

Abstract
Modified vaccinia virus Ankara (MVA) is being widely investigated as a safe smallpox vaccine and as an expression vector to produce vaccines against other infectious diseases and cancer. MVA was isolated following more than 500 passages in chick embryo fibroblasts and suffered several major deletions and numerous small mutations resulting in replication defects in human and most other mammalian cells as well as severe attenuation of pathogenicity. Due to the host range restriction, primary chick embryo fibroblasts are routinely used for production of MVA-based vaccines. While a replication defect undoubtedly contributes to safety of MVA, it is worth considering whether host range and attenuation are partially separable properties. Marker rescue transfection experiments resulted in the creation of recombinant MVAs with extended mammalian cell host range. Here, we characterize two host-range extended rMVAs and show that they (i) have acquired the ability to stably replicate in Vero cells, which are frequently used as a cell substrate for vaccine manufacture (ii) are severely attenuated in immunocompetent and immunodeficient mouse strains following intranasal infection, (iii) are more pathogenic than MVA but less pathogenic than the ACAM2000 vaccine strain at high intracranial doses, (iv) do not form lesions upon tail scratch in mice in contrast to ACAM2000 and (v) induce protective humoral and cell-mediated immune responses similar to MVA. The extended host range of rMVAs may be useful for vaccine production.
Keywords: attenuated live vaccines, recombinant vaccinia virus, virus vectors, virus pathogenesis
1. Introduction
Modified vaccinia virus Ankara (MVA) is a host-range restricted, highly attenuated vaccine strain that was obtained by passaging chorioallantoic vaccinia virus Ankara (CVA) >500 times in primary chick embryo fibroblasts (CEF) [1–4]. In addition to serving as an attenuated smallpox vaccine, MVA has potential as a safe vector for recombinant vaccines against other microbial pathogens and cancer [5–8]. Despite the current interest in MVA and the availability of the complete genome sequence [9], the basis for the host range restriction is incompletely understood. Although MVA replication is deficient in human and most other mammalian cells, growth can occur in baby hamster kidney 21 cells [10, 11] and fruit bat cells [12]. Unlike other vaccinia virus (VACV) host range mutants [13], the replication defect of MVA is manifested by formation of aberrant non-infectious virus particles with no impairment of viral protein synthesis in non-permissive cells [5]. Severe attenuation coupled with robust gene expression has made MVA an extremely useful vector.
There are six major deletions in the MVA genome, comprising about 15% of the total DNA [141, 15] as well as numerous smaller mutations [9]. The region of the MVA genome responsible for the host range defect was interrogated by carrying out homologous recombination with a panel of cosmids prepared from a replication-competent VACV strain and assessing plaque formation in African green monkey BS-C-1 cells, which are marginally permissive for MVA [16]. Recombinant MVAs (rMVAs) derived from three overlapping cosmids, each containing approximately 40 kbp of DNA near the left end of the VACV genome, exhibit enhanced MVA replication in monkey, human and rabbit cells. Two of the host-range extended viruses, rMVA 51.1 and rMVA 44/47.1, were derived by recombination with one and two cosmids, respectively, and share some newly acquired DNA [16]. In another approach, DNA sequences corresponding to the six major deletions of MVA were removed from the parental CVA [17]. However, these deletions failed to confer mammalian host range restriction or strong attenuation, indicating that other genetic changes are responsible for the MVA phenotype. On the other hand, introducing a deletion corresponding to one near the left end of MVA into the Lister strain of VACV reduced replication in human cells and a combination of multiple MVA deletions provided attenuation [18]. Further work is needed to determine the genetic basis for MVA host restriction and the contribution of this property to attenuation.
While the inability of MVA to replicate in human and most other mammalian cells can account for the absence of pathogenicity, MVA contains mutations of immune evasion and other genes that could contribute to attenuation even in host-range extended rMVAs. Thus, we were interested in determining the degree of attenuation of rMVA 51.1 and rMVA 44/47.1 in mice. In addition, the host range restriction of MVA limits the cell substrates that can be used for vaccine manufacture. Vero cells, which are widely used to make vaccines, would be an attractive alternative to primary CEF for production of MVA-based vaccines. The abilities of rMVA 44/47.1 and rMVA 51.1 to replicate in BS-C-1 cells [16] encouraged us to consider that they would also replicate in Vero cells. Here we demonstrate that rMVA 44/47.1 and rMVA 51.1 can be propagated in Vero cells, are greatly attenuated in immunocompetent and immunodeficient mice, and induce protective humoral and cell mediated immune responses similar to those of MVA.
2. Materials and methods
2.1. Cells and viruses
Primary chick embryo fibroblasts (CEF) and continuous cell lines were maintained at 37°C with 5% CO2 in modified Eagle minimal essential medium (Quality Biologicals, Inc., Gaithersburg, MD) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 10 units of penicillin/ml and 10 μg of streptomycin/ml. VACV strains ACAM2000, Western Reserve (WR) and “Ankara” were grown, purified and titrated as described previously [19, 20]. ACAM2000 (Lot number: VV04-003-A) was grown and titrated on BS-C-1 cells. VACV Ankara, used to construct the cosmid library for marker rescue and formation of rMVAs was originally understood to be the parent strain of MVA [16]. However, sequence analysis (J. Mendez-Rios, personal communication) indicates that VACV Ankara does not correspond to the sequence of CVA [21], the actual progenitor of MVA, and may be related to the Copenhagen vaccine strain. However, for consistency the name Ankara has been retained for this report. Generation and purification of rMVA 44/47.1 and rMVA 51.1 were described previously [16]. Titers of MVA, rMVAs and Ankara were determined by immunostaining in CEF as described [10, 16]. All procedures with viruses were performed in a registered BSL-2 laboratory.
2.2. Virus replication in Vero cells
Vero cells (5 × 105) were infected with 0.1 plaque forming units (pfu)/cell in 12-well plates. After 1 h, the monolayers were washed twice and overlaid with fresh medium. At various times post-infection, cells from triplicate wells were harvested individually in 1 ml of medium and stored at −80°C. The cells were then lysed and sonicated; cell-associated virus yields were determined by assay on CEF for MVA and rMVAs and on BS-C-1 cells for ACAM2000.
