

Abstract
Many pharmaceutical companies worldwide specialize in oncology drug development and marketing. Among them, we have continued to take up the challenge of understanding the metabolism of pyrimidines as essential components of deoxyribonucleic acid for many years, and have provided unique products such as UFT® and TS-1 for cancer patients. Using our cumulative experience and knowledge, we are currently developing novel agents such as TAS-114, a dual inhibitor of deoxyuridine triphosphatase and dihydropyrimidine dehydrogenase, and TAS-102, a unique pyrimidine derivative inducing deoxyribonucleic acid dysfunction in cancer cells. Regarding molecular-targeted drugs, we have made huge efforts to establish ideal drug discovery platforms for the last several years. For kinase inhibitors, we established three core platforms such as a kinase-directed chemical library, a kinase assay panel and a target selection informatics system. The core platforms were further combined with peripheral technologies to measure essential parameters such as physicochemical properties, pharmacokinetics, efficacy and toxicities. Unique drug candidates have been identified at an early stage by assessing all important parameters. Several promising programs are proceeding simultaneously in the clinical or preclinical development stage such as TAS-115, a dual inhibitor of c-Met and vascular endothelial growth factor receptor, TAS-2104, a selective Aurora A inhibitor, TAS-117, an allosteric Akt inhibitor, TAS-2985, an irreversible fibroblast growth factor receptor inhibitor and TAS-2913, a T790M mutant selective epidermal growth factor receptor inhibitor. Other than kinase inhibitors, another drug discovery engine was established based on the fragment-based drug discovery technology. TAS-116, a new class of Hsp-90α/β inhibitor, is one of the products. Taiho's final goal is to provide innovative anticancer drugs together with companion diagnostics that are truly beneficial for patients.
Keywords: GI medicine, lung-Med, new technology/instruments, immunology, breast medicine
INTRODUCTION
Taiho Pharmaceutical Co., Ltd. primarily specializes in oncology drug development and marketing as an affiliate of Otsuka Holdings Co., Ltd. Immunology and urology are other areas of our research specialization. Taiho's ∼40-year history of oncology drug development has dealt with the challenges of understanding the metabolism of pyrimidines as essential components of nucleic acids, and modulating their incorporation to eradicate cancer cells which have higher proliferative potential and more active metabolism than normal cells. Using our cumulative experience and knowledge, we are currently pursuing a development of a completely new modulator of pyrimidine metabolism, as well as a unique pyrimidine derivative in order to induce DNA dysfunction in cancer cells (Fig. 1). Development of these classes of compounds, which might be categorized as ‘conventional cytotoxic’ drugs, is generally of less interest for the majority of major pharmaceutical companies as well as small biotechnology companies. However, cytotoxic drugs such as antimetabolites, taxanes, platinum and topoisomerase inhibitors are still commonly used as basic drugs in most clinical regimens in various tumor types. We believe that the importance of cytotoxic drugs in the clinic will not change dramatically for at least several decades, and hence, have taken on the challenge to develop new more efficacious drugs in this field.
Figure 1.

Development of pyrimidine derivatives and its modulators in Taiho.
On the other hand, the human genome has now been completely decoded, and the analysis of genes involved in the pathogenesis, progression and malignant alteration of cancer has also rapidly advanced. This has resulted in various molecular-targeted drugs, having been developed and introduced into the clinic, permitting high specificity against particular cancers while causing relatively few side effects. Clearly, molecular-targeted drugs are essential for the future cancer treatment. Thus, beginning with a partnership with Sugen, Inc. in 1998, we have committed large research efforts, especially in the last couple of years, to the development of this category of compounds.
This paper describes Taiho's approach for anticancer drug development from the past to the present and also touches on our philosophy regarding future research directions and new challenges.
THE CHALLENGE OF DEVELOPING PYRIMIDINE DERIVATIVES
Clinically Available Pyrimidine Derivatives
5-Fluorouracil (5-FU) has played a central role over the last 50 years in the treatment of gastrointestinal cancer. Even today, it maintains considerable importance. The antitumor effects of 5-FU are primarily induced by the depletion of the thymidine 5′-triphosphate (dTTP) pool which is required for DNA synthesis. The depletion of the dTTP pool depends on thymidylate synthase (TS) inhibition caused by the formation of a covalent ternary complex consisting of TS, 5,10-methylenetetrahydrofolate and 5-fluoro-2′-deoxyuridine, 5′-monophosphate (FdUMP), an active metabolite of 5-FU. In addition, not only RNA dysfunction induced by 5-fluorouridine-5′-triphosphate (FUTP) incorporation into RNA, but also DNA damages induced by 2′-deoxyuridine, 5′-triphosphate (dUTP) and 5-fluoro-2′-deoxyuridine, 5′-triphosphate (FdUTP) partially contribute to the antitumor effect (1). Our research approach for many years was to maximize the antitumor effect of 5-FU, and to reduce the side effects of 5-FU, by using combinations of drugs consisting of oral pyrimidine derivatives and their modulators (Fig. 2).
