

Abstract
Cytochromes P450 (CYPs) represent a diverse group of heme-thiolate proteins found in almost all organisms. CYPs share a common protein fold but differ in substrate selectivity and catalyze a wide variety of monooxygenation reactions via activation of molecular oxygen. Among 57 human P450s, the 3A4 isoform (CYP3A4) is the most abundant and the most important because it metabolizes the majority of the administered drugs. A remarkable feature of CYP3A4 is its extreme promiscuity in substrate specificity and cooperative substrate binding, which often leads to undesirable drug-drug interactions and toxic side effects. Owing to its importance in drug development and therapy, CYP3A4 has been the most extensively studied mammalian P450. In this review we provide an overview on recent progress and remaining problems in the CYP3A4 research.
1. Introduction
Cytochromes P450 (CYPs) are a superfamily of heme enzymes found across all kingdoms of life.1 Based on amino acid sequence similarity, CYPs are arranged into families (>40% identity) and subfamilies (>55% identity). Presently, there are over 270 different CYP families with more than 12,000 individual genes.2 CYPs participate in vital life processes by oxidizing endogenous or exogenous compounds (e.g., drugs, chemicals, pollutants, steroids, bile and fatty acids, vitamin D and other natural products and xenobiotics). Prokaryotic CYPs are soluble proteins, whereas eukaryotic counterparts are attached to the endoplasmic reticulum or inner mitochondrial membrane. Most CYPs require two electrons, delivered by a flavin- or [2Fe-2S]-cluster-containing redox partner, and molecular oxygen per catalytic cycle. The general P450 monooxygenation cycle is complex and schematically shown in Figure 1. Briefly, binding of a substrate to the low-spin ferric enzyme leads to displacement of water as the sixth ligand to the heme iron and a high spin shift. The substrate-bound complex has a greater reduction potential and more easily accepts an electron from the redox partner. After the first reduction, the heme iron binds oxygen, forming an oxy-heme complex. This complex receives the second electron from the redox partner and, after two subsequent protonations and heterolysis of the O-O bond, an oxygenated product is formed and the heme returns to the resting low-spin state.
Figure 1.
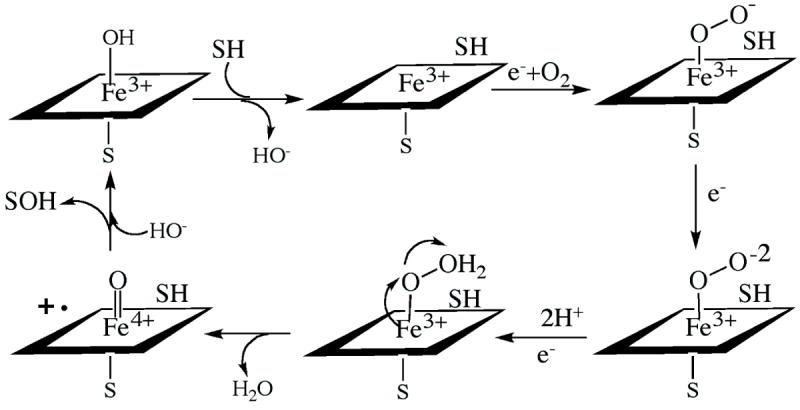
Cytochrome P450 catalytic cycle.
Humans have 57 CYP genes and 33 pseudogenes arranged into 18 families and 42 subfamilies.3 The 3A4 isoform (CYP3A4) is the most abundant and is mainly expressed in the liver and gastrointestinal tract. CYP3A4 is highly promiscuous in substrate specificity and can accommodate and metabolize compounds diverse in size and chemical structure. This allows CYP3A4 to oxidize over half of all administered drugs.4 Through fast degradation, CYP3A4 can lower bioavailability and therapeutic efficiency of pharmaceuticals and vice versa, drug plasma levels can be increased if CYP3A4 is inhibited. In addition to detoxification and clearance of xenobiotics, CYP3A4 exerts genotoxic effects via activation of procarcinogens,5 and contributes to adverse drug-drug interactions because of the ability to accommodate in the active site two or more molecules of the same or different type. Owing to these important properties, CYP3A4 is of practical interest to the pharmaceutical industry and has been studied more extensively than any other human P450 isoform. There is a number of excellent reviews on general properties and substrate cooperativity in CYP3A4.6-12 Here we will discuss only recently published data with an emphasis on the structural aspects that extend our views on this remarkable enzyme.
2. Substrate binding and cooperativity in CYP3A4
One of the earliest observations on CYP3A4 was atypical (non-Michaelis-Menten) steady state kinetics with testosterone, progesterone, α-naphthoflavone and a few other substrates, manifested as a sigmoidal or biphasic plot of reaction velocity vs. substrate concentration.4 It is widely accepted now that such allosteric behavior is due to simultaneous binding of more than one substrate molecule within/near the active site of CYP3A4 that can either stimulate or inhibit turnover. The key residues that influence substrate binding, cooperativity and regioselectivity of metabolism were first identified via site-directed mutagenesis based on a three-dimensional homology model of CYP3A4 and include Phe108, Ser119, Ile120, Leu211, Asp214, Ile301, Phe304, Ala305, Thr309, Ala370 and Leu373.6,13-15 These studies suggested that the substrate and effector binding sites are adjacent and lay within a large cavity. Determination of the first x- ray structures of CYP3A416,17 showed that most of the predicted residues indeed do comprise the active site or the substrate access channel (Fig. 2).
Figure 2.
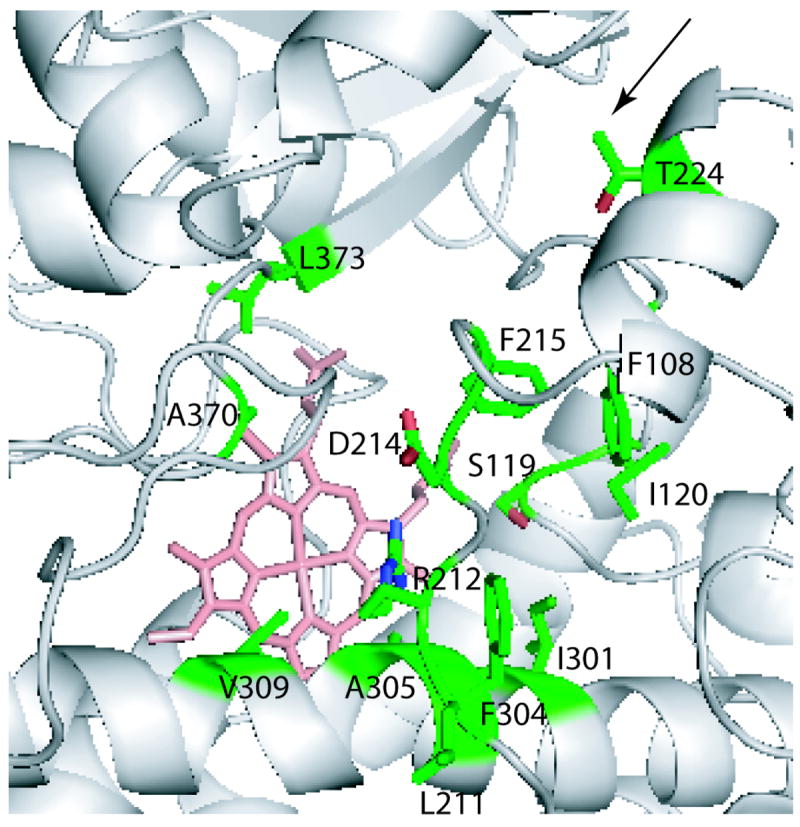
CYP3A4 residues experimentally shown to influence substrate binding, cooperativity and regioselectivity of metabolism. All residues except Leu211 are located in the active site or the substrate access channel (indicated by an arrow). Leu211 is a surface residue adjacent to the Phe-cluster.