2.3. Mouse pathogenicity studies
Viruses were diluted in phosphate-buffered saline (PBS) containing 2% fetal bovine serum, and the virus concentration of each dilution used in animals was verified by plaque assay on the same day. Groups of 4 to 10, 5–6 weeks-old BALB/c, C57BL/6 or ICR-SCID female mice (Taconic Biotechnology, Germantown, NY) were inoculated with viruses by intranasal or intracranial routes. For intranasal infections, mice were anesthetized by inhalation of isoflurane and 20 μl (106 pfu) of virus was introduced into one nostril. For intracranial infections, mice were anesthetized with a mixture of ketamine (75 mg/kg) and xylazine (7.5 mg/kg) in PBS. A volume of 30 μl (104–108 pfu) of virus was injected with a syringe connected to a half-inch 27-gauge needle, which was inserted through the parietal bone of the skull at an angle of about 90° above the horizontal. A limiting tubing device was attached allowing the needle to penetrate not more than 2 mm under the bone. Animals were weighed and observed three to seven times per week for up to 47 days post-infection for ICR-SCID mice or 21 days for BALB/6 or C57BL/6. Animals that lost 30% or more of their starting weight were euthanized in accordance with NIAID Animal Care and Use protocols. Experiments were performed in an ABSL-2 facility with approval of the NIAID Animal Care and Use Committee.
2.4. Immunogenicity studies
Intramuscular vaccination of 6 weeks-old mice with 105–108 pfu of virus and tail scratch with 104–108 pfu were carried out as described [22]. For intramuscular vaccination, the total 100 μl dose was divided and 50 μl was injected into the gastrocnemius muscles of each back leg. Blood collection from the mandibular plexus into serum collection tubes (BD Biosciences, San Jose, CA) was carried out at 3 weeks after vaccination. Serum was isolated from clotted blood samples by centrifugation as directed by the manufacturer. Challenge with 107 pfu of VACV Western Reserve (WR) by the intranasal route was performed as described above. Mock-infected control animals were inoculated with an equivalent volume of diluent.
2.5. Intracellular cytokine staining of mouse splenocytes
Procedures for preparation and staining of splenocytes were modified from the method previously described [23] as follows. Mouse P815 mastocytoma target cells, at a concentration of 107 cells/ml in RPMI medium were infected with 10 pfu/cell of MVA for 90 min at 37°C, brought up to 106 cells/ml in RPMI medium and incubated 4 to 5 h at 37°C. After washing, cells were suspended at 2.5×106 cells/ml. Splenocytes were prepared from individual mice one week post-vaccination. For stimulation, 1.5×106 splenocytes were mixed with 2.5 ×105 MVA-infected P815 cells. After 4 h at 37°C, brefeldin A (Sigma, St. Louis, MO) was added to a concentration of 10 μg/ml, and the incubation was continued for 10–15 h. Cells were incubated for 10 min with Fc block (anti-CD16/CD32, clone 2.4G2, a gift from J. Bennink, Laboratory of Viral Diseases), and then stained with peridinin chlorophyll-a protein (PerCP) conjugated anti-CD8 (clone 53.6–7) for 30 min at room temperature. After fixation and permeabilization, the cells were stained with allophycocyanin (APC)-conjugated anti-gamma interferon (IFN-γ) (clone XMG 1.2), fluorescein isothiocyanate (FITC)-conjugated anti-interleukin-2 (IL-2) (clone JES6-5H4) and R-phycoerythrin (PE)-conjugated anti-tumor necrosis factor (TNFα) (clone MP6-XT22) for 1 h. Cells were then washed and suspended in 2% paraformaldehyde. All staining reagents were purchased from BD Biosciences (San Jose, CA). At least 100,000 cells were acquired on a FACSCalibur cytometer using CellQuest software (BD Biosciences) and analyzed with FlowJo software (TreeStar, Cupertino, CA).
2.6. VACV enzyme-linked immunosorbent assay (ELISA)
Microtiter plates (96-well, Thermo Labsystems, Franklin, VA) were coated with 107 pfu/ml of sucrose gradient purified MVA in CB1 bicarbonate buffer (Immunochemistry Technologies, Bloomington, MN) at 37°C overnight. Virus was inactivated by incubation with 2% paraformaldehyde for 10 min at 4°C. The assay was performed as described [24] except that the times of incubation of sera and secondary antibodies were increased to overnight and 5 h, respectively, and peroxidase-conjugated anti-mouse IgG (Roche Applied Science, Indianapolis, IN) was used.
2.7. Statistical analysis
Statistical differences between groups of mice were assessed by two-way RM ANOVA, Bonferroni multiple comparison for morbidity or Gehan-Breslow-Wilcoxon test with a Bonferroni correction for mortality, using Prism software (Graph Pad, San Diego, CA). Geometric mean antibody titer and the geometric standard deviation was calculated using Microsoft Excel software.
3. Results
3.1. Replication of rMVAs in mammalian cells
The rMVA 44/47.1 and rMVA 51.1 were previously shown to have enhanced replication relative to MVA in African green monkey BS-C-1, human MRC-5, human HeLa and rabbit RK-13 cells [16]. We extended this analysis to African green monkey kidney Vero cells because of their important use for vaccine production. Plaque sizes of rMVAs 44/47.1 and 51.1 were compared to those of MVA, Ankara (the strain used to make the cosmid library for derivation of the rMVAs), and ACAM2000 ([25], a licensed replication-competent vaccine). Ankara and ACAM2000 formed the largest plaques on BS-C-1 and Vero cells, next largest were produced by rMVA 44/47.1 and then by rMVA 51.1 (Fig. 1A). MVA produced pin-point foci (Fig. 1A). In contrast, MVA and the two rMVAs formed similar size plaques at 48 h in CEFs (not shown).
Fig. 1.