Figure 2.

Modes of action of 5-fluorouracil (5-FU) and the deoxyuridine triphosphatase (dUTPase) inhibitor, TAS-114 in pyrimidine nucleotide metabolism. 5-FU is metabolized in the pyrimidine nucleotide synthesis pathway and exerts its antitumor action as fluoronucleotides. 5-Fluoro-2′-deoxyuridine, 5′-monophosphate (FdUMP) inhibits thymidylate synthase by forming a covalent ternary complex with methylene tetrahydrofolate (THF). Another active metabolite, 5-fluorouridine-5′-triphosphate (FUTP) causes RNA dysfunction by misincorporation into RNA instead of uridine-5′-triphosphate (UTP). On the other hand, 5-FU is catabolized by dihydropyrimidine dehydrogenase (DPD) mainly in liver. dUTPase selectively catalyzes dUTP and 5-fluoro-2′-deoxyuridine, 5′-triphosphate (FdUTP) hydrolysis and plays an important role in restricting dUTP or FdUTP misincorporation into DNA instead of dTTP. TAS-114, a dUTPase inhibitor, promotes 5-FU and uracil misincorporation into DNA and enhances 5-FU-mediated cytotoxicity. TAS-114 also has a moderate DPD-inhibitory activity in addition to its potent dUTPase inhibition, resulting in the effective utilization of 5-FU. FU, 5-fluorouracil; DPD, Dihydropyrimidine dehydrogenase; UP, uridine phosphorylase; UK, uridine kinase; OPRT, orotate phosphoribosyltransferase; TP, thymidine phosphorylase; RNR, ribonucleotide reductase; TK, thymidine kinase; TS, thymidylate synthase; dUTPase, deoxyuridine triphophatase; methylene THF, methylene tetrahydrofolate.
Futraful® (tegafur), a prodrug of 5-FU, was originally synthesized by Dr S.A. Hiller of Riga Institute of the Republic of Latvia in 1966. 5-FU induces maximum antitumor effects when a sustained blood concentration is maintained with continuous infusion. The advantage of Futraful® is its oral availability, which achieves the maintenance of a blood level similar to continuous infusion of 5-FU. Futraful® was introduced clinically in Japan in 1974 as the first oral anticancer drug in the world, with good efficacy against cancer and enhanced compliances due to the increased convenience for patients.
5-FU loses its antitumor effect through degradation by dihydropyrimidine dehydrogenase (DPD). Therefore, the next approach was directed at a DPD inhibitor, which induces a stronger antitumor effect by increasing intratumoral 5-FU concentration. UFT® is a combination drug containing tegafur and uracil. Using uracil as a moderate DPD inhibitor enabled to approximately doubling the utilization efficiency of 5-FU (2,3). Specifically, adequate 5-FU blood levels were maintained by UFT®, although the amount of tegafur in UFT® was reduced by approximately half compared with tegafur monotherapy. From a quality of life (QOL) view point, the reduction of tegafur was abeneficial because it lowered the amount of negative effect of 5-FU which include hand-foot syndrome (HFS), neurotoxicity and possibly cardiovascular toxicity. The concept called DPD-inhibitory pyrimidine (DIF) was established as a result (4). UFT® has various indications such as colorectal, non-small-cell lung, breast, gastric and liver cancers and is widely used in Japan. It has also demonstrated efficacy as an adjuvant chemotherapy in specific groups of patients with colorectal, non-small-cell lung and gastric cancers (5–7). UFT® is also used in Asia and the EU with limited indications.
The concept of DIF was reinforced by TS-1 (S-1), which is an oral combination drug consisting of tegafur, gimeracil and oteracil potassium at a molar ratio of 1:0.4:1. By creating the new, more potent DPD inhibitor gimeracil, which can increase the utilization efficiency of 5-FU up to approximately 10 times, the amount of tegafur in S-1 could be reduced to approximately one-tenth compared with tegafur monotherapy. Oteracil was developed as a countermeasure against side effects. It remains within the intestinal wall as it is not easily absorbed into the blood stream, and reduces diarrhea that could possibly be caused by the activation of 5-FU in the intestine by orotate phosphoribosyltransferase (OPRT) (8) (Figs 2 and 3). S-1 demonstrated its efficacy in combination with cis-diamminedichloroplatinum in the first-line treatment of advanced gastric cancer (9). S-1 has been widely used not only in Japan, but also in other Asian countries as a standard drug for advanced gastric cancer. The use of S-1 has also started in EU countries for the same indication. S-1 has proven its efficacy in the adjuvant chemotherapy of gastric cancer as well (10,11). Other than gastric cancer, S-1 also showed significant effectiveness on non-small-cell lung cancers in combination with carboplatin (12) and on colorectal cancers in combination with irinotecan (13). S-1 has also been used in the treatment of various types of cancers such as pancreatic, breast, liver and biliary cancers in Japan. It should be kept in mind, however, that S-1 causes some side effects such as diarrhea and stomatitis in Western countries. As an unexpected event, lacrimal stenosis/obstruction has been recently reported. Reduction of these side effects is one clear direction for the development of new fluoropyrimidine derivatives.
