

Abstract
β-Elemene is a promising new plant-derived drug with broad-spectrum anticancer activity. It also increases cisplatin cytotoxicity and enhances cisplatin sensitivity in resistant human carcinoma cells. However, little is known about the mechanism of its action. To explore the potential therapeutic application of β-elemene as a drug-resistance modulator, this study investigated the underlying mechanism of β-elemene activity in cisplatin-resistant ovarian cancer cells. β-Elemene enhanced cisplatin sensitivity to a much greater extent in chemoresistant A2780/CP70 and MCAS human ovarian carcinoma cells compared to the chemosensitive parental cell line A2780. The dose-modifying factors for cisplatin were between 35 and 60 for A2780/CP70 cells and between 1.6 and 2.5 for A2780 cells. In the cisplatin-resistant ovarian carcinoma cells, β-elemene abrogated cisplatin-induced expression of excision repair cross-complementation group-1 (ERCC-1), a marker gene in the nucleotide excision repair pathway that repairs cisplatin-caused DNA damage. In addition, β-elemene not only reduced the level of X-linked inhibitor of apoptosis protein (XIAP), but also downregulated cisplatin-mediated XIAP expression in chemoresistant cells. Furthermore, β-elemene blocked the cisplatin-stimulated increase in the level of phosphorylated c-Jun NH2-terminal kinase (JNK) in these cells. These novel findings suggest that the β-elemene enhancement of cisplatin sensitivity in human chemoresistant ovarian cancer cells is mediated at least in part through the impairment of DNA repair activity and the activation of apoptotic signaling pathways, thereby making resistant ovarian cancer cells susceptible to cisplatin-induced cell death.
Keywords: apoptosis, cisplatin resistance, β-elemene, Chinese medicine, ovarian cancer, excision repair cross-complementation group-1, X-linked inhibitor of apoptosis protein, c-Jun NH2-terminal kinase
Introduction
Human ovarian carcinoma remains a major cause of mortality and morbidity in the United States (1). Roughly 80% of patients will present with advanced-stage disease, but current chemotherapy is able to produce significant response rates and even long-term remission in only 20% of these women. cis-Diamminedichloroplatinum (II) (cisplatin) is one of the most effective anticancer drugs in the treatment of human ovarian cancer and other tumors (2–4). However, the efficacy of cisplatin is hampered because cancer cells acquire resistance to its cytotoxicity. Although the mechanism of cisplatin resistance in vivo is not clearly defined, laboratory studies with tumor tissues and cell lines suggest that enhanced nucleotide excision repair (NER) of cisplatin-caused DNA damage and impaired cisplatin-induced apoptosis play crucial roles in the development of the cisplatin-resistance phenotype (4–6). The expression of DNA repair genes such as excision repair cross-complementation group-1 (ERCC-1) and xeroderma pigmentosum complementation group A (XPA) is reported to be strongly associated with a poor prognosis in ovarian carcinoma and other tumors (7,8). Therefore, compounds that can circumvent cisplatin resistance and augment the effects of chemotherapy are needed.
One candidate drug to fulfill this role is β-elemene (β-1-methyl-1-vinyl-2,4-di-isopropenyl-cyclohexane). β-Elemene (Fig. 1), a natural anticancer plant-derived drug, was approved by the Chinese Food and Drug Administration for the treatment of human cancers. The major advantages of β-elemene as an anticancer drug are: i) it has broad-spectrum antitumor effects in many types of cancer, including drug-resistant tumors; ii) it does not direct multidrug resistance and can reverse the resistance to other drugs; and iii) it has low toxicity and is therefore well tolerated and accepted by patients with cancer. The effect of β-elemene on other drug sensitivity in human tumors is unknown. We have recently reported that β-elemene increases sensitivity to cisplatin and augments cisplatin-induced apoptosis in chemoresistant human ovarian cancer cells and other tumor cells (9–20). These novel findings indicate that β-elemene may be efficacious in the treatment of cisplatin-resistant tumors.
Figure 1.

The chemical structure of β-elemene.
We hypothesized that β-elemene enhancement of cisplatin sensitivity in human chemoresistant ovarian cancer cells is mediated at least in part through the regulation of DNA repair activity and apoptotic death signaling. To test this hypothesis and elucidate the mechanism underlying the effect of β-elemene on cisplatin cytotoxicity, we investigated whether β-elemene promotes apoptotic cell death in cisplatin-treated resistant ovarian cancer cells by downregulating the expression of ERCC-1 and X-linked inhibitor of apoptosis protein (XIAP) and inactivating c-Jun NH2-terminal kinase (JNK). This research is significant because β-elemene may be useful therapeutically as a modulator of platinum drug resistance in human malignancies. Our results provide a framework for exploiting the combination of β-elemene and cisplatin as a potentially effective chemotherapy regimen to overcome cisplatin drug resistance in ovarian cancer and other tumors.
Materials and methods
Chemicals and immunoreagents
The (−)-β-elemene (98% purity) was obtained from Yuanda Pharmaceuticals Ltd. Inc. (Dalian, China). Cisplatin, dimethylsufoxide and other chemicals were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Antibodies against XIAP and β-actin, peroxidase-labeled anti-rabbit immunoglobulin G (IgG), Blotto B, and the ECL western blot analysis system were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Anti-phospho-JNK1 (Thr183/Tyr185) antibody was purchased from Cell Signaling Technology (Beverly, MA, USA), as described previously (21,22).