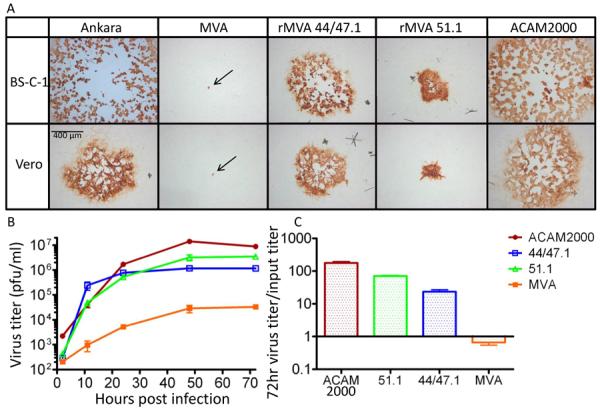
Replication of rMVAs. (A) Cell-to-cell virus spread. Monolayer of BS-C-1 and Vero cells were infected with MVA, rMVA 44/47.1 and rMVA 51.1, Ankara and ACAM2000 viruses. At 48 h after infection, the cells were fixed and immunostained with broadly reactive anti-VACV antibody followed by horseradish peroxidase conjugated to anti-rabbit immunoglobulin. Arrows point to the pin-point foci formed by MVA. (B) Growth curve. Vero cells were infected with 0.1 pfu/cell of MVA, rMVA 44/47.1, rMVA 51.1 or ACAM2000 and harvested at the indicated times post infection. Cell-associated viruses were titrated on CEF monolayers in the case of MVA and rMVAs and BS-C-1 cells for ACAM2000. Standard error bars shown. (C) The ratios of virus output to virus input are plotted with standard error bars.
The abilities of MVA, the two rMVAs, and ACAM2000 to replicate in Vero cells were determined. In order to calculate the virus yields, plaque titrations were carried out in both CEF and BS-C-1 cells. ACAM2000 plaqued poorly in CEF and therefore the BS-C-1 titers are shown in Fig. 1B. For opposite reasons, the MVA titer was determined in CEF. The titers in CEF are also shown for the rMVAs (Fig. 2B), although the titers determined by plaque assay in BS-C-1 cells were about 2-fold higher in each case. We noted that the titer of MVA increased from 2 to 72 h indicating that it is not totally replication incompetent (Fig. 1B) similar to results previously reported [26]. Nevertheless, the output of MVA was slightly less than the input in Vero cells (Fig. 1C). As predicted, the rMVAs grew to higher titers in Vero cells than MVA. The rMVA 51.1 reproducibly attained higher titers than the rMVA 44/47.1, despite the larger plaque size of the latter. By 72 h the titer of rMVA 51.1 approached that of ACAM2000. The yields of ACAM2000 and rMVA 51.1 in Vero cells increased over input by about 2 logs, whereas the rMVA 44/47.1 increased less (Fig. 1C). In CEFs, the parental MVA and rMVAs grew to similar titers, which were about 3-fold higher than that of the rMVAs in Vero cells.
Fig. 2.

Comparison of virulence in the intranasal challenge model.
Groups (n=5) of 5- to 6-weeks-old female mice (BALB/c C57BL/6 and ICR-SCID) were infected intranasally with 106 PFU of MVA, rMVA 44/47.1 and rMVA 51.1, Ankara and ACAM2000 viruses. The uninfected group was mock infected with diluents. Animals were monitored daily for weight loss and death for 47 days although only 14 are shown in the figure. Error bars indicate standard error. Asterisks in BALB/c and 57BL/6 groups indicate numbers of mice infected with Ankara that were sacrificed due to weight loss >30%. In ICR-SCID mice group infected with Ankara, the asterisks refer to two deaths and three sacrificed mice. One ACAM2000-infected SCID mouse was sacrificed due to weight loss >30% at day 47. The postmortem was positive for ACAM2000 virus.
The rMVA 44/47.1 and rMVA 51.1 were passed seven times consecutively in Vero cells to evaluate their replicative stability. After each round the rMVAs were titered by plaque assay in CEF and Vero cells. The titers remained constant regardless of which cell line was used for titering (Table 1). In addition, the slightly higher yields of 51.1 compared to 44/47.1 and their distinctive plaque morphologies were preserved.
Table 1.
Stable replication of rMVAs in Vero cells
| 51.1 (pfu × 106) | 44/47.1 (pfu × 106) | |||
|---|---|---|---|---|
|
| ||||
| Passage No. | CEF | Vero | CEF | Vero |
| 1 | 3.2 | NT | 1.2 | NT |
| 2a | 5.0 | 5.2 | 2.4 | 6.5 |
| 2b | 9.0 | 9.6 | NT | NT |
| 3a | 13 | 16 | 1.3 | 3.9 |
| 3b | 6.4 | 6.5 | NT | NT |
| 4a | 4.8 | 5.3 | 0.9 | 1.2 |
| 4b | 6.2 | 5.4 | 0.6 | 1.8 |
| 5a | 9.5 | 8.4 | 4.6 | 6.7 |
| 5b | 20 | 20 | 1.1 | 5.4 |
| 6a | 6.2 | 12 | 2.0 | 3.8 |
| 6b | 6.5 | 5.6 | 1.4 | 4.1 |
| 7a | 5.6 | 8.0 | 3.2 | 8.0 |
| 7b | 8.5 | 13 | 1.6 | 2.8 |
a and b represent independent serial passages.
Passage 1: cells infected with 0.1 pfu/cell.
Passage 2: cells infected with 0.1 ml of previous.
Passage 3 – 7: cells infected with 0.05 ml of previous.
Cells harvested at 48 h after each passage.
NT: not titered
3.2. Comparison of MVA and rMVA virulence in upper respiratory challenge model
We were interested in determining whether increased virulence was associated with the extended host range of the rMVAs. Immunocompetent BALB/c and C57BL/6 mice and immunodeficient ICR-SCID mice were challenged by the intranasal route with 106 pfu of MVA, rMVA 44/47.1, rMVA 51.1, vaccine strain ACAM2000, and Ankara viruses. Morbidity and mortality were monitored for up to 47 days after infection. Severe weight loss and 100% mortality occurred in all mouse strains infected with Ankara (Fig. 2A,B,C). In contrast, there was no weight loss with MVA, rMVA 44/47.1, rMVA 51.1 or ACAM2000 in either C57BL/6 or ICR-SCID mice (Fig. 2B,C). In BALB/c mice, there was slightly greater weight loss in the animals infected with ACAM2000 than in those infected with rMVAs though the differences were not statistically significant (Fig. 2A). Although the BALB/c mice did not lose weight after infection with MVA or the rMVAs, the weight gain observed in the controls did not occur (Fig. 2A). Thus, in this intranasal model, the extended host range of rMVA 44/47.1 and rMVA 51.1 was not accompanied by enhanced virulence.