Figure 3.

Mode of action of TAS-114S. S-1(TS-1), an orally available fixed combination 5-FU-based drug composed of tegafur, gimeracil and oteracil potassium (molar ratio; 1:0.4:1), achieves selective accumulation of FdUMP and dUMP in tumors. TAS-114, a dUTPase inhibitor, promotes FdUTP and dUTP misincorporation into DNA and enhances the antitumor activity of S-1 selectively in tumors. GI, gastrointestinal; Tox, toxicity; HFS, hand-foot syndrome.
New Oral Pyrimidines Under Clinical Investigation
We are currently focusing on an enzyme called deoxyuridine triphosphatase (dUTPase), which is a gatekeeper enzyme limiting the incorporation of dUTP, a metabolite of uracil, into DNA by converting it back to dUMP. Generally, excessive incorporation of uracil into DNA is lethal for eukaryotic cells (14) and therefore, its misincorporation must be prevented. As regards 5-FU, dUTPase inhibits the incorporation of FdUTP as a metabolite of 5-FU into DNA by converting it to FdUMP, and limits the antitumor potency of 5-FU (Fig. 2). It has also been reported that tumors with high expression of dUTPase are resistant to 5-FU compared with tumors with low expression of dUTPase (15,16), suggesting a reverse correlation between sensitivity to 5-FU and expression of the enzyme. Therefore, dUTPase expression in a tumor is considered to be one of the potential predictive factors for 5-FU and a specific inhibitor of this enzyme could be a good and specific target for treating cancer. We tried to design a specific inhibitor of dUTPase using the fragment-based drug discovery (FBDD) technology which will be described later, and finally succeeded to create the world's first and selective dUTPase inhibitor TAS-114. Dramatic improvement in the antitumor activity of 5-FU was observed using it in conjunction with 5-FU or a 5-FU prodrug (17). The mechanism of enhancement was thought to be as follows: FdUTP, an active metabolite of 5-FU, is incorporated into DNA in a large amount by the inhibition of dUTPase. In addition, dUTP, a metabolite of uracil, which normally would not be incorporated into DNA, was also incorporated into DNA. Incorporation of both FdUTP and dUTP induces DNA dysfunction, resulting in tumor death. Significant enhancement in antitumor activity was observed, especially when TAS-114 was combined with a 5-FU oral prodrug such as S-1 (18) (Fig. 3) or capecitabine (19) (Fig. 4) which can be administered orally. Therefore, these combinations were considered to be exciting approaches for clinical development. In addition, TAS-114 is not only a dUTPase inhibitor, but also a DPD inhibitor. Combination with capecitabine can realize the concept of DIF by inhibiting DPD, which can effectively use 5-FU, the primary metabolite of capecitabine. The dose of capecitabine used in combination could be reduced significantly compared with that of capecitabine monotherapy (19). In terms of side effects, it is expected to significantly reduce the incidence of HFS, a main side effect of capecitabine. From an efficacy standpoint, a significant enhancement above that of capecitabine monotherapy has been observed in preclinical models, due to DNA dysfunction induced by DNA incorporation of FdUTP in addition to the inhibition of TS. In S-1 combination (18), the DPD-inhibitory effect of TAS-114 is negligible because it is masked by the strong DPD inhibitor gimeracil, which is one of the subcomponents of S-1. Phase I clinical trials of TAS-114 in combination with S-1 (TAS-114S) or capecitabine (TAS-114C) are in progress.
Figure 4.
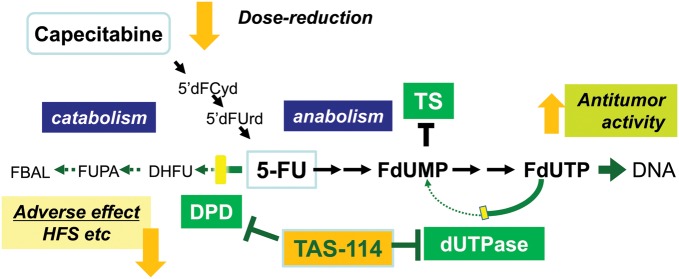
Mode of action of TAS-114C. 5-FU is rapidly degraded by DPD. Therefore, 5-FU-based drugs without a DPD inhibitor (i.e. capecitabine) need massive dosing for cancer treatment although this may cause a higher incidence of HFS. Combination with TAS-114 could reduce the dose of capecitabine by DPD inhibition while maintaining adequate tumor 5-FU levels. On the other hand, the antitumor activity of capecitabine may increase by dUTPase inhibition.