Substrate binding to CYP3A4 has been extensively studied by a variety of techniques,18 leading to the following conclusions: (i) substrates are not locked in the CYP3A4 active site and may dissociate and rebind during different stages of the catalytic cycle; (ii) owing to a loose fit, the binding affinity and turnover of one substrate can be modulated by another molecule present in the active site; (iii) substrate association may involve a step that does not induce changes in the heme Soret spectrum and thus cannot be detected by absorbance spectroscopy (‘absorbance-silent’); and (iv) before moving into the active site cavity, CYP3A4 ligands may interact with a peripheral binding site. Still under debate is whether or not the main reason for atypical kinetics is multiple substrate binding19,20 or the pre- existence of kinetically different conformers21-23 (‘induced fit’ vs. ‘conformational selection’ mechanism).
The number of molecules simultaneously bound to an enzyme and exhibiting negative or positive cooperativity is traditionally estimated based on the Hill coefficient (nH) derived from steady-state kinetics or equilibrium substrate binding titrations. The nH values reported for various CYP3A4 substrates never exceed 2.0, including those for testosterone. 4,24 However, there is experimental data suggesting that three testosterone molecules can simultaneously bind to CYP3A4.20,25 More recently, this was confirmed by global analysis of equilibrium substrate binding and steady-state kinetics of NADPH consumption during CYP3A4-dependent testosterone hydroxylation.26 This study found that only binding of the second testosterone molecule leads to significant product formation, whereas the third binding event improves the coupling efficiency between electron consumption and metabolite formation. In contrast, electron paramagnetic resonance analysis of the ligand-dependent ferric spin state equilibrium in CYP3A427 limited the testosterone stoichiometry to 2:1 and indicated that the first testosterone molecule binds with a higher affinity but has a negligible effect on the spin equilibrium (a non-productive mode) and makes binding of the second testosterone less energetically favorable. The second binding event, however, increases the probability of generating the high spin form (positive cooperativity) although the high spin shift is not complete. Interestingly, the coupling free energies estimated in the latter study strongly disfavored not only the second testosterone binding but also ligation of the water molecule to the heme.
Another approach, a ‘ligand exclusion’ method, was used to accurately estimate the stoichiometry of 7-benzyloxyquinoline (7BQ) binding.28 Investigation of the effects of several large inhibitors on the Hill coefficient for 7BQ debenzylation has shown that at least four 7BQ molecules can simultaneously bind to CYP3A4 (nH = 3.7 in the presence of bromoergocryptine). According to computer modeling, no structural rearrangement is required to place three molecules of 7BQ in the core of the active site and two molecules in the substrate access channel of the 1W0E ligand-free structure.28
Longitudinal T1 nuclear magnetic resonance relaxation and molecular docking were utilized to probe the structural basis for heterotropic cooperativity between midazolam and carbamazepine.29 The results indicated that two midazolam molecules or one midazolam and one carbamazepine molecule can adopt a stacked configuration within the CYP3A4 active site. Cooperativity of ligand binding through direct interaction between the stacked molecules was proposed to be a common motif for homotropic and heterotropic cooperativity in P450s. The same laboratory investigated allosteric effects on branch points within the CYP3A4 catalyzed sequential metabolism of a tertiary amine by isotope dilution analysis.30 Nile Red was used as a fluorescent ligand and substrate, and α-naphthoflavone as a heterotropic activator. It was found that α-naphthoflavone not only accelerates the sequential metabolism of Nile Red and increases the rates of its two metabolites formation, but also perturbs the metabolite ratio by affecting the substrate off rate and coupling efficiency. These observations indicate that α-naphthoflavone and possibly other heterotropic activators can exert allosteric effects by modulating specific branch points in the P450 cycle.
There is more experimental evidence for the existence of a peripheral ligand binding site as well. Our group investigated the kinetics of CYP3A4 association with bromoergocryptine (BEC), a large substrate that does not display binding cooperativity, by monitoring a low-to-high spin conversion in the heme iron.31 The reaction was biphasic, with a non-hyperbolic dependence of kfast vs. [BEC], where the kfast value did not change at sub-equimolar substrate concentrations but increased proportionally when the CYP3A4:BEC ratio exceeded unity. This was consistent with a three step binding model proposed earlier based on the fluorescence studies.32 According to this model, BEC binds first outside at the peripheral site without perturbing the heme spectrum and then translocates into the catalytic cavity. A similar non-hyperbolic dependence of the binding rate constant vs. ligand concentration was observed for the inhibitor ritonavir.33 The fact that the break in the V-shaped plot of kfast vs. [ritonavir] was also observed when the CYP3A4:BEC ratio reached unity led us to hypothesize that the large and highly hydrophobic ritonavir molecule also binds first at the peripheral docking area and, after it becomes saturated, enters the active site.
The x-ray structure of the CYP3A4-progesterone complex17 provides a glimpse at what this peripheral docking site may look like. In this crystal structure, progesterone is bound in a surface cleft above the phenylalanine cluster, a unique feature of CYP3A4 (Fig. 3A). The substrate molecule is well ordered, has low B-factors (indicative of low thermal motion), and forms stacking interactions with Phe219 and a hydrogen bond with Asp214. Furthermore, it is positioned close to Leu211 and Phe213 which together with Asp214 are implicated in progesterone-dependent cooperativity. Such a mode of interaction implies that the progesterone binding site is not a crystallization artifact but rather represents a docking area where this and other molecules could bind prior to translocation into the catalytic cavity.17
Figure 3.
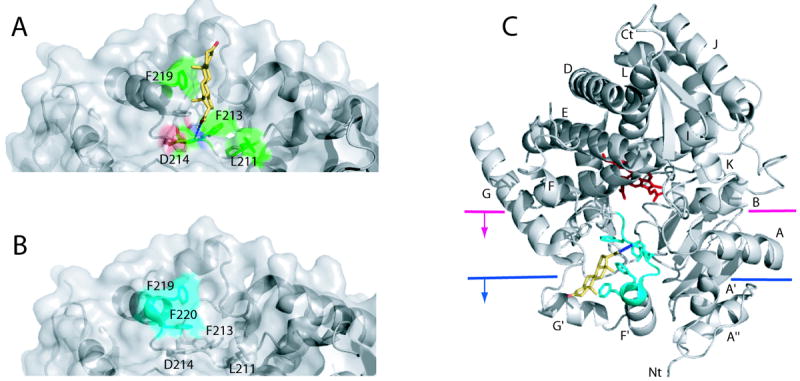
Peripheral ligand binding site in CYP3A4. A, A progesterone binding site identified in the 1WOF crystal structure.17 The progesterone molecule (shown in yellow) is located above the Phe-cluster (side chains are displayed) and forms a hydrogen bond with the amide nitrogen of Asp214 (depicted as a dotted line) and hydrophobic interactions with Leu211, Phe213 and Phe219 (shown in green). B, A high affinity binding site for Fluorol-7GA identified with FRET34 is located on the surface near residues 217-220 (shown in cyan) and overlaps with the progesterone binding site. C, A cartoon diagram of CYP3A4 with two possible levels of incorporation into the membrane bilayer.35,36 The heme is shown in red sticks, progesterone in yellow, and the progesterone/Fluorol-7GA binding site in cyan.
A fluorescence resonance energy transfer (FRET) technique was utilized to more precisely locate the peripheral ligand binding site in CYP3A4.34 Differences in the binding affinity of a model substrate, Fluorol-7GA, were measured by FRET from a thiol-reactive fluorescent probe, pyrene iodoacetamide, position of which was varied via specific attachment to native and genetically introduced surface cysteine residues. Data analysis suggested that (i) CYP3A4 has at least two widely separated binding sites for Fluorol-7GA, (ii) the first substrate binding event takes place outside of the active site pocket, and (iii) the high affinity peripheral site is located near residues 217-220 (Fig. 3B). Since association of Fluorol-7GA with the peripheral site is accompanied by a high spin shift, the identified area is postulated to represent a putative allosteric site, binding to which leads to a conformational change necessary for the substrate binding in the active site.