3.3. Comparison of MVA and rMVA virulence in an intracranial mouse model
To investigate neurovirulence differences, BALB/c mice were infected with MVA, rMVA 44/47.1, rMVA 51.1 and ACAM2000 viruses via the intracranial route. Groups of mice inoculated with 104 or 105 pfu did not show signs of morbidity regardless of the virus inoculated (data not shown). Significant differences between the virus strains occurred with the 106 pfu inoculation groups (Fig. 3A). Mice infected by ACAM2000 exhibited greater weight loss than by MVA, rMVA 44/47.1 and rMVA 51.1 with p < 0.01, <0.05 and <0.05, respectively, during the first week. However, the lower weight loss caused by MVA relative to the two rMVAs also reached significance (p<0.01) with the 106 pfu dose. At 107 pfu, severe weight loss and deaths occurred in each of the groups including MVA (Fig. 3B). Although the three groups of mice exhibited only slight weight differences (Fig. 3B), the MVA-infected mice except one managed to stay above the 30% weight loss cut off requiring sacrifice whereas all of the ACAM2000-infected mice fell below that level and were sacrificed (Fig. 3C). For rMVA51.1 and rMVA 44/47.1 the survival rates were 33% and 7% respectively. The differences between the survival of MVA and the other viruses were statistically significant (p<0.001). However, the difference in survival between rMVA 51.1 and ACAM2000 was also significant (p<0.01). At 108 pfu, all of the mice except for one in the rMVA 51.1 group died or were sacrificed. For comparison, the neurovirulent WR strain of VACV has an LD50 of only 10 pfu by the intracranial route [27].
Fig. 3.

Comparison of virulence in intracranial challenge model. (A) Five- to six-weeks-old female BALB/c mice were infected intracranially with 106 PFU of MVA (n=10), rMVA 44/47.1 (n=15) and rMVA 51.1 (n=15) and ACAM2000 (n=15) viruses. The weight control group was mock infected with diluents. Animals were monitored daily for weight loss for 21 days with no deaths. The data shown are combined from two independent experiments. Error bars indicate standard error. (B) Experimental details are the same as in panel A except that the mice were infected with 107 pfu of MVA (n=15), rMVA 44/47.1 (n=15) and rMVA 51.1 (n=15) and ACAM2000 (n=15) viruses. (C) All 15 mice infected with ACAM2000, 14 mice infected with rMVA 44/47.1, 10 mice infected with rMVA 51.1 and 1 mouse infected with MVA were sacrificed on days 4 – 5 due to >30% loss of starting weight.
3.4. Formation of skin lesions
The abilites of MVA, rMVAs and ACAM2000 viruses to produce skin lesions upon tail scratch inoculation of mice were tested. Mice scarified with ACAM2000 (2×105 pfu) had a lesion at the site of inoculation on the tail and developed a scab by day 12, whereas only redness was detected after scarification with a much higher (108 pfu) dose of MVA, rMVA 44/47.1 or rMVA 51.1. Thus the greater replication of the rMVAs in cell culture was not accompanied by increased skin lesion size.
3.5. Induction of humoral and cellular immunity
Skin scarification is the preferred route for administering smallpox vaccine such as ACAM2000 to humans, whereas the intramuscular route has usually been used for MVA. However, Liu et al. [28] reported that skin scarification of mice is also superior to other routes for administration of MVA. We therefore used both tail scratch and intramuscular routes for MVA and rMVAs and compared them to tail scratch with ACAM2000. During the three weeks post vaccination, antibody and CTL production were measured. Similar antibody levels were induced in mice vaccinated with MVA, rMVA 44/47.1, or rMVA 51.1 (Fig. 5A). Dose-dependence occurred in each case except for tail scratch inoculation of MVA. ACAM2000 gave higher antibody titers than MVA or rMVA administered by tail scratch. However, we did not observe an advantage of tail scratch inoculation as the antibody titers for MVA and rMVAs were higher following intramuscular inoculation (Fig. 5A). Moreover, at the highest intramuscular dose (108 pfu), the antibody titers were similar to that induced by tail scratch with 2 × 105 pfu of ACAM2000.
Fig. 5.

Vaccine-induced immune responses. (A) IgG antibodies. Groups of mice (n=4) were vaccinated intramuscularly or by tail scratch with 105, 106, 107 and 108 pfu of MVA, rMVA 44/47.1 or rMVA 51.1 intramuscularly or by tail scarification. Four mice were inoculated by tail scratch (t.s.) with 2×105 pfu of ACAM2000. The intramuscular vaccination with 105 pfu was repeated with 10 mice for each virus strain and the data combined with that of the initial experiment. Animals were bled 3 weeks post vaccination. Serum ELISA IgG titers were determined using 96-well plates coated with purified MVA. Geometric mean titers (GMT) were determined. The error bars represent geometric standard deviations. In some cases the values were so close that the error bars were not resolved. (B) CD8+ T cells. Mice (n=5) were inoculated intramuscularly (i.m) with 106 pfu of MVA, rMVA 44/47.1 or rMVA 51.1 or by tail scratch (t.s.) with 2 ×105 pfu of ACAM2000. After seven days, splenocytes were processed as described in Materials and methods and the percentage of CD8+ splenocytes that were positive for IFNγ, TNFα, and IL-2+ was determined by flow cytometry.
One week after intramuscular vaccination, the CD8 responses to MVA and rMVAs were determined in splenocytes harvested from individual animals. The CD8+IFN-γ+ and CD8+TNF+ responses induced by MVA, rMVA 44/47.1 and rMVA 51.1 were similar and higher than the responses induced by tail scratch inoculation of ACAM2000 (Fig. 5B). The CD8+IL-2+ response pattern was similar but lower than to the other cytokines (Fig. 5B). Wyatt et al (2004) had previously shown that the induction of CD8+ T cells by MVA was dose dependent and that at high doses the response was greater than with Dryvax, from which ACAM2000 was derived.
