

Abstract
Sponsored by the New York Academy of Sciences and with support from the National Institute of Mental Health, the Life Technologies Foundation, and the Josiah Macy Jr. Foundation, “Advancing Drug Discovery for Schizophrenia” was held March 9–11 at the New York Academy of Sciences in New York City. The meeting, comprising individual talks and panel discussions, highlighted basic, clinical, and translational research approaches, all of which contribute to the overarching goal of enhancing the pharmaceutical armamentarium for treating schizophrenia. This report surveys work by the vanguard of schizophrenia research in such topics as genetic and epigenetic approaches; small molecule therapeutics; and the relationships between target genes, neuronal function, and symptoms of schizophrenia.
Keywords: schizophrenia, genetics, GWAS, neuronal function, small molecules, therapeutics
Background and perspectives: keynote lectures
Session chairs: Stephen R. Marder and Bryan Roth
Though pioneering pharmacotherapy for the treatment of schizophrenia occurred in the 1950s with the introduction of antipsychotics, there have been decidedly few major innovations in the intervening years. Indeed, the current classes of drugs available largely involve the same mechanisms of action and the same neurobiological targets. Compounding the lack of progress has been the systemic pull-back of the pharmaceutical industry from research and development of new treatments for schizophrenia.
With the advent of successful methods for identifying anomalies at the genetic level, schizophrenia and other neuropsychiatric disorders stand to benefit immensely from incorporating these approaches and broadening our understanding of the pathogenesis of these illnesses.
“Advancing Drug Discovery for Schizophrenia” convened with an opening session of keynote lectures moderated by session chairs Stephen R. Marder (University of California, Los Angeles) and Bryan Roth (University of North Carolina School of Medicine). Marder called for a change in the way schizophrenia treatment has previously been conceptualized by moving away from focusing only on treating symptoms in individuals who are in the acute phase of the illness. Innovative drugs may emerge from improving our understanding of the pathologic processes that occur early in the development of the illness, thus providing strategies for altering the course of the disorder. Importantly, genetic and epigenetic approaches present a very promising means by which to accomplish this goal.
Genetic and epigenetic approaches to studying schizophrenia
Patrick F. Sullivan (University of North Carolina at Chapel Hill) introduced the advantages of genetic approaches to schizophrenia by listing several benefits: the temporality of exposure to genetic risk factor preceding disease can be reasonably assumed; attempts at unbiased searches are possible; and, in the past two years, there has been substantial progress in uncovering the genetic basis of schizophrenia. He suggested that the purpose of employing genetic techniques is to look both for genetic variance that confers risk or prevention and for whether multiple genes work in concert to form pathways to an illness. To illustrate this point, he offered the analogy of a match to a forest fire—genetic studies seek to identify the initiating event in a disorder, or in other words, the “match” that sparks the fire.
This line of work has begun to make progress.1,2 Sullivan noted that there are novel and robust findings that include common variation in the major histocompatibility complex, the region encoding the microRNA miR-137, and four predicted targets of miR-137. Copy number variants that are rare but have high penetrance with robust results include regions on 1q21.1, 15q13.3, 16p11.2, 22q11.2, and NRX1. At this point, up to 15 possible genetic loci for schizophrenia have been identified. Further, Sullivan stressed that the accumulated data suggest that schizophrenia is relatively highly polygenic, which may be a general feature of complex traits. For example, various genes may actually code at the symptom level versus the disorder level (Table 1).
Table 1.
Genetic characteristic of SCZ
| SCZ is highly polygenic |
|---|
|
The results to date are notable, Sullivan underscored, in that the novelty of findings correspond relatively poorly to prior ideas about the etiology of schizophrenia. Empirical data suggest that both common and rare variations are involved in the etiology of schizophrenia. Rare variant–only models have been evaluated and are not consistent with the data. Sullivan suggested that with genome-wide association studies (GWAS), we can, however, estimate that increasing sample sizes are likely to produce more significant loci and considerably better pathway analyses. He likened GWAS to a powerful centrifuge that allows for the separation of specific genes. Most heartening, he suggested, is the likelihood that the discovery profile for schizophrenia has a linear trajectory—increasingly larger samples will lead to an increasing number of loci identified.
Genetic architecture of psychotic illness
Edward M. Scolnick (Broad Institute of MIT and Harvard University) began by suggesting the great importance of unraveling the full genetic architecture of schizophrenia and bipolar disorder as the critical step in establishing their underlying neurobiology. While the overarching theme of the conference pointed to a general stagnation in the field of schizophrenia treatment, Scolnick lauded the significant strides made recently. In other words, the field has become “unstuck.” This movement could also be attributed to the application of three new technologies: human genetics, optogenetic methods to manipulate specific circuits in the brain, and induced pluripotent stem cells.
Scolnick noted the apparent disparity between developments in schizophrenia versus cancer treatments. Bolstering this swell in cancer drug development is the increased understanding of the genetic and molecular aspects of various forms of cancer, something that, given the limited knowledge of its genetic basis, is not yet possible for schizophrenia.
In the past three years, however, using detailed maps of the human genome and doing association studies, a number of risk genes for schizophrenia and bipolar illness have been identified. CNVs (copy number variants) and genomic associations have been identified with a high degree of statistical certainty; some of the CNVs are de novo and some are inherited. Scolnick referred to characterizing diseases by the frequency of variants in addition to penetrance. For example, Mendelian diseases possess high penetrance and are deterministic of the development of the disease. However, many diseases posses a mixture of high- and low-frequency variation, indicated Scolnick. Increasingly, GWAS methodology is being used to investigate the penetrance of diseases and is appropriate for application to schizophrenia. Pathways involving synaptic proteins, the WNT signaling pathway, as well as miR-137 have been identified, which now provide clues about the biology of some illnesses. In addition, it has become clear that schizophrenia and autism may share some risk genes in common. Though the outcomes are very different, it may be possible, Scolnick suggested, that causative DNA variants may someday be used diagnostically.