Regarding our drug discovery efforts for fluoropyrimidines as anticancer agents, we have different approaches from 5-FU-based programs as described above.
For example, TAS-102 is a novel oral anticancer agent consisting of α,α,α-trifluorothymidine (FTD), as the antitumor component and a thymidine phosphorylase inhibitor (TPI) which prevents the degradation of FTD at a molar ratio of 1:0.5 (20) (Fig. 5). Co-administration of the TPI enables adequate serum levels of FTD in humans, who have high endogenous thymidine phosphorylase activity. By phosphorylation of FTD with thymidine kinase (TK), F3dTMP is formed and inhibits TS. However, the inhibitory process of TS differs from that of FdUMP of 5-FU. It appears that TS inhibition by F3dTMP of FTD is reversible (21), and disappears after drug washout in a cancer cell line (22). Thus, we believe that TS inhibition has only minor, if any, antitumor effect. F3dTMP is further phosphorylated into F3dTTP and then incorporated into DNA where it remains for protracted periods (20). Therefore, the majority of antitumor effect is thought to be induced by DNA dysfunction. The uptake and retention of FTD in DNA might be high in tumor cells due to aggressive growth and decreased DNA repair (23). When the antitumor effect was examined in human tumor xenograft models, unique effects were observed (Fig. 6). The tumor growth inhibition during drug administration was not particularly strong compared with other cytotoxic agents such as paclitaxel and irinotecan, but the inhibition of tumor growth continued long after the end of drug administration. This unique profile was observed more clearly in a survival model where TAS-102 significantly prolonged the survival time compared with other agents such as irinotecan. The unique antitumor effect was thought to be induced by DNA dysfunction due to prolonged retention of F3dTTP in DNA. In clinical trials, a significant prolongation of overall survival was observed in a placebo-controlled, double-blinded, randomized PII trial in third/fourth line metastatic colorectal cancer patients who failed 5-FU-based standard chemotherapy (TAS-102, n = 112; placebo, n = 57; median OS, 9.0 vs 6.6 months; HR, 0.56; P = 0.0011) (24) (Fig. 7). Hematological toxicity was the main toxicity observed. Diarrhea, which affects drug intake compliance, was light, and other toxicities related to the QOL of patients were relatively weaker as expected for this class of agent. A global PIII trial is currently in progress in a similar population of the patients in the PII trial. TAS-102 is an attractive new fluoropyrimidine with a primarily different mechanism of action compared with 5-FU.
Figure 5.

Mode of action of TAS-102. α,α,α-Trifluorothymidine (FTD) is converted by TK to its monophosphate form. F3dTMP is further phosphorylated to its triphosphate form, F3dTTP, which is well incorporated into DNA. TAS-102 exerts antitumor activity primarily via FTD incorporation into DNA. Thymidine phosphorylase inhibitor (TPI) inhibits the degradation of FTD, which is catalyzed by thymidine phosphorylase (TP) to FTY (trifluorothymine).
Figure 6.
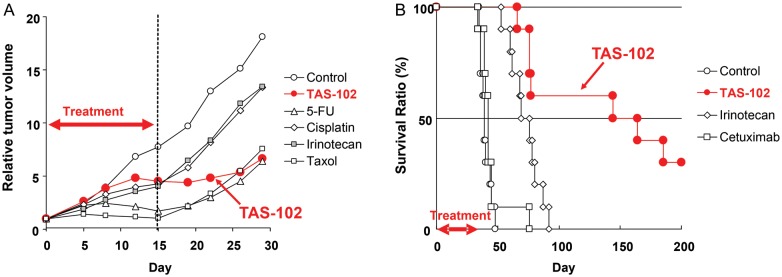
Antitumor effects of TAS-102 in preclinical xenograft models. TAS-102 treatment demonstrated the sustained tumor growth inhibition and survival benefit in preclinical studies. (A) KM20C xenograft model in mice; drugs were administered to mice subcutaneously implanted with a human colon cancer line, KM20C. Dosing schedules were as follows: TAS-102 (150 mg/kg/day, p.o., Days 1–14), 5-FU (15 mg/kg/day, c.i., Days 1–15), cisplatin (7 mg/kg/day, i.v., Days 1,8), irinotecan (40 mg/kg/day, i.v., Days 1 and 8), taxol (30 mg/kg/day, i.v., Days 1 and 8). (B) KM20C survival model in mice; Drugs were administered to mice intraperitoneally implanted with KM20C. Dosing schedules were as follows: TAS-102 (200 mg/kg/day, p.o., five consecutive days followed by 2 days rest a week for 6 weeks), irinotecan (100 mg/kg/day, i.v., once a week for 6 weeks) and cetuximab (40 mg/kg/day, i.p., twice a week for 6 weeks).