3.6. Protection from lethal challenge with a pathogenic strain of VACV
At four weeks after intramuscular or tail scratch vaccination of BALB/c mice with 105, 106 or 107 pfu of MVA, rMVA 44/47.1 or rMVA 51.1 or 2 × 105 pfu of ACAM2000 by tail scratch, the mice were challenged by intranasal infection with 107 pfu of VACV WR. This dose of VACV WR was a log higher than routinely used in order to provide a more rigorous challenge that might allow better comparisons. Unvaccinated mice rapidly lost weight and died by day six, whereas mice immunized with ACAM2000 lost weight during the first four days but then all gradually recovered (Fig. 6A). Mice vaccinated with 107 pfu of MVA, rMVA 44/47.1 or rMVA 51.1 by tail scratch survived the challenge with weight losses slightly greater than with ACAM2000 (Fig. 6A). At a vaccination dose of 105 and 106 pfu, however, there were some deaths in the MVA and rMVA groups. However, all mice that were immunized with MVA or rMVAs by the intramuscular route survived. Although the challenged mice that had received intramuscular vaccination with 105 and 106 pfu of MVA and rMVAs appeared to lose slightly more weight than those vaccinated with ACAM2000 (Fig. 6A), the small number of animals (n=4) in this initial experiment precluded meaningful statistical analysis. Therefore, we repeated the intramuscular vaccination with 105 pfu using 10 mice in each group. Following challenge, the weight losses between the groups were not statistically different and the survival for mice immunized with ACAM2000, MVA, rMVA 44/47.1 and rMVA 51.1 was 90%, 80%, 90% and 100%, respectively (Fig. 6B).
Fig. 6.
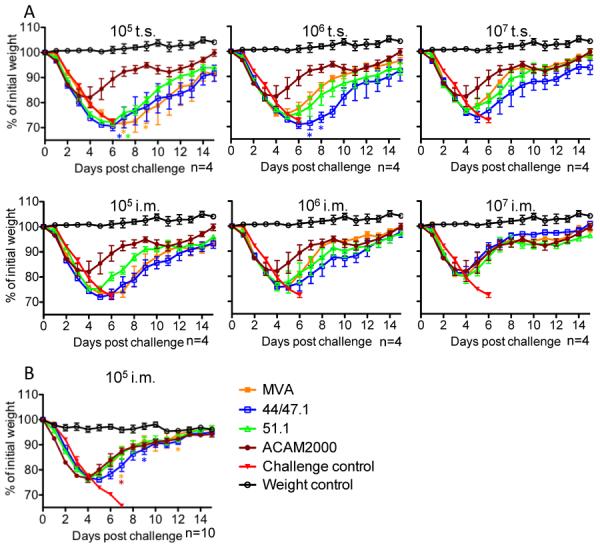
Protection of BALB/c mice against lethal VACV WR challenge. (A) Groups (n=4) of 6-week-old female BALB/c mice were vaccinated intramuscularly (i.m.) or by tail scrach (t.s.) with MVA, rMVA 44/47.1 and rMVA 51.1 at 105 to 107 pfu or with 2×105 pfu of ACAM2000 by tail scratch. Challenge control group (n=4) was mock vaccinated intramuscularly with diluents. Four weeks later, the animals were challenged intranasally with 107 pfu of VACV WR, and monitored for 21 days for weight loss and death. All of the challenged mice in the control group died. Asterisks in 105 pfu t.s. groups represent individual mice that were sacrificed. The asterisk in 106 pfu t.s. group on day 7 represented a death and on day 8 a sacrifice (B) Groups (n=10) of mice were vaccinated intramuscularly with 105 pfu of MVA, rMVA 44/47.1 or rMVA 51.1 or with 2.5 × 105 pfu of ACAM2000 and challenged with VACV WR as described in panel A. The challenged controls were found dead, whereas asterisks in the vaccinated groups indicate sacrifice due to loss of 30% of starting weight.
3.7. Discussion
MVA is currently being deployed as a vector for candidate vaccines against numerous microbial pathogens and cancer. While MVA appears to be exceptionally safe, efforts are being made to improve immunogenicity and vaccine production. Two main approaches have been taken to improve immune responses by MVA: deletion of remaining host defense or other genes [29–33] and insertion of cytokine genes [34, 35]. Efforts to improve vaccine production include the replacement of primary chick embryo fibroblasts with avian suspension cell lines [36, 37]. An alternative approach to facilitate vaccine production would be to extend the host range of MVA to an existing mammalian cell line that is approved for vaccine production. We envisioned that increased replication of a host-range extended MVA might also provide a more robust immune response. However, the host-range extension would need to be accomplished without compromising safety. In this regard, an attempt was made to increase the host-range and properties of MVA by replacement of mutated K1L and F11L open reading frames with full-length versions. Expression of F11L increased the motility of infected cells and expression of K1L increased virus yield in rabbit cells but neither one alone or the two together increased the size of plaques or virus yield in human, monkey or murine cells and neither safety nor immunogenicity was tested [38]. Mayr [39] succeeded in modifying the host range of MVA by repeatedly passaging the virus in Vero cells.
As a starting point, we took advantage of rMVAs that had been produced by transfection with one or two cosmids containing DNA from replication-competent strain of VACV [16]. The rMVAs were screened for their ability to replicate in African green monkey BS-C-1 cells. Two rMVAs, 51.1 and 44/47.1, were selected for the present study. We found that the host range of rMVA 44/47.1 and rMVA 51.1 included Vero cells, a widely used substrate for vaccine production. The yield of rMVA 51.1 was nearly 2-logs higher than the input and that of the parent MVA and approached that of ACAM2000, a vaccine strain that is normally propagated on Vero cells. It was surprising that the yield of rMVA 51.1 was higher than that of rMVA 44/47.1 since the latter formed larger plaques. One explanation may be that rMVA 44/47.1 is more cytotoxic (unpublished data of LSW). The replicative stability of both rMVA 51.1 and rMVA 44/47.1 was established by passing the viruses consecutively seven times in Vero cells. The sequences of the parental Ankara used to construct the cosmid library, rMVA 51.1, rMVA 44/47.1 and the genes contributing to enhanced replication and virus spread are currently being analyzed. There are notable genetic and phenotypic differences between the two rMVAs [16] and the altered MVA strain subsequently described by Mayr [39]. The altered strain lost DNA during passaging in Vero cells, did not acquire enhanced replication in human cells and suffered decreased replication in CEF. In contrast the rMVAs obtained additional DNA through marker rescue, acquired the ability to replicate in human cells as well as African green monkey cells, and retained efficient replication in CEF.
