

Abstract
The filamentous bacterium Streptomyces coelicolor modulates polar growth and branching by phosphorylating the cytoskeletal protein DivIVA. Previous MALDI-TOF analysis of DivIVA showed that a large 7.2 kDa tryptic peptide was multiply phosphorylated. To aid localization of the phosphorylation sites, we introduced additional tryptic cleavage sites into DivIVA, and the resulting phosphopeptides were analyzed by LC–MS/MS. Phosphopeptide isomers could be separated chromatographically, but because of overlapping elution and spectrum quality, site assignment by standard software tools was ambiguous. Because fragment ions carrying the phosphate group are essential for confident localization, large numbers of spectra were collected using targeted LC–MS/MS, and a special script was developed for plotting the elution of site-determining fragments from those spectra under the XIC of the parent ions. Where multiple phosphopeptide isomers were present, the elution of the site-determining y-ions perfectly coincided with the elution of the corresponding phosphopeptide isomer. This method represents a useful tool for user inspection of spectra derived from phosphopeptide isomers and significantly increases confidence when defining phosphorylation sites. In this way, we show that DivIVA is phosphorylated in vivo on five sites in the C-terminal part of the protein (T304, S309, S338, S344, and S355). The data have been deposited to the ProteomeXchange Consortium with identifier PXD000095.
Keywords: Ascore, DivIVA, phosphopeptides, phosphorylation site localization, ScaffoldPTM, targeted LC−MS/MS
Introduction
We describe the use of targeted LC–MS/MS and different software tools to determine the phosphorylation sites in the Streptomyces coelicolor cytoskeletal element DivIVA.
Apical growth in the filamentous bacteria Streptomyces is directed by a polarisome-like complex involving the essential coiled-coil protein DivIVA.1−3 This bacterial polarization machinery is regulated by a Ser/Thr protein kinase, AfsK, which localizes to hyphal tips and phosphorylates DivIVA, thereby modulating growth polarity and hyphal branching. AfsK-mediated phosphorylation of DivIVA is induced by antibiotics that arrest cell wall synthesis, such as bacitracin.1 It will be vital to unambiguously identify the amino acid residues that are phosphorylated by AfsK to mechanistically dissect the role of phosphorylation of the cell polarity determinant DivIVA.
Previous MALDI-TOF analysis of DivIVA immunoprecipitated from S. coelicolor following bacitracin treatment showed that a large 7.2 kDa tryptic peptide (TPATASLPPSPAPSMAPAGA SAPSYGGNQSMGGGPGQSGP SYGGQQQMSPAMTQPMAPVR PQGPSPMGQAPSPMR) that contains most of the C-terminal region of DivIVA (see Figure 4) was multiply phosphorylated.1 The identification of the phosphorylation sites is necessary so that they can be mutagenised to unphosphorylatable (Ala) or phosphomimetic (Asp) residues to analyze the effect of phosphorylation on DivIVA activity further. To this end, two additional tryptic cleavage sites were introduced into the 7.2 kDa fragment by mutagenesis.
Figure 4.

Protein sequence of DivIVA: The “7.2 kDa” peptide is highlighted in yellow; the phosphorylated tryptic peptide located N-terminal to it (Peptide 1) is highlighted in blue. Peptides 2 and 3 are labeled with arrows. The arginine residues introduced by mutagenesis (originally glutamine residues Q343 and Q360) are labeled in green. Sites of phosphorylation determined in this paper are labeled in red.
The confident determination of the site of phosphorylation can be challenging and depends mainly on the generation of site-determining fragments carrying the phosphate group.4 It becomes even more complicated if the phosphate group can be localized at different sites within the sequence (isomers). In the initial analysis of the resulting peptides potential phosphopeptide isomers were observed. Phosphopeptide isomers are easily missed during data-dependent LC–MS/MS acquisitions if they elute closely or together because the mass spectrometer is usually set up to implement a dynamic exclusion window to avoid repetitive fragmentation of peptides. This means, however, that data for closely eluting isomers may not even be collected. Furthermore, search tools may not give a clear result when a mixed spectrum of coeluting isomers needs to be interpreted, or indeed the user may even ignore less probable results in error, choosing to believe only a top hit and ignoring lesser scoring results. To improve the analysis, we applied targeted LC–MS/MS without dynamic exclusion to collect spectra continuously over the whole LC peak of the parent ions. A software tool was then developed to generate plots of site-determining fragment ions from those spectra. Those plots were then compared with the elution of parent ions. The retention time of the specific ions coincided with the retention times of the phosphopeptides. This method is very specific and useful for targeted LC–MS/MS analysis of phosphopeptide isomers to localize the different phosphorylation sites.