Figure 7.

Result of the PII trial of TAS-102 in third/fourth line colorectal cancer. Patients were randomly assigned in a 2:1 ratio to either TAS-102 plus best supportive care (BSC) or placebo plus BSC. A dose of 35 mg/m2 TAS-102 was taken orally twice a day. TAS-102 or placebo was taken in a 28-day cycle: a 2-week cycle of 5 days of treatment followed by a 2-day rest, and then a 14-day rest.
Building on the findings of the mechanism of TAS-102, we are confident in developing new-generation products in the near future. Even though most drug companies have chosen not to pursue this category of anticancer drugs, Taiho will continue in this challenging area based on our long cultivated experience and knowledge.
THE CHALLENGE OF DEVELOPING MOLECULAR-TARGETED DRUGS
We have had various alliances and collaborations with biotechnology companies, research institutes and universities with the purpose of obtaining novel-targeted molecules, novel lead compounds, drug discovery technologies and/or the latest research infrastructures. To fully integrate such information and technology, we felt the need for a firm policy and extreme improvement in our own research capability. We executed this idea by consolidating all of our drug discovery functions into Tsukuba Research Institute in May 2011. Since then, much effort was devoted to molecular-targeted drug development, mainly focusing on the three types of targets: (i) protein kinase families, (ii) protein homeostasis and (ii) transcription and regulation of gene expression (Table 1).
Table 1.
Taiho's molecular-targeting early-stage pipeline
| Product | Target | Stage | Profile |
|---|---|---|---|
| TAS-115 | Met/VEGFR | Phase I | Dual inhibitor of HGF/VEGF signaling |
| TAS-116 | Hsp90α/β | Preclinical |
|
| TAS-117 | Akt | Preclinical | Highly potent and selective Akt inhibitor |
| TAS-2104 | Aurora A | Late Preclinical |
|
| TAS-2913 | EGFR T790M | Preclinical | Mutant selective EGFR inhibitor |
| TAS-2985 | FGFR | Preclinical | Highly potent, irreversible FGFR inhibitor |
VEGFR, vascular endothelial growth factor receptor; EGFR, epidermal growth factor receptor; FGFR, fibroblast growth factor receptor; HGF, hepatocyte growth factor; VEGF, vascular endothelial growth factor.
Development of Kinase Inhibitors
Abnormalities or mutations in kinase genes can be the drivers in the onset or progression of cancers. It is, however, generally difficult to generate highly selective kinase inhibitors because the catalytic domain of the kinase families shows basically a high-degree homology.
To overcome this issue, we established a platform to generate a kinase-directed chemical library based on the proprietary technologies for structure-based inhibitor design and high-speed library synthesis. Such a library was subjected to a system which can comprehensively analyze all the interactions between each compound and many types of kinases, thus permitting us to efficiently identify and optimize selective inhibitors for several target kinases in parallel.
At first, this approach was employed to explore ATP-binding pockets of target kinases and developed two ATP-competitive inhibitors: TAS-115, a dual inhibitor of c-Met and vascular endothelial growth factor-receptor tyrosine kinase (RTK) (25–27), and TAS-2104, a selective Aurora A inhibitor (28,29).
Recently, it is well recognized that mutation in the ATP-binding pocket could be a gate-keeper mutation for drug resistance. Because this becomes an obstacle for ATP-competitive drugs, different inhibitory mechanisms such as allosteric kinase inhibition and covalent-binding kinase inhibition might be required. To strengthen our kinase drug discovery engine in this field, we constructed specially designed chemical libraries for allosteric or covalent target inhibition. Moreover, we carefully established a specialized drug metabolism and pharmacokinetics platform to optimize unique pharmacokinetic parameters of covalent-binding kinase inhibitors. As a consequence, we succeeded in developing an allosteric inhibitor of Akt, TAS-117 (30,31), as well as two covalent-binding kinase inhibitors, TAS-2985 and TAS-2913. TAS-2985 is a selective inhibitor of fibroblast growth factor-RTK (32,33), and TAS-2913 is a third-generation epidermal growth factor (EGF)-RTK inhibitor (34), which aims to overcome a T790M gatekeeper mutation but does not yet affect wild-type EGF-RTK.