Scolnick concluded by posing the idea that the full-scale application of genomics is now limited only by money and time to unravel the full genetic architecture of these illnesses. Further, given the burden of these illnesses, he reasoned that there is “no excuse” for NIH and NIMH funds not to be made available to perform the needed genetic studies and to finally enlighten their etiologies. He continued by saying that until we understand fully the genes that cause these illnesses the field will remain a “backwater of medicine.” Funding for neuroscience is certainly needed to understand their pathophysiology, but without an understanding of the genetic bases, he cautioned, scientists will continue to guess at what to work on to elucidate the illness and discover new targets for treatment.
What can we learn from animal models of schizophrenia?
Applying genetic methodologies to psychiatric disorders presents unique challenges, suggested Eric J. Nestler (Mount Sinai Medical Center). These include that they are categorically defined by behavioral abnormalities and that the genetic risks involved are likely to be a combination of multiple genetic variations. Thus, progress in understanding the etiology and pathophysiology of schizophrenia has been frustratingly slow, as has been the discovery of significant, novel therapeutic mechanisms. The exceedingly challenging neurobiology of higher brain function and the ethical and practical difficulties of examining the living human brain have no doubt contributed to this relatively slow progress. Another important factor is the extremely challenging nature of modeling schizophrenia in laboratory animals.3 This is due to the inaccessibility, in animals, of many of the key symptoms of schizophrenia, the subjective nature of the symptoms, the lack of objective biomarkers, and the early state of knowledge of the underlying genetics and neurobiology of the syndrome.
What we have learned from genetic studies of schizophrenia thus far, Nestler argued, is especially complicating vis-à-vis animal models, because most genes of relatively strong effect implicated in schizophrenia are also implicated in bipolar disorder and even autism. Moreover, Nestler proposed that what can be learned from placing genes of very small effect into laboratory animals is itself questionable. Indeed, Nestler continued by saying that most available animal models of schizophrenia have significant limitations, ranging from weak validation to poor predictive power for drug efficacy in humans.
Illustrating the complications that arise from translating models of human disorders in, for example, murine models, Nestler explored difficulties for establishing construct validity. For example, though genes may be associated with schizophrenia, abnormalities in these specific genes may cause schizophrenia but may also cause other disorders. Moreover, there is difficulty in making animal models as complicated as human models, and this too may require experimentally manipulating multiple genes.
Nestler also discussed two other forms of validity in respect to the limitations of animal models. Perhaps the most obvious limitation is the lack of face validity—there are simply features of psychiatric disorders that are not accessible in animals. Second, Nestler referenced the lack of predictive validity of animal models. Not only are there no new medications to test for efficacy in animals, but we must also interpret any response to drugs with great caution. In contrast, there may be better face validity for some of the negative symptoms associated with schizophrenia (e.g., social withdrawal and anhedonia), which have been modeled successfully, but may not be unique to this illness.
Concluding his talk, Nestler remained optimistic that, with time, the genes that confer risk or resilience in mental illness will be determined, citing advances in autism as support. The generation of convincing and useful animal models of schizophrenia thus represents a major set of challenges that will not have easy answers. On the other hand, given current limitations of clinical studies, Nestler concluded that it is hard to imagine significant progress in schizophrenia pathophysiology or therapeutics without good animal models.
Session II: Genetic and epigenetic approaches
Session chair: Mark A. Geyer
A novel therapeutic target for psychosis
The discovery of genes associated with risk for schizophrenia holds promise for identifying therapeutic targets based on etiopathogenesis. Daniel R. Weinberger (National Institute of Mental Health) reported recent work from his lab documenting an association with schizophrenia to the hERG family potassium channel gene, KCNH2.4 Risk-associated variants predict expression of a novel brain and primate specific isoform, KCNH2 3.1, which both has unique physiological properties and is upregulated in schizophrenia prefrontal and hippocampal cortices. Because many antipsychotic drugs bind to KCNH2 channels, accounting in some cases for QT prolongation, and atypical agents such as clozapine are particularly potent KCNH2 inhibitors, Weinberger and coworkers hypothesized that activity at KCNH2 channels may be a therapeutic mechanism of antipsychotic drugs. As a result, targeting the 3.1 isoform not expressed in the heart would have a particularly favorable therapeutic-to-toxic ratio. Weinberger stated, “We have tested the first hypothesis in two independent samples of patients who underwent placebo controlled studies of antipsychotic therapy, including an inpatient sample from the CBDB/NIMH experimental therapeutics unit (n = 59) and Caucasian subjects treated with olanzapine as part of the CATIE trial from whom we also had drug clearance data (total N = 89).” In both groups, Weinberger and colleagues found that the KCNH2 genotype predicted response to treatment (e.g., positive symptoms P < 0.0007, thought disturbance P < 0.0004 in the CBDB sample, positive symptoms P < 0.04 in CATIE). Currently, Weinberger’s group is testing the second hypothesis in human and model cell lines that over-express KNCH2 3.1 and also in a mouse model that overexpresses KCNH2 3.1 in cortex and shows predicted abnormalities in K+ channel kinetics and in working memory.
