knowledge of the mechanisms underlying maintenance of chamber identity and the importance of Nkx genes in this process. Moreover, we propose that the functions of Nkx genes in preserving chamber-specific traits may help to explain the molecular, cellular and electrophysiological phenotypes observed in patients carrying NKX2-5 mutations.
MATERIALS AND METHODS
Zebrafish transgenes and mutations
We used zebrafish carrying the following transgenes: Tg(-5.1myl7:nDsRed2)f2 (Mably et al., 2003), Tg(myl7:EGFP)twu277 (Huang et al., 2003), Tg(myl7:DsRedt4)sk74 (Garavito-Aguilar et al., 2010) and Tg(myl7:kaede)sd22 (de Pater et al., 2009). We identified point mutations in nkx2.5 and nkx2.7 through targeting induced local lesions in genomes (TILLING) (Draper et al., 2004; Solnica-Krezel et al., 1994). The nkx2.5vu179 allele contains a G→A transition at position 564 of the open reading frame leading to a nonsense mutation that is predicted to cause truncation of the protein within the homeodomain (supplementary material Fig. S1A). The nkx2.7vu413 allele contains a C→A transversion at position 321 of the open reading frame leading to a nonsense mutation that is predicted to cause truncation of the protein prior to the homeodomain (supplementary material Fig. S1B). Both mutations are recessive lethal. We have not detected developmental abnormalities in either heterozygote, suggesting that these point mutations have loss-of-function, rather than dominant-negative, effects. All zebrafish work followed Institutional Animal Care and Use Committee (IACUC)-approved protocols.
Injection
Embryos were injected at the one-cell stage with 2 ng of an anti-nkx2.7 morpholino (MO) (5′-TGGAGGTCACAGGACTCGGAAGCAT-3′) (Targoff et al., 2008) along with 2 ng of an anti-p53 (tp53 - Zebrafish Information Network) MO (5′-GCGCCATTGCTTTGCAAGAATTG-3′) (Robu et al., 2007). For mosaic cell-labeling experiments, embryos were injected at the one-cell stage with 150 pg of Tg(myl7:EGFP) plasmid.
In situ hybridization
Whole-mount in situ hybridization for myl7 (ZDB-GENE-991019-3), vmhc (ZDB-GENE-991123-5), amhc (myh6; ZDB-GENE-031112-1), scl (tal1; ZDB-GENE-980526-501), hand2 (ZDB-GENE-000511-1), irx4a (ZDB-GENE-040426-1825), hey2 (ZDB-GENE-000526-1) and ltbp3 (ZDB-GENE-060526-130) was performed as previously described (Yelon et al., 1999). Fluorescence in situ hybridization was performed using a variation on a published protocol (Schoenebeck et al., 2007). Probes were labeled with digoxigenin and fluorescein and were detected by deposition of TSA Plus fluorescein solution (Perkin Elmer) and Alexa Fluor 555 tyramide (Invitrogen). Embryos were washed in PBT (0.1% Tween-20 in PBS) and cleared in glycerol prior to imaging.
Immunofluorescence
Whole-mount immunofluorescence was performed as previously described (Alexander et al., 1998), using primary monoclonal antibodies against sarcomeric myosin heavy chain (MF20) and atrial myosin heavy chain (S46). MF20 and S46 were obtained from the Developmental Studies Hybridoma Bank maintained by the Department of Biological Sciences, University of Iowa, under contract NO1-HD-2-3144 from the National Institute of Child Health and Human Development (NICHD).
Imaging
Images were captured with Zeiss M2Bio and Axioplan microscopes and a Zeiss AxioCam digital camera and were processed with Zeiss AxioVision and Adobe Creative Suite software. Confocal imaging was performed with Zeiss LSM510 and Leica SP5 confocal microscopes, and z-stacks were processed with Imaris software (Bitplane).
Cardiomyocyte counting
We counted cardiomyocytes in embryos carrying the transgene Tg(-5.1myl7:nDsRed2), using immunofluorescence to detect DsRed in cardiomyocyte nuclei and to detect atrial myosin heavy chain (Amhc) in atrial cells, as previously described (Targoff et al., 2008). Embryos at 36 and 52 hours post-fertilization (hpf) were imaged with an Axioplan microscope, and embryos at 26 hpf were imaged with a Zeiss LSM510 confocal microscope. Student’s t-test (homoscedastic, two-tail distribution) was used to analyze the differences between the means of cell number data sets.
Genotyping
PCR genotyping was performed on genomic DNA extracted from individual embryos following in situ hybridization, immunofluorescence, or live imaging. Detection of nkx2.5vu179 was executed with a derived cleaved amplified polymorphic sequences (dCAPS) assay (Neff et al., 1998) using primers 5′-TCACCTCCACACAGGTGAAGATCTA-3′ and 5′-AAACGCAGGGTAGGTGTTGT-3′ to generate a 224-bp fragment. Digestion of the mutant PCR product with Tsp509I creates 26-bp and 198-bp fragments. Detection of nkx2.7vu413 was performed using primers 5′-CTTTTTCAGGCATGTGTCCA-3′ and 5′-AAAGCG TCTTTCCAGCTCAA-3′ to generate a 146-bp fragment. Digestion of the mutant PCR product with MseI creates 35-bp and 111-bp fragments.
Photoconversion
Photoconversion of Kaede was achieved by exposing a region of interest to a 405-nm laser using a Leica SP5 confocal microscope (Dixon et al., 2012; Huang et al., 2012). Successful cell labeling was confirmed in each embryo through acquisition of z-stacks immediately post-photoconversion and documentation of a transition from green to red fluorescence. Embryos were recovered, transferred into fresh media, and protected from light without any evidence of generalized toxicity.
RESULTS
Mutation of nkx2.5 disrupts cardiac chamber formation
Using morpholinos (MOs) to inhibit nkx2.5 and nkx2.7 function, our prior studies have indicated important roles of Nkx genes in controlling cardiomyocyte number and morphogenesis in each cardiac chamber (Targoff et al., 2008). However, in our previous work, it was difficult to decipher whether Nkx genes guide ventricular and atrial cell fate decisions, owing to the inherent variability and incomplete degree of MO-mediated gene knockdown. In order to develop a more durable tool for loss of nkx2.5 function, we generated a nonsense mutation in the zebrafish nkx2.5 gene through TILLING (supplementary material Fig. S1A) (Draper et al., 2004; Solnica-Krezel et al., 1994). Embryos homozygous for this mutation appear morphologically intact (Fig. 1A,B), with the prominent exception of their disrupted cardiac morphology (Fig. 1C,D). The nkx2.5 mutant embryo exhibits a diminutive, narrowed ventricular chamber and a bulbous, dilated atrial chamber (Fig. 1C-F). This mutant phenotype mimics the underdeveloped ventricle and globular atrium achieved through injection of a combination of anti-nkx2.5 and anti-nkx2.7 MOs in our previous studies (Targoff et al., 2008). Moreover, the nkx2.5 mutant demonstrates a stronger phenotype than that of our anti-nkx2.5 MO-injected embryos and underscores the incomplete knockdown in gene function attained with our individual MOs (Targoff et al., 2008). Thus, the nkx2.5 mutant provides a powerful tool for examining how loss of nkx2.5 function results in striking enlargement of the atrial chamber and foreshortening of the ventricular chamber.
Fig. 1.

Mutation of nkx2.5 disrupts cardiac morphogenesis. (A,B) Lateral views of live zebrafish embryos, anterior to the left, at 52 hpf. Other than cardiac defects and pericardial edema, the nkx2.5 mutant embryo (B) appears morphologically normal. (C,D) Lateral views of live embryos, anterior to the top, at 52 hpf. In contrast to the wild-type heart (C), the nkx2.5 mutant heart (D) is unlooped and has striking defects in both ventricular and atrial morphology. (E,F) MF20/S46 immunofluorescence distinguishes ventricular myocardium (red) from atrial myocardium (yellow). Frontal views, anterior to the top, at 52 hpf. In comparison to the wild-type heart (E), the nkx2.5 mutant heart (F) has a diminutive ventricle and an enlarged atrium. (G-L) In situ hybridization depicts expression of myl7 (G,H), vmhc (I,J) and amhc (K,L) in wild-type (G,I,K) and nkx2.5 mutant (H,J,L) embryos. Dorsal views, anterior to the top, at 26 hpf. (G,H) nkx2.5 mutant embryos exhibit subtle defects in heart tube extension, including a broad inflow region and a compact outflow region. In nkx2.5 mutants, the ventricular portion of the heart tube is abnormally short and wide (I,J) and the atrial portion of the heart tube has a splayed appearance (K,L).