Development of Molecular-targeted Drugs Aimed at the Maintenance of Protein Homeostasis
FBDD is a novel approach for drug discovery that starts with the screening of simple and small molecules (fragments). Highly sensitive biophysical screening technologies are required because such fragments show only low-affinity interaction with the target protein. Hit fragments are converted into high-affinity drug-like molecules through a cycle of optimization process based on our proprietary bioinformatics as well as chemoinformatic platforms.
FBDD engine generated TAS-116, a new class of Hsp-90α/β inhibitor that binds not only to the conventional-binding pockets as existing Hsp-90 inhibitors, but also to a novel-binding pocket (35,36). Such a unique binding mode makes TAS-116 highly specific for Hsp-90α/β without inhibiting other Hsp-90 family proteins such as GRP94 in endoplasmic reticulum or TRAP-1 in mitochondria. TAS-116 showed superior efficacy in several xenograft models, and relatively wide safety margins compared with the existing Hsp-90 inhibitors (37).
FUTURE PROSPECTIVES
Anticancer drug discovery and development has become very competitive because not only major multinational pharmaceutical companies but also many small biotechnology companies have been putting in large efforts in this area. Taiho has been consistently working on cancer drug discovery mainly via fluoropyrimidines. Several oral pyrimidine derivatives produced by us might distinguish us from other companies, and we will keep this policy furthermore. Furthermore, the uniqueness of our molecular-targeting strategy to create novel drugs for cancer patients throughout the world will be harnessed.
In the clinic, detailed clinical regimens have been determined for individual tumor types based on the evidence from clinical studies, and those regimens are being adjusted based on the new data from clinical trials of new drugs or combinations of various anticancer drugs. Since anticancer drug development requires >7–10 years for the clinical trial, it is important to accurately predict how therapeutic regimens and medical standards will change during this period. In addition, even as early as the preclinical stage, we have to commit to a direction of clinical development by analyzing and assessing the selection of target tumors and the lines of therapy, monotherapy or combination therapy, and potential partner drugs for combination, etc. The application of biomarkers also becomes an important issue. It is, therefore, essential to share information with, and obtain feedback from, key opinion leaders and clinical experts, especially when the strategy of clinical development is being finalized for individual programs. We will continue to promote this cooperation and collaboration.
Funding
Funding to pay the Open Access publication charges for this article was provided by Taiho Pharmaceutical Co., Ltd.
Conflict of interest statement
Teruhiro Utsugi, PhD, is the Executive Director of the Tsukuba Research Institute and is a member of the Board of Taiho Pharmaceutical Co., Ltd.
Acknowledgements
I have written this manuscript as a representative of research division of Taiho Pharmaceutical Co., Ltd. All the work was contributed by colleagues and predecessors in Taiho. I also thank clinicians who have kindly supported Taiho's drug development. The author also thank professor J. Patrick Barron and Y. Taguchi, MBA remunerated editors of Taiho.
References
- 1.Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–8. doi: 10.1038/nrc1074. doi:10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 2.Fujii S, Ikenaka K, Fukushima M, Shirasaka T. Effect of uracil and its derivatives on antitumor activity of 5-fluorouracil and 1-(2-tetrahydrofuryl)-5-fluorouracil. Gann. 1978;69:763–72. [PubMed] [Google Scholar]
- 3.Takiuchi H, Ajani JA. Uracil-tegafur in gastric carcinoma: a comprehensive review. J Clin Oncol. 1998;16:2877–85. doi: 10.1200/JCO.1998.16.8.2877. [DOI] [PubMed] [Google Scholar]
- 4.Omura K. Clinical implications of dihydropyrimidine dehydrogenase (DPD) activity in 5-FU-based chemotherapy: mutations in the DPD gene, and DPD inhibitory fluoropyrimidines. Int J Clin Oncol. 2003;8:132–8. doi: 10.1007/s10147-003-0330-z. doi:10.1007/s10147-003-0330-z. [DOI] [PubMed] [Google Scholar]