The fact that the progesterone and Fluorol-7GA binding sites overlap (compare Figs. 3A and B) does not necessarily mean that this surface area represents a specific substrate/effector binding site. The 214-220 stretch comprising the F’ helix is part of the F-G region predicted to be embedded into the membrane in native CYP3A4 (Fig. 3C). 35,36 According to molecular dynamics simulations, large-scale movements in the F-F’-G’-G helices occur upon interaction with the lipid bilayer.36 These movements allow opening and closing of the substrate access channel which, in turn, is necessary for substrate binding and product release. Such conformational dynamics and low substrate specificity of CYP3A4 argue against the possibility that the 214-220 fragment serves as a specific site designed to interact with a limited number of substrate or effector molecules. It is more likely that this area nonspecifically associates with and helps to direct hydrophobic compounds (that may or may not be metabolized by the enzyme) from the membrane into the active site, which would enable CYP3A4 to effectively sample and, when possible, oxidize and neutralize toxins to which the organism has been exposed. Such complexity may simply be the consequence of the requirement that CYP3A4 be a promiscuous enzyme with a larger and flexible active site that enables interactions with substrates far from the heme. Alternatively, functional cooperativity without specific molecular recognition could provide a detoxification advantage.37 At low to moderate substrate concentrations of a toxic substrate, homotropic allosteric control ensures a slow production of toxic products, whereas at high concentrations a more rapid turnover eliminates the toxic substrate more rapidly. This, of course, comes at the cost of more toxic product but is basically the best the organism can do when presented with large amounts of a toxic substrate.
It also should be mentioned that based on a comparative analysis of all available crystal structures of CYPs, it was suggested that the acquisition of the F’-G’ insertion by eukaryotic P450s and their incorporation into the membrane led to selective repositioning of the beta-domain.36 Compared to the soluble bacterial counterparts, the beta-domain in eukaryotic CYPs is shifted toward the heme plane and, as a result, the heme A propionate side chain orients towards the proximal side of the heme plane (Fig. 4). This extends the volume of the catalytic cavity and opens a substrate access/product escape channel directly connected to the lipid bilayer.
Figure 4.
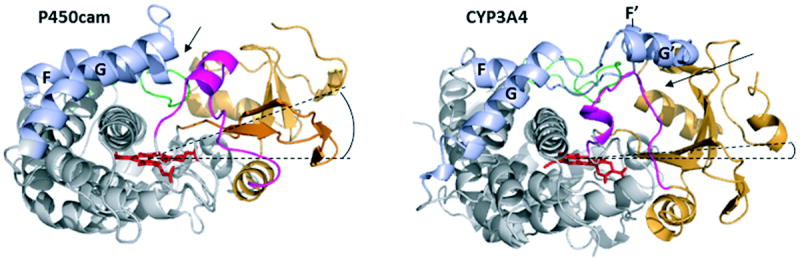
Comparison of the x-ray structures of soluble bacterial CYP101 (P450cam, PDB ID 1DZ4) and human CYP3A4 (1TQN). The beta-domain is depicted in orange, the B-B’ loop and B’ helix in magenta, the F, F’, G’ and G helices and connecting loops in light blue, the C-terminal loop in green, and the heme cofactor in red sticks. Dashed lines pass through the heme plane and the center of the beta-domain. The F’-G’ helix/loop insertion in membrane-bound mammalian CYPs shifts the beta-domain toward the heme plane36 (compare angles between the dashed lines), which opens a channel located between the B-B’ loop and the β1 and β3 sheets of the beta-domain (shown by an arrow). In contrast, in CYP101 and other soluble P450s substrates access the active site primarily through a channel formed by the F-G loop and B’ helix (indicated by an arrow).
3. Crystal structures of CYP3A4 with substrates bound to the active site
In addition to the CYP3A4-progesterone complex17 discussed above, two more crystal structures of CYP3A4 have been determined where a substrate is bound to the active site. The x-ray model of the CYP3A4-erythromycin complex was determined to 2.75 Å resolution (2JOD).38 Erythromycin is a macrolide antibiotic and a weak type I ligand of CYP3A4 (Kd = 125 μM, Fig. 5A) that induces <20% of a high spin shift.24,39 The drug is metabolized by N-demethylation of the D-desosamine group, which is positioned 17 Å from the heme in the crystal structure (Fig. 5B). This implies that erythromycin is bound to CYP3A4 in a non-productive orientation. There are no specific polar interactions between the erythromycin molecule and the protein, but a close proximity of four phenylalanine side chains suggests that the complex is partially stabilized via hydrophobic interactions. Residual electron density observed in the active site was considered as an indication of different binding modes for erythromycin.38 The low affinity and loose binding may in part explain why erythromycin was absent in the active site of the second CYP3A4 molecule in the 2JOD asymmetric unit38 and in the 1TQN structure.16
Figure 5.
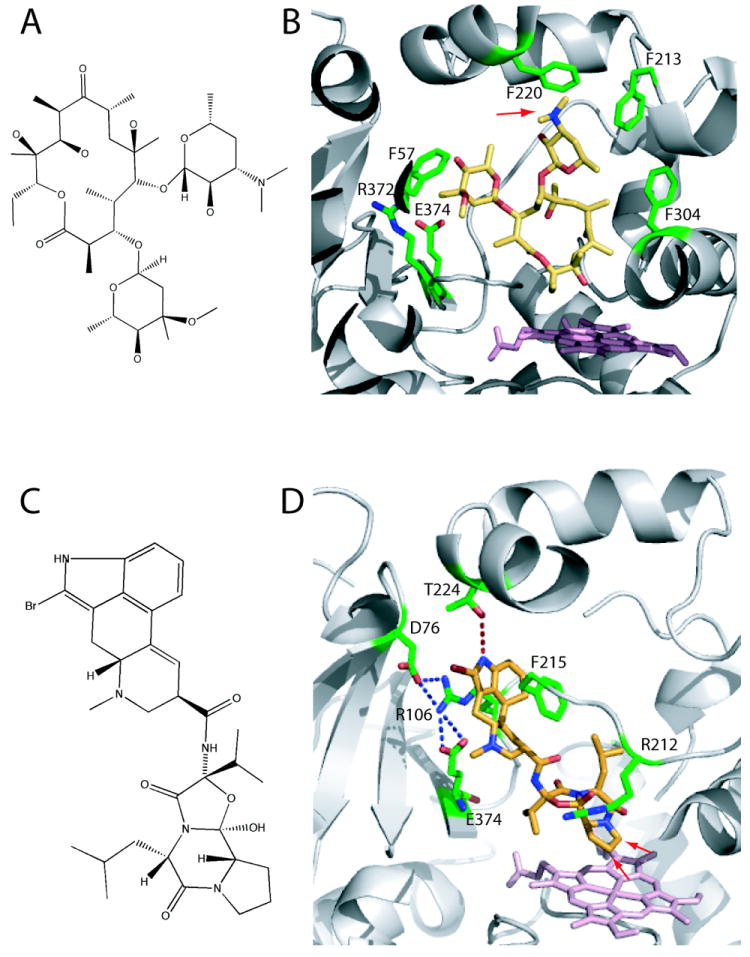
A, Chemical structure of erythromycin. B, Erythromycin bound to the active site of CYP3A4 (2JOD).38 The heme is in pink and residues within the van der Waals distance are in green. Erythromycin is bound in a non-productive mode because its demethylation site (indicated by an arrow) is 17 Å away from the heme. C, Chemical structure of BEC. D, BEC bound to the active site of CYP3A4 in a productive mode (3UA1 structure).31 The tripeptide moiety of BEC approaches the heme, with the two primary oxidations sites (indicated by arrows) being ~ 4 Å away from the heme iron. To allow BEC to access the heme, Arg212 adopts a different conformer. The lysergic group is sandwiched between the side chains of Arg106 and Phe215, and is hydrogen bonded to Thr224.
Besides low affinity (Kd for typical CYP3A4 substrates is within the 5-150 μM range), poor aqueous solubility is another factor that impedes crystallization of the functional enzyme-substrate complexes. We conducted trials with a number of substrates but succeeded only in co-crystallization of CYP3A4 with BEC. BEC is an ergot alkaloid that consists of lysergic acid and a proline-containing cyclic tripeptide connected via an amide bond (Fig. 5C). The drug acts as a dopamine receptor agonist and binds to CYP3A4 with a high affinity (Kd of 0.4 μM) causing a near complete low-to-high spin shift. The most important feature of the crystallographic CYP3A4-BEC complex, determined to 2.15 Å resolution,31 is that BEC is oriented in a productive mode. The 8’ and 9’ carbons of the proline ring, the primary and secondary sites of oxidation, are closest to the heme and only 3.7-4.1 Å away from the iron (Fig. 5D). The tripeptide group establishes multiple van der Waals contacts and is next to Arg212, which shifts aside to allow access to the heme. The lysergic moiety is sandwiched between the side chains of Arg106 and Phe215 and hydrogen bonded to Thr224 via the N1 atom. Side directed mutagenesis showed that both Arg212 and Thr224 define affinity and facilitate BEC binding,31 which further proves the biological relevance of the x-ray model. Based on the structural and experimental data, it was hypothesized that interaction of the lysergic group with Thr224 directs the tripeptide moiety toward the heme, while the following conformational readjustments in Arg212 modulate spin equilibrium.