Cells and cell culture conditions
The human cisplatin-resistant ovarian cancer cell lines A2780/CP70 and MCAS and the parental sensitive ovarian cancer cell line A2780 have been studied extensively by our laboratory (15,17,18). The cells were cultured in monolayers using RPMI-1640 medium (Invitrogen, Life Technologies, Gaithersburg, MD, USA) supplemented with 10% (v/v) fetal bovine serum, 50 U/ml penicillin, and 50 μg/ml streptomycin (Invitrogen), and were grown in logarithmic growth at 37°C in a humidified atmosphere of 5% CO2 and 95% air. The cells were routinely tested for mycoplasma infection using a commercial assay system (MytoTect; Invitrogen), and new cultures were established monthly from frozen stocks. All media and reagents contained <0.1 ng/ml endotoxin as determined by a Limulus polyphemus amebocyte lysate assay (Whittaker Bioproducts, Walkersville, MD, USA). Before starting the experiments, the cells were sub-cultured and grown to 70–80% confluence. Cisplatin was initially dissolved at 5 mM in phosphate-buffered saline (PBS) without Ca2+ or Mg2+. Cisplatin and β-elemene were serially diluted, respectively, in culture medium to obtain the desired concentrations.
Cell growth inhibition assay
The antiproliferative effects of β-elemene alone, cisplatin alone, and cisplatin plus β-elemene were assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Promega Corp., Madison, WI, USA) according to the manufacturer’s instructions. In brief, the cells were evenly distributed in 96-well plates (5×103 cells/well), grown overnight and then treated for 24, 48, 72 and 96 h with β-elemene alone (0, 20, 40, 60, 80, 100, 120, 140, 160, 180 and 200 μg/ml), cisplatin alone (0, 1.0, 2.0, 4.0, 8.0, 16.0, 32.0, 64.0, 128.0, 256.0 and 512.0 μM), or a combination of cisplatin (at the above concentrations) plus β-elemene (40 μg/ml). After incubation, 20 μl of CellTiter 96 Aqueous One Solution reagent were added to each well of the assay plates containing treated and untreated cells in 100 μl of culture medium, and the plates were incubated at 37°C and 5% CO2 for 1–4 h. The optical density at 590 nm was determined using a 96-well Opsys MR™ microplate reader (Thermo Labsystems, Chantilly, VA, USA). Proliferation rates were calculated from the optical density of drug-treated cells relative to that of cells with no added drug (control value, 100%), as follows: percentage cell viability = [(OD with drug - blank) ÷ (OD without drug - blank)] × 100. The half-maximal inhibitory concentration (IC50) was determined from the dose-response curves. The dose-modifying factor (DMF) was calculated as the IC50 for cisplatin without β-elemene divided by the IC50 for cisplatin with β-elemene: DMF = IC50 (cisplatin) ÷ IC50 (cisplatin + β-elemene).
Generation of ERCC-1 antiserum
Polyclonal anti-peptide antiserum was generated by Bio-Synthesis Inc. (Lewisville, TX, USA). A synthetic peptide containing the carboxy-terminus of ERCC-1 was coupled to keyhole limpet hemocyanin using m-maleimidobenzoyl-N-hydroxysuccinimide ester as a cross-linker. This was used to immunize New Zealand white female rabbits, which were bled at regular intervals to obtain serum containing the antibodies. The undiluted antiserum was used in western blot analyses, as described previously (23,24).
Protein extraction and western blot analysis
Ovarian tumor cells treated with β-elemene, cisplatin or their combination were harvested by trypsinization, washed with ice-cold PBS, and lysed on ice for 30 min in mammalian cell lysis buffer (Quality Biological Inc., Gaithersburg, MD, USA) containing 10 μl/ml 200 mM phenylmethylsulfonyl fluoride, 10 μl/ml 100 mM sodium orthovanadate and 10 μg/ml aprotinin. Lysates were clarified by centrifugation at 13,000 × g for 30 min at 4°C, and the protein concentrations in the supernatants were determined by Bradford assay (Bio-Rad, Richmond, CA, USA). Proteins (40 to 60 μg) from whole-cell lysates were mixed 1:1 with 2X sodium dodecyl sulfate (SDS) gel solution (Quality Biological Inc.), heated for 5 min at 95°C, separated by 10% SDS-polyacrylamide gel electrophoresis, and transferred to nitrocellulose membranes (Schleicher & Schuell BioScience Inc., Keene, NH, USA). After blocking in Blotto B for 1 h at room temperature, the membranes were incubated overnight at 4°C with specific primary antibodies (diluted 1:100–1:300). The membranes were washed with TBS/0.1% Tween-20 solution, incubated with anti-rabbit peroxidase-conjugated secondary antibody (diluted 1:10,000), and washed again. Immunoreactive bands were detected with enhanced chemiluminescence substrate according to the manufacturer’s instructions and visualized using X-ray film (Eastman Kodak, Rochester, NY, USA). All blots shown are representative of three independent experiments.
Statistical analysis
All quantitative values are presented as means ± SD. Data were analyzed using two-way analysis of variance (ANOVA) for comparison among groups. Student’s t-test was used to analyze the significance of differences between untreated and treated groups. All p-values were determined using a two-sided t-test, and p-values <0.05 were considered to indicate significance.