Linking DNA structural variation to brain dysfunction and schizophrenia
Structural variation in the genome in the form of copy-number variants (CNVs; submicroscopic gains or losses of segments of DNA) has now been identified as a major contributing genetic factor in schizophrenia.
Maria Karayiorgou (Columbia University) discussed the importance of examining low-frequency alleles with moderate effects, as they are easier to characterize and potentially more likely to contribute to the development of the disorder. Further, exploring the genetic variation in the landscape de novo may have great utility in delineating sporadic versus inherited types of schizophrenia. Importantly, Karayiorgou suggested that inherited CNVs are more disrupting than those that are de novo.
Karayiorgou focused on one such CNV on chromosome 22q11.2. Recent studies are beginning to paint a clearer and more consistent picture of the impairments in psychological and cognitive competencies that are associated with microdeletions in chromosome 22q11.2. In contrast to previous talks stressing the uncertainty of associating schizophrenia risk with any specific gene or CNV, Karayiourgou maintained that 30% of individuals with a deletion on 22q11 develop schizophrenia or schizoaffective disorder. Indeed, parallel studies in humans and animal models have begun to uncover the complex genetic and neural substrates altered by this microdeletlon. For example, Krayiorgou illustrated studies that have begun to identify CNVs that may explicate some of the working memory-related processing deficits seen in schizophrenia, because they occur in regions that oversee microRNA, thus affecting prefrontal cortex neuronal synaptic transmission and plasticity. In addition to offering a deeper understanding of the effects of this genetic lesion, Krayiorgou ended her presentation with the hope that these findings may guide analysis of other CNVs associated with psychiatric disorders.
Targeting the neuro-epigenome in the prevention and treatment of psychosis
Alessandro Guidotti (University of Illinois at Chicago) began by stating that the efficacy of current drugs is limited and that, without new developments, there is little hope for progress in the treatment of schizophrenia. An area of research that holds promise is exploring the role of the fundamental interaction between GABAergic interneurons and pyramidal neurons and how this function is compromised in schizophrenia and bipolar disorders. Schizophrenia and bipolar disorder patients show a downregulation of GAD67, reelin, and other genes expressed in telencephalic GABAergic neurons, likely associated with promoter hypermethylation mediated by an overexpression of DNA methyltransferase (DNMT).5 The inhibitory action of DNMT on gene expression may also occur through formation of chromatin repressor complexes that include histone deacetylases (HDAC) or histone methyltransferases, thereby shifting chromatin from a conformation permissive for transcription to one that is repressive. Guidotti has concentrated on a pharmacological strategy to reduce the hypermethylation of GABAergic promoters and to induce a permissive chromatin conformation. This strategy is to administer drugs such as the HDAC inhibitor valproate (VPA), which induces DNA demethylation when administered at doses that facilitate chromatin remodeling.6
Studies employing this strategy in mice, as presented by Guidotti, suggest that when combined with VPA, clinically relevant doses of clozapine elicit a synergistic potentiation of VPA-induced GABAergic promoter demethylation (Fig. 1). Olanzapine and quetiapine (two similar atypical antipsychotic drugs) also facilitate chromatin remodeling, but at doses higher than used clinically, whereas haloperidol and risperidone are inactive. Hence, Guidotti suggested the synergistic potentiation of VPA’s action on chromatin remodeling by clozapine appears to be a unique property of the dibenzepines and is independent of their action on catecholamine or serotonin receptors. Furthermore, the administration of clozapine in conjunction with VPA reverses the downregulation of the GAD67 expression induced in mice by seven days of methionine administration.5
Figure 1.
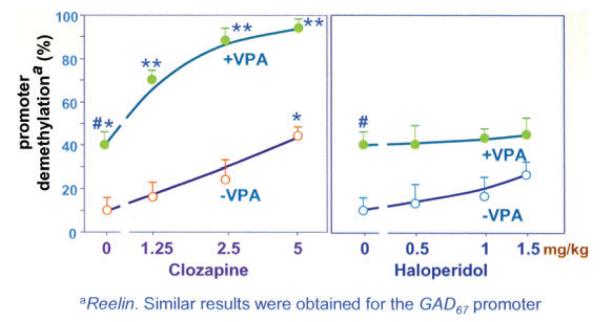
Clozapine but not haloperidol, alone or in combination with VPA, induces reelin promoter demethylation. Mice were pretreated for seven days with methionine to hypermethylate reelin and GAD67 promoters. After methionine withdrawal, various doses of clozapine or haloperidol alone or combined with valproate (VPA) (0.7 mg/kg/sc) were administered twice a day for three days. Open circles denote mice that did not receive VPA. Filled circles denote mice that received VPA. The data represent the mean ± SE of three mice. P < 0.05 when VEH + VPA-treated mice were compared with VEH-treated mice; P < 0.05 when VEH + clozapine-treated mice were compared to VEH-treated mice; and P < 0.02 when VEH + VPA-treated mice were compared with clozapine + VPA-treated mice.7
Guidotti concluded by positing that chromatin remodeling mechanisms may be altered in the brains of schizophrenia and bipolar disorder patients. Thus, by activating DNA demethylation, the association of clozapine or its derivatives with VPA or other more potent and selective HDAC inhibitors could be considered a promising prevention and treatment strategy to normalize the GABAergic promoter hypermethylation and GABAergic gene expression downregulation detected in the postmortem brain of schizophrenia and bipolar patients.