Increased atrial and decreased ventricular cell numbers are first evident in nkx2.5 mutants following heart tube extension
To improve our understanding of the impressive discrepancies in chamber size in the nkx2.5 mutant, we investigated the cellular origins of these defects. First, we examined the formation of the lateral plate mesoderm and determined that nkx2.5, nkx2.7, scl and hand2 are all expressed normally in nkx2.5 mutant embryos during early somitogenesis stages (supplementary material Fig. S2; data not shown). These findings suggest that the initial establishment of the bilateral heart fields occurs appropriately despite the loss of nkx2.5 gene function.
Next, we employed chamber-specific markers that permit examination of ventricular and atrial cardiomyocyte populations prior to heart tube formation. Expression of an early marker of myocardial differentiation, myl7, and early markers of ventricular and atrial cardiomyocytes, ventricular myosin heavy chain (vmhc) and atrial myosin heavy chain (amhc), reveals normal differentiation of bilateral cardiac precursor populations and normal cardiac fusion in nkx2.5 mutants (supplementary material Fig. S3A,B,D,E; data not shown). However, morphological anomalies become apparent in nkx2.5 mutants during heart tube extension (Fig. 1G,H). The ventricular portion of the mutant heart tube fails to lengthen normally (Fig. 1I,J), and the atrial portion exhibits a subtle expansion and widening (Fig. 1K,L). Thus, nkx2.5 mutant embryos first develop subtle morphological aberrations as the heart tube elongates, foreshadowing the later phenotype of a ballooned atrium and a shrunken ventricle.
By counting cardiomyocytes in wild-type and nkx2.5 mutant embryos, we examined whether the numbers of cells in each chamber reflect quantitatively the emerging discrepancies in chamber size. At 26 hpf, we found that the initial numbers of differentiated ventricular and atrial cardiomyocytes in nkx2.5 mutant embryos are indistinguishable from those in wild-type embryos (Fig. 2A,B,G). These findings are reminiscent of the ventricular elongation defect that occurs independently of cell number alterations in embryos injected with anti-nkx2.5 and anti-nkx2.7 MOs (Targoff et al., 2008). At 36 hpf, we observed a prominent increase in the number of atrial cardiomyocytes in nkx2.5 mutant embryos: they exhibit ∼50% more atrial cells than wild-type embryos do at this stage (Fig. 2C,D,H). Furthermore, there is a corresponding decrement in ventricular cells at the same time point and no statistically significant difference in the total number of cardiomyocytes (Fig. 2H). Thus, a dramatic shift in ventricular and atrial cell numbers occurs within a narrow developmental window in nkx2.5 mutant embryos.
Fig. 2.

Increased atrial and decreased ventricular cell numbers are first evident in nkx2.5 mutants following heart tube extension. (A-F) Immunofluorescence indicates expression of the transgene Tg(-5.1myl7:nDsRed2) (red) in both cardiac chambers facilitating cardiomyocyte counting at 26 hpf (A,B), 36 hpf (C,D) and 52 hpf (E,F). Atria are labeled with the anti-Amhc antibody S46 (green). (G-I) Bar graphs indicate numbers of atrial and ventricular cardiomyocyte nuclei, as well as the total number of cardiomyocytes; mean and s.e.m. of each data set are shown, and asterisks indicate statistically significant differences from wild type (P<0.001). (G) At 26 hpf, we find no statistically significant difference in cell numbers in wild-type (n=20) and nkx2.5 mutant (n=7) embryos. (H) At 36 hpf, comparison of wild-type (n=16) and nkx2.5 mutant (n=10) embryos reveals an increase in atrial cell number and a decrease in ventricular cell number in nkx2.5 mutants. (I) At 52 hpf, comparison of wild-type (n=16) and nkx2.5 mutant (n=11) embryos reveals an increase in atrial cell number and a decrease in ventricular cell number in nkx2.5 mutants.
Given the acute, inversely correlated shift in ventricular and atrial cardiomyocyte numbers observed at 36 hpf, we investigated whether persistence of the quantitative differences in chamber-specific populations could explain the morphogenetic defects seen in the cardiac chambers of nkx2.5 mutants at later stages (Fig. 1E,F). Indeed, at 52 hpf, nkx2.5 mutants continue to exhibit a severe collapse in the number of ventricular cardiomyocytes and a substantial surplus of atrial cardiomyocytes (Fig. 2E,F,I). Exacerbation of the nkx2.5 mutant phenotype at this stage is such that the rudimentary ventricle is composed of ∼40% of the number of wild-type ventricular cells and the atrial cell number is reciprocally expanded. Yet, the total number of cardiomyocytes in nkx2.5 mutant embryos is only slightly decreased relative to that of wild-type embryos (P=0.025) (Fig. 2I). Thus, the bulbous atrium and contracted ventricle in the nkx2.5 mutant embryo reflect a substantial influence of nkx2.5 on ventricular and atrial cell number and morphogenesis between the time of heart tube extension and the process of chamber emergence.
Loss of nkx2.7 function in nkx2.5 mutants yields a more prominent ventricular deficiency
Given previous studies in mouse and zebrafish demonstrating the overlapping roles of co-expressed Nkx genes (Tanaka et al., 2001; Targoff et al., 2008; Tu et al., 2009), we sought to disrupt the function of the other NKX2-5 ortholog expressed in the zebrafish heart, nkx2.7 (Lee et al., 1996). Prior to our identification of an nkx2.7 mutation (supplementary material Fig. S1B), we performed initial studies by injecting a previously characterized anti-nkx2.7 MO (Targoff et al., 2008). Knockdown of nkx2.7 causes subtle morphological defects in wild-type embryos (Fig. 3A,D). Furthermore, injection of anti-nkx2.7 MO into nkx2.5 heterozygotes produces mild dilation of the atrium (Fig. 3B,E). More dramatically, in nkx2.5 homozygotes injected with anti-nkx2.7 MO (hereafter referred to as Nkx-deficient embryos), the atrial myosin heavy chain Amhc is found throughout the entirety of the globular heart (Fig. 3C,F). In contrast to the numbers of cardiomyocytes in wild-type embryos (Fig. 2I; 110±11 ventricular cells, 96±17 atrial cells, 206±26 total cells, n=11), Nkx-deficient embryos (0±0 ventricular cells, 174±14 atrial cells, n=9) have no detectable ventricular cells, a strikingly increased number of atrial cells (P<0.001) and a slightly decreased total number of cardiomyocytes (P=0.003). This phenotype suggests that all cardiomyocytes take on an atrial identity in the absence of nkx2.5 and nkx2.7 function (Fig. 3F). Our findings highlight the essential and shared roles of nkx2.5 and nkx2.7 genes in regulating formation of a morphologically and molecularly distinct ventricular chamber.
Fig. 3.

Loss of nkx2.7 function in nkx2.5 mutants yields a more prominent ventricular deficiency. (A-F) Frontal views, anterior to the top, of MF20/S46 immunofluorescence, which distinguishes ventricular myocardium (red) from atrial myocardium (yellow), at 52 hpf. In comparison to wild type (A), injection of anti-nkx2.7 MO (D) causes subtle abnormalities in chamber morphology. (B,E) In the nkx2.5 heterozygote, mild atrial enlargement is observed following injection of anti-nkx2.7 MO. (C,F) Injection of anti-nkx2.7 MO into nkx2.5 mutants yields severe loss of ventricular tissue and dramatic expansion of Amhc (S46, yellow) throughout the heart. Arrow indicates ventricular remnant that includes Amhc localization.