Results
β-Elemene suppresses cell proliferation and augments cisplatin-induced cytotoxicity in resistant and sensitive human ovarian cancer cells
We first examined the in vitro antitumor activity of β-elemene in human ovarian carcinoma cells, as determined by the MTT assay. β-Elemene at concentrations of 20–200 μg/ml dose-dependently inhibited the growth and proliferation of both A2780 and A2780/CP70 cells at 24, 48, 72 and 96 h, with IC50 values at 24, 48, 72 and 96 h ranging between 60 and 65 μg/ml for A2780 cells and between 65 and 80 μg/ml for A2780/CP70 cells (Table I). Similarly, the IC50 values of β-elemene for MCAS cells were between 60 and 78 μg/ml. The IC50 values were not significantly different between the cisplatin-sensitive (A2780) and cisplatin-resistant (A2780/CP70 and MCAS) cell lines (p>0.05), indicating that β-elemene has a similar antitumor activity toward both sensitive and resistant ovarian cancer cells in vitro. Thus, cisplatin-resistant ovarian tumor cells are still sensitive to β-elemene.
Table I.
β-Elemene increases cisplatin cytotoxicity and enhances cisplatin sensitivity in human ovarian carcinoma cells, as determined by MTT assay.
| IC50 | ||||
|---|---|---|---|---|
|
|
||||
| Drug | 24 h | 48 h | 72 h | 96 h |
| A2780 cells | ||||
| β-Elemene (μg/ml) | 65 | 65 | 65 | 60 |
| Cisplatin (μM) | 6.2 | 1.75 | 1.6 | 1.5 |
| β-Elemene + cisplatin (μM) | 3.8 | 0.8 | 0.75 | 0.6 |
| Dose-modifying factor | 1.6 | 2.2 | 2.1 | 2.5 |
| A2780/CP70 cells | ||||
| β-Elemene (μg/ml) | 80 | 70 | 68 | 65 |
| Cisplatin (μM) | 95 | 66 | 65 | 60 |
| β-Elemene + cisplatin (μM) | 2.5 | 1.9 | 1.85 | 1.0 |
| Dose-modifying factor | 38 | 34.7 | 35.1 | 60 |
The results reveal that β-elemene increased cisplatin cytotoxicity by 34.7- to 60-fold in A2780/CP70 cells, but only 1.6- to 2.5-fold in A2780 cells. The IC50 (half maximal inhibitory concentration) value is defined as the concentration of β-elemene or cisplatin needed for 50% inhibition of cell growth and proliferation. The dose-modifying factor (DMF) was calculated as the IC50 for cisplatin without β-elemene divided by the IC50 for cisplatin with β-elemene: DMF = IC50 (cisplatin) ÷ IC50 (cisplatin + β-elemene). MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
Next, we assessed the enhancing effect of β-elemene on cisplatin cytotoxicity in human ovarian tumor cells using the MTT assay. A2780 and A2780/CP70 cells were exposed to cisplatin alone or in combination with β-elemene (40 μl/ml) for 24, 48, 72 and 96 h, and the inhibition of cell growth was measured in vitro. At concentrations of 0 to 512.0 μM, cisplatin caused a dose-dependent inhibition of A2780 and A2780/CP70 cell proliferation at all four time points. The IC50 values of cisplatin alone for chemoresistant A2780/CP70 cells at 24, 48, 72 and 96 h were 95.0, 66.0, 65.0, and 60.0 μM, respectively, and decreased strikingly to 2.5, 1.9, 1.85 and 1.0 μM, respectively, when cisplatin was combined with β-elemene (Table I; p<0.01); the dose-modifying factors (DMFs) for cisplatin in A2780/CP70 cells ranged from 35 to 60. Similarly, the IC50 of cisplatin alone for MCAS cells was 38.0 μM and was reduced markedly to 6.5 μM when cisplatin was combined with β-elemene (p<0.01); the DMF in this cell line was 5.85. Although the IC50 values of cisplatin alone for chemosensitive A2780 cells (6.2, 1.75, 1.6 and 1.5 μM at 24, 48, 72 and 96 h, respectively) decreased significantly when cisplatin was combined with β-elemene (3.8, 0.8, 0.75 and 0.6 μM, respectively) (Table I; p<0.05), the DMFs for cisplatin in A2780 cells, which ranged from 1.6 to 2.5, were significantly lower than those in A2780/CP70 and MCAS cells (p<0.05). These results suggest that β-elemene and cisplatin may act synergistically to enhance cytotoxicity in both chemoresistant and chemosensitive ovarian carcinoma cells, but that β-elemene has a greater effect on cisplatin sensitivity in chemoresistant ovarian tumor cells.
The effect of β-elemene on the protein level of ERCC-1, phospho-JNK1 and XIAP in chemoresistant human ovarian carcinoma cells
Enhanced DNA repair capacity contributes to cisplatin resistance in human tumors, and NER is responsible for the repair of platinum-DNA adducts in human cells. Furthermore, high levels of ERCC-1 protein support the efficient DNA repair capacity of resistant cancer cells and ERCC-1 is a marker gene for the NER mechanism. Thus, to investigate whether the mechanism by which β-elemene reverses drug resistance to cisplatin involves, at least in part, the inhibition of DNA repair activity, we tested the effect of β-elemene on cisplatin-upregulated ERCC-1 expression in resistant ovarian cancer cells. β-Elemene significantly attenuated the cisplatin-induced increase in the ERCC-1 protein level in A2780/CP70 ovarian cancer cells (Fig. 2), indicating that the reduction of DNA repair activity by β-elemene is positively associated with increased cisplatin cytotoxicity in resistant ovarian tumor cells.