Advanced paternal age alters complex behaviors and brain DNA methylation in offspring
Prompted by epidemiological research that shows advanced paternal age increases risk for schizophrenia in offspring,a Jay Gingrich (Columbia University Medical School) discussed potential epigenetic mechanisms behind this effect. With each round of sperm production, epigenetic marks are erased and then reprogrammed—a process that may become degraded with advanced paternal age. To explore this, Gingrich and colleagues compared the offspring from older mouse fathers to those from young mouse fathers. Several behaviors differed between the two, including measures of an open-field ambulatory test, startle responses, and paired pulse inhibition. These mice showed distinct methylation patterns on their genomes at 0.4 % of the methylation sites. At these sites, the offspring of older males were less methylated than those of the younger males in introns, exons, and promoters. These findings suggest that differences inboth the promoter activity and alternative splicing patterns may drive some of the changes observed in the mice. Notably, hotspots of altered DNA methylation could be found in schizophrenia-related genes, though, Gingrich noted, the researchers didn’t select the mice for disease-related behaviors.
Neuronal epigenome mapping in developing and diseased prefrontal cortex
Alterations in chromatin structure and function, including changes in levels or distribution of histone lysine methylation markings and other epigenetic regulators of gene expression, could affect neuronal signaling in schizophrenia and other major psychiatric disease.8 However, to date, relatively nothing is known about the regulation of neuronal and other cell type–specific epigenomes in the diseased brain. Schahram Akbarian (University of Massachusetts) opened his talk by pointing out the potential for neuroepigenome mapping approaches to advancing the epigenetic field through focusing on the postmortem human brain. Specifically, he discussed work exploring the genome-wide distribution of trimethylated histone H3K4 (H3K4me3), an epigenetic histone mark associated with actual or potential transcription, in neuronal nuclei collected postmortem from prefrontal cortex (PFC) across a wide age range (0.5–70 years).
Neuronal nuclei from postmortem prefrontal cortex were immunotagged with NeuN antibody, NeuN+ and NeuN− nuclei were sorted separately via fluorescence-activated “cell” (nuclei) sorting (FACS), and purified mononuclesomes were enriched for H3K4me3 analyzed by massively parallel sequencing using an Illumina Solexa platform.9 Akbarian’s group has identified 16,000–22,000 H3K4me3-enriched regions (peaks), the majority located proximal to (within 2 kb of) the transcription start sites (TSS) of annotated genes. These included signatures specific to neurons as well as signatures specific to individual subjects (Fig. 2). Preliminary analyses presented by Akbarian provided evidence for a large-scale remodeling of histone methylation landscapes in immature PFC neurons during early infancy, with comparatively minor changes during subsequent periods of maturation and aging. Further, he predicted that epigenome mapping in defined cell populations of the diseased human brain, in conjunction with RNA and transcriptome profilings, could reveal important information about gene expression alterations at specific loci. These approaches, he stressed, will also be important to unravel the genetic and epigenetic risk architectures of specific cases and, furthermore, reveal insight about developmental regulation of chromatin structures in specific cell populations.
Figure 2.

Cell type–specific epigenome mapping in human brain. Left: schematic presentation of the nucleosome as the elementary unit of chromatin, composed of a histone octamer (blue) around which 146 bp of DNA (black) is wrapped. Histones and the genomic DNA are subject to various types of covalent modifications. Right: flow chart starting with extraction of nuclei from postmortem brain tissue for subsequent immunotagging with neuron nuclei–specific marker (NeuN), fluorescence-activated separation and sorting of neuronal and nonneuronal nuclei, and preparation of chromatin with enzyme-based digestion (micrococcal nuclease, MNase) into mononucleosomes. This is followed by immunoprecipitation with site- and modification-specific antihistone antibody (for example, antitrimethyl-histone H3-lysine 4) and massively parallel sequencing. Sequence tags from the immunoprecipitates (ChIP-seq) are then uploaded into the genome browser in order to visualize histone metylation landscapes at selected loci, as shown here for the GAD1/GAD67 promoter.
Session III: New molecular targets and approaches to small molecule therapeutics
Session chair: Robert W. Buchanan
Development of novel antipsychotic drugs: new approaches to old problems
Dopamine D2 receptor antagonism is a unifying property of all antipsychotic drugs in use for schizophrenia. Jeffrey A. Lieberman (Columbia University and New York State Psychiatric Institute) stressed during his talk that, as a result, while these drugs can be effective at ameliorating psychosis, they are largely ineffective at treating negative and cognitive symptoms, can produce serious side effects involving different organ systems, and may even contribute to gray matter volume reduction. The differences between the majority of the current drugs, Lieberman suggested, are primarily due to side-effect profiles, with pronounced side-effects often outweighing the therapeutic potential; additionally, considerable heterogeneity in response to treatment exists.
For years, psychiatric researchers and pharmaceutical companies have sought to develop mechanistically novel antipsychotic drugs based on targets other than dopamine receptors. For example, Lieberman highlighted the considerable amount of work that has been devoted to precedented targets and mechanisms (D-2, D-3 dopamine, and 5-HT2A serotonin antagonism); novel mechanisms (partial agonism, functional selectivity); and novel targets (glutamatergic, cholinergic, GABAergic), including intracellular signaling mechanisms (AKT, GSK). Unfortunately, drugs targeting many of these mechanisms were abandoned before they were tested in clinical trials.