Following recovery of the nkx2.7vu413 mutant allele from our TILLING efforts (supplementary material Fig. S1B), we took advantage of this more stable reagent to validate our conclusion that nkx2.5 and nkx2.7 collaborate to promote ventricle formation. The nkx2.7 homozygous mutant embryos exhibit subtle defects in cardiac chamber morphogenesis (Fig. 4C-F); their overall body morphology appears otherwise normal (Fig. 4A,B). Yet, with decreasing dosage of nkx2.7 in the nkx2.5 mutant background, we observe a subtle decrement in ventricular tissue and slightly enhanced extension of Amhc localization into the ventricular territory (Fig. 4G,H). Moreover, nkx2.5-/-;nkx2.7-/- double mutant embryos exhibit a dramatic exacerbation of the phenotype with apparent loss of the ventricular chamber and widespread presence of Amhc throughout the heart (Fig. 4I). The ability to phenocopy the Nkx-deficient embryo with the nkx2.5-/-;nkx2.7-/- double mutant (compare Fig. 4I with Fig. 3F) demonstrates the reliability of the anti-nkx2.7 MO and, moreover, emphasizes the shared roles of Nkx genes and the importance of Nkx gene dosage in preserving the morphology and molecular identity of the ventricle.
Fig. 4.
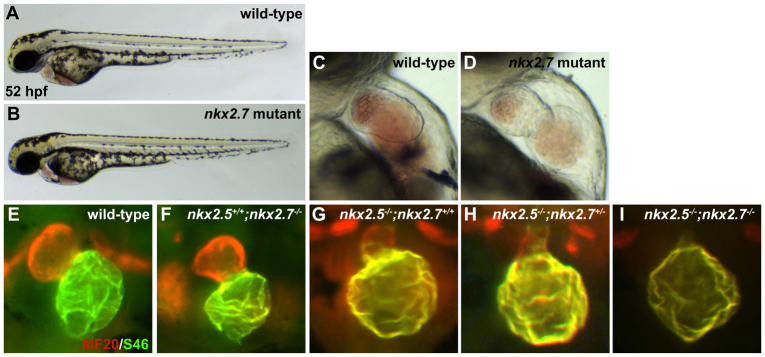
nkx2.5-/-;nkx2.7-/- double mutant demonstrates the synergistic requirement for Nkx genes in maintenance of ventricular identity. (A,B) Lateral views of live embryos, anterior to the left, at 52 hpf. General embryonic morphology appears similar in wild-type (A) and nkx2.7 mutant (B) embryos. (C,D) Lateral views of live embryos, anterior to the top, at 52 hpf. The nkx2.7 mutant heart (D) exhibits subtle defects in ventricular and atrial morphology. (E-I) Frontal views, anterior to the top, of MF20/S46 immunofluorescence, which distinguishes ventricular myocardium (red) from atrial myocardium (yellow), at 52 hpf. In comparison to wild-type embryos (E), nkx2.7 mutants (F) exhibit subtle abnormalities in chamber size and shape whereas nkx2.5 mutants (G) demonstrate an impressive size discrepancy between the ventricular and atrial chambers. (H) In the nkx2.5 mutant, loss of a single allele of nkx2.7 leads to increased localization of Amhc (S46, yellow) within the ventricle. (I) Exacerbation of the ventricular deficiency is evident in the nkx2.5-/-;nkx2.7-/- mutants, and Amhc is present throughout the entire cardiac structure.
Ventricular cardiomyocyte identity in the Nkx-deficient heart is eliminated gradually over time
Our analysis of the influence of Nkx genes on cardiac chamber size (Fig. 4E-I) suggests that nkx2.5 and nkx2.7 could work together to maintain the size of the ventricle and to restrain the size of the atrium. To examine how these genes influence chamber size over time, we analyzed the impact of loss of nkx2.5 and nkx2.7 function on the relative contributions of ventricular and atrial cardiomyocytes at earlier stages. In order to perform these experiments efficiently, we focused the majority of our analysis on Nkx-deficient embryos (as in Fig. 3F), because we could generate large numbers of these embryos more easily than was feasible for nkx2.5-/-;nkx2.7-/- double mutants (as in Fig. 4I).
As morphogenetic defects are evident in the heart tube of nkx2.5 mutant embryos (Fig. 1G,H) and differentiation of bilateral cardiac precursor populations occurs normally in nkx2.5-/-;nkx2.7-/- double mutants (supplementary material Fig. S3C,F), we initiated our examination of vmhc and amhc expression in Nkx-deficient embryos at 26 hpf. Injection of anti-nkx2.7 MO into wild-type embryos yields a disorganized and wider atrial component and a shorter, narrowed ventricular component of the heart tube (Fig. 5A,C,E,G). These results indicate early errors in morphogenesis, reminiscent of the phenotypes observed in nkx2.5 mutant heart tubes (Fig. 5B,D). Although the phenotypic trends are consistent in both scenarios, we do observe individual variability in the morphogenetic abnormalities of the nkx2.5 mutant and the anti-nkx2.7 MO-injected embryos. However, when injecting the anti-nkx2.7 MO into nkx2.5 homozygotes, we observe a more noteworthy and consistent phenotype: the atrial portion of the heart tube is sprawling and the ventricular portion is substantially shorter and wider than its wild-type counterpart (Fig. 5B,D,F,H). These data indicate that early disruptions in heart tube formation in the Nkx-deficient embryo foreshadow later, more dramatic chamber shape defects, suggesting that the loss of Nkx gene function has a gradual effect on chamber morphology over time. In addition to the morphological phenotypes that are present at 26 hpf, we find that the ventricular genes irx4a (Lecaudey et al., 2005) and hey2 (Winkler et al., 2003; Zhong et al., 2000) are progressively downregulated with increased loss of Nkx gene dosage in nkx2.5 mutants and nkx2.5-/-;nkx2.7-/- double mutants (supplementary material Figs S4, S5). Together, these findings highlight the shifts in morphology and chamber-specific gene expression that are initiated at the heart tube stage following the loss of Nkx gene function.
Fig. 5.
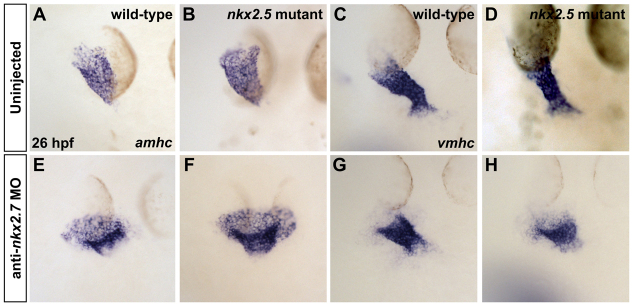
Loss of nkx2.7 function in nkx2.5 mutants exacerbates defects during heart tube extension. (A-H) In situ hybridization depicts expression of amhc (A,B,E,F) and vmhc (C,D,G,H) in wild-type embryos (A,C), nkx2.5 mutants (B,D), wild-type embryos injected with anti-nkx2.7 MO (E,G) and Nkx-deficient embryos (F,H). Dorsal views, anterior to the top, at 26 hpf. The nkx2.5 mutant embryos exhibit subtle defects in heart tube extension, including a broader atrial region (A,B) and a slightly shorter ventricular region (C,D). Following MO injection, wild-type embryos demonstrate a spread of atrial cells and a compact coalescence of the ventricular cells (E,G). MO injection into nkx2.5 mutants leads to an exacerbated phenotype with a sprawling, widened atrial portion and a stunted ventricular portion of the heart tube (F,H).
In order to track the progression of the Nkx-deficient phenotype following heart tube assembly, we examined embryos at 36 hpf and observed dynamic changes in the expression of vmhc and amhc (Fig. 6A-H). In the nkx2.5 mutant embryo, the ventricular chamber is diminished in size, yet vmhc expression is robust (Fig. 6C,D). By contrast, we see that vmhc expression fades in the morphological ventricle of the Nkx-deficient heart (Fig. 6D,H) and that amhc expression is enhanced in this region (Fig. 6F). The extension of amhc expression into the minute ventricular chamber of the Nkx-deficient heart is a striking contrast to the constrained expression of amhc within the morphological atrium of the nkx2.5 mutant heart (Fig. 6B,F). The coincident downregulation of vmhc expression and upregulation of amhc expression within ventricular territory suggest a gradual destabilization of chamber identity in Nkx-deficient embryos.
Fig. 6.