Experimental Section
Generation of Tryptic Cleavage Sites
Upon digestion with trypsin or other common enzymes, the DivIVA protein could not be cleaved into suitable peptides. Experiments were therefore performed with limited digests using Proteinase K, but the results were not reproducible and still did not cover the complete sequence. Therefore, two artificial cleavage sites for trypsin were introduced into the 7.2 kDa peptide region. Two glutamine residues (Q343 and Q360, see Figure 4) were chosen for mutagenesis to arginine residues. These two residues lie in a low-complexity region that is predicted to be disordered, according to the DISOPRED2 Disorder Prediction Server, suggesting that these changes would have minimal structural effects. In brief, divIVA was amplified with primers KF478 (ctggttaacccatatggactacaaggacgacgatgacaagatgccgttgacccccgaggac) and KF86 (ggtcgacggcgagacggtca), which introduced an N-terminal FLAG-tag and cloned in pUC19. Site-directed mutagenesis was performed to introduce the two additional tryptic cleavage sites using primers AH13 (ggtcgatgggcggcggcccgggc)/AH14 (ggttgccgccgtaggacggagc) and AH15 (ggcagatgtcgcccgcgatgacc)/AH16 (gctggccgccgtaggacggacc). The FLAG-divIVA (Q343R Q360R) allele was subsequently subcloned into pIJ10550, which is a derivative of the vector pIJ69025 in which the apr resistance gene has been exchanged for the viomycin resistance gene vio (Hempel, unpublished). The FLAG-divIVA (Q343R Q360R) allele is placed directly downstream of a thiostrepton-inducible promoter tipAp. The modified DivIVA protein was expressed in S. coelicolor and immunoprecipitated after the cultures were exposed to bacitracin to induce high-level DivIVA phosphorylation.1
Mass Spectrometric Analysis of Phosphorylation Sites
Protein samples were digested with TPCK Immobilized Magnetic Trypsin (Clontec, Saint-Germain-en-Laye, France) for 60 or 90 min. The digestions were stopped by removing the magnetic beads and addition of TFA to a final concentration of 0.5%. Sample aliquots were analyzed by LC–MS/MS on an LTQ-ORBITRAP mass spectrometer (Thermo Fisher, Hemel Hempstead, U.K.) coupled to a nanoAcquity (Waters, Manchester, U.K.) UPLC-system using a 25 cm long column (BEH C18, 1.7 μm, 75 μm × 250 mm, Waters). For detection and analysis of phosphopeptides, multistage activation was used with neutral loss m/z of 48.99 and 32.66 (for 2+ and 3+ charged ions). Targeted LC–MS/MS was performed using an inclusion list for just the m/z values of the phosphopeptides of interest using the most abundant charge state (2+ or 3+) observed in the previous experiments. Data acquisition was performed without chromatographic peak detection and without dynamic exclusion. In this way, multiple MS2 spectra were acquired across any eluting peaks of the correct mass. A detailed description of the MS procedures can be found in the Supporting Information Methods.
Data Processing
Raw files were processed with MaxQuant version 1.3.0.5 (http://maxquant.org)6 to generate recalibrated peaklist-files that were used for a database search using an in-house Mascot 2.4 Server (Matrix Science Limited, London, U.K.). Mascot-mgf files were generated from MaxQuant apl-files using a perl script written in-house (available on request). MaxQuant output was also used for quantification of peptide abundances and retention times.