In addition, Lieberman offered the observation that increasing attention is being focused on the complex genetics of schizophrenia as well as the signaling pathways implicated in its pathophysiology. Lieberman’s presentation reviewed the limitations of existing therapeutic agents and development strategies, including several of the major genetic findings that have identified signaling pathways representing potential targets for novel pharmacological intervention—for example, genes in the 22qll locus, DISC1, Neuregulin1/ ErbB4, and components of the Akt/GSK-3 pathway.
Lieberman concluded that it may be critical both to consider the necessity of multiple medications that treat different aspects (or domains) of schizophrenia and to renew commitment to research and development of pharmacotherapy for the illness.
Allosteric modulators of metobotropic glutamate receptor 5 for the treatment of schizophrenia
P. Jeffrey Conn (Vanderbilt University) presented work demonstrating the path between hypotheses and actual treatments of schizophrenia. The glutamate/NMDA receptor function hypofunction hypothesis has garnered wide attention because of its significant potential in targeting the range of symptoms presented in schizophrenia, including positive, negative, and cognitive symptoms. Further supporting this hypothesis, a large number of cellular and behavioral studies suggest that selective agonists of the metabotropic glutamate receptors (mGluRS) could provide a novel approach to treatment of schizophrenia.10 Unfortunately, it has been difficult to develop compounds that act as selective orthosteric agonists of mGluRS and that have properties likely to be suitable for development of therapeutic agents. Conn presented work from his group, which has been successful in implementing a novel approach to activation of these receptors by developing highly selective allosteric potentiators of mGluR5.These compounds do not activatemGluR5 directly, but rather potentiate the response of these receptors to the endogenous agonist. Thus, these allosteric potentiators offer high selectivity for the targeted receptor and provide an exciting new approach to development of novel selective activators of mGluR5 and other G protein–coupled receptor (GPCR) subtypes.10
In vivo studies reveal that these compounds have robust effects in animal models, which have been used to predict efficacy of novel antipsychotic agents. Conn showed findings that demonstrate that introduction of these compounds reduces amphetamine-induced motor hyperactivity, a putative animal model of limbic hyperactivity and positive symptoms of schizophrenia. In addition, mGluR5-positive allosteric modulators (PAMs) enhance multiple forms of synaptic plasticity in the central nervous system (CNS) and have cognition-enhancing effects in animal models.11 These studies, therefore, provide an exciting new approach for discovery of novel, highly selective activators of specific GPCR subtypes for treatment of schizophrenia and other CNS disorders. In conclusion, Conn suggested that recent advances in the discovery of a broad range of mGluR5 allosteric modulators with different functional profiles will allow the development of a more complete understanding of the properties of individual mGluR5-PAMs with the hope of determining which may be most suitable for development as novel therapeutic agents.
Chemical genomic studies of DISC1/GSK3–mediated signaling in neuropsychiatric disorders
A critical need exists for gaining insight into the underlying etiology and pathophysiology of schizophrenia and other neuropsychiatric disorders in order to advance the development of new types of targeted therapeutic interventions. Surveying recent work, Stephen J. Haggarty (Harvard Medical School, Broad Institute of MIT and Harvard University) presented studies of the Disrupted in Schizophrenia 1 (DISC1) gene, which is disrupted by a balanced chromosomal translocation t(1;11)(q42.1;q14.3) in a single extended Scottish family with a high incidence of schizophrenia, major depression, and bipolar disorder. Studies have revealed a key role for DISC1-mediated signaling in diverse aspects of neuroplasticity. One of the direct binding targets of DISC1 is the multifunctional serine/threonine kinase GSK3, which is known to play a key role in the regulation of neurogenesis and synaptic function.12 GSK3 has also been shown to be inhibited in vivo directly, and indirectly, by the mood stabilizer lithium and indirectly through the activity of antipsychotics at various GPCRs.13
On the basis of these findings, as well as emerging human genetic studies implicating other components of the GSK3 signaling pathway in the etiology of schizophrenia, Haggarty discussed the advent of chemical genomic approaches to identify small molecule probes that target known and novel components of DISC1/GSK3 signaling.
Using pharmacological and viral-mediated gene expression to modulate AKT kinase activity, Haggarty presented work demonstrating a key role for AKT kinase activity in determining the cellular and mood-related behavioral effects of lithium.13,14 In contrast, selective and direct GSK3 inhibition by an ATP-competitive, highly selective, brain penetrant GSK3 inhibitor (CHIR-99021) was found to bypass the requirement for AKT activation (Fig. 3).14 In order to try to develop pathway-selective inhibitors of GSK3 and to develop small molecule probes that enable the selective targeting of individual DISC1 domains, Haggarty’s group has used small molecule microarray screening to measure the interaction of DlSC1 variants in a high throughput manner.
Figure 3.

DISC1/GSK3 signaling involved in regulating neuroplasticity and its regulation. While lithium inhibits GSK3 both indirectly and directly, CHIR-99021 is an ATP competitive inhibitor of GSK3 that mimics the cellular and behavioral effects of lithium. AKTI-17 is an allosteric inhibitor of AKT that blocks the ability of lithium to modulate TCF/LEF–mediated transcription and behavior but not the effect of CHIR-99021.
Finally, Haggarty presented efforts to use induced pluripotent stem cell (iPSC)–derived human neurons for use in high-throughput screening for small molecule probes of DISC1/GSK3 signaling. Haggarty showed findings that demonstrated the ability to identify known and novel small molecules that regulate TCF/LEF–dependent transcription that is under control of GSK3 signaling in iPSC-derived human neurons. Collectively, he noted, through the use of genetically accurate cell models and in using iPSC-derived human neurons, these studies will enable a better understanding of the role of DISC1/GSK3 in regulating signaling pathways implicated in schizophrenia, as well as potentially lead to the identification of new targets and improved treatments.