Gradual elimination of ventricular gene expression and expansion of atrial gene expression in the Nkx-deficient heart. In situ hybridization illustrates expression of amhc (A,B,E,F,I,J,M,N) and vmhc (C,D,G,H,K,L,O,P) in wild-type embryos (A,C,I,K), nkx2.5 mutants (B,D,J,L), wild-type embryos injected with anti-nkx2.7 MO (E,G,M,O) and Nkx-deficient embryos (F,H,N,P). (A-H) Dorsal views, anterior to the bottom, at 36 hpf. The nkx2.5 mutants display a dilated atrium (A,B) and shortened ventricle (C,D). Following MO injection, wild-type embryos demonstrate a swollen atrium and widened ventricle (E,G). In nkx2.5 mutants, MO injection leads to distinct expansion of amhc and fading of vmhc in the ventricular remnant (F,H; arrows). (I-P) Ventral views, anterior to the top, at 52 hpf. Phenotypes are exacerbated at 52 hpf, both in terms of morphological defects and expression pattern changes. Most dramatically, in the Nkx-deficient embryo, amhc is expressed throughout the entire heart whereas vmhc expression is effectively abolished (N,P).
Examination of Nkx-deficient embryos at 52 hpf demonstrated that their reciprocal shifts in vmhc and amhc expression patterns become exacerbated over time (Fig. 6I-P). In Nkx-deficient embryos, amhc expression intensifies and spreads throughout the heart, ultimately encompassing the entire myocardium (Fig. 6N). In contrast to the nkx2.5 mutant, in which vmhc expression is persistent within a small ventricle (Fig. 6K,L), both vmhc expression and any morphologically evident ventricular chamber disappear in Nkx-deficient embryos (Fig. 6P).
To confirm the dynamics of the vmhc and amhc expression patterns in Nkx-deficient embryos, we employed two-color fluorescence in situ hybridization to enhance the sensitivity of detecting both transcripts within an individual embryo (Fig. 7). Consistent with our other data (Fig. 6), our fluorescence in situ hybridization results highlight the encroachment of ectopic amhc expression into the nkx2.5 mutant (Fig. 7D-F,M-O) and Nkx-deficient (Fig. 7G-I,P-R) ventricle. In addition, these results identify a small patch of nearly undetectable vmhc expression at the arterial pole of the single chamber of the Nkx-deficient heart (Fig. 7P-R). Similar observations in nkx2.5-/-;nkx2.7-/-double mutant embryos (Fig. 8) reinforce the interpretation that nkx2.5 and nkx2.7 work together to maintain vmhc expression while also preventing acquisition of ectopic amhc expression. Overall, the gradual loss of vmhc expression, gradual loss of a morphologically identifiable ventricle and gradual expansion of amhc expression that result from loss of nkx2.5 and nkx2.7 function support the notion that Nkx genes play an important role in maintaining the molecular and morphological characteristics that define the ventricle.
Fig. 7.
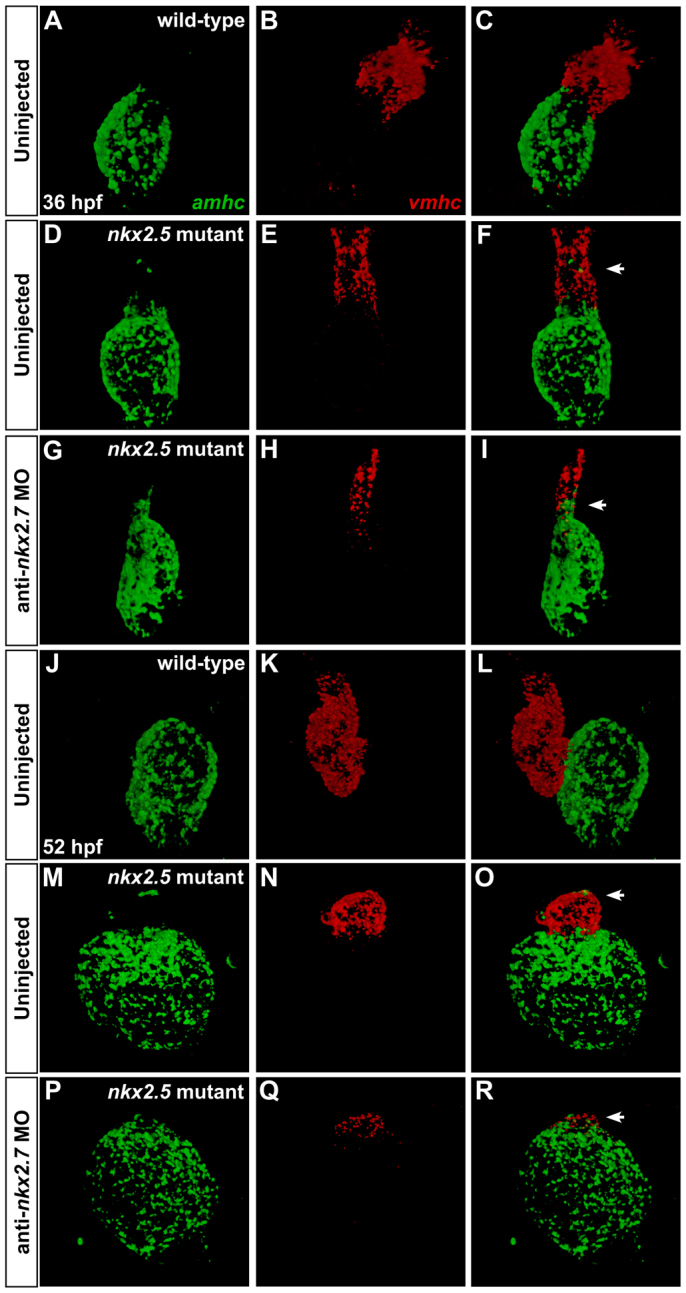
Dynamic expression patterns highlight gradual transitions in cardiomyocyte identity. Two-color fluorescence in situ hybridization facilitates sensitive detection of amhc (green) and vmhc (red) expression. Confocal projections of fixed, dissected wild-type (A-C,J-L), nkx2.5 mutant (D-F,M-O) and Nkx-deficient hearts (G-I,P-R); ventral views, anterior to the top. (A-I) At 36 hpf, ectopic amhc-expressing cells are found in the ventricle in the nkx2.5 mutant heart (D-F; arrowhead) and, more extensively, in the Nkx-deficient heart (G-I; arrowhead). (J-R) At 52 hpf, ectopic amhc-expressing cells are visualized in the dramatically diminished nkx2.5 mutant ventricle (M-O; arrowhead) whereas only a tuft of residual vmhc expression remains at the pole of the Nkx-deficient heart (P-R; arrowhead).
Fig. 8.
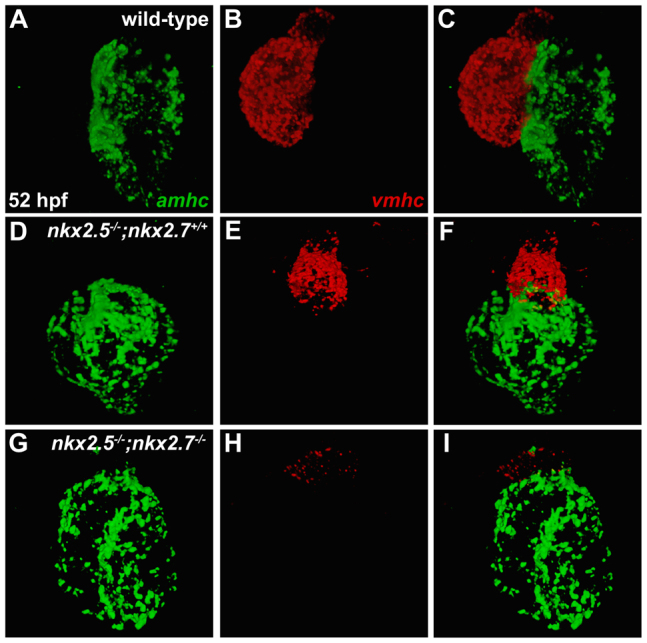
nkx2.5 and nkx2.7 work together to maintain vmhc expression and to limit amhc expression. (A-I) Fluorescence in situ hybridization depicts expression patterns of amhc (green) and vmhc (red) in wild-type (A-C), nkx2.5 mutant (D-F) and nkx2.5-/-;nkx2.7-/- double mutant (G-I) hearts at 52 hpf. Compared with robust expression of vmhc in both the wild-type (B) and nkx2.5 mutant (E) hearts, encroachment of ectopic amhc expression is demonstrated alongside dwindling vmhc expression in the ventricular remnant of the nkx2.5-/-;nkx2.7-/- mutant heart (G-I).