Mascot searches were performed on the Streptomyces coelicolor protein sequences downloaded from Uniprot (www.uniprot.org, 8093 sequences, 04042013) to which the mutated DivIVA sequence had been added, or for simplifying searches on a small database containing the target sequences (wild-type and mutated DivIVA) in a background of 100 random E. coli sequences using 5 ppm precursor tolerance, 0.5 Da fragment tolerance, and oxidation (M) as well as phosphorylation (STY) as variable modifications. Mascot search results were imported into Scaffold 3.6.3 including an X!Tandem search for evaluation and further into ScaffoldPTM (version 2.0, proteomesoftware.com, Portland, OR) for analysis of phosphorylation sites using the Ascore algorithm implemented in ScaffoldPTM.
A Perl script was developed searching for scans in the MS2 peak list file (MaxQuant/apl and Mascot Distiller/mgf peak list formats are supported) in which the mass of the fragment ion of interest is present, allowing for a user-defined tolerance window. It retrieves the corresponding intensity that can then be plotted against the scan number.
Data Sets Available
The mass spectrometry proteomics data (including the Perl script for plotting of fragments) have been deposited with the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository7 with the data set identifier PXD000095 and DOI 10.6019/PXD000095 (PRIDE accessions 29619–29621).
Results and Discussion
Standard LC–MS/MS for Peptide Identification
Initially, two runs were performed with different tryptic digests of a DivIVA protein preparation using a data-dependent (top 5) mode of acquisition including dynamic exclusion and chromatographic peak detection. The data were processed and merged in one Mascot search. When searched on the complete S. coelicolor Uniprot database, the DivIVA protein was identified with high significance as a top match with the highest score and the highest emPAI among 37 S. coelicolor proteins. (For details, see the Data Sets Available section.) At an FDR of 0.85%, 37 peptides (including modified peptides) were assigned to DivIVA, resulting in a sequence coverage of 48% and a Mascot protein score of 9121. All three tryptic peptides generated as a result of the introduced tryptic cleavage sites were detected with high confidence. The assigned peptides included three potential phosphopeptides.
All further searches were performed on a small custom database containing the target sequences (wild-type and mutated DivIVA) in a background of 100 random E. coli sequences. On the basis of this, this initial experiment identified three peptides (numbered 1 to 3 according to their occurrence in the protein sequence, see Figure 4) carrying potentially phosphorylated residues (Supporting Information Table 1). Peptide 1 (QLETQADDSLAPPR) is not derived from the 7.2 kDa fragment but lies immediately N-terminal to it in the primary amino acid sequence of DivIVA. Peptide 2 (TPATASLPPSPAPSMAPAGASAPSYGGNR) and Peptide 3 (SMGGGPGQSGPSYGGQR) were derived from the parental 7.2 kDa fragment and were generated as a consequence of the two introduced tryptic cleavage sites. All three peptides were also detected in their unphosphorylated form (see later).
The Mascot search results for Peptide 1 showed that both residues T304 or S309 can be phosphorylated. Localization probabilities of ≥99% (based on Mascot Delta Score) were observed for the eight spectra detected. Three of them indicated phosphorylation of T304, and four spectra pointed to S309 with scan numbers indicating separate elution of those two isomers.
Three MS2 spectra were obtained for Peptide 2, all with probabilities below 50% (including some rank 2 to 4 matches). Spectra with the highest probabilities (∼48%) pointed to S338 or Y339 as phosphorylated residues.
For Peptide 3, a total of seven matching spectra were obtained in Mascot, and for two of those spectra Mascot gave probabilities around 99%, indicating unambiguous localization for phosphorylation at S344. All other spectra gave probabilities in the range of 40–80% (including some rank 2 to 4 matches) pointing to all potential sites.
This is a typical situation in the analysis of post-translational modifications requiring special targeted analysis combined with user inspection of details of spectra. To accomplish this detailed analysis, we chose targeted LC–MS/MS (as described for phosphopeptides previously)8 to acquire as many MS2 spectra of each peptide as possible and have developed a script allowing for the detection and plotting of characteristic fragment ions present in all acquired spectra (in silico SRM).