Despite the large size (655 Da) and complex chemical structure (Fig. 5C), BEC fits into the active site without significant structural perturbations. This is in contrast to the CYP3A4-erythromycin complex, where conformational rearrangements in the F-F’-G’-G helix-loop region, I-helix and the C-terminal loop take place.38 Such dissimilarity might be due to an elongated shape and a higher conformational flexibility of the BEC molecule. The amide bond allows rotational movements of the lysergic and tripeptide groups, giving rise to a number of BEC conformers that can enter the active site. As the x-ray structure demonstrated (Fig. 5D), one of the conformers binds to CYP3A4 tightly and in a mode suitable for oxidation.
The CYP3A4-BEC structure supports the resonance Raman spectroscopy data obtained on a nanodisk-incorporated CYP3A4.40 The low frequency resonance Raman spectra of ferric substrate-free CYP3A4 exhibit several features associated with the ‘propionate bending’ and ‘vinyl bending’ modes in the heme prosthetic group. BEC binding causes spectral changes consistent with the out-of-plane macrocycle distortion and heme peripheral group disposition. A comparison of the ligand-free and BEC-bound structures showed that, indeed, association of BEC with CYP3A4 leads to distortions in the heme plane and vinyl group conformation.31
4. Interaction of CYP3A4 with inhibitors
Chemicals that inhibit P450 catalysis are usually type II ligands that cause a red shift in the Soret band (from ~416 to 420-424 nm) upon direct ligation to the heme iron via nitrogen provided by imidazole, pyridine or primary amino groups. Such interaction lowers the redox potential of P450 and prevents both an electron flow from a redox partner and oxygen binding. Comparison of the absorbance spectra of substrate-free and ligand-bound P450 can provide some insights into the manner of the type II ligand binding (e.g., estimate dissociation constants and the percentage of a low spin shift). While the absolute P450 spectra mostly depend on the electronic properties of the nitrogen ligand, the difference spectra were shown to be more sensitive to the steric properties of the ligand.41
Many commonly used drugs can act as moderate-to-potent inhibitors of CYP3A4 but not necessarily through nitrogen ligation. For example, CYP3A4 can be inhibited by macrolide antibiotics, antidepressants, steroids and several other types of drugs via reversible or irreversible binding and mechanism-based inhibition.42 CYP3A4 inactivation frequently causes unfavorable drug-drug interactions and might lead to altered drug disposition, efficacy or toxicity. Here we will primarily focus on the mechanism of CYP3A4 interaction with two most potent inhibitors, ketoconazole and ritonavir.
4.1 Ketoconazole
Ketoconazole is a widely used antifungal drug (Fig. 6A) that ligates to the P450 heme via the azole nitrogen. Ketoconazole is not highly selective and in addition to CYP3A4 inhibits several other isoforms from the 1B, 2B and 2C family.43 The crystal structure of the ketoconazole-bound CYP3A4 has been determined to 2.8 Å resolution (2VOM).38 The most interesting feature of this structure is the presence of two drug molecules bound to the active site in an antiparallel tandem configuration (Fig. 6B). To accommodate two large ketoconazole molecules, the protein undergoes a significant conformational change, especially in the F-G region and around the Phe-cluster (residues 213, 215, 219, 220, 241 and 304). Positioning of the I-helix and the C-terminal loop are also altered. The crystallographic complex is stabilized through hydrogen bonding between the keto groups of both ketoconazole molecules and the Ser119 and Arg372 side chains, as well as via π-stacking interactions between the chlorobenzyl ring of the ligated drug molecule and the Phe-304 ring (Fig. 6B).
Figure 6.
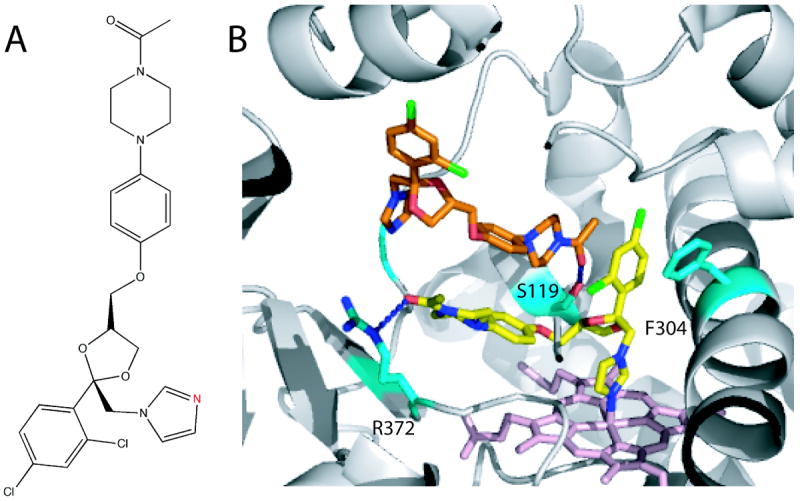
A, Chemical structure of ketoconazole. The nitrogen atom through which ketoconazole ligates to the heme is shown in red. B, Two ketoconazole molecules bound to the active of CYP3A4 in an antiparallel tandem fashion (2VOM structure).38 The ligating ketoconazole (yellow) is hydrogen bonded to Arg372 and forms π-stacking interactions with Phe304. The non-ligating ketoconazole (orange) is hydrogen bonded to Ser119.
The 2VOM structure raised questions regarding the plausibility of the observed ketoconazole binding mode and the chain of events that could lead to such an orientation. Recent biochemical and molecular dynamics (MD) simulation studies addressed these problems and clarified the mechanism of drug association. A surface plasmon resonance analysis44 resolved two kinetically different binding events for ketoconazole, likely reflecting association in a slowly dissociating inhibitory mode (the ligating azole group in) and a catalytically productive mode (the non-ligating group in). The pre-steady state kinetics of ketoconazole binding to CYP3A4, in turn, showed that this process is complex, displays cooperativity (nH = 1.4), involves at least three consecutive steps, and likely starts with an association to a peripheral site.45 Although the latter kinetic data were consistent with the presence of a single molecule in the active site, the possibility of the multiple ligand binding could not be ruled out. To explore the physicochemical origin for the positive cooperativity in ketoconazole binding, MD simulations in conjunction with free-energy calculations have been conducted.46 The computational study established that shape complementarity through nonpolar van der Waals interactions is the main driving force for the ketoconazole binding, and that association of the first drug molecule increases affinity for the binding of the second molecule by 5 kJ mol-1, which explains the experimentally observed cooperativity. Each simulated CYP3A4 system (ligand-free, ketoconazole 1- or ketoconazole 2-bound) was capable of binding the inhibitor in only a few selected conformations, thereby supporting the conformational selection mechanism. Further, although electrostatic interactions disfavor the overall binding process, CYP3A4 might be electrostatically preorganized to bind ketoconazole in a heme-ligating position regardless of the occupancy of the active site, which could contribute to the tight binding. Taken together, the experimental and theoretical studies suggest that the CYP3A4-ketoconazole structure is not a crystallization artifact but reflects a biologically relevant enzyme-inhibitor complex.
4.2 Ritonavir
Ritonavir (Fig. 7A) is a peptidomimetic drug originally designed as an HIV protease inhibitor,47 but it can also act as a potent and specific inactivator of the CYP3A subfamily48 and, in particular, CYP3A4.49 Owing to its strong CYP3A4 inhibiting ability, ritonavir is currently administered as a booster for enhancement of pharmacokinetics of other HIV protease inhibitors that otherwise are quickly metabolized by CYP3A4.48
Figure 7.