Transformation from ventricular to atrial identity in the absence of Nkx gene function
Our data indicate that loss of Nkx gene function interferes with the maintenance of an appropriate number of ventricular cells; also, over the same developmental time frame, loss of Nkx gene function causes a marked increase in the size of the atrium. These reciprocal shifts between the ventricular and atrial populations could be unrelated; in essence, Nkx genes may play one role in promoting ventricular cell number and also play another independent role in limiting atrial cell number. Alternatively, the inverse correlation between the ventricular and atrial phenotypes in Nkx-deficient embryos might indicate inter-related roles of Nkx genes in preserving ventricular identity and repressing atrial identity.
As an initial step towards distinguishing between these possibilities, we explored the cellular basis for the cardiac chamber defects in nkx2.5 mutant embryos. For example, to examine whether increased proliferation could account for the increased number of atrial cardiomyocytes in nkx2.5 mutants, we used mosaic labeling to follow individual cardiomyocytes between 26 and 52 hpf (supplementary material Fig. S6 and Table S1). These experiments failed to reveal enhanced proliferation of atrial cells and also did not indicate any alterations in patterns of cell death; most tracked cells remained stable between 26 and 52 hpf, rather than disappearing or dividing. Similarly, our mosaic labeling experiments did not provide evidence for defects in proliferation or cell death that could account for the loss of ventricular cardiomyocytes in nkx2.5 mutant embryos (supplementary material Fig. S6 and Table S1). Consistent with these results, our previous studies of anti-nkx2.5 and anti-nkx2.7 MO-injected embryos had failed to demonstrate any contribution of altered proliferation or apoptosis to the observed defects in cardiac chamber size (Targoff et al., 2008).
Next, we investigated whether the ventricular deficiency in nkx2.5 mutant embryos could be caused by a failure to recruit second heart field (SHF)-derived cells to the arterial pole of the heart (de Pater et al., 2009; Hami et al., 2011; Lazic and Scott, 2011; Zhou et al., 2011). To address this, we used a developmental timing assay that relies upon the differential timing of visualization of DsRed and EGFP in embryos expressing both Tg(-5.1myl7:nDsRed2) and Tg(myl7:EGFP) (de Pater et al., 2009). Our data provide clear evidence for the accumulation of late-differentiating cells at the arterial pole of the nkx2.5 mutant heart (supplementary material Fig. S7), indicating the presence of SHF-derived cardiomyocytes in the ventricle. However, downregulated expression of ltbp3, an SHF marker (Zhou et al., 2011), in nkx2.5 mutant embryos (supplementary material Fig. S8A,C) suggests that there could be diminished contribution from the SHF, consistent with a recent report demonstrating reduced ltbp3 expression and reduced production of SHF-derived cardiomyocytes in embryos injected with an anti-nkx2.5 MO (Guner-Ataman et al., 2013). Moreover, ltbp3 expression is absent in nkx2.5-/-;nkx2.7-/- double mutants (supplementary material Fig. S8E), further highlighting the impact of Nkx gene function on regulation of ltbp3. Altogether, these data suggest that reduced SHF contributions could be responsible for a portion of the ventricular phenotype in nkx2.5 mutant embryos. However, as the ventricular myocardium is derived from both the first heart field (FHF) and the SHF (de Pater et al., 2009; Hami et al., 2011; Lazic and Scott, 2011; Zhou et al., 2011), defective SHF recruitment is inadequate to account for the complete loss of ventricular cells in Nkx-deficient embryos.
Because we did not find that independent, chamber-specific mechanisms can fully explain the ventricular and atrial phenotypes in nkx2.5 mutants, we turned our attention towards the possibility that the ventricular and atrial defects are inter-related and represent the result of a fate transformation. To test the hypothesis that Nkx genes function to prevent ventricular cells from taking on an atrial identity, we employed the transgene Tg(myl7:kaede) (de Pater et al., 2009) to mark cardiomyocytes in the ventricular portion of the linear heart tube at 26-30 hpf and to track the position of these labeled cells at 48-52 hpf (Fig. 9). In order to avoid marking cells near the atrioventricular boundary, we only selected cells in relatively arterial positions within the vmhc-expressing territory of the heart tube (supplementary material Fig. S9). When using this strategy in wild-type embryos, photoconversion of Kaede in ventricular cardiomyocytes within the heart tube (Fig. 9A-C) always led to the detection of labeled cells within the ventricle, and not the atrium (Fig. 9D-F). By contrast, ventricular cardiomyocytes labeled in the nkx2.5 mutant heart tube (Fig. 9G-I) were later found both in the ventricular rudiment and extending into the atrium (Fig. 9J-L). Thus, in nkx2.5 mutants, cells that are initially within the vmhc-expressing portion of the heart tube can become embedded into the amhc-expressing atrial chamber as development proceeds (Fig. 9G-L; supplementary material Fig. S9). Finally, we conducted parallel studies in which we photoconverted ventricular cardiomyocytes within the Nkx-deficient heart tube (Fig. 9M-O). At later stages, after vmhc expression had disappeared and no morphologically distinct ventricle was evident (supplementary material Fig. S9), photoconverted cells were detected within the enlarged, amhc-expressing atrium of the Nkx-deficient heart (Fig. 9P-R; supplementary material Fig. S9). Taken together, our photoconversion experiments suggest that loss of Nkx gene function can cause ventricular cardiomyocytes to transform into atrial cardiomyocytes. Therefore, our data indicate the importance of Nkx genes for safeguarding ventricular characteristics and highlight the malleable nature of cardiomyocyte identity.
Fig. 9.

Ventricular-to-atrial transformation occurs in the absence of Nkx gene function. Confocal projections of live zebrafish hearts carrying Tg(myl7:kaede) are depicted immediately following photoconversion at 26-30 hpf (A-C,G-I,M-O) and after later visualization at 48-52 hpf (D-F,J-L,P-R); arterial pole to the top. Green fluorescence of Kaede (A,G,M) is converted to red fluorescence (B,H,N) following UV exposure of the region of interest; merged views (C,I,O) indicate that labeled cells are at the arterial pole of the heart tube. At later time points, retained red fluorescence (E,K,Q) is visualized together with the green Kaede (D,J,P) that is continually produced by the transgene; merged views (F,L,R) indicate the locations of the labeled cells. (A-F) In wild-type embryos, labeling of ventricular cells near the arterial pole of the heart tube (A-C) yields labeled cells within the ventricular chamber at 48 hpf (D-F) (n=17/17). (G-L) By contrast, comparable photoconversion of ventricular cardiomyocytes at the arterial pole of the nkx2.5 mutant heart tube (G-I) generates labeled cells both within the diminutive ventricular chamber and expanding into the atrial chamber (J-L, arrowhead in L) (n=10/12). (M-R) In Nkx-deficient embryos, labeled cells from the vmhc-expressing portion (supplementary material Fig. S9N) of the heart tube (M-O) are later detected within the single, amhc-expressing (supplementary material Fig. S9P) chamber of the heart (P-R) (n=2/2).
DISCUSSION
Our studies uncover a novel role for Nkx genes in protecting chamber-specific characteristics. Following initial patterning of ventricular and atrial cardiomyocytes, nkx2.5 and nkx2.7 are essential to enforce the stability of chamber-specific expression patterns and to preserve normal proportions of cardiomyocytes in the ventricle and atrium. Thus, our findings reveal an intriguing function of Nkx genes in maintaining ventricular character while repressing atrial character. Furthermore, our studies illuminate the flexible nature of chamber identity and draw attention to the importance of active maintenance of chamber-specific traits within differentiated myocardium.
Through demonstration of the requirement for zebrafish Nkx genes in preservation of ventricular identity, our work adds a new facet to our understanding of Nkx gene function that both complements and extends previous studies of Nkx2-5 in mouse. Although many roles of mouse Nkx genes in cardiac specification and morphogenesis have already been clearly articulated, their functions in regulating chamber identity have not been fully elucidated. In Nkx2.5-/- mutant (Lyons et al., 1995; Tanaka et al., 1999), Nkx2.5-/-;Nkx2.6-/- double mutant (Tanaka et al., 2001) and Nkx2-5-/-;Hand2-/- double mutant (Yamagishi et al., 2001) mice, ventricular dysgenesis and reduction of Mlc2v expression have previously been suggested to illustrate defects in maturation and/or survival of the ventricular myocardium, as well as defects in progenitor specification and proliferation (Prall et al., 2007). Our data suggest a new possibility underlying the observed ventricular deficiencies and alterations in gene expression in these mice: some of the mutant ventricular cells might undergo transdifferentiation from ventricular to atrial identities. Synthesizing the mouse and zebrafish data sets, it seems that Nkx genes have multiple influences on chamber proportions: in addition to their roles in regulating specification, proliferation, recruitment, maturation and survival of ventricular and atrial cardiomyocytes, these genes are also important for maintaining chamber identity.