Figure 2.
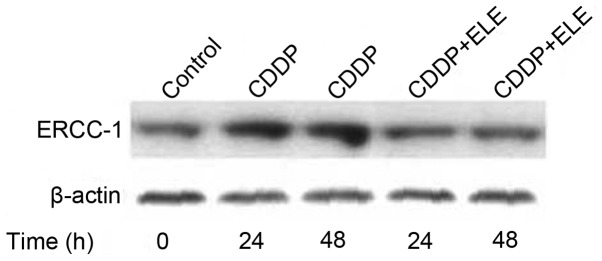
β-Elemene blocks the cisplatin-induced increase in ERCC-1 protein in human ovarian tumor cells. A2780/CP70 cells were exposed to 40 μM cisplatin (CDDP) alone or cisplatin plus β-elemene (ELE) at the IC20 for 24 or 48 h. The cells were harvested and lysed. Cell lysates containing 40 μg of protein were analyzed on western blots using ERCC-1 antiserum and antibody against β-actin (loading control), as described previously (23,24).
The upregulation of ERCC-1 gene expression is mediated by activator protein 1 (AP-1) transcriptional activity, which is activated by a JNK phosphorylation cascade. In A2780/CP70 cells, β-elemene inhibited the cisplatin-induced increase in JNK phosphorylation (Fig. 3), suggesting that β-elemene may suppress ERCC-1 expression via a phosphatidylinositol 3-kinase (PI3K)/JNK signaling pathway.
Figure 3.
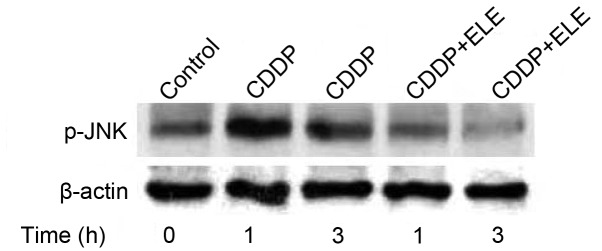
β-Elemene abrogates cisplatin-induced JNK1 protein phosphorylation in human ovarian tumor cells. A2780/CP70 cells were exposed to 40 μM cisplatin alone or cisplatin plus β-elemene (ELE) at the IC20 for 1 or 3 h. The cells were harvested and lysed. Cell lysates containing 60 μg of protein were analyzed on western blots using antibodies against phospho-JNK1 (Thr183/Tyr185) and β-actin (loading control), as described previously (21,22).
Activation of the PI3K/protein kinase B (Akt) pathway leads to the upregulated expression of XIAP, which modulates death signaling pathways and is a determinant of cisplatin resistance in ovarian cancer cells. β-Elemene not only reduced the XIAP protein level (Fig. 4) but also abrogated cisplatin-induced XIAP expression in resistant ovarian tumor cells (Fig. 5), indicating the involvement of XIAP in the mechanism of β-elemene action in resistant ovarian cancer cells.
Figure 4.
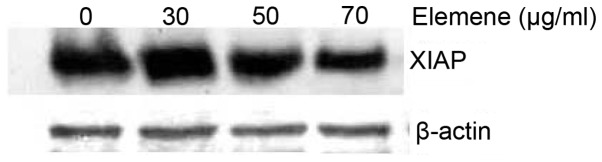
β-Elemene downregulates XIAP protein levels in human ovarian tumor cells. A2780/CP70 cells were exposed to β-elemene at the indicated concentrations for 24 h. The cells were harvested and lysed. Cell lysates containing 60 μg of protein were analyzed on western blots using antibodies against XIAP and β-actin (loading control), as described previously (16).
Figure 5.
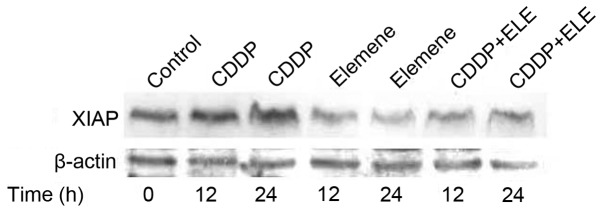
The effect of β-elemene and cisplatin on XIAP protein levels in human ovarian tumor cells. A2780/CP70 cells were exposed to 30 μM cisplatin (CDDP) alone, 70 μg/ml β-elemene alone, or cisplatin plus β-elemene (ELE) at the IC20 for 12 or 24 h. The cells were harvested and lysed. Cell lysates containing 40 μg of protein were analyzed on western blots using antibodies against XIAP and β-actin (loading control).
Discussion
The greatest limitation to the successful treatment of ovarian cancer is the development of clinical resistance to cisplatin. Although the mechanism of cisplatin resistance in vivo is not clearly understood, laboratory studies on tumor tissues and cell lines suggest that resistance to cisplatin is multifactorial (25,26). These factors include impaired cellular uptake of cisplatin (27), increased intracellular detoxification by glutathione and metallothionein systems (28), altered patterns of DNA platination, impaired cisplatin-induced apoptosis and enhanced repair of platinum-damaged DNA (4–8,26,27,29,30).