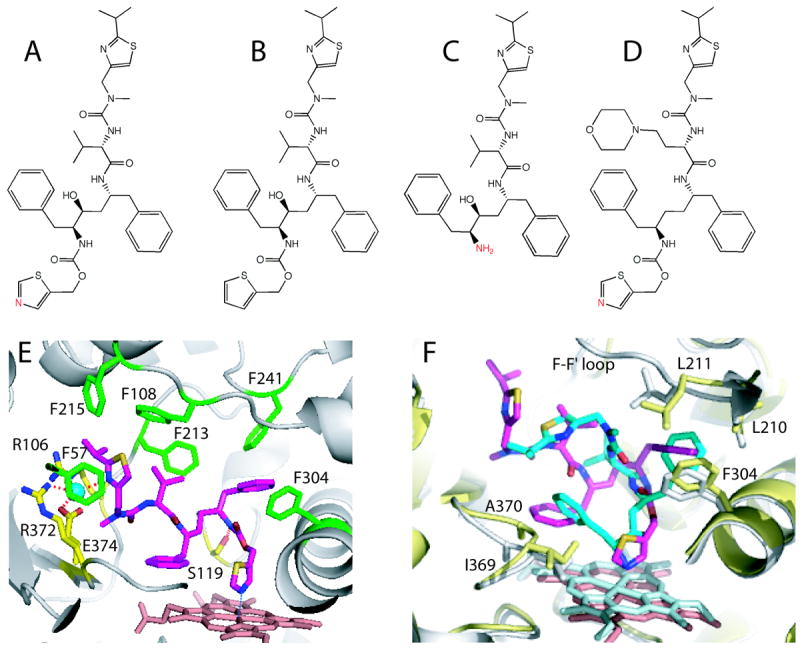
A-D, Chemical structures of ritonavir, deaza-ritonavir, DTMCR, and cobicistat, respectively. The heme-ligating primary amino group and thiazole nitrogens are shown in red. E, Ritonavir (magenta) bound to the active site of CYP3A4 (3NXU structure).54 Phenylalanine residues surrounding ritonavir are in green, whereas residues comprising the polar ‘umbrella’, a cluster of charged residues connected to the isopropyl moiety via a water molecule (cyan sphere), and the H-bond forming Ser119 are in yellow. F, Superposition of the ritonavir-(magenta, pink and gray) and DTMCR-bound (cyan, light cyan and yellow; 3TJS)33 structures of CYP3A4. To optimize hydrophobic interactions via phenyl side groups, DTMCR rotates by 180° relative to ritonavir. Since DTMCR is shorter than ritonavir, it cannot interact with the polar ‘umbrella’ and F-F’ loop. As a result, the F-F’ loop becomes disordered and the active site is solvent accessible in the DTMCR-bound structure. Also, DTMCR has differently oriented phenyl groups and does not clash with the 369-370 peptide. This is in contrast to the CYP3A4-ritonavir complex, where the heme shifts downwards and the Fe-N bond is slightly elongated because of steric hindrance with the 369-370 peptide.
Until recently, ritonavir was thought to be a mechanism-based inhibitor of CYP3A449,50 or act as a competitive, noncompetitive or mixed competitive-noncompetitive inactivator. 51-53 In our study on recombinant CYP3A4,54 we showed that ritonavir is a type II ligand that inhibits catalysis via strong, irreversible thiazole nitrogen ligation and by lowering the heme redox potential which, in turn, precludes an electron flux from a redox partner, cytochrome P450 reductase (CPR). Owing to the tight binding, the drug can easily replace other molecules (substrates and inhibitors) bound to the active site.33,54 The kinetics of CYP3A4-ritonavir association was later reexamined using a wider range of inhibitor:CYP3A4 ratios.33 As mentioned in section 2, the V-shaped dependence of kfast vs. ritonavir concentration was attributed to the association of ritonavir with a peripheral binding site prior to translocating into the active site pocket. This type of interaction may contribute to the biphasicity of the CYP3A4-ritonavir binding reaction and explain a large difference between the equilibrium and kinetic dissociation constants observed previously (50 and 840 m, respectively).54
The x-ray structure of the CYP3A4-ritonavir complex was solved to 2.0 Å, the highest resolution for a ligand-bound CYP3A4 reported thus far.54 The drug fits well into the active site pocket, establishes extensive hydrophobic and van der Waals interactions with the K-β loop region and Phe-cluster via the side groups, and is fully sequestered from solvent (Fig. 7E). In addition, ritonavir is anchored to the active site via a direct hydrogen bond with Ser119 and a water-mediated bridge between the terminal isopropyl thiazole group and polar ‘umbrella’ (residues Tyr53, Asp61, Asp36, Arg106, Arg372, and Glu374). The ritonavir-bound structure is most similar to the CYP3A4-ketoconazole complex, in that it has a similar bending in the I-helix and the F-F’ loop conformation, with Arg212 facing outside.38 In the CYP3A4-ritonavir complex, however, the F-F’ loop conformation is stabilized by a hydrogen bond between the Arg212 guanidinium group and the carbonyl oxygen of Lys208. Site-directed mutagenesis showed that the R212A mutation has no significant effect on the affinity or amplitude of spectral changes induced by ritonavir but decreases the limiting binding rate by 20%.33 This suggests that Arg212 is not critical for the inhibitor association but may assist the binding process by stabilizing the optimal conformer.
4.3 Ritonavir analogs
To elucidate the relative importance of non-bonded interactions during ritonavir association, we investigated CYP3A4 interaction with two analogs, deaza-ritonavir and desthiazolylmethyloxycarbonyl ritonavir (DTMCR), that lack the thiazole nitrogen and the entire thiazole group, respectively (Fig. 7B and C).33 Comparative analyses showed that elimination of the heme-ligating nitrogen converts ritonavir to a type I ligand and prevents co-crystallization with CYP3A4. DTMCR, on the other hand, ligates to the heme via the terminal primary amino group with a 70-fold lower affinity than ritonavir but is retained in the active site during crystallization. The 2.25 Å crystal structure of the CYP3A4-DTMCR complex revealed that, in order to optimize interactions with the protein and place the phenyl rings into hydrophobic pockets, DTMCR rotates by 180° (Fig. 7F). It was concluded, therefore, that association of ritonavir-like molecules is driven by heme coordination, whereas hydrophobic interactions provided by the side groups define the binding mode. Furthermore, comparison of the ritonavir- and DTMCR-bound structures showed (Fig. 7F) that the fit of ritonavir to the CYP3A4 active site can be further improved. In particular, replacement of the phenyl ring closest to Ala370 with a smaller hydrophobic moiety would eliminate steric clashing and strengthen the N-Fe bond and, consequently, could increase the inhibitory potency of the drug.
One highly undesirable side effect of prolonged use of ritonavir is development of drug resistance in HIV. Cobicistat is a new pharmacoenhancer that does not have such an adverse effect.55 This ritonavir-like compound lacks the hydroxyl group required for the HIV protease binding and has a morpholine ring instead of the valine side group (Fig. 7D). As a result, cobicistat is devoid of any anti-HIV activity but can selectively and potently inhibit CYP3A4 (Ki of 0.9 μM in hepatic microsomes vs. 0.3 μM for ritonavir). Other advantages of the new drug are reduced capability for adverse drug interactions, improved tolerability, and higher aqueous solubility.55
4.4 Seco-pancratistatin analogs
Derivatives of amaryllidaceae alkaloids represent a novel class of CYP3A4 inhibitors.56 Since some amaryllidaceae constituents possess the CYP3A4 inhibitory activity while others do not, it was possible to identify the pharmacophore responsible for the interaction with CYP3A4 and, through structure-activity studies, synthesize compounds that inhibit CYP3A4 as potently as ketoconazole and ritonavir.57,58 Notably, neither of the two most potent seco-pancratistatin analogs (Ki of 30-70 nM for recombinant CYP3A4) has a nitrogen atom that can ligate to the heme (Fig. 8). It is of great interest, therefore, to elucidate the inhibitory mechanism and the interaction mode of these compounds.
Figure 8.