Over the long term, it will be valuable to determine how the influence of Nkx genes on chamber identity interfaces with their many other functions. Although our work does not suggest that significant defects in cardiomyocyte proliferation or survival underlie the Nkx-deficient phenotype, we are unable to rule out the possibility that these mechanisms play subtle roles in the dramatic ventricular loss and atrial gain. Additionally, our data, together with recent analyses of embryos injected with an anti-nkx2.5 MO (Guner-Ataman et al., 2013), suggest that Nkx genes may act to promote SHF contributions to the ventricle during the same time frame when they act to enforce ventricular identity. Thus, our results are consistent with a model in which Nkx genes are required to maintain ventricular identity, potentially in both FHF-derived and SHF-derived cells, while also being essential to drive contribution of SHF-derived cells to the arterial pole. Future studies will help to elucidate the distinctions between the molecular mechanisms underlying these seemingly distinct roles of Nkx genes in FHF-derived and SHF-derived populations.
It is interesting to consider whether Nkx factors play direct roles in maintaining ventricular gene expression and repressing atrial gene expression. Direct regulation of chamber-specific genes by Nkx factors has been implied by recent analyses of the upstream regulatory regions of the zebrafish gene vmhc (Jin et al., 2009; Zhang and Xu, 2009). In these studies, deletion of Nkx-binding sites in transgenic reporters has suggested positive regulation of vmhc by Nkx factors in the ventricular chamber and negative regulation of vmhc by Nkx factors in the atrial chamber. Alternatively, Nkx factors might have a more indirect influence on chamber identity. Studies in mouse have demonstrated that Bmp2 acts downstream of Nkx2-5 to restrict specification in the cardiac crescent and to promote proliferation of late-differentiating cardiomyocytes (Prall et al., 2007). Perhaps a similar relationship between Nkx factors and BMP signaling could be responsible for the impact of Nkx genes on chamber identity maintenance. Consistent with this idea, overexpression of bmp2b in zebrafish results in ectopic expression of amhc along with a small ventricular chamber (de Pater et al., 2012), mimicking aspects of the Nkx-deficient and nkx2.5-/-;nkx2.7-/- phenotypes.
Regardless of the precise molecular mechanisms through which Nkx factors influence chamber identity, it is intriguing that our studies heighten awareness of the malleability of the character of differentiated cardiomyocytes, building on early work in chick that demonstrated the plasticity of chamber-specific fates in certain contexts (Yutzey et al., 1995; Yutzey et al., 1994). Although a network of signals, including components of the retinoic acid (Hochgreb et al., 2003; Waxman et al., 2008; Xavier-Neto et al., 1999), bone morphogenetic protein (Marques and Yelon, 2009), fibroblast growth factor (Marques et al., 2008) and Nodal (Keegan et al., 2004; Reiter et al., 2001) pathways, are known to influence the initial assignment of chamber fates, little is known about what additional inputs are required to maintain these decisions. Our work suggests that Nkx factors could act upstream of irx4a and hey2 in enforcing ventricular identity, consistent with the established functions of mammalian Irx4 (Bao et al., 1999; Bruneau et al., 2001a) and Hey2 (Koibuchi and Chin, 2007; Xin et al., 2007) in the repression of atrial gene expression in ventricular cardiomyocytes. Notably, loss of either Irx4 or Hey2 in mice (Bruneau et al., 2001a; Koibuchi and Chin, 2007; Xin et al., 2007) does not reveal the apparent degree of transdifferentiation observed in Nkx-deficient and nkx2.5-/-;nkx2.7-/- double mutant zebrafish embryos, suggesting that Nkx factors might act near the top of a transcriptional hierarchy controlling ventricular identity maintenance. Future elucidation of the pathways downstream of Nkx2.5 and Nkx2.7 will help to solidify the relationships between these transcriptional regulators; moreover, it will be important to develop new reagents that can facilitate a higher-resolution understanding of the Nkx-deficient and nkx2.5-/-;nkx2.7-/- phenotypes. Identification of additional molecular markers of ventricular and atrial identity will help to resolve whether ventricular specification is fully normal and whether transformation to an atrial identity is complete when Nkx gene function is lost. Although our data clearly indicate the plasticity of ventricular characteristics, we have not yet found any evidence in our studies suggesting that a reciprocal flexibility exists in differentiated atrial cells. In this regard, it is fascinating to note the finding that the atria in mice lacking myocardial expression of the atrial transcription factor gene COUP-TFII (Nr2f2 - Mouse Genome Informatics) exhibit ventricular, rather than atrial, features (Wu et al., 2013). Perhaps both the atrial and ventricular gene expression programs require active repression in the reciprocal chamber in order to preserve chamber-specific characteristics; it will be exciting for future work to illuminate the parallels between the mechanisms that reinforce ventricular and atrial identities. Finally, a recent report describing atrial-to-ventricular cardiomyocyte conversion in response to ventricular-specific ablation in the zebrafish embryo further underscores the malleable nature of cardiac chamber identity (Zhang et al., 2013). Altogether, insights regarding the molecular mechanisms mediating the establishment and preservation of cardiac chamber-specific identities hold robust potential for therapeutic advances in cardiac regenerative medicine.
Importantly, our understanding of the ventricular-to-atrial transformation that occurs following the loss of Nkx gene function opens doors for a greater appreciation of congenital heart disease pathology. The maintenance of chamber identity is fundamental to the structural and morphological foundations of cardiac architecture. Thus, inability to preserve the characteristics of ventricular and atrial cardiomyocytes can result in problems with cardiac form and function. Mutations in NKX2-5 have been recognized in patients with septal defects, outflow tract abnormalities (double outlet right ventricle and Tetralogy of Fallot) and hypoplastic left heart syndrome (Benson et al., 1999; Elliott et al., 2003; McElhinney et al., 2003; Schott et al., 1998). We speculate that defects in maintaining ventricular identity in these patients could yield an underdeveloped ventricle as observed in Tetralogy of Fallot or hypoplastic left heart syndrome. Additionally, patients with NKX2-5 mutations often exhibit progressive atrioventricular conduction defects into adulthood (Jay et al., 2003), and failure to maintain chamber identity might also contribute to electrophysiological malfunction associated with loss of fast conducting properties and defects in specification of the ventricular conduction system lineage (Moskowitz et al., 2007; Pashmforoush et al., 2004).
In conclusion, the potential for cardiomyocyte plasticity following depletion of Nkx gene function may help to explain fundamental morphogenetic errors during embryogenesis and to decipher the etiologies of the defects in patients with NKX2-5 mutations. In addition, we envisage that a deeper understanding of the mechanisms through which nkx2.5 and nkx2.7 sustain ventricular identity might ultimately improve our ability to modify fate assignment as a means to replenish deficient populations in a diseased heart. Altogether, our data offer new directions towards paradigms for preserving and directing differentiation of ventricular cardiomyocytes and for understanding congenital cardiac pathology.
Supplementary Material
Acknowledgments
We thank members of the Yelon, Torres-Vázquez, Knaut and Targoff laboratories for thoughtful input.