Despite evidence for enhanced replication in cultured mammalian cells, the rMVAs were highly attenuated in the mouse model. Following intranasal inoculation of 106 pfu of the rMVAs, neither immunocompetent BALB/c and C57BL/6 mice nor immunodeficient SCID mice showed signs of morbidity or mortality, in contrast to the deaths of all mice infected with the Ankara strain from which the DNA used for insertion into MVA was derived and one SCID mouse infected with ACAM2000. In order to provide a more lethal challenge model, we inoculated mice intracranially. A wide dose range of 104 to 108 pfu of MVA and rMVAs and 104 to 107 pfu of ACAM 2000 were tested. Since all mice inoculated with 104 or 105 pfu exhibited neither weight loss nor deaths and all mice inoculated with 108 pfu except one in the rMVA 51.1 group died or were sacrificed, only those inoculated with 106 and 107 pfu provided useful comparisons. At 106 pfu of virus there was some weight loss but no deaths. Based on extent of weight loss, MVA was significantly more attenuated than either of the rMVAs or ACAM2000. However, both rMVAs were significantly more attenuated than ACAM2000. Deaths occurred at the higher dose of 107 pfu and again MVA was the least virulent but mortality associated with rMVA 51.1 was less than with ACAM2000 and this was also statistically significant. The higher morbidity of the rMVAs compared to MVA could be due to cytoxicity or low replication in the brain. Whereas tail scratch inoculation of ACAM2000 caused skin lesions, a 500-fold higher inoculation of rMVAs and MVA did not. These studies indicated that considerable attenuation is retained even after the host-range of MVA is extended in cell culture. In addition to extending host range and maintaining attenuation, we hoped that rMVAs 51.1 and 44/47.1 would provide a greater immune response. However, specific antibody and CD8+ T cell induction by MVAs and rMVAs were similar to each other and they provided equivalent protection against an intranasal challenge by a pathogenic strain of VACV. These data suggest that the rMVAs did not replicate efficiently in mice when inoculated by tail scratch or intramuscularly. It may be useful, however, to test rMVAs in additional animal models including non-human primates to further evaluate attenuation and immunogenicity.
Some related studies have been carried out with NYVAC, a highly attenuated strain of VACV created by deletion of 18 open reading frames, including the C7L and K1L host range genes, from the Copenhagen strain [40]. NYVAC is able to replicate in African green monkey cells, but not in human cells; however, addition of the VACV C7L host range gene into NYVAC restored replication in human and murine cells without enhancing virulence by nasal inoculation of BALB/c mice [41]. The immunogenicity of a NYVAC-C7L vector expressing HIV proteins was enhanced in mice [42] and a NYVAC with a deletion of the B19 type 1 interferon-binding protein and insertions of C7L and K1L had improved innate and adaptive immunostimulatory properties [43, 44].
Modified vaccinia virus Ankara (MVA) is severely host-range restricted
Recombinant host-range extended MVAs can replicate in Vero and other mammalian cells
Host-range extended MVAs remain severely attenuated in mouse models
Immunogenicity of host-range extended MVAs is similar to MVA
Extended host range may be beneficial for vaccine production
Fig. 4.
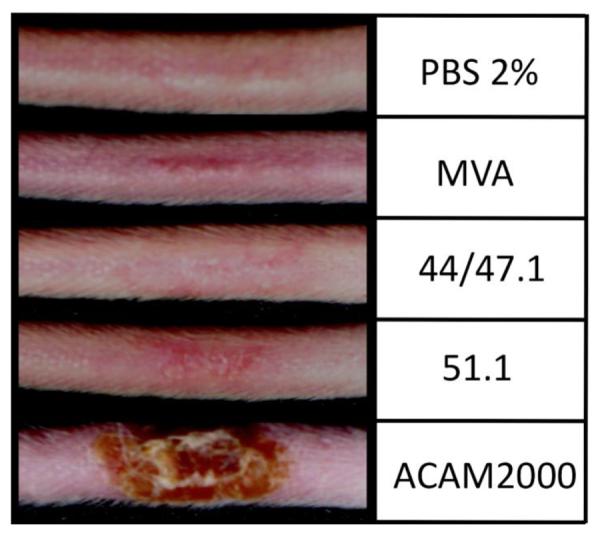
Lesion formation following tail scarification. Groups (n=3) of 5-weeks-old BALB/c mice were inoculated by tail scratch with 108 PFU of MVA, rMVA 44/47.1 or rMVA 51.1 or with 2×105 pfu of ACAM2000 or PBS containing 2% fetal bovine serum. Mice were inspected for lesion formation. Images were made on day 12.