Software Tool for Visualization of Site-Determining MS2 ions from Targeted LC–MS/MS
To confirm and improve the initial results described above and to determine if multiple phosphopeptide isomers were present (especially for Peptides 2 and 3), we performed targeted LC–MS/MS using an inclusion list for just the m/z values of the phosphopeptides of interest using the most abundant charge state (2+ or 3+) observed in the previous experiments. Data acquisition was performed without chromatographic peak detection and without dynamic exclusion. In this way, multiple MS2 spectra were acquired across any eluting peaks of the correct mass. Data from two independent runs of different tryptic digests were processed as described above, and the merged Mascot search results were imported into Scaffold 3.6.3 (including an X!Tandem search) and further into ScaffoldPTM 2.0 (Proteome Software) for evaluation of the phosphorylation sites using the Ascore4 probabilistic algorithm and scoring technique incorporated into ScaffoldPTM (Supporting Information, Tables Peptide 1 to Peptide 3).
Starting from the initial results (see previous), we were mainly interested in the targeted analysis of Peptide 3 (SMGGGPGQSGPSYGGQR) with an m/z = 830.3333 for [M+2H]2+ of the monophosphorylated version. From this peptide, 89 spectra with localization probabilities ≥95% were obtained after ScaffoldPTM analysis. For 49 of these spectra, the phosphorylation site was assigned by Ascore to S355, and for 40 spectra the site was assigned to S344. On the basis of the scan numbers the two groups seemed to elute at slightly different but overlapping retention times. However, because of the large number of spectra and the lack of intensity values, this was not straightforward to analyze, leaving the software results still difficult to interpret.
At this stage, one option would have been to optimize the LC-gradient for better separation of possible isomers. However, this would need to be done specifically for each peptide/isomer, and it is therefore not a practical option, especially with a high-throughput nanoUPLC system, limited sample amounts, and complex samples containing many phosphopeptides of interest.
Therefore, we developed a tool to analyze the elution of specific fragment ions of the peptide isomers. Spectra were inspected visually and compared with the theoretical fragment ion series to find specific site-determining fragment ions for the two potential phosphorylation sites. For the potential S355 isomer, the y8-ion was unique, and for the S344 isomer, the y9-ion was unique, and their intensities were plotted against retention time (Figure 1) using a Perl script written in house. The script searches for scans in the MS2 peak list file in which the mass of the fragment ion of interest is present and retrieves the corresponding intensity, allowing for plotting the elution of those fragments over retention time/scan number. Because the analysis was performed in the ion trap (IT) of the LTQ Orbitrap, a tolerance window of 0.5 Da was used. The correct assignment of the site-determining ions could clearly be further improved using instruments, allowing for fast high-accuracy mass measurements and smaller tolerance windows.
Figure 1.
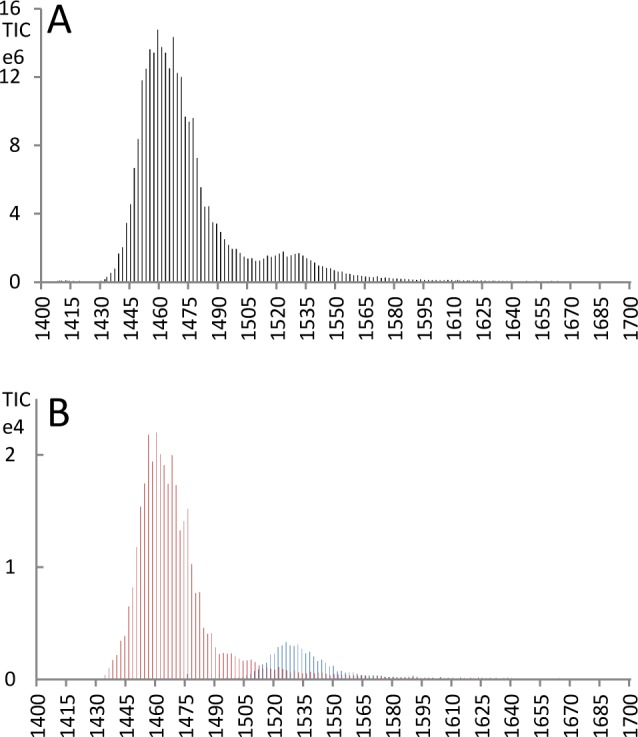
(A) XIC of m/z = 830.3327 corresponding to monophosphorylated doubly charged Peptide 3 (SMGGGPGQSGPSYGGQR) generated in Qual Browser/Xcalibur 2.1 software (Thermo) and exported for scan-based plotting. (B) Scan-based plot of the elution of the y8-ion (m/z = 901.36) of the S355 isomer (red) and the y9-ion (m/z = 908.42) of the S344 isomer (blue) generated with the in-house developed tool (mass tolerance 0.5 Da). The elution of the two fragment ions coincides exactly with the two peaks of the parent phosphopeptide (A).