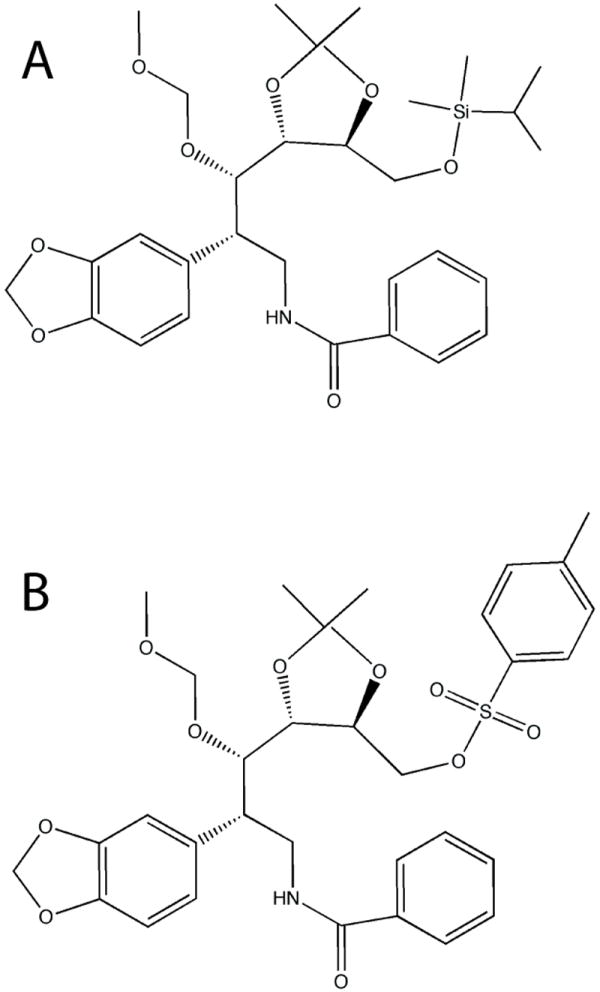
Seco-pancrastatin analogs. Compounds A and B inhibit CYP3A4 in vitro with Ki of 30 and 70 nM, respectively,57,58 via an unknown mechanism.
Considering recent advances in structure/function studies on CYP3A4, it is highly possible that ritonavir analogs and other types of inhibitors with a more favorable balance of physicochemical and pharmacochemical properties could be developed.
5. Computational studies on CYP3A4
Determination of the CYP3A4 x-ray structures triggered a number of theoretical studies that provided deeper insights into the catalytic mechanism and substrate/inhibitor binding, and made possible a more accurate prediction of small-molecule association to CYP3A4.59-68 The first MD simulations were conducted by Park et al.59 to clarify the roles of solvent dynamics and the active site loops in the ligand binding process and heme iron coordination. They showed that the malleability of the F-F’ loop (residues 211-218) defines the broad substrate specificity of CYP3A4, and that its complex with the inhibitor metyrapone is stabilized not only via direct coordination to the heme iron but also through a hydrogen bond between the metyrapone carbonyl oxygen and the Ser119 hydroxyl. The latter interaction is not observed in the crystal structure of metyrapone-bound CYP3A4 (1WOG)17 but quickly forms (within 10 ps) and remains stable during the entire simulation course. The side chain of Ser119 also is predicted to form an H-bonding interaction with the carbonyl oxygen (O20) of progesterone. The computational studies, thus, identify Ser119 as an important active site residue that can stabilize the inhibitor and substrate binding via a hydrogen bond formation.
Fishelovitch et al.62 performed MD simulations to understand factors that determine cooperative binding and carbon abstraction in diazepam. Cooperative binding of diazepam could be observed in the 1WOE but not 1TQN conformer because of a different positioning of Arg212. It was suggested that when Arg212 faces the active site (1TQN structure) it may strongly interact with and lower accessibility of Phe304, predicted to play a key role in cooperative binding. According to the simulations, addition of the effector molecule decreases the flexibility of Phe304 and its surroundings, and shortens the distance between the site of hydrogen abstraction and the oxygen atom of the catalytic oxyferryl species (compound I). In the following study, the same group explored features of compound I using quantum mechanics (QM)/ molecular mechanics calculations on CYP3A4 models containing one or two diazepam molecules in the active site.61 The major finding was that in the absence of a substrate compound I is destabilized, in that it has an elongated Fe-S bond and the radical localizes on sulfur, which disfavors substrate oxidation. In contrast, when the effector molecule is present, it positions the substrate close to compound I, which stabilizes the Fe-S bond and prevents the sulfur radical formation, thereby giving preference to substrate oxidation. Again, the compound I stabilization by the effector molecule was observed only in the 1WOE conformer.
Several investigations were aimed at identifying preferred solvent and substrate access/exit channels in CYP3A4 and associated gating mechanisms. Using MD and steered MD simulations, Fishelovitch et al.63 identified six egress pathways for two small product molecules, temazepam and testosterone-6βOH, and concluded that the product shape and orientation determine the preferred exit channel. The two investigated products, for instance, preferably exit through the channel formed by the B-C loop rather than a putative substrate channel, the largest channel located between the F-F’ loop and the β1 sheet (channels 2e and 2a, respectively, according to classification of Cojocaru et al).69 Notably, in most of the identified channels, the gate keepers are one or two π-stacked phenylalanine residues, whose hydrophobic interactions need to be broken to allow the product to pass the gate. Metyrapone also exits from the CYP3A4 active site via channel 2e, as it requires only a small rupture force and small displacement of residues.70 Ketoconazole molecules, on the other hand, can use both 2a and 2e channels as egress routes, with Ser119 being a critical bottleneck residue.65
MD simulations also clarified the role of CPR in activating water channels in CYP3A4.64 Passing of water molecules via the channel located on the proximal side of the heme was found to be gated by Arg375, a conserved residue forming a salt bridge with one of the heme propionates. This channel region overlaps with the CPR binding site. MD simulations showed that the presence of the FMN domain, the electron donating moiety of CPR, promotes expulsion of water molecules from the active site and assists the substrate binding. The CPR-mediated control of the proximal solvent channel is thought to be adopted by other P450 isoforms as well, because such a mechanism could regulate the proton delivery and productivity of the catalytic cycle.
Computer simulation and modeling studies have also been undertaken to predict and analyze sites of oxidation in a number of CYP3A4 substrates. Shahrokh et al.68 used QM calculations and MD simulations with improved heme parameters to probe the reactivity of 4-hydroxy-tamoxifen (4OHT), a primary metabolite of the anti-cancer drug tamoxifen, for dehydrogenation and to generate representative CYP3A4 and substrate configurations. This approach helped to identify the 4OHT binding modes consistent with the in vitro metabolism. Since the R212A mutant produced half of the dehydrogenated product as wild type, Arg212 was proposed to be a key residue that affects 4OHT dehydrogenation by imposing both steric and electrostatic inhibition of the rebound process, but only when it is oriented toward the active site. Thus, the 1TQN but not 1WOE conformer could produce the catalytically productive mode favoring the 4OHT dehydrogenation. Another requirement for the 4OHT dehydrogenation reaction was the opening of solvent channel 4, formed by the F-G loop, which is closed in the starting 1TQN model.
In contrast, the 1WOE ligand-free model was more useful for predicting the sites of metabolism in raloxifene,66 whereas the ritonavir-bound 3NXU structure was the only one suitable for docking of a large anti-obesity drug dirlotapide in a catalytically relevant orientation.71 According to the latter study, the ligand-induced conformational change is a determining factor for prediction of sites of metabolism and molecular interactions during structure-based lead optimization. It was concluded also that the binding configuration and chemical reactivity are equally important for catalysis of bigger molecules, and that enthalpic and entropic energies of the substrate-bound complexes can influence the flexibility of the P450 active site.