Acknowledgments
We thank Jeffrey Americo and Catherine Cotter for excellent technical assistance and Patricia Earl for advice. This work was supported by the Intramural Program of the National Institutes of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hochstein-Mintzel V, Huber HC, Stickl H. Virulenz und immunogenität eines modifizierten vaccinia-virus (Stamm MVA) [Virulence and immunogenicity of a modified vaccinia virus (strain MVA) Z Immun-Forsch. 1972;144:140–5. [PubMed] [Google Scholar]
- [2].Stickl H, Hochstein-Mintzel V, Mayr A, Huber HC, Schäfer H, Holzner A. MVA-stufenimpfung gegen pocken. klinische erprobung des attenuierten pocken-lebendimpfstoffes, stamm MVA (MVA vaccination against smallpox: clinical trials of an attenuated live vaccinia virus strain (MVA) Dtsch Med Wschr. 1974;99:2386–92. doi: 10.1055/s-0028-1108143. [DOI] [PubMed] [Google Scholar]
- [3].Mayr A, Hochstein-Mintzel V, Stickl H. Abstammung, eigenschaften und verwendung des attenuierten vaccinia-stammes MVA (Passage history, properties, and applicability of the attenuated vaccinia virus strain MVA) Infection. 1975;3:6–14. [Google Scholar]
- [4].Mayr A, Stickl H, Müller HK, Danner K, Singer H. Der pockenimpfstamm MVA: marker, genetische struktur, erfahrungen mit der parenteralen schutzimpfung und verhalten im abwehrgeschwächten organismus (The smallpox vaccination strain MVA: marker, genetic structure, experience gained with parenteral vaccination and behavior in organisms with debilitated defense mechanism. Zbl Bakt Hyg. 1978;167:375–90. [PubMed] [Google Scholar]
- [5].Sutter G, Moss B. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc Natl Acad Sci USA. 1992;89:10847–51. doi: 10.1073/pnas.89.22.10847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sutter G, Wyatt LS, Foley PL, Bennink JR, Moss B. A recombinant vector derived from the host range-restricted and highly attenuated MVA strain of vaccinia virus stimulates protective immunity in mice to influenza virus. Vaccine. 1994;12:1032–40. doi: 10.1016/0264-410x(94)90341-7. [DOI] [PubMed] [Google Scholar]
- [7].Drexler I, Staib C, Sutter G. Modified vaccinia virus Ankara as antigen delivery system: how can we best use its potential? Curr Opin Biotech. 2004;15:506–12. doi: 10.1016/j.copbio.2004.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gomez CE, Najera JL, Krupa M, Perdiguero B, Esteban M. MVA and NYVAC as Vaccines against Emergent Infectious Diseases and Cancer. Curr Gene Ther. 2011;11:189–217. doi: 10.2174/156652311795684731. [DOI] [PubMed] [Google Scholar]
- [9].Antoine G, Scheiflinger F, Dorner F, Falkner FG. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology. 1998;244:365–96. doi: 10.1006/viro.1998.9123. [DOI] [PubMed] [Google Scholar]
- [10].Carroll M, Moss B. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: propagation and generation of recombinant viruses in a nonhuman mammalian cell line. Virology. 1997;238:198–211. doi: 10.1006/viro.1997.8845. [DOI] [PubMed] [Google Scholar]
- [11].Drexler I, Heller K, Wahren B, Erfle V, Sutter G. Highly attenuated modified vaccinia virus Ankara replicates in baby hamster kidney cells, a potential host for virus propagation, but not in various human transformed and primary cells. J Gen Virol. 1998;79:347–52. doi: 10.1099/0022-1317-79-2-347. [DOI] [PubMed] [Google Scholar]
- [12].Jordan I, Horn D, Oehmke S, Leendertz FH, Sandig V. Cell lines from the Egyptian fruit bat are permissive for modified vaccinia Ankara. Virus Res. 2009;145:54–62. doi: 10.1016/j.virusres.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Moss B. Poxviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 2905–46. [Google Scholar]
- [14].Altenburger W, Süter C-P, Altenburger J. Partial deletion of the human host range gene in the attenuated vaccinia virus MVA. Arch Virol. 1989;105:15–27. doi: 10.1007/BF01311113. [DOI] [PubMed] [Google Scholar]
- [15].Meyer H, Sutter G, Mayr A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J Gen Virol. 1991;72:1031–8. doi: 10.1099/0022-1317-72-5-1031. [DOI] [PubMed] [Google Scholar]
- [16].Wyatt LS, Carroll MW, Czerny C-P, Merchlinsky M, Sisler JR, Moss B. Marker rescue of the host range restricted defects of modfied vaccinia virus Ankara. Virology. 1998;251:334–42. doi: 10.1006/viro.1998.9397. [DOI] [PubMed] [Google Scholar]
- [17].Meisinger-Henschel C, Spath M, Lukassen S, Wolferstatter M, Kachelriess H, Baur K, et al. Introduction of the six major genomic deletions of modified vaccinia virus Ankara (MVA) into the parental vaccinia virus is not sufficient to reproduce an MVA-like phenotype in cell culture and in mice. J Virol. 2010;84:9907–19. doi: 10.1128/JVI.00756-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dimier J, Ferrier-Rembert A, Pradeau-Aubreton K, Hebben M, Spehner D, Favier AL, et al. Deletion of major nonessential genomic regions in the vaccinia virus Lister strain enhances attenuation without altering vaccine efficacy in mice. J Virol. 2011;85:5016–26. doi: 10.1128/JVI.02359-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Earl PL, Moss B, Wyatt LS, Carroll MW. Generation of recombinant vaccinia viruses. Curr Protoc Mol Biol. 2001;Chapter 16:16/7/1–7.9. doi: 10.1002/0471142727.mb1617s43. [DOI] [PubMed] [Google Scholar]
- [20].Earl PL, Cooper N, Wyatt LS, Moss B, Carroll MW. Preparation of cell cultures and vaccinia virus stocks. Curr Protoc Mol Biol. 2001;43:16..1–3. doi: 10.1002/0471142727.mb1616s43. [DOI] [PubMed] [Google Scholar]
- [21].Meisinger-Henschel C, Schmidt M, Lukassen S, Linke B, Krause L, Konietzny S, et al. Genomic sequence of chorioallantois vaccinia virus Ankara, the ancestor of modified vaccinia virus Ankara. J Gen Virol. 2007;88:3249–59. doi: 10.1099/vir.0.83156-0. [DOI] [PubMed] [Google Scholar]
- [22].Melamed S, Paran N, Katz L, Ben-Nathan D, Israely T, Schneider P, et al. Tail scarification with vaccinia virus Lister as a model for evaluation of smallpox vaccine potency in mice. Vaccine. 2007;25:7743–53. doi: 10.1016/j.vaccine.2007.09.023. [DOI] [PubMed] [Google Scholar]