Targeting the PI3K/AKT pathway: novel therapeutic options for schizophrenia
Neuregulin 1 (NRG1) and ErbB4, critical neurodevelopmental genes, are implicated in genetic risk for schizophrenia, but the biological mechanisms are unknown. Amanda J. Law (National Institute of Mental Health) presented work testing the hypotheses that aberrant PI3K/AKT signaling represents a pathogenic consequence of schizophrenia-associated genetic variation in ErbB4 and that pharmacological manipulation of this pathway may represent a novel therapeutic avenue for the treatment of psychosis. She explained that her group has used an “integrative translational neuroscience approach” to test the hypothesis, incorporating data from patient-derived Iymphoblastoid B cells, human brain mRNA expression profiling, clinical genetics, and pharmacological studies in rodents. These results pinpoint a genetically regulated pathway associated with schizophrenia and with ErbB4 genetic risk variation involving upregulation of a Pl3K-linked ErbB4 receptor CYT-l and of a specific phosphoinositide 3-kinase (PI3K) subunit. Law presented intracellular signaling data collected in human Iymphoblastoid cells showing that NRG1-mediated phosphatidyl-inositol,3,4,5-triphosphate [PI(3,4,5)P3] production (and cell migration) is associated with ErbB4 risk genotype and PI3K enzyme levels and is significantly impaired in patients with schizophrenia. In the human brain, the association of ErbB4 genotype and PI3K subunit expression was confirmed, and antipsychotic drug administration was found to downregulate expression of the PI3K subunit, implicating the PI3K pathway as a novel therapeutic target. Specific inhibition of the PI3K subunit using a small molecule inhibitor blocks the effects of amphetamine in a mouse pharmacological model of psychosis. Law posited that these studies provide novel insight into how ErbB4 may contribute to the pathophysiology of schizophrenia, reveal a previously unidentified link between NRG1-ErbB4 and PI3K signaling in the disorder, and suggest that a specific PI3K merit further consideration as a molecular target for rationally designed drugs for the treatment of psychiatric disorders.
Evaluation of potent, selective, and peripherally available kynurenine aminotransferase II inhibitors for the treatment of cognitive deficits in schizophrenia
Representing work conducted in the private sector, Brian Campbell (Pfizer) discussed recent work on a naturally occurring NMDA agonist, kynurenic acid (KYNA). KYNA is a biologically active byproduct of tryptophan metabolism that is reported to act as an endogenous antagonist of NMDA receptors and may also interfere with nicotinic α7 receptor function. Since KYNA levels are elevated in schizophrenic patients, it has been hypothesized that this may contribute to the cognitive deficits observed in schizophrenia. Though inhibitors of kynurenine aminotransferase 11 (KAT II), the primary enzyme involved in brain KYNA synthesis, have been proposed as targets to treat symptom domains in schizophrenia, research into this hypothesis has been hampered by lack of potent, selective, and brain penetrant tools. Campbell reported on the discovery and pharmacological characterization of a new class of KAT II inhibitors with nanomolar potency, >1000-fold selectivity over other kynurenine pathway enzymes, and excellent brain penetration. Systemic administration rapidly decreased KYNA by as much as 80% in brain dialysates in a dose-dependent manner. In addition, Campbell detailed the profiling of these compounds through a range of animal models relevant for domains of schizophrenia, which revealed efficacy in models of working memory in both rodents and non-human primates and also in a rat model of attention (i.e., a sustained attention task). Furthermore, Campbell and the group at Pfizer have found activity in an anhedonia model (chronic mild stress), which may suggest therapeutic utility for the negative symptoms of schizophrenia. These compounds are inactive in classical models of antipsychotic activity, suggesting that KAT II inhibitors may be suitable as adjunctive agents, given along with antipsychotics, to treat the cognitive and negative symptoms associated with schizophrenia that are so poorly managed today.
Genes to function to symptoms
Session chair: John H. Krystal
Large-scale neuronal analysis as a functional endophenotype for schizophrenia
Bita Moghaddam (University of Pittsburgh) opened the final session of the meeting. Dissatisfied with how simple circuit versions of the glutamate hypothesis of schizophrenia fail to deliver for the whole brain, she proposed an alternative way of thinking about the disorder. Instead of seeing the main clues we have for schizophrenia—NMDA receptor hypofunction and decreased GAD67, an enzyme that synthesizes GABA—as factors that contribute to the disorder, she suggested that decreased GAD67 may instead reflect a compensation for abnormal levels of glutamate. If markers of disease like these are compensatory, rather than primary, then trying to correct them would give opposite effects than expected. With this in mind, she cited a tightly regulated GABA shunt in mitochondria that recycles GABA molecules from glutamate. Moghaddam suggested that imbalances in GABA and glutamate levels could stem from improper mitochondrial function—something with repercussions for excitatory and inhibitory signaling throughout the brain.
Translational neuroscience for schizophrenia
Seeking to better simulate the molecular processes underway in neurons in disease, Akira Sawa (Johns Hopkins University) reported on the “garden of human neural cells” growing in his lab. He has had some success in maintaining olfactory neurons from humans obtained through a relatively noninvasive nasal biopsy,15 deriving human neurons from iPSCs,16 and deriving neurons directly from human skin cells.17 After two weeks of treatment in media, these latter neurons, called induced neurons (iN), showed neuron-specific markers and fired action potentials.