Extracting these two ions clearly deconvolutes the spectra to a main peak and an overlapping shoulder matching the XIC of the parent ion. It shows that the S355 isomer is present under the main peak and the S344 isomer elutes under the shoulder of that peak (Figure 1). The maxima of the peaks are separated by ∼30 s. The phosphorylation of S355 is strongly supported by the phosphosite prediction tool NetPhos 2.0 (http://www.cbs.dtu.dk/services/NetPhos/); a probability of 0.961 is predicted for S355 in the wild-type DivIVA sequence. Residue S344 is immediately adjacent to the residue modified to generate the tryptic cleavage site. NetPhos predicts 0.238 for phosphorylation of QS344 (wild-type) and 0.577 for the modified sequence motif RS344. Another prediction tool, KinasePhos2 (http://kinasephos2.mbc.nctu.edu.tw/index.html), predicting probabilities for specific kinases finds increased phosphorylation probability of RS344 for only one specific kinase (Aurora related kinase), while six other kinases remain virtually unchanged and two are reduced (data not shown). It is formally possible that the phosphorylation observed on residue S344 might arise as an artifact of the adjacent Q343R substitution. However, a triply phosphorylated species of the naturally occurring 7.2 kDa peptide was observed in previous MALDI-TOF analysis,1 supporting the phosphorylation of S344 in addition to the other two residues (S355, S338) reported in this paper. (A summary of the localization of peptides and phosphorylation sites is given in Figure 4.)
For Peptide 2 (TPATASLPPSPAPSMAPAGASAPSYGGNR, m/z = 921.0932 for [M+3H]3+ of monophosphorylated version), 70 spectra were acquired in total (data not shown). After ScaffoldPTM analysis, only 19 spectra were left with localization probabilities ≥75%. Eleven of those spectra had probabilities ≥95% (including one spectrum with 100% probability), assigning the phosphorylation site to S338. In addition, there were eight spectra with Ascore scores >0 and probabilities in the range of 75–92% pointing to Y339 (Supporting Information Tables). No special separation in retention time (scan numbers) seemed to occur for the two potential sites.
For more detailed analysis, the elution profiles of two y-ions, y5 and y7, specific for phosphorylation of S338, were analyzed using the method described above for Peptide 3. The y6-ion specific for S338 phosphorylation was not present, but the y5-ion excludes phosphorylation of Y339, and if y5 is present, then the y7-ion is specific for S338 phosphorylation and excludes all other potential residues of the peptide.
Notably, the spectra were dominated by y-ions. Further, all of the site-determining ions selected were y-ions, avoiding the potential dangers of sequence scrambling associated with the cyclization of b-ions.9
As shown in Figure 2A, the monophosphorylated, triply charged peptide eluted under a single broad peak. The y7-ion occurred in 64 of 70 acquired spectra and eluted exactly under the XIC peak of the peptide (Figure 2B). The y-5 ion occurred in 29 of the 70 spectra, and eluted in the same range as the XIC peak and the y5-ion (Figure 2C). These results support the finding that phosphorylation occurs at residue S338. This is also supported by the phosphosite prediction tool NetPhos 2.0, which predicts a higher probability for S338 (0.979) than for Y339 (0.838).
Figure 2.

(A) XIC (generated as in Figure 1A) of m/z = 921.0932 corresponding to the monophosphorylated, triply charged version of Peptide 2 (TPATASLPPSPAPSMAPAGASAPSYGGNR). (B,C) Plot of the elution of the y7-ion (m/z = 830.32) occurring in 64 out of 70 spectra (B) and the y5-ion (m/z = 566.27) occurring in 29 out of 70 spectra (C) generated as in Figure 1B. The scan numbers of the detected y-ions are in the same range in both plots and coincide with the scan numbers of the XIC in panel A.