- 5.Lembersky BC, Wieand HS, Petrelli NJ, et al. Oral uracil and tegafur plus leucovorin compared with intravenous fluorouracil and leucovorin in stage II and III carcinoma of the colon: results from national surgical adjuvant breast and bowel project protocol C-06. J Clin Oncol. 2006;24:2059–64. doi: 10.1200/JCO.2005.04.7498. doi:10.1200/JCO.2005.04.7498. [DOI] [PubMed] [Google Scholar]
- 6.Kato H, Ichinose Y, Ohta M, et al. A randomized trial of adjuvant chemotherapy with uracil-tegafur for adenocarcinoma of the lung. N Engl J Med. 2004;350:1713–21. doi: 10.1056/NEJMoa032792. doi:10.1056/NEJMoa032792. [DOI] [PubMed] [Google Scholar]
- 7.Nakajima T, Kinoshita T, Nashimoto A, et al. Randomized controlled trial of adjuvant uracil-tegafur versus surgery alone for serosa-negative, locally advanced gastric cancer. Br J Surg. 2007;94:1468–76. doi: 10.1002/bjs.5996. doi:10.1002/bjs.5996. [DOI] [PubMed] [Google Scholar]
- 8.Shirasaka T, Nakano K, Takechi T, et al. Antitumor activity of 1 M tegafur-0.4 M 5-chloro-2,4-dihydroxypyridine-1 M potassium oxonate (S-1) against human colon carcinoma orthotopically implanted into nude rats. Cancer Res. 1996;56:2602–6. [PubMed] [Google Scholar]
- 9.Koizumi W, Narahara H, Hara T, et al. S-1 plus cisplatin versus S-1 alone for first-line treatment of advanced gastric cancer (SPIRITS trial): a phase III trial. Lancet Oncol. 2008;9:215–21. doi: 10.1016/S1470-2045(08)70035-4. doi:10.1016/S1470-2045(08)70035-4. [DOI] [PubMed] [Google Scholar]
- 10.Sakuramoto S, Sasako M, Yamaguchi T, et al. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med. 2007;357:1810–20. doi: 10.1056/NEJMoa072252. doi:10.1056/NEJMoa072252. [DOI] [PubMed] [Google Scholar]
- 11.Sasako M, Sakuramoto S, Katai H, et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol. 2011;29:4387–93. doi: 10.1200/JCO.2011.36.5908. doi:10.1200/JCO.2011.36.5908. [DOI] [PubMed] [Google Scholar]
- 12.Okamoto I, Yoshioka H, Morita S, et al. Phase III trial comparing oral S-1 plus carboplatin with paclitaxel plus carboplatin in chemotherapy-naïve patients with advanced Non-small-cell lung cancer: results of a west japan oncology group study. J Clin Oncol. 2010;28:5240–6. doi: 10.1200/JCO.2010.31.0326. doi:10.1200/JCO.2010.31.0326. [DOI] [PubMed] [Google Scholar]
- 13.Muro K, Boku N, Shimada Y, et al. Irinotecan plus S-1 (IRIS) versus fluorouracil and folinic acid plus irinotecan (FOLFIRI) as second-line chemotherapy for metastatic colorectal cancer: a randomized phase 2/3 non-inferiority study (FIRIS study) Lancet Oncol. 2010;11:853–60. doi: 10.1016/S1470-2045(10)70181-9. doi:10.1016/S1470-2045(10)70181-9. [DOI] [PubMed] [Google Scholar]
- 14.Gadsden MH, McIntosh EM, Game JC, Wilson PJ, Haynes RH. dUTP pyrophosphatase is an essential enzyme in Saccharomyces cerevisiae. EMBO J. 1993;12:4425–31. doi: 10.1002/j.1460-2075.1993.tb06127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ladner RD, Lynch FJ, Groshen S, et al. dUTP nucleotidohydrolase isoform expression in normal and neoplastic tissues: association with survival and response to 5-fluorouracil in colorectal cancer. Cancer Res. 2000;60:3493–503. [PubMed] [Google Scholar]
- 16.Nobili S, Napoli C, Landini I, et al. Identification of potential pharmacogenomic markers of clinical efficacy of 5-fluorouracil in colorectal cancer. Int J Cancer. 2011;128:1935–45. doi: 10.1002/ijc.25514. doi:10.1002/ijc.25514. [DOI] [PubMed] [Google Scholar]
- 17.Yano W, Yokogawa T, Fukuoka M, et al. In vitro characterization of a novel dUTPase inhibitor, TAS-114, as a fluoropyrimidine enhancer. 24th EORTC-NCI-AACR symposium 2012; (Abstract 67) [Google Scholar]
- 18.Yokogawa T, Wakasa T, Yano W, et al. TAS-114 enhances S-1 activity in vivo when used in combination. 24th EORTC-NCI-AACR symposium 2012; (Abstract 65) [Google Scholar]
- 19.Wakasa T, Yamamura K, Tsuji S, et al. TAS-114 in combination, with capecitabine-based chemotherapy may represent a novel therapeutic strategy. 24th EORTC-NCI-AACR symposium 2012; (Abstract 68) [Google Scholar]
- 20.Emura T, Nakagawa F, Fujioka A, et al. An optimal dosing schedule for a novel combination antimetabolite, TAS-102, based on its intracellular metabolism and its incorporation into DNA. Int J Mol Med. 2004;13:249–55. [PubMed] [Google Scholar]