To more accurately predict small molecule-binding to CYP3A4, Lill et al.60 developed a computational model that combines flexible docking with multidimensional quantitative structure–activity relationship (QSAR) technique. An inhibitory potential for 48 structurally diverse compounds was evaluated based on the scoring functions that included the terms for hydrophobicity, hydrogen-bonding, topological adaptation, and entropy change. Analysis of the energetic contributions suggested that in addition to hydrophobic interactions, which dominated for most of the investigated compounds, induced protein fit and water-bridged protein-ligand contacts were also essential for small-molecule binding. The developed method allows screening various compounds not only for the CYP3A4 binding probability but also for possible toxic metabolites and drug-drug interactions, which is critical for the drug development process. Finally, it was reported recently that the inhibitor binding to CYP3A4 can be predicted fairly well based on the total binding free energies calculated for the inhibitor-heme model complexes.67
6. Concluding remarks
Determination of the first x-ray structures of CYP3A4 was a major breakthrough in understanding the enzyme’s function. These structures provided the first insights into the architecture of the active site, identified flexible elements, and gave clues on how the ligand binding and allosteric regulation may occur. The more recent, higher resolution crystallographic data gave a clearer picture of the ligand-associated CYP3A4 and revealed the first productive mode for the substrate binding. Utilization of advanced biophysical techniques has expanded our views on the mechanism of homotropic and heterotropic cooperativity, and raised the possibility that substrate association and catalysis can be regulated both from within and outside of the active site. Advances in theoretical studies, in turn, helped to create better models for computational calculations and develop techniques for predicting the binding ability and inhibitory potency of known and hypothetical drugs.
However, the mechanistic basis for the atypical kinetics of substrate oxidation by CYP3A4 is still a matter of debate. Currently, there is experimental evidence supporting both the ligand-induced cooperativity and conformational selection mechanisms. While more attention is now being paid to the peripheral ligand binding site(s), it remains unclear whether this is a putative effector binding site or a nonspecific ligand-docking area, from which hydrophobic molecules could be transported into the active site for possible metabolism. The exact location of the effector binding site(s) within the catalytic cavity also remains unknown, and the co-substrate-activator structures of CYP3A4 are yet to be determined. Another remaining question is the relative role of hydrophilic and polar interactions in the ligand binding. Although the dominance of hydrophobic forces in the CYP3A4-substrate/inhibitor association has been well documented and is widely accepted, recent studies suggest that polar contacts provided by Ser119, Arg212, and Thr224 may also be crucial. This field needs to be further explored to unravel and fully understand all mechanisms controlling the substrate and inhibitor binding in CYP3A4, which is vital for rationalization and more accurate prediction of protein-drug and drug-drug interactions.
Acknowledgments
Financial support from the United States National Institutes of Health (Grant GM33688) and the California Center for Antiviral Drug Discovery is gratefully acknowledged.
Abbreviations
- 4OHT
4-hydroxy-tamoxifen
- 7BQ
7-benzyloxyquinoline
- BEC
Bromoergocryptine
- CPR
Cytochrome P450 reductase
- CYP
Cytochrome P450
- CYP3A4
Cytochrome P450, 3A4 isoform
- FRET
Fluorescence resonance energy transfer
- QM
Quantum mechanics
References
- 1.Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. Kluwer Academic/Plenum Publishers; New York: 2005. [Google Scholar]
- 2.Nelson DR, Nebert DW. eLS. 2011 [Google Scholar]
- 3.Nebert DW, Russell DW. Lancet. 2002;360:1155–1162. doi: 10.1016/S0140-6736(02)11203-7. [DOI] [PubMed] [Google Scholar]
- 4.Guengerich FP. Annu Rev Pharmacol Toxicol. 1999;39:1–17. doi: 10.1146/annurev.pharmtox.39.1.1. [DOI] [PubMed] [Google Scholar]
- 5.Rendic S, Guengerich FP. Chem Res Toxicol. 2012;25:1316–1383. doi: 10.1021/tx300132k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szklarz GD, Halpert JR. Drug Metab Dispos. 1998;26:1179–1184. [PubMed] [Google Scholar]
- 7.Tang W, Stearns RA. Curr Drug Metab. 2001;2:185–198. doi: 10.2174/1389200013338658. [DOI] [PubMed] [Google Scholar]
- 8.Atkins WM. Annu Rev Pharmacol Toxicol. 2005;45:291–310. doi: 10.1146/annurev.pharmtox.45.120403.100004. [DOI] [PubMed] [Google Scholar]
- 9.Niwa T, Murayama N, Yamazaki H. Curr Drug Metab. 2008;9:453–462. doi: 10.2174/138920008784746364. [DOI] [PubMed] [Google Scholar]
- 10.Sligar SG, Denisov IG. Drug Metab Rev. 2007;39:567–579. doi: 10.1080/03602530701498521. [DOI] [PubMed] [Google Scholar]
- 11.Denisov IG, Frank DJ, Sligar SG. Pharmacol Ther. 2009;124:151–167. doi: 10.1016/j.pharmthera.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denisov IG, Sligar SG. Arch Biochem Biophys. 2012;519:91–102. doi: 10.1016/j.abb.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fowler SM, Riley RJ, Pritchard MP, Sutcliffe MJ, Friedberg T, Wolf CR. Biochemistry. 2000;39:4406–4414. doi: 10.1021/bi992372u. [DOI] [PubMed] [Google Scholar]
- 14.Fowler SM, Taylor JM, Friedberg T, Wolf CR, Riley RJ. Drug Metab Dispos. 2002;30:452–456. doi: 10.1124/dmd.30.4.452. [DOI] [PubMed] [Google Scholar]
- 15.Khan KK, He YQ, Domanski TL, Halpert JR. Mol Pharmacol. 2002;61:495–506. doi: 10.1124/mol.61.3.495. [DOI] [PubMed] [Google Scholar]
- 16.Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF. J Biol Chem. 2004;279:38091–38094. doi: 10.1074/jbc.C400293200. [DOI] [PubMed] [Google Scholar]
- 17.Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- 18.Isin EM, Guengerich FP. Anal Bioanal Chem. 2008;392:1019–1030. doi: 10.1007/s00216-008-2244-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harlow GR, Halpert JR. Proc Natl Acad Sci U S A. 1998;95:6636–6641. doi: 10.1073/pnas.95.12.6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He YA, Roussel F, Halpert JR. Arch Biochem Biophys. 2003;409:92–101. doi: 10.1016/s0003-9861(02)00484-8. [DOI] [PubMed] [Google Scholar]
- 21.Koley AP, Buters JT, Robinson RC, Markowitz A, Friedman FK. J Biol Chem. 1997;272:3149–3152. doi: 10.1074/jbc.272.6.3149. [DOI] [PubMed] [Google Scholar]
- 22.Atkins WM, Wang RW, Lu AY. Chem Res Toxicol. 2001;14:338–347. doi: 10.1021/tx0002132. [DOI] [PubMed] [Google Scholar]
- 23.Davydov DR, Halpert JR, Renaud JP, Hui Bon Hoa G. Biochem Biophys Res Commun. 2003;312:121–130. doi: 10.1016/j.bbrc.2003.09.247. [DOI] [PubMed] [Google Scholar]
- 24.Farooq Y, Roberts GC. Biochem J. 432:485–493. doi: 10.1042/BJ20100744. [DOI] [PubMed] [Google Scholar]
- 25.Baas BJ, Denisov IG, Sligar SG. Arch Biochem Biophys. 2004;430:218–228. doi: 10.1016/j.abb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 26.Denisov IG, Baas BJ, Grinkova YV, Sligar SG. J Biol Chem. 2007;282:7066–7076. doi: 10.1074/jbc.M609589200. [DOI] [PubMed] [Google Scholar]