For Peptide 1, QLETQADDSLAPPR (m/z = 810.8669 for [M+2H]2+ of monophosphorylated version), 27 MS2 spectra with 100% localization probability were obtained after ScaffoldPTM analysis, confirming the initial Mascot results. For 24 spectra the phosphorylation site was assigned to S309, while in the 3 remaining spectra the site was assigned to T304. An XIC showed the occurrence of two peaks of the expected m/z separated by 3 min of retention time (Figure 3). After assigning the identified scans to the retention time it became clear that the S309 isomer eluted earlier than the T304 isomer. The phosphorylation probabilities predicted by NetPhos were 0.93 for S309 and 0.256 for T304.
Figure 3.

XIC of m/z = 810.8669 corresponding to the monophosphorylated, doubly charged version of Peptide 3 (QLETQADDSLAPPR) generated in Qual Browser/Xcalibur 2.1 software (Thermo) showing the elution of two phosphopeptide isomers separated by 3 min. Numbers on top of peaks indicate scan numbers allowing for the assignment of QLETQADDpSLAPPR to the first peak and QLEpTQADDSLAPPR to the second peak.
Unphosphorylated and Phosphorylated Phosphopeptide Isomers
The separation of the two isomers of Peptide 1 is much more distinct (3 min) compared with the isomers of Peptide 3 (30 s). Similar behavior of different phosphopeptide isomers, including overlapping elution, has been previously described.8,10 In our study the unphosphorylated versions of phospho-Peptides 1 to 3 have also been detected. (For details, see the Data Sets Available section). Table 1 shows a comparison of abundance and retention time of the unphosphorylated and phosphorylated isomer versions of the peptides. The values were extracted from the MaxQuant output (allpeptides.txt), and the results indicate that high levels of phosphorylation occurred after bacitracin induction, confirming previous observations based on Pro-Q Diamond staining.1 The rate was in the range of 38–48% for Peptides 2 and 3, while only ∼0.6% was observed for Peptide 1. Phosphorylation predominantly causes a clear reduction of elution time, although a slight increase can also occur.10 As shown in Table 1, all but one of the five observed phosphopeptide isomers had an enhanced retention time with differences in the range of +0.6 to +3 min. The highest increase was observed for peptide QLEpTQADDSLAPPR. The only reduction was observed for peptide QLETQADDpSLAPPR.
Table 1. Comparison of Intensities and Retention Times (RT, min) of Nonphosphorylated Peptides and Their Phosphorylated Counterparts (Isomers).
| peptide | phosphopeptide isomer Sequence | intensitya | ratio (P) | RTa |
|---|---|---|---|---|
| Peptide 1 | QLETQADDSLAPPR | 4.63 × 109 | 28.31 | |
| Peptide 1 | QLETQADDpSLAPPR | 2.64 × 107 | 0.005699 | 28.03 |
| Peptide 1 | QLEpTQADDSLAPPR | 2.67 × 106 | 0.000577 | 31.27 |
| Peptide 2 | TPATASLPPSPAPSMAPAGASAPSYGGNR | 1.92 × 107 | 33.02 | |
| Peptide 2 | TPATASLPPSPAPSMAPAGASAPpSYGGNR | 7.28 × 106 | 0.37996 | 33.63 |
| Peptide 3 | SMGGGPGQSGPSYGGQR | 7.91 × 108 | 22.04 | |
| Peptide 3 | SMGGGPGQSGPpSYGGQR | 3.82 × 108 | 0.48319 | 22.80 |
| Peptide 3 | pSMGGGPGQSGPSYGGQR | nd | 0.06b | 23.25b |
Values were extracted from the MaxQuant output table allpeptides.txt. Intensities were calculated as the sum and retention times as the average from two independent runs including both 2+ and 3+ charged peptides.
Peak of the pSMGGGPGQSGPSYGGQR isomer of Peptide 3 was not detected in MaxQuant; instead the values were estimated from the XIC generated in Xcalibur (see Figure 1).