Footnotes
Funding
This work was supported by grants from the National Institutes of Health (NIH) [R01 HL069594 and R01 HL108599 to D.Y.; R01 HD076585 to L.S.-K.; K12 HD043389 and K08 HL088002 to K.L.T.]; the American Heart Association [D.Y.]; and the March of Dimes [D.Y.]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Author contributions
K.L.T., T.S. and D.Y. designed these studies; K.L.T. performed the majority of the experiments, with additional contributions from S.C. and V.G; K.L.T. and D.Y. analyzed the data; S.-H.K. and L.S.-K. generated the nkx2.5 and nkx2.7 mutations; and K.L.T. and D.Y. wrote the manuscript with input from all authors.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.095562/-/DC1
References
- Alexander J., Stainier D. Y., Yelon D. (1998). Screening mosaic F1 females for mutations affecting zebrafish heart induction and patterning. Dev. Genet. 22, 288–299 [DOI] [PubMed] [Google Scholar]
- Azpiazu N., Frasch M. (1993). tinman and bagpipe: two homeo box genes that determine cell fates in the dorsal mesoderm of Drosophila. Genes Dev. 7 7B, 1325–1340 [DOI] [PubMed] [Google Scholar]
- Bao Z. Z., Bruneau B. G., Seidman J. G., Seidman C. E., Cepko C. L. (1999). Regulation of chamber-specific gene expression in the developing heart by Irx4. Science 283, 1161–1164 [DOI] [PubMed] [Google Scholar]
- Benson D. W., Silberbach G. M., Kavanaugh-McHugh A., Cottrill C., Zhang Y., Riggs S., Smalls O., Johnson M. C., Watson M. S., Seidman J. G., et al. (1999). Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J. Clin. Invest. 104, 1567–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdougo E., Coleman H., Lee D. H., Stainier D. Y., Yelon D. (2003). Mutation of weak atrium/atrial myosin heavy chain disrupts atrial function and influences ventricular morphogenesis in zebrafish. Development 130, 6121–6129 [DOI] [PubMed] [Google Scholar]
- Biben C., Weber R., Kesteven S., Stanley E., McDonald L., Elliott D. A., Barnett L., Köentgen F., Robb L., Feneley M., et al. (2000). Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2-5. Circ. Res. 87, 888–895 [DOI] [PubMed] [Google Scholar]
- Bodmer R. (1993). The gene tinman is required for specification of the heart and visceral muscles in Drosophila. Development 118, 719–729 [DOI] [PubMed] [Google Scholar]
- Bruneau B. G., Bao Z. Z., Tanaka M., Schott J. J., Izumo S., Cepko C. L., Seidman J. G., Seidman C. E. (2000). Cardiac expression of the ventricle-specific homeobox gene Irx4 is modulated by Nkx2-5 and dHand. Dev. Biol. 217, 266–277 [DOI] [PubMed] [Google Scholar]
- Bruneau B. G., Bao Z. Z., Fatkin D., Xavier-Neto J., Georgakopoulos D., Maguire C. T., Berul C. I., Kass D. A., Kuroski-de Bold M. L., de Bold A. J., et al. (2001a). Cardiomyopathy in Irx4-deficient mice is preceded by abnormal ventricular gene expression. Mol. Cell. Biol. 21, 1730–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau B. G., Nemer G., Schmitt J. P., Charron F., Robitaille L., Caron S., Conner D. A., Gessler M., Nemer M., Seidman C. E., et al. (2001b). A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 106, 709–721 [DOI] [PubMed] [Google Scholar]
- Chen J. N., Fishman M. C. (1996). Zebrafish tinman homolog demarcates the heart field and initiates myocardial differentiation. Development 122, 3809–3816 [DOI] [PubMed] [Google Scholar]
- de Pater E., Clijsters L., Marques S. R., Lin Y. F., Garavito-Aguilar Z. V., Yelon D., Bakkers J. (2009). Distinct phases of cardiomyocyte differentiation regulate growth of the zebrafish heart. Development 136, 1633–1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pater E., Ciampricotti M., Priller F., Veerkamp J., Strate I., Smith K., Lagendijk A. K., Schilling T. F., Herzog W., Abdelilah-Seyfried S., et al. (2012). Bmp signaling exerts opposite effects on cardiac differentiation. Circ. Res. 110, 578–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon G., Elks P. M., Loynes C. A., Whyte M. K., Renshaw S. A. (2012). A method for the in vivo measurement of zebrafish tissue neutrophil lifespan. ISRN Hematol. 2012, 915868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper B. W., McCallum C. M., Stout J. L., Slade A. J., Moens C. B. (2004). A high-throughput method for identifying N-ethyl-N-nitrosourea (ENU)-induced point mutations in zebrafish. Methods Cell Biol. 77, 91–112 [DOI] [PubMed] [Google Scholar]
- Elliott D. A., Kirk E. P., Yeoh T., Chandar S., McKenzie F., Taylor P., Grossfeld P., Fatkin D., Jones O., Hayes P., et al. (2003). Cardiac homeobox gene NKX2-5 mutations and congenital heart disease: associations with atrial septal defect and hypoplastic left heart syndrome. J. Am. Coll. Cardiol. 41, 2072–2076 [DOI] [PubMed] [Google Scholar]
- Garavito-Aguilar Z. V., Riley H. E., Yelon D. (2010). Hand2 ensures an appropriate environment for cardiac fusion by limiting Fibronectin function. Development 137, 3215–3220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grow M. W., Krieg P. A. (1998). Tinman function is essential for vertebrate heart development: elimination of cardiac differentiation by dominant inhibitory mutants of the tinman-related genes, XNkx2-3 and XNkx2-5. Dev. Biol. 204, 187–196 [DOI] [PubMed] [Google Scholar]
- Guner-Ataman B., Paffett-Lugassy N., Adams M. S., Nevis K. R., Jahangiri L., Obregon P., Kikuchi K., Poss K. D., Burns C. E., Burns C. G. (2013). Zebrafish second heart field development relies on progenitor specification in anterior lateral plate mesoderm and nkx2.5 function. Development 140, 1353–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hami D., Grimes A. C., Tsai H. J., Kirby M. L. (2011). Zebrafish cardiac development requires a conserved secondary heart field. Development 138, 2389–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W., Jia Y., Takimoto K. (2009). Interaction between transcription factors Iroquois proteins 4 and 5 controls cardiac potassium channel Kv4.2 gene transcription. Cardiovasc. Res. 81, 64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochgreb T., Linhares V. L., Menezes D. C., Sampaio A. C., Yan C. Y., Cardoso W. V., Rosenthal N., Xavier-Neto J. (2003). A caudorostral wave of RALDH2 conveys anteroposterior information to the cardiac field. Development 130, 5363–5374 [DOI] [PubMed] [Google Scholar]
- Huang C. J., Tu C. T., Hsiao C. D., Hsieh F. J., Tsai H. J. (2003). Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev. Dyn. 228, 30–40 [DOI] [PubMed] [Google Scholar]
- Huang P., Xiong F., Megason S. G., Schier A. F. (2012). Attenuation of Notch and Hedgehog signaling is required for fate specification in the spinal cord. PLoS Genet. 8, e1002762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay P. Y., Berul C. I., Tanaka M., Ishii M., Kurachi Y., Izumo S. (2003). Cardiac conduction and arrhythmia: insights from Nkx2.5 mutations in mouse and humans. Novartis Found. Symp. 250, 227–238; discussion 238-241, 276-229 [PubMed] [Google Scholar]
- Jin D., Ni T. T., Hou J., Rellinger E., Zhong T. P. (2009). Promoter analysis of ventricular myosin heavy chain (vmhc) in zebrafish embryos. Dev. Dyn. 238, 1760–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan B. R., Meyer D., Yelon D. (2004). Organization of cardiac chamber progenitors in the zebrafish blastula. Development 131, 3081–3091 [DOI] [PubMed] [Google Scholar]
- Koibuchi N., Chin M. T. (2007). CHF1/Hey2 plays a pivotal role in left ventricular maturation through suppression of ectopic atrial gene expression. Circ. Res. 100, 850–855 [DOI] [PubMed] [Google Scholar]
- Lazic S., Scott I. C. (2011). Mef2cb regulates late myocardial cell addition from a second heart field-like population of progenitors in zebrafish. Dev. Biol. 354, 123–133 [DOI] [PubMed] [Google Scholar]
- Lecaudey V., Anselme I., Dildrop R., Rüther U., Schneider-Maunoury S. (2005). Expression of the zebrafish Iroquois genes during early nervous system formation and patterning. J. Comp. Neurol. 492, 289–302 [DOI] [PubMed] [Google Scholar]