- 27.Roberts AG, Campbell AP, Atkins WM. Biochemistry. 2005;44:1353–1366. doi: 10.1021/bi0481390. [DOI] [PubMed] [Google Scholar]
- 28.Kapelyukh Y, Paine MJ, Marechal JD, Sutcliffe MJ, Wolf CR, Roberts GC. Drug Metab Dispos. 2008;36:2136–2144. doi: 10.1124/dmd.108.021733. [DOI] [PubMed] [Google Scholar]
- 29.Roberts AG, Yang J, Halpert JR, Nelson SD, Thummel KT, Atkins WM. Biochemistry. 2011;50:10804–10818. doi: 10.1021/bi200924t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woods CM, Fernandez C, Kunze KL, Atkins WM. Biochemistry. 2011;50:10041–10051. doi: 10.1021/bi2013454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sevrioukova IF, Poulos TL. J Biol Chem. 2012;287:3510–3517. doi: 10.1074/jbc.M111.317081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Isin EM, Guengerich FP. J Biol Chem. 2006;281:9127–9136. doi: 10.1074/jbc.M511375200. [DOI] [PubMed] [Google Scholar]
- 33.Sevrioukova IF, Poulos TL. Arch Biochem Biophys. 2012;520:108–116. doi: 10.1016/j.abb.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davydov DR, Rumfeld JAO, Sineva EV, Fernando H, Davydova NY, Halpert JR. J Biol Chem. 2012;287:6797–6809. doi: 10.1074/jbc.M111.325654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao Y, White MA, Muralidhara BK, Sun L, Halpert JR, Stout CD. J Biol Chem. 2006;281:5973–5981. doi: 10.1074/jbc.M511464200. [DOI] [PubMed] [Google Scholar]
- 36.Denisov IG, Shih AY, Sligar SG. J Inorg Biochem. 2012;108:150–158. doi: 10.1016/j.jinorgbio.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Atkins WM, Lu WD, Cook DL. J Biol Chem. 2002;277:33258–33266. doi: 10.1074/jbc.M204425200. [DOI] [PubMed] [Google Scholar]
- 38.Ekroos M, Sjogren T. Proc Natl Acad Sci U S A. 2006;103:13682–13687. doi: 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Das A, Grinkova YV, Sligar SG. J Am Chem Soc. 2007;129:13778–13779. doi: 10.1021/ja074864x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mak PJ, Denisov IG, Grinkova YV, Sligar SG, Kincaid JR. J Am Chem Soc. 2011;133:1357–1366. doi: 10.1021/ja105869p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Locuson CW, Hutzler JM, Tracy TS. Drug Metab Dispos. 2007;35:614–622. doi: 10.1124/dmd.106.012609. [DOI] [PubMed] [Google Scholar]
- 42.Zhou SF. Curr Drug Metab. 2008;9:310–322. doi: 10.2174/138920008784220664. [DOI] [PubMed] [Google Scholar]
- 43.von Moltke LL, Greenblatt DJ, Schmider J, Wright CE, Harmatz JS, Shader RI. Biochem Pharmacol. 1998;55:113–122. doi: 10.1016/s0006-2952(97)00239-6. [DOI] [PubMed] [Google Scholar]
- 44.Pearson JT, Hill JJ, Swank J, Isoherranen N, Kunze KL, Atkins WM. Biochemistry. 2006;45:6341–6353. doi: 10.1021/bi0600042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Isin EM, Guengerich FP. J Biol Chem. 2007;282:6863–6874. doi: 10.1074/jbc.M610346200. [DOI] [PubMed] [Google Scholar]
- 46.Bren U, Oostenbrink C. J Chem Inf Model. 2012;52:1573–1582. doi: 10.1021/ci300118x. [DOI] [PubMed] [Google Scholar]
- 47.Kempf DJ, Marsh KC, Denissen JF, McDonald E, Vasavanonda S, Flentge CA, Green BE, Fino L, Park CH, Kong XP, et al. Proc Natl Acad Sci U S A. 1995;92:2484–2488. doi: 10.1073/pnas.92.7.2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kempf DJ, Marsh KC, Kumar G, Rodrigues AD, Denissen JF, McDonald E, Kukulka MJ, Hsu A, Granneman GR, Baroldi PA, Sun E, Pizzuti D, Plattner JJ, Norbeck DW, Leonard JM. Antimicrob Agents Chemother. 1997;41:654–660. doi: 10.1128/aac.41.3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koudriakova T, Iatsimirskaia E, Utkin I, Gangl E, Vouros P, Storozhuk E, Orza D, Marinina J, Gerber N. Drug Metab Dispos. 1998;26:552–561. [PubMed] [Google Scholar]
- 50.von Moltke LL, Durol AL, Duan SX, Greenblatt DJ. Eur J Clin Pharmacol. 2000;56:259–261. doi: 10.1007/s002280000125. [DOI] [PubMed] [Google Scholar]
- 51.Iribarne C, Berthou F, Carlhant D, Dreano Y, Picart D, Lohezic F, Riche C. Drug Metab Dispos. 1998;26:257–260. [PubMed] [Google Scholar]
- 52.Eagling VA, Back DJ, Barry MG. Br J Clin Pharmacol. 1997;44:190–194. doi: 10.1046/j.1365-2125.1997.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zalma A, von Moltke LL, Granda BW, Harmatz JS, Shader RI, Greenblatt DJ. Biol Psychiatry. 2000;47:655–661. doi: 10.1016/s0006-3223(99)00176-6. [DOI] [PubMed] [Google Scholar]
- 54.Sevrioukova IF, Poulos TL. Proc Natl Acad Sci U S A. 2010;107:18422–18427. doi: 10.1073/pnas.1010693107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu L, Liu H, Murray B, Callebaut C, Lee MS, Hong A, Strickley RG, Tsai LK, Stray KM, Wang Y, Rhodes GR, Desai MC. ACS Med Chem Lett. 2010;1:209–213. doi: 10.1021/ml1000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McNulty J, Nair JJ, Singh M, Crankshaw DJ, Holloway AC. Bioorg Med Chem Lett. 2009;19:5607–5612. doi: 10.1016/j.bmcl.2009.08.032. [DOI] [PubMed] [Google Scholar]
- 57.McNulty J, Nair JJ, Singh M, Crankshaw DJ, Holloway AC, Bastida J. Bioorg Med Chem Lett. 2009;19:3233–3237. doi: 10.1016/j.bmcl.2009.04.086. [DOI] [PubMed] [Google Scholar]
- 58.McNulty J, Nair JJ, Singh M, Crankshaw DJ, Holloway AC. Bioorg Med Chem Lett. 2010;20:2335–2339. doi: 10.1016/j.bmcl.2010.01.157. [DOI] [PubMed] [Google Scholar]
- 59.Park H, Lee S, Suh J. J Am Chem Soc. 2005;127:13634–13642. doi: 10.1021/ja053809q. [DOI] [PubMed] [Google Scholar]
- 60.Lill MA, Dobler M, Vedani A. Chem Med Chem. 2006;1:73–81. doi: 10.1002/cmdc.200500024. [DOI] [PubMed] [Google Scholar]
- 61.Fishelovitch D, Hazan C, Hirao H, Wolfson HJ, Nussinov R, Shaik S. J Phys Chem B. 2007;111:13822–13832. doi: 10.1021/jp076401j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fishelovitch D, Hazan C, Shaik S, Wolfson HJ, Nussinov R. J Am Chem Soc. 2007;129:1602–1611. doi: 10.1021/ja066007j. [DOI] [PubMed] [Google Scholar]
- 63.Fishelovitch D, Shaik S, Wolfson HJ, Nussinov R. J Phys Chem B. 2009;113:13018–13025. doi: 10.1021/jp810386z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fishelovitch D, Shaik S, Wolfson HJ, Nussinov R. J Phys Chem B. 2010;114:5964–5970. doi: 10.1021/jp101894k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krishnamoorthy N, Gajendrarao P, Thangapandian S, Lee Y, Lee KW. J Mol Model. 2009;16:607–614. doi: 10.1007/s00894-009-0571-5. [DOI] [PubMed] [Google Scholar]
- 66.Moore CD, Shahrokh K, Sontum SF, Cheatham TE, 3rd, Yost GS. Biochemistry. 2010;49:9011–9019. doi: 10.1021/bi101139q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee JY, Kang NS, Kang YK. Biopolymers. 2012;97:219–228. doi: 10.1002/bip.22009. [DOI] [PubMed] [Google Scholar]
- 68.Shahrokh K, Cheatham TE, 3rd, Yost GS. Biochim Biophys Acta. 2012;1820:1605–1617. doi: 10.1016/j.bbagen.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cojocaru V, Winn PJ, Wade RC. Biochim Biophys Acta. 2007;1770:390–401. doi: 10.1016/j.bbagen.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 70.Li W, Liu H, Luo X, Zhu W, Tang Y, Halpert JR, Jiang H. Drug Metab Dispos. 2007;35:689–696. doi: 10.1124/dmd.106.014019. [DOI] [PubMed] [Google Scholar]
- 71.Sun H, Bessire AJ, Vaz A. Bio Med Chem Lett. 2012;22:371–376. doi: 10.1016/j.bmcl.2011.10.121. [DOI] [PubMed] [Google Scholar]