We have been investigating the mechanisms of cisplatin drug resistance, focusing on the relationship between DNA repair and cisplatin resistance in ovarian cancer and other tumors (21–24,31–36). Increasing evidence indicates that NER is responsible for the repair of platinum-caused DNA damage (4–8,37). Repair-defective cells are hypersensitive to cisplatin (38,39), and enhanced DNA repair has been implicated in the cisplatin-resistance phenotype (27,30,40). Furthermore, increased repair of cisplatin-caused interstrand cross-links and intrastrand adducts is associated with resistance in human ovarian cancer cells (41) and in laboratory-derived cisplatin-resistant lines (27). ERCC-1 is a key DNA repair protein in the NER process and a useful biomarker for NER activity in human cells. The overexpression of ERCC-1 and other NER genes has been associated with the repair of cisplatin-induced DNA damage (30,37,41) and clinical resistance to cisplatin (42,43). The expression levels of ERCC-1 in cisplatin hypersensitive, repair-deficient cells are 50- to 30-fold lower than those in resistant cells. These observations indicate that enhanced DNA repair capacity contributes to the development of cisplatin resistance in human cancers. In the present study, we showed that β-elemene increased the sensitivity to cisplatin and blocked cisplatin-induced ERCC-1 protein expression in resistant human ovarian cancer cells, suggesting that β-elemene enhances cisplatin sensitivity in resistant ovarian cancer cells by decreasing the proficiency of repair of cisplatin-induced DNA damage.
A number of studies have reported that NER gene expression is regulated by the JNK/AP-1 pathway in response to cisplatin in vitro. The AP-1 family of transcription factors is a heterodimeric protein composed of proteins belonging to the c-Fos, c-Jun, ATF and JDP families, and is responsible for the activation of a wide variety of genes in different cell types and tissues. AP-1 binding sites (5′-TGAG/CTCA-3′) are frequently found in promoters or enhancers of genes that are inducible by cisplatin. Evidence has shown that cisplatin induces the expression of c-fos/c-jun(21,22,32) and activates JNK (21,22,32,44,45) in ovarian cancer cells. Therefore, the activation of AP-1 and subsequent overexpression of AP-1-regulated NER genes may enhance DNA repair capacity in affected cells and contribute to decreased chemosensitivity in human ovarian cancer cells (37,41).
This hypothesis is supported by several lines of evidence. We have previously demonstrated that cisplatin exposure activates an AP-1-mediated increase in ERCC-1 expression in human ovarian tumor cells (23,32,33). Treatment with phorbol ester, an AP-1 agonist, also induced increases in ERCC-1 mRNA and protein levels in human ovarian carcinoma cells in vitro(23,24,34). AP-1 may be a common activator of NER genes (46). The overexpression of wild-type c-Jun is associated with cisplatin resistance (44). In contrast, the inhibition of AP-1 activity in cells modified by inhibition of Gli1 with a specific short-hairpin RNA downregulates c-Jun activity and NER gene (ERCC-1 and XPD) expression, blocks platinum-DNA adduct repair, and results in supra-additive cell killing with cisplatin (44,47). Furthermore, cells in which AP-1 has been genetically inactivated are hypersensitive to genotoxic insults, including anticancer agents. These findings suggest that AP-1 may play a prominent role in modulating DNA repair processes in both physiological and pathophysiological conditions. AP-1-dependent DNA repair activities may provide a stress-protective function in cells by effectively reducing the cytotoxic, mutagenic, and carcinogenic consequences of DNA damage. This may also explain, at least in part, the observed increase in mRNA levels of ERCC-1 and other NER genes in clinical platinum-resistant specimens (6,42,43,48).
Given that the promoters of ERCC-1 and other NER repair genes contain AP-1 binding sites, signal transduction pathways that modulate AP-1 may be important in the regulation of DNA repair. JNK, a member of the MAP kinase family and Ras pathway, is responsible for the phosphorylation of c-Jun protein at serine residues 63 and 73 in the NH2-terminal domain, which results in greatly enhanced AP-1 binding to regulated genes and subsequent transcriptional regulation (49). Recent work has shown that cellular damage induced by DNA damaging agents, including cisplatin (44,50), activates the JNK pathway involving AP-1. This response has been reported to protect against cisplatin-induced DNA damage by allowing DNA repair and survival following cisplatin treatment (44). Inhibition of this pathway in cells modified by the overexpression of a dominant-negative mutant of c-Jun blocks DNA repair and leads to decreased viability following treatment with cisplatin (44). These observations suggest that the Ras/JNK pathway may mediate a physiological response to DNA damage. We have previously shown that upon stimulation with cisplatin, JNK may directly phosphorylate c-Jun at serine residues 63 and 73 to activate ERCC-1 transcription via AP-1 in A2780/CP70 ovarian cancer cells (32), which would place ERCC-1 under the influence of the JNK/Ras/AP-1 signal transduction pathway. However, the upstream signaling cascade leading to the activation of JNK in response to cisplatin-induced DNA damage remains to be further elucidated.
PI3K is a heterodimer composed of one regulatory subunit (p85) and one catalytic subunit (p110) (51). The catalytic subunit of PI3K phosphorylates phosphatidylinositol (PI) at the 3′ position of the inositol sugar ring, generating PI 3-phosphate, PI 3,4-bisphosphate, and PI 3,4,5-trisphosphate (51). Experiments with PI3K inhibitors, constitutively active PI3K mutants, and dominant-negative PI3K mutants have established an important role for PI3K in apoptosis (51,52). The best-known downstream target of PI3K is the serine-threonine kinase Akt, which transmits survival signals from growth factors (53). However, PI3K also has many other targets, including NF-κB (54), BAD (55) and JNK (56), and is involved in cell growth, proliferation, differentiation and survival. Therefore, β-elemene may affect DNA repair activity in these cells by regulating the PI3K/JNK/AP-1 signaling pathway, leading to the downregulated expression of ERCC-1 and other NER genes, and cell death.