Conclusions
DivIVA is a cytoskeletal protein that plays a key role in directing polar growth and branching in the filamentous bacteria Streptomyces. The activity of this polarity protein is controlled by the Ser/Thr kinase AfsK, which extensively phosphorylates DivIVA on its C-terminus.1 Here we define the phosphorylation sites in DivIVA. Because of poor fragmentation and closely eluting phosphopeptide isomers the exact localization of phosphorylation sites can be challenging, and results from automated software tools are often ambiguous. To overcome this problem we first collected a large number of MS2 spectra for the peptides of interest by using an inclusion list and repeated MS fragmentation. We then developed and applied a script to extract and plot the retention time of site-determining fragment ions. Plots of different ions were aligned with the XIC of the parent ion, demonstrating the differential elution of phosphopeptide isomers. This method retrieves data from the mass spectrometry acquisition and visualizes information otherwise difficult to recognize. It is limited only by the acquired data and clearly increases the confidence in otherwise ambiguous results. Using this method, the phosphorylation sites in the DivIVA protein were determined. Our results demonstrate that DivIVA is phosphorylated in vivo on 5 out of 16 potential phosphorylation sites in the C-terminal part of the protein (T304, S309, S338, S344, and S355). Defining these sites means that in the future they can be mutagenised to unphosphorylatable (Ala) or phosphomimetic (Asp) residues to permit a detailed analysis of the effects of phosphorylation on DivIVA function.
Acknowledgments
This study was supported by BBSRC Institute Strategic Programme Grant BB/J004561/1, by the John Innes Foundation, and by a John Innes Centre PhD studentship to A.M.H. The assistance of the PRIDE Team with depositing the data with the ProteomeXchange Consortium is acknowledged.
Supporting Information Available
Methods for the analysis of phosphorylation sites in the DivIVA protein and supporting tables. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ A.M.H.: Biozentrum, University of Basel, Klingelbergstrasse 50/70, 4056 Basel, Switzerland.
Author Present Address
∥ M.J.N.: Proteomics & Mass Spectrometry Facility, Donald Danforth Plant Science Center, 975 North Warson Road, St. Louis, MO 63132, United States.
Author Contributions
⊥ G.S. and A.M.H. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Hempel A. M.; Cantlay S.; Molle V.; Wang S.-B.; Naldrett M. J.; Parker J. L.; Richards D. M.; Jung Y. G.; Buttner M. J.; Flärdh K. The Ser/Thr protein kinase AfsK regulates polar growth and hyphal branching in the filamentous bacteria Streptomyces. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, E2371–E2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards D. M.; Hempel A. M.; Flärdh K.; Buttner M. J.; Howard M. Mechanistic basis of branch-site selection in filamentous bacteria. PLoS Comp. Biol. 2012, 8, e1002423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flärdh K.; Richards D. M.; Hempel A. M.; Howard M.; Buttner M. J. Regulation of apical growth and hyphal branching in Streptomyces. Current Opin. Microbiol. 2012, 15, 737–743. [DOI] [PubMed] [Google Scholar]
- Beausoleil S. A.; Villén J.; Gerber S. A.; Rush J.; Gygi S. P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, 1285–1292. [DOI] [PubMed] [Google Scholar]
- Huang J.; Shi J.; Molle V.; Sohlberg B.; Weaver D.; Bibb M. J.; Karoonuthaisiri N.; Lih C. J.; Kao C. M.; Buttner M. J.; Cohen S. N. Cross-regulation among disparate antibiotic biosynthetic pathways of Streptomyces coelicolor. Mol. Microbiol. 2005, 58, 1276–1287. [DOI] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Côté R.; Reisinger F.; Barsnes H.; Foster J. M.; Rameseder J.; Hermjakob H.; Martens L. The Proteomics Identifications database: 2010 update. Nucleic Acids Res. 2010, 38, D736–D742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courcelles M.; Bridon G.; Lemieux S.; Thibault P. Occurrence and detection of phosphopeptide isomers in large-scale phosphoproteomics experiments. J. Proteome Res. 2012, 11, 3753–3765. [DOI] [PubMed] [Google Scholar]
- Harrison A. G. To b or not to b: the ongoing saga of peptide b ions. Mass Spectrom. Rev. 2009, 28, 640–654. [DOI] [PubMed] [Google Scholar]
- Kim J.; Petritis K.; Shen Y.; Camp D. G. II; Moore R. J.; Smith R. D. Phosphopeptide elution times in reversed-phase liquid chromatography. J. Chromatogr., A 2007, 1172, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.