- Lee K. H., Xu Q., Breitbart R. E. (1996). A new tinman-related gene, nkx2.7, anticipates the expression of nkx2.5 and nkx2.3 in zebrafish heart and pharyngeal endoderm. Dev. Biol. 180, 722–731 [DOI] [PubMed] [Google Scholar]
- Lyons I., Parsons L. M., Hartley L., Li R., Andrews J. E., Robb L., Harvey R. P. (1995). Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 9, 1654–1666 [DOI] [PubMed] [Google Scholar]
- Mably J. D., Mohideen M. A., Burns C. G., Chen J. N., Fishman M. C. (2003). heart of glass regulates the concentric growth of the heart in zebrafish. Curr. Biol. 13, 2138–2147 [DOI] [PubMed] [Google Scholar]
- Marques S. R., Yelon D. (2009). Differential requirement for BMP signaling in atrial and ventricular lineages establishes cardiac chamber proportionality. Dev. Biol. 328, 472–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques S. R., Lee Y., Poss K. D., Yelon D. (2008). Reiterative roles for FGF signaling in the establishment of size and proportion of the zebrafish heart. Dev. Biol. 321, 397–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhinney D. B., Geiger E., Blinder J., Benson D. W., Goldmuntz E. (2003). NKX2.5 mutations in patients with congenital heart disease. J. Am. Coll. Cardiol. 42, 1650–1655 [DOI] [PubMed] [Google Scholar]
- Moorman A. F., Christoffels V. M. (2003). Cardiac chamber formation: development, genes, and evolution. Physiol. Rev. 83, 1223–1267 [DOI] [PubMed] [Google Scholar]
- Moskowitz I. P., Kim J. B., Moore M. L., Wolf C. M., Peterson M. A., Shendure J., Nobrega M. A., Yokota Y., Berul C., Izumo S., et al. (2007). A molecular pathway including Id2, Tbx5, and Nkx2-5 required for cardiac conduction system development. Cell 129, 1365–1376 [DOI] [PubMed] [Google Scholar]
- Neff M. M., Neff J. D., Chory J., Pepper A. E. (1998). dCAPS, a simple technique for the genetic analysis of single nucleotide polymorphisms: experimental applications in Arabidopsis thaliana genetics. Plant J. 14, 387–392 [DOI] [PubMed] [Google Scholar]
- Ng S. Y., Wong C. K., Tsang S. Y. (2010). Differential gene expressions in atrial and ventricular myocytes: insights into the road of applying embryonic stem cell-derived cardiomyocytes for future therapies. Am. J. Physiol. 299, C1234–C1249 [DOI] [PubMed] [Google Scholar]
- Pashmforoush M., Lu J. T., Chen H., Amand T. S., Kondo R., Pradervand S., Evans S. M., Clark B., Feramisco J. R., Giles W., et al. (2004). Nkx2-5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell 117, 373–386 [DOI] [PubMed] [Google Scholar]
- Prall O. W., Menon M. K., Solloway M. J., Watanabe Y., Zaffran S., Bajolle F., Biben C., McBride J. J., Robertson B. R., Chaulet H., et al. (2007). An Nkx2-5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell 128, 947–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter J. F., Verkade H., Stainier D. Y. (2001). Bmp2b and Oep promote early myocardial differentiation through their regulation of gata5. Dev. Biol. 234, 330–338 [DOI] [PubMed] [Google Scholar]
- Robu M. E., Larson J. D., Nasevicius A., Beiraghi S., Brenner C., Farber S. A., Ekker S. C. (2007). p53 activation by knockdown technologies. PLoS Genet. 3, e78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohr S., Otten C., Abdelilah-Seyfried S. (2008). Asymmetric involution of the myocardial field drives heart tube formation in zebrafish. Circ. Res. 102, e12–e19 [DOI] [PubMed] [Google Scholar]
- Schoenebeck J. J., Keegan B. R., Yelon D. (2007). Vessel and blood specification override cardiac potential in anterior mesoderm. Dev. Cell 13, 254–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott J. J., Benson D. W., Basson C. T., Pease W., Silberbach G. M., Moak J. P., Maron B. J., Seidman C. E., Seidman J. G. (1998). Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 281, 108–111 [DOI] [PubMed] [Google Scholar]
- Solnica-Krezel L., Schier A. F., Driever W. (1994). Efficient recovery of ENU-induced mutations from the zebrafish germline. Genetics 136, 1401–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M., Chen Z., Bartunkova S., Yamasaki N., Izumo S. (1999). The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development 126, 1269–1280 [DOI] [PubMed] [Google Scholar]
- Tanaka M., Schinke M., Liao H. S., Yamasaki N., Izumo S. (2001). Nkx2.5 and Nkx2.6, homologs of Drosophila tinman, are required for development of the pharynx. Mol. Cell. Biol. 21, 4391–4398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Targoff K. L., Schell T., Yelon D. (2008). Nkx genes regulate heart tube extension and exert differential effects on ventricular and atrial cell number. Dev. Biol. 322, 314–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu C. T., Yang T. C., Tsai H. J. (2009). Nkx2.7 and Nkx2.5 function redundantly and are required for cardiac morphogenesis of zebrafish embryos. PLoS ONE 4, e4249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman J. S., Keegan B. R., Roberts R. W., Poss K. D., Yelon D. (2008). Hoxb5b acts downstream of retinoic acid signaling in the forelimb field to restrict heart field potential in zebrafish. Dev. Cell 15, 923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler C., Elmasri H., Klamt B., Volff J. N., Gessler M. (2003). Characterization of hey bHLH genes in teleost fish. Dev. Genes Evol. 213, 541–553 [DOI] [PubMed] [Google Scholar]
- Wu S. P., Cheng C. M., Lanz R. B., Wang T., Respress J. L., Ather S., Chen W., Tsai S. J., Wehrens X. H., Tsai M. J., et al. (2013). Atrial identity is determined by a COUP-TFII regulatory network. Dev. Cell 25, 417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier-Neto J., Neville C. M., Shapiro M. D., Houghton L., Wang G. F., Nikovits W., Jr, Stockdale F. E., Rosenthal N. (1999). A retinoic acid-inducible transgenic marker of sino-atrial development in the mouse heart. Development 126, 2677–2687 [DOI] [PubMed] [Google Scholar]
- Xin M., Small E. M., van Rooij E., Qi X., Richardson J. A., Srivastava D., Nakagawa O., Olson E. N. (2007). Essential roles of the bHLH transcription factor Hrt2 in repression of atrial gene expression and maintenance of postnatal cardiac function. Proc. Natl. Acad. Sci. USA 104, 7975–7980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi H., Yamagishi C., Nakagawa O., Harvey R. P., Olson E. N., Srivastava D. (2001). The combinatorial activities of Nkx2.5 and dHAND are essential for cardiac ventricle formation. Dev. Biol. 239, 190–203 [DOI] [PubMed] [Google Scholar]
- Yelon D., Horne S. A., Stainier D. Y. (1999). Restricted expression of cardiac myosin genes reveals regulated aspects of heart tube assembly in zebrafish. Dev. Biol. 214, 23–37 [DOI] [PubMed] [Google Scholar]
- Yutzey K. E., Rhee J. T., Bader D. (1994). Expression of the atrial-specific myosin heavy chain AMHC1 and the establishment of anteroposterior polarity in the developing chicken heart. Development 120, 871–883 [DOI] [PubMed] [Google Scholar]
- Yutzey K., Gannon M., Bader D. (1995). Diversification of cardiomyogenic cell lineages in vitro. Dev. Biol. 170, 531–541 [DOI] [PubMed] [Google Scholar]
- Zhang R., Xu X. (2009). Transient and transgenic analysis of the zebrafish ventricular myosin heavy chain (vmhc) promoter: an inhibitory mechanism of ventricle-specific gene expression. Dev. Dyn. 238, 1564–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R., Han P., Yang H., Ouyang K., Lee D., Lin Y. F., Ocorr K., Kang G., Chen J., Stainier D. Y., et al. (2013). In vivo cardiac reprogramming contributes to zebrafish heart regeneration. Nature 498, 497–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong T. P., Rosenberg M., Mohideen M. A., Weinstein B., Fishman M. C. (2000). gridlock, an HLH gene required for assembly of the aorta in zebrafish. Science 287, 1820–1824 [DOI] [PubMed] [Google Scholar]
- Zhou Y., Cashman T. J., Nevis K. R., Obregon P., Carney S. A., Liu Y., Gu A., Mosimann C., Sondalle S., Peterson R. E., et al. (2011). Latent TGF-β binding protein 3 identifies a second heart field in zebrafish. Nature 474, 645–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.