A proposed mechanism that is consistent with the evidence is presented in Fig. 6. In this model, cisplatin increases PI3K activity, which activates JNK, and JNK activates AP-1. Activated AP-1 upregulates NER gene expression, thereby increasing DNA repair activity and cell survival. This mechanism may be responsible for reduced cellular sensitivity to cisplatin. β-Elemene may sensitize resistant ovarian cancer cells to cisplatin by blocking the activation of the PI3K/JNK signaling pathway, which would reduce DNA repair activity and enhance cisplatin cytotoxicity. In our previous study, cisplatin increased Raf-1 and c-Fos expression in human ovarian carcinoma cells (22). PI3K and Akt regulate the effect of Raf on gene expression. Therefore, there are also three alternative PI3K/Akt/Raf signaling pathways that may link cisplatin to AP-1 activation. In all three pathways, cisplatin-activated PI3K acts through a phosphorylation cascade to activate Akt and Raf, which can then trigger three different pathways to activate AP-1: i) Raf → MEKK1 → MKK4 → JNK → c-Jun; ii) Raf → MEK1/2 → ERK1/2 → c-Jun; and iii) MEK1/2 → ERK1/2 → p90RSK → CREB → c-Fos. Phosphorylated c-Jun and/or c-Fos then augment AP-1 activity, which results in upregulated NER gene expression, enhanced DNA repair capacity and increased cell survival.
Figure 6.

Proposed molecular mechanism for the effect of β-elemene on cisplatin chemosensitivity in human ovarian carcinoma cells. In the proposed mechanism shown here, β-elemene mediates altered DNA repair activity and cisplatin sensitivity in human ovarian carcinoma cells by blocking cisplatin-induced PI3K/JNK and PI3K/Akt activation, thereby preventing from the activation of the downstream signaling components such as AP-1 and NF-κB. This leads to decreased DNA-repair activity and increased caspase activity, respectively, which both confer sensitization of resistant ovarian cancer cells to the chemotherapeutic agent cisplatin. See text for details.
The major goal of cancer chemotherapy is to commit tumor cells to death or apoptosis following exposure to anticancer agents. Considerable evidence collected during the past decades indicates that cisplatin kills cells through the induction of cell apoptosis (57). The mechanisms of cisplatin-induced apoptosis are complex and involve many regulators (58). Caspase cascades are activated in response to cisplatin exposure, and this activation leads to an irreversible commitment to apoptotic cell death (25,59,60). Caspases are held in check, in part, by protein-protein interactions with inhibitor of apoptosis proteins (IAPs). The IAPs such as XIAP, cellular IAP-1, and cellular IAP-2 bind directly to caspases such as caspase-3, -7 and -9, and inhibit their enzymatic activities. In mammalian cells, two major regulatory pathways have been proposed for caspase cascades, an extrinsic pathway and an intrinsic pathway (61).
In the extrinsic pathway, the Fas receptor is activated by the binding of an extracellular ligand such as Fas ligand (FasL), and this induces the assembly of a death-inducing signaling complex, which includes the Fas-associated death domain protein as an adaptor and procaspase-8 (or procaspase-10). The procaspase is activated and in turn initiates the activation of two effector caspases, caspase-3 and -7 (62). The Fas/FasL-activated caspase-8/caspase-3 pathway may be involved in tumor cell response to cisplatin (58,63–65). Alterations in this apoptotic signaling pathway, such as defects in the expression of CD95L or CD95, or defects in caspase-8 or caspase-3 activation, may contribute to cisplatin-resistance. Conversely, increased caspase levels may restore sensitivity to cisplatin chemotherapy in tumor cells (64,65). The intrinsic pathway of apoptosis is mediated by members of the Bcl-2 family, which destabilize the mitochondrial membrane, causing the release of cytochrome c from mitochondria. In the presence of ATP and cytochrome c, apoptotic protease-activating factor-1 (Apaf-1) activates caspase-9, which in turn activates caspase-3 (62,66). Although the exact mechanism whereby Bcl-2 family members regulate mitochondrial damage remains under debate, Bcl-2 and Bcl-XL are thought to exert anti-apoptotic effects by stabilizing the mitochondrial membrane potential and preventing the release of apoptosis-inducing molecules such as cytochrome c(66–68). Cisplatin may cause mitochondrial release of cytochrome c and activation of caspase-3 by acting through Bcl-2 family proteins (59,69). Cisplatin has been shown to induce the expression of Bax and/or the cleavage of Bcl-2 to increase the Bax:Bcl-2 ratio and activate the apoptotic cascade (59,70). Moreover, antisense oligonucleotides targeting Bcl-2 and Bcl-XL sensitized tumor cells to the cytostatic effect of cisplatin (68).
Failure of the apoptotic pathways may lead to cisplatin resistance. Recent evidence has demonstrated that the failure to upregulate FasL in response to cisplatin exposure is associated with chemoresistance in ovarian cancer cells (71). In one study, cisplatin decreases the XIAP content in cisplatin-sensitive, but not cisplatin-resistant, human ovarian cancer cells (72). Other researchers have confirmed this finding, showing that the acquisition of cisplatin resistance is associated with the ability of cisplatin-treated ovarian tumor cells to upregulate XIAP expression (73). These observations indicate that impaired cisplatin-induced apoptosis may account for the chemoresistance to cisplatin therapy in ovarian tumors.