As an example of the dividends these approaches may provide, Sawa showed a comparison of mRNA profiles in olfactory neurons between people with schizophrenia and controls. The groups differed in genes related to actin binding, the NF-κB protein complex involved in DNA transcription, intracellular protein transport, and immune and stress responses. Consistent with these changes, olfactory neurons from patients with schizophrenia show abnormalities in association with cytoskeletons and oxidative stress (Table 2).
Table 2.
Translational neuroscience approaches to schizophrenia
| Advantage | Disadvantage | |
|---|---|---|
| Olfactory neurons |
|
|
| iPS cells—derived neurons |
|
|
| iN cells (induced neuronal cells) |
|
|
| Autopsied brains |
|
|
Behavioral selectivity of β-arrestin: dependent signaling of dopamine D2 receptor in the CNS
The last three speakers discussed whether there might be ways to refine dopamine signaling in the brain to treat schizophrenia more effectively and with fewer side effects. Starting with D2Rs, the target of all antipsychotics, Marc Caron (Duke University) noted that D2Rs engage both a G protein–coupled pathway and one involving the scaffolding protein β-arrestin-2, which in turn activates the AKT/GSK3β signaling independently associated with schizophrenia.
To probe how much this β-arrestin-2 pathway mediates psychosis-like behaviors in mice, he disabled it by removing GSK3β in D2R-containing neurons. This interfered with apomorphine-induced rearing as well as amphetamine-induced hyperlocomotion and prepulse inhibition, but not cognitive tasks. Removing GSK3β’s target, β-catenin, from D1R-containing neurons induced more amphetamine-induced psychosis, whereas removing it from D2R-containing neurons induced less. Together, the results implicate this pathway in psychosis-like behaviors and antipsychotic response, and the extent of its contribution may be further delineated with D2R mutants engineered to selectively signal through either the G protein or the β-arrestin-2 pathway.
The schizophrenia risk gene CAV1 is both propsychotic and required for antipsychotic drug activity at 5-HT2A serotonin receptors in vivo
In a later talk (see below), John Allen (University of North Carolina) reported new efforts to discover D2R ligands that selectively activate the β-arrestin-2 pathway but not the G protein pathway. In a chemical biology effort in Bryan Roth’s laboratory (University of North Carolina), the group screened hundreds of new compounds and came up with two that are biased for their signaling activity at the D2R-β-arrestin-2 pathway. When administered to mice, these arrestin-biased compounds decreased PCP- or amphetamine-induced hyperlocomotion without increasing catalepsy. These results suggest that selective D2R-β-arrestin-2 signaling and recruitment may contribute to antipsychotic activity without motor side effects.
The dopamine D1-D2 receptor heteromer: novel signaling complex with potential role in schizophrenia
In another twist for dopamine signaling, Susan George (University of Toronto) presented her evidence for a novel dopamine receptor made up of a D1R and a D2R. This combination receptor offers a new mode of signaling for dopamine because it activates a G protein pathway that is not induced when either receptor is activated alone. The D1-D2 heteromers occur endogenously in the brain and are enriched in the nucleus accumbens and globus pallidus of rats. Using a competitive binding assay to detect a high-affinity state for a D2 agonist in the D1-D2 heteromer, George found this state is enhanced in rats treated with amphetamine and in the globus pallidus of the postmortem brain in schizophrenia. The work suggests both that abnormal coupling between D1 and D2 could be a molecular marker for pathological states in the brain and that this coupling may be a new treatment target—something that has already been explored in mouse models of depression.
Serotonin signaling is also thought to contribute to schizophrenia symptoms, and the atypical antipsychotics have been designed to also target 5-HT2A serotonin receptors, following suggestions that this activity set clozapine apart from the typical antipsychotics. Prompted by a rare CNV found in the caveolin-1 (CAV1) gene in a case of schizophrenia,18 John Allen explored the state of serotonin signaling in knockout mice missing CAV1. CAV1 encodes a scaffolding protein involved in clustering diverse signaling molecules together, including 5-HT2A receptors.19 Loss of CAV1 resulted in an increased sensitivity to PCP-induced hyperlocomotion and disrupted prepulse inhibition, suggesting a propsychotic-like effect in the mice. Loss of the CAV1 gene also attenuated the activity of atypical antipsychotics and 5-HT2A antagonists in behavioral studies (Fig. 4). Similarly, these mice made fewer head twitches in response to a hallucinogenic 5-HT2A agonist and also disrupted 5-HT2A signalingin cortical neurons. The number of 5-HT2A receptors was normal in these mice, which suggests that loss of the CAV1 scaffold mislocalized 5-HT2A receptors and their downstream effector molecules, compromising their function and resulting in compromised antipsychotic activity.20
Figure 4.