- 21.Langenbach RJ, Danenberg PV, Heidelberger C. Thymidylate synthetase: mechanism of inhibition by 5-fluoro-2′-deoxyuridylate. Biochem Biophys Res Commun. 1972;48:1565–71. doi: 10.1016/0006-291x(72)90892-3. doi:10.1016/0006-291X(72)90892-3. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka N, Fujioka A, Yamamura K, et al. Trifluorothymidine incorporation into DNA is the primary mechanism of action of TAS-102, and leads to markedly prolonged survival in a mouse model. 24th EORTC-NCI-AACR symposium 2012; (Abstract 66) [Google Scholar]
- 23.Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8:180–92. doi: 10.1038/nrc2344. doi:10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- 24.Yoshino T, Mizunuma N, Yamazaki K, et al. TAS-102 monotherapy for pretreated metastatic colorectal cancer: a double-blind, randomized, placebo-controlled phase 2 trial. Lancet Oncol. 2012;13:993–1001. doi: 10.1016/S1470-2045(12)70345-5. doi:10.1016/S1470-2045(12)70345-5. [DOI] [PubMed] [Google Scholar]
- 25.Fujita H, Miyadera K, Kato M, et al. TAS-115; a highly potent c-Met+VEGFRs dual inhibitor with prominent safety profile. American Association for Cancer Research Annual Meeting 2011; (Abstract 3577) [Google Scholar]
- 26.Ochiiwa H, Fujita H, Miyadera K, et al. The prominent safety profile of TAS-115, a novel c-Met+VEGFR dual kinase inhibitor can lead to potentiate the efficacy via durable inhibitor of angiogenesis. American Association for Cancer Research Annual Meeting 2011; (Abstract 3600) [Google Scholar]
- 27.Fujita H, Kato M, Miyadera K, et al. MET/VEGFR dual inhibition and prominent safety profiles of TAS-115 are favorable for the combination with chemotherapeutic drugs. American Association for Cancer Research Annual Meeting 2012; (Abstract 1785) [Google Scholar]
- 28.Sootome H, Masuko N, Ishihara K, Mitsuya M, Hirai H, Utsugi T. Highly potent and selective Aurora A inhibitor, TAS-2104 enhanced antitumor activity of taxanes in vitro. 24th EORTC-NCI-AACR symposium 2012; (Abstract 251) [Google Scholar]
- 29.Ishihara K, Masuko N, Sootome H, et al. TAS-2104, a highly potent and effective Aurora A inhibitor, enhanced antitumor activity of taxanes in vivo. 24th EORTC-NCI-AACR symposium 2012; (Abstract 252) [Google Scholar]
- 30.Abe T, Ichikawa K, Fujita R, et al. Characterization of TAS-117, a novel, highly potent and selective inhibitor of AKT. 24th EORTC-NCI-AACR symposium 2012; (Abstract 356) [Google Scholar]
- 31.Ichikawa K, Abe T, Fukushima H, et al. A novel highly potent AKT inhibitor TAS-117 demonstrated synergistic antitumor activity in combination with paclitaxel through enhancement of apoptosis induction. 24th EORTC-NCI-AACR symposium 2012; (Abstract 5) [Google Scholar]
- 32.Sootome H, Fujita H, Ochiiwa H, et al. Identification & biological characterization of a highly potent, irreversible inhibitor of FGFR, TAS-2985. 24th EORTC-NCI-AACR symposium 2012; (Abstract 380) [Google Scholar]
- 33.Nakatsuru Y, Fujita H, Sootome H, et al. Significant in vivo antitumor activity by a highly potent, irreversible FGFR Inhibitor, TAS-2985. 24th EORTC-NCI-AACR symposium 2012; (Abstract 383) [Google Scholar]
- 34.Miyadera K, Kato M, Takahashi I, et al. TAS-2913 is a mutant selective EGFR inhibitor for NSCLC: characterization against EGFR T790M in cell and xenograft models. 24th EORTC-NCI-AACR symposium 2012; (Abstract 142) [Google Scholar]
- 35.Okubo S, Muraoka H, Kanoh A, et al. TAS-116, a novel, orally bioavailable HSP90a/ß selective inhibitor demonstrates highly potent antitumor activity in preclinical models with a favorable PK profile. 24th EORTC-NCI-AACR symposium 2012; (Abstract 291) [Google Scholar]
- 36.Yoshimura C, Kitade M, Oshiumi H, et al. Evolution of highly selective HSP90a/ß inhibitors with unique binding mode. 24th EORTC-NCI-AACR symposium 2012; (Abstract 292) [Google Scholar]
- 37.Kodama Y, Hitotsumachi H, Shibata Y, et al. TAS-116, a novel, orally bioavailable highly potent HSP90a/ß selective inhibitor demonstrates favorable tissue distribution properties which lead to minimized ocular toxicity. 24th EORTC-NCI-AACR symposium 2012; (Abstract 290) [Google Scholar]