In the present study, β-elemene suppressed XIAP expression and blocked cisplatin-induced XIAP upregulation in resistant ovarian tumor cells. PI3K activates Akt and NF-κB, which block apoptosis by upregulating the expression of IAP family of proteins, thereby inhibiting the activities of caspase-3, -7 and -9. Thus, β-elemene may enhance cisplatin sensitivity in resistant ovarian cancer cells by abrogating cisplatin-induced PI3K activity and the PI3K/Akt signaling pathway, resulting in decreased IAP expression and increased apoptotic cell death. In a proposed mechanism that is consistent with these findings, cisplatin-induced activation of the PI3K/Akt pathway causes the phosphorylation of IKKα and subsequent activation of the transcription factor NF-κB, which upregulates IAP expression to inhibit caspase activation and promote cell survival: cisplatin → PI3K → Akt → IKKα → NF-κB → IAP expression → caspase inhibition → cell survival (Fig. 6). Our previous demonstration of cisplatin-enhanced Raf-1 activity in human ovarian cancer cells (22) suggests that IAP expression may also be upregulated by two alternative signaling pathways involving Raf: i) cisplatin → Ras → PI3K → PDK → Akt → Raf → MEKK1 → IKKα → NF-κB → IAP expression; and ii) cisplatin → Ras → Raf → MEKK1 → IKKα → NF-κB → IAP expression. This presents the possibility of crosstalk between the cisplatin-activated PI3K/PDK/Akt/IKK signal transduction pathway and the cisplatin-activated Ras/Raf/MEKK1/IKK signaling pathway in the activation of NF-κB and IAP expression, and the inhibition of caspase activity. These pathways collaboratively lead to cell survival in cisplatin-resistant ovarian cancer cells.
On the basis of the evidence obtained in our studies and those of other groups, we propose that cisplatin increases PI3K activity, which increases JNK and AP-1 activation; activated AP-1 upregulates NER gene expression and increases DNA repair activity. This mechanism may reduce cellular sensitivity to cisplatin. In the current study, β-elemene promoted cisplatin cytotoxicity and apoptosis by blocking cisplatin-induced increases in the levels of ERCC-1 and XIAP in resistant ovarian tumor cells. Based on these observations, we propose that β-elemene alters DNA repair activity and cisplatin sensitivity in human ovarian carcinoma cells by preventing the cisplatin-induced activation of PI3K/JNK and PI3K/Akt, consequently blocking the activation of the downstream transcription factors AP-1 and NF-κB. This results in reduced DNA repair activity, enhanced caspase activity, and increased apoptotic cell death in cisplatin-resistant ovarian cancer cells. Fig. 6 illustrates the proposed signaling pathways responsible for chemotherapeutic resistance to cisplatin and a possible mechanism by which β-elemene may act as a drug-resistance modulator to enhance the antitumor activity of cisplatin in resistant human ovarian cancer cells.
Taken altogether, we showed in this study that β-elemene increases sensitivity to cisplatin; decreases cisplatin-induced expression of ERCC-1, a DNA repair gene, through blocking a JNK/AP-1 pathway; and augments cisplatin-induced cell death by downregulating XIAP expression in resistant ovarian cancer cells. These results suggest that β-elemene enhances susceptibility to cisplatin in resistant tumor cells through the regulation of DNA repair activity and apoptotic death signaling in human ovarian cancer. These novel findings indicate that β-elemene may be efficacious as a drug-resistance modulator for cisplatin-resistant carcinomas. This information provides a better understanding of the mechanisms of modulation of cisplatin sensitivity and assists in the design of potentially effective β-elemene-based chemotherapy regimens to overcome cisplatin resistance in human ovarian cancer and other tumor types.
Acknowledgements
This publication was made possible by grants from the Natural Science Foundation of Science and Technology Department of Guangxi Province (no. 0991294); the Guangxi Scientific Research and Technological Development Program (no. 200901059); and by grants from the National Institutes of Health (nos. P20RR16440-010003, P20RR16440-020003, P20RR16440-030003, P20RR16440-040003) and a West Virginia University School of Medicine Research Grant (to Q.Q.L.).