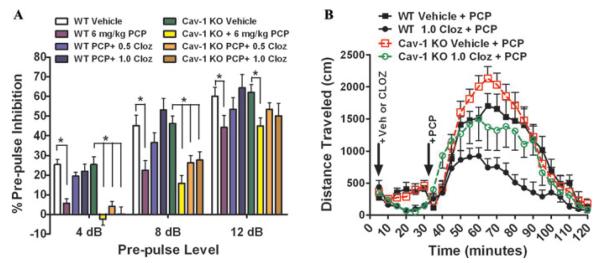
Genetic deletion of schizophrenia risk gene caveolin-1 in mice annenuates antipsychotic drug activity. (A) Littermate wildtype (WT) and caveolin-1 knockout (Cav-1 KO) mice were injected with vehicle, 0.5, or 1.0 mg/kg clozapine (CLOZ) and were followed 30 min later by 6 mg/kg phencyclidine (PCP) and immediately placed into acoustic startle response chambers. Clozapine-normalized PCP-disrupted prepulse inhibition (PPI) in WT but not in Cav-1 KO mice at the 4 dB and 8 dB prepulse levels (mean ± SEM, n = 12 littermate pairs; *, P < 0.05 versus WT Saline or Cav-1 KO Saline at each prepulse level). (B) Littermate WT and Cav-1 KO mice received vehicle or 1.0 mg/kg CLOZ and were placed into open field locomotion chambers and allowed to acclimate; 30 min later, mice were injected with PCP and the total distance traveled (cm) was determined. Clozapine was less effective at suppressing hyperlocomotion in Cav-1 KO mice (mean ± SEM, n = 12 littermate pairs).20
Although the crowd was dwindling, energy remained high at the end of the talks, with several people saying that they felt optimistic about the prospects for discovering truly innovative drugs for schizophrenia. One participant raised the issue of specificity, asking how disturbances to intracellular signaling pathways available to all cells can produce the malfunctions in specific brain circuits observed in schizophrenia. Moghaddam suggested that patterns of metabolic activity, combined with aberrant signaling, somehow target certain circuits for dysfunction. John Krystal (Yale University) suggested a genetic explanation, noting that susceptible brain regions in schizophrenia are the most recently evolved and thus could be the most genetically labile. Either way, a challenge will be to deliver treatment to ailing brain circuits without disrupting those that are functioning normally.
In his closing remarks, Krystal called the meeting “a next-generation conference” with research beginning to meet the urgent need for mechanistically novel compounds. “What’s exciting is not how far we’ve come, but the possibility that we might be getting to the point where we can use science to guide psychiatry.
Footnotes
See the SRF hypothesis: http://www.schizophreniaforum.org/for/curr/Malaspina/default.asp
References
- 1.Sullivan PF. Schizophrenia Psychiatric Genome-Wide Association Study Consortium: genome-wide association study of schizophrenia identifies five novel loci. Submitted.
- 2.Sullivan PF. The Psychiatric GWAS Consortium: big science comes to psychiatry. Neuron. 2010;68:182–186. doi: 10.1016/j.neuron.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nature Neurosci. 2010;13:1161–1169. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huffaker SJ, Chen J, Nicodemus KK, et al. A primate-specific, brain isoform of KCNH2 affects cortical physiology, cognition, neuronal repolarization and risk of schizophrenia. Nature Med. 2009;15:509–518. doi: 10.1038/nm.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guidotti A, Auta J, Chen Y, et al. Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology. 2011;60:1007–1016. doi: 10.1016/j.neuropharm.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guidotti A, Dong E, Kundakovic M, et al. Characterization of the action of antipsychotic subtypes on valproate-induced chromatin remodeling. Trends. Pharmacol. Sci. 2009;30:55–60. doi: 10.1016/j.tips.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 7.Dong E, Grayson DR, Guidotti A, Costa E. Antipsychotic subtypes can be characterized by differences in their ability to modify GABAergic promoter methylation. Epigenomics. 2009;1:201–211. doi: 10.2217/epi.09.2. [DOI] [PubMed] [Google Scholar]
- 8.Cheung I, Shulha HP, Jiang Y, et al. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc. Natl. Acad. Sci. USA. 2010;107:8824–8829. doi: 10.1073/pnas.1001702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peter CJ, Akbarian S. Balancing histone methylation activities in psychiatric disorders. Trends. Mol. Med. 2011;17:372–379. doi: 10.1016/j.molmed.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conn PJ, Lindsley CW, Jones CK. Activation of metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia. Trends Pharmacol. Sci. 2009;30:25–31. doi: 10.1016/j.tips.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ayala JE, Chen Y, Banko JL, et al. mGluR5 positive allosteric modulators facilitate both hippocampal LTP and LTD and enhance spatial learning. Neuropsychopharmacol. 2009;34:2057–71. doi: 10.1038/npp.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mao Y, Ge X, Frank CL, et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell. 2009;136:1017–1031. doi: 10.1016/j.cell.2008.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu. Rev. Pharmacol. Toxicol. 2009;49:327–247. doi: 10.1146/annurev.pharmtox.011008.145634. [DOI] [PubMed] [Google Scholar]
- 14.Pan JQ, Lewis MC, Ketterman JK, et al. AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacol. 2011;36:1397–1411. doi: 10.1038/npp.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sawa A, Cascella NG. Peripheral olfactory system for clinical and basic psychiatry: a promising entry point to the mystery of brain mechanism and biomarker identification in schizophrenia. Am. J. Psychiatry. 2009;166:137–139. doi: 10.1176/appi.ajp.2008.08111702. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 17.Pang ZP, Yang N, Vierbuchen T, et al. Induction of human neuronal cells by defined transcription factors. Nature. 476:220–223. doi: 10.1038/nature10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walsh T, McClellan JM, McCarthy SE, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 19.Allen JA, Halverson-Tamboli RA, Rasenick MM. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 2007;8:128–140. doi: 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- 20.Allen JA, Y. P., Setola V, Farrell M, Roth BL. Schizophrenia risk gene CAV1 is both pro-psychotic and required for atypical antipsychotic drug actions in vivo. Transl. Psychiatr. 2011:e33. doi: 10.1038/tp.2011.35. doi:10.1038/tp.2011.35. Published online 16 August 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]