- [23].Earl PL, Cotter C, Moss B, Vancott T, Currier J, Eller LA, et al. Design and evaluation of multi-gene, multi-clade HIV-1 MVA vaccines. Vaccine. 2009;27:5885–95. doi: 10.1016/j.vaccine.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Earl PL, Americo JL, Wyatt LS, Eller LA, Montefiori DC, Byrum R, et al. Recombinant modified vaccinia virus Ankara provides durable protection against disease caused by an immunodeficiency virus as well as long-term immunity to an orthopoxvirus in a non-human primate. Virology. 2007;366:84–97. doi: 10.1016/j.virol.2007.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Monath TP, Caldwell JR, Mundt W, Fusco J, Johnson CS, Buller M, et al. ACAM2000 clonal Vero cell culture vaccinia virus (New York City Board of Health strain) - a second-generation smallpox vaccine for biological defense. Int J Infect Dis. 2004;8:S31–S44. doi: 10.1016/j.ijid.2004.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Okeke MI, Nilssen O, Traavik T. Modified vaccinia virus Ankara multiplies in rat IEC-6 cells and limited production of mature virions occurs in other mammalian cell lines. J Gen Virol. 2006;87:21–7. doi: 10.1099/vir.0.81479-0. [DOI] [PubMed] [Google Scholar]
- [27].Buller RML, Smith GL, Cremer K, Notkins AL, Moss B. Decreased virulence of recombinant vaccinia virus expression vectors is associated with a thymidine kinase-negative phenotype. Nature. 1985;317:813–5. doi: 10.1038/317813a0. [DOI] [PubMed] [Google Scholar]
- [28].Liu LZ, Zhong Q, Tian T, Dubin K, Athale SK, Kupper TS. Epidermal injury and infection during poxvirus immunization is crucial for the generation of highly protective T cell-mediated immunity. Nature Med. 2010;16:224–7. doi: 10.1038/nm.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Garcia-Arriaza J, Najera JL, Gomez CE, Tewabe N, Sorzano COS, Calandra T, et al. A candidate HIV/AIDS vaccine (MVA-B) lacking vaccinia virus gene C6L enhances memory HIV-1-specific T-cell responses. PLoS One. 2011;6 doi: 10.1371/journal.pone.0024244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Falivene J, Zajac MPD, Pascutti MF, Rodriguez AM, Maeto C, Perdiguero B, et al. Improving the MVA vaccine potential by deleting the viral gene coding for the IL-18 binding protein. PLoS One. 2012;7 doi: 10.1371/journal.pone.0032220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Perdiguero B, Gomez CE, Najera JL, Sorzano COS, Delaloye J, Gonzalez-Sanz R, et al. Deletion of the viral anti-apoptotic gene F1L in the HIV/AIDS vaccine candidate MVA-C enhances immune responses against HIV-1 antigens. PLoS One. 2012;7 doi: 10.1371/journal.pone.0048524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Garber DA, O'Mara LA, Zhao J, Gangadhara S, An I, Feinberg MB. Expanding the repertoire of modified vaccinia Ankara-based vaccine vectors via genetic complementation strategies. PLoS One. 2009;4 doi: 10.1371/journal.pone.0005445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Garber DA, O'Mara LA, Gangadhara S, McQuoid M, Zhang X, Zheng R, et al. Deletion of specific immune-modulatory genes from modified vaccinia virus Ankara-based HIV vaccines engenders improved immunogenicity in Rhesus macaques. J Virol. 2012;86:12605–15. doi: 10.1128/JVI.00246-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chavan R, Marfatia KA, An IC, Garber DA, Feinberg AB. Expression of CCL20 and granulocyte-macrophage colony-stimulating factor, but not Flt3-L, from modified vaccinia virus Ankara enhances antiviral cellular and humoral immune responses. J Virol. 2006;80:7676–87. doi: 10.1128/JVI.02748-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Perera LP, Waldmann TA, Mosca JD, Baldwin N, Berzofsky JA, Oh SK. Development of smallpox vaccine candidates with integrated interleukin-15 that demonstrate superior immunogenicity, efficacy, and safety in mice. J Virol. 2007;81:8774–83. doi: 10.1128/JVI.00538-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jordan I, Vos A, Beilfuss S, Neubert A, Breul S, Sandig V. An avian cell line designed for production of highly attenuated viruses. Vaccine. 2009;27:748–56. doi: 10.1016/j.vaccine.2008.11.066. [DOI] [PubMed] [Google Scholar]
- [37].Lohr V, Rath A, Genzel Y, Jordan I, Sandig V, Reichl U. New avian suspension cell lines provide production of influenza virus and MVA in serum-free media: Studies on growth, metabolism and virus propagation. Vaccine. 2009;27:4975–82. doi: 10.1016/j.vaccine.2009.05.083. [DOI] [PubMed] [Google Scholar]
- [38].Zwilling J, Sliva K, Schwantes A, Schnierle B, Sutter G. Functional F11L and K1L genes in modified vaccinia virus Ankara restore virus-induced cell motility but not growth in human and murine cells. Virology. 2010;404:231–9. doi: 10.1016/j.virol.2010.05.008. [DOI] [PubMed] [Google Scholar]
- [39].Mayr A. Altered strain of the modified vaccinia virus Ankara (MVA) USA patent 6,682,743 B2. 2004
- [40].Tartaglia J, Perkus ME, Taylor J, Norton EK, Audonnet JC, Cox WI, et al. NYVAC - A highly attenuated strain of vaccinia virus. Virology. 1992;188:217–32. doi: 10.1016/0042-6822(92)90752-b. [DOI] [PubMed] [Google Scholar]
- [41].Najera JL, Gomez CE, Domingo-Gil E, Gherardi MM, Esteban M. Cellular and biochemical differences between two attenuated poxvirus vaccine candidates (MVA and NYVAC) and role of the C7L gene. J Virol. 2006;80:6033–47. doi: 10.1128/JVI.02108-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Najera JL, Gomez CE, Garcia-Arriaza J, Sorzano CO, Esteban M. Insertion of Vaccinia Virus C7L Host Range Gene into NYVAC-B Genome Potentiates Immune Responses against HIV-1 Antigens. PLoS One. 2010;5 doi: 10.1371/journal.pone.0011406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Quakkelaar ED, Redeker A, Haddad EK, Harari A, McCaughey SM, Duhen T, et al. Improved innate and adaptive immunostimulation by genetically modified HIV-1 protein expressing NYVAC vectors. PLoS One. 2011;6:e16819. doi: 10.1371/journal.pone.0016819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kibler KV, Gomez CE, Perdiguero B, Wong SM, Huynh T, Holechek S, et al. Improved NYVAC-based baccine bectors. PLoS One. 2011;6 doi: 10.1371/journal.pone.0025674. [DOI] [PMC free article] [PubMed] [Google Scholar]