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Reed E. Cisplatin, carboplatin, and oxaliplatin. In: Chabner BA, Longo DL, editors. Cancer Chemotherapy and Biotherapy: Principles and Practice. 4th edition. Lippincott, Williams and Wilkins; Philadelphia, PA: 2006. pp. 332–343. [Google Scholar]
- 3.Reed E. Cisplatin and platinum analogs. In: DeVita VT, Rosenberg SA, Lawrence TS, editors. Cancer Principles and Practice of Oncology. 8th edition. Lippincott, Williams and Wilkins; Philadelphia, PA: 2008. pp. 419–426. [Google Scholar]
- 4.Reed E. Platinum-DNA adduct, nucleotide excision repair and platinum based anticancer chemotherapy. Cancer Treatment Rev. 1998;24:331–344. doi: 10.1016/s0305-7372(98)90056-1. [DOI] [PubMed] [Google Scholar]
- 5.Reed E. DNA damage and repair in clinical oncology: an overview. Clin Cancer Res. 2010;16:4511–4516. doi: 10.1158/1078-0432.CCR-10-0528. [DOI] [PubMed] [Google Scholar]
- 6.Reed E. Nucleotide excision repair and anticancer chemotherapy. Cytotechnol. 1998;27:187–201. doi: 10.1023/A:1008016922425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reed E. ERCC1 and clinical resistance to platinum-based therapy. Clin Cancer Res. 2005;11:6100–6102. doi: 10.1158/1078-0432.CCR-05-1083. [DOI] [PubMed] [Google Scholar]
- 8.Reed E. ERCC1 measurements in clinical oncology. N Engl J Med. 2006;355:1054–1055. doi: 10.1056/NEJMe068162. [DOI] [PubMed] [Google Scholar]
- 9.Wang G, Li X, Huang X, Zhao J, Ding H, Cunningham C, Coad J, Flynn D, Reed E, Li QQ. Antitumor effect of β-elemene in non-small cell lung cancer cells is mediated via induction of cell cycle arrest and apoptotic cell death. Cell Mol Life Sci. 2005;62:881–893. doi: 10.1007/s00018-005-5017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li X, Wang G, Zhao J, Ding H, Cunningham C, Chen F, Flynn DC, Reed E, Li QQ. Antiproliferative effect of β-elemene in chemoresistant ovarian carcinoma cells is mediated through arrest of the cell cycle at the G2-M phase. Cell Mol Life Sci. 2005;62:894–904. doi: 10.1007/s00018-005-5027-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao J, Li QQ, Zou B, Wang G, Li X, Kim JE, Cuff CF, Huang L, Reed E, Gardner K. In vitro combination characterization of the new anticancer plant drug β-elemene with taxanes against human lung carcinoma. Int J Oncol. 2007;31:241–252. [PubMed] [Google Scholar]
- 12.Li QQ, Wang G, Zhang M, Cuff CF, Huang L, Reed E. β-elemene, a novel plant-derived antineoplastic agent, increases cisplatin chemosensitivity of lung tumor cells by triggering apoptosis. Oncol Rep. 2009;22:161–170. doi: 10.3892/or_00000420. [DOI] [PubMed] [Google Scholar]
- 13.Li QQ, Wang G, Reed E, Huang L, Cuff CF. Evaluation of cisplatin in combination with beta-elemene as a regimen for prostate cancer chemotherapy. Basic Clin Pharmacol Toxicol. 2010;107:868–876. doi: 10.1111/j.1742-7843.2010.00592.x. [DOI] [PubMed] [Google Scholar]
- 14.Li QQ, Wang G, Huang F, Banda M, Reed E. Antineoplastic effect of beta-elemene on prostate cancer cells and other types of solid tumour cells. J Pharm Pharmacol. 2010;62:1018–1027. doi: 10.1111/j.2042-7158.2010.01135.x. [DOI] [PubMed] [Google Scholar]
- 15.Lee RX, Li QQ, Reed E. β-elemene effectively suppresses the growth and survival of both platinum-sensitive and -resistant ovarian tumor cells. Anticancer Res. 2012;32:3103–3113. [PMC free article] [PubMed] [Google Scholar]
- 16.Li QQ, Lee RX, Liang H, Zhong Y. Anticancer activity of β-elemene and its synthetic analogs in human malignant brain tumor cells. Anticancer Res. 2013;33:65–76. [PMC free article] [PubMed] [Google Scholar]
- 17.Li QQ, Lee RX, Liang H, Zhong Y, Reed E. Enhancement of cisplatin-induced apoptosis by β-elemene in resistant human ovarian cancer cells. Med Oncol. 2013;30:424–436. doi: 10.1007/s12032-012-0424-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zou B, Li QQ, Zhao J, Li JM, Cuff CF, Reed E. β-elemene and taxanes synergistically induce cytotoxicity and inhibit proliferation in ovarian cancer and other tumor cells. Anticancer Res. 2013;33:929–940. [PubMed] [Google Scholar]
- 19.Li QQ, Wang G, Huang F, Li JM, Cuff CF, Reed E. Sensitization of lung cancer cells to cisplatin by β-elemene is mediated through blockade of cell cycle progression: antitumor efficacies of β-elemene and its synthetic analogs. Med Oncol. 2013;30:488–498. doi: 10.1007/s12032-013-0488-9. [DOI] [PubMed] [Google Scholar]
- 20.Li QQ, Wang G, Liang H, Li JM, Huang F, Agarwal PK, Zhong Y, Reed E. β-elemene promotes cisplatin-induced cell death in human bladder cancer and other carcinomas. Anticancer Res. 2013;33:1421–1428. [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong X, Li QQ, Reed E. SU5416 sensitizes ovarian cancer cells to cisplatin through inhibition of nucleotide excision repair. Cell Mol Life Sci. 2003;60:794–802. doi: 10.1007/s00018-003-3002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong X, Li X, Wang G, Zhu Y, Hu G, Zhao J, Neace C, Ding H, Reed E, Li QQ. Mechanisms underlying the synergistic effect of SU5416 and cisplatin on cytotoxicity in human ovarian tumor cells. Int J Oncol. 2004;25:445–451. [PubMed] [Google Scholar]
- 23.Li Q, Ding L, Yu JJ, Mu C, Tsang B, Bostick-Bruton F, Reed E. Cisplatin and phorbol ester independently induce ERCC-1 protein in human ovarian tumor cells. Int J Oncol. 1998;13:987–992. doi: 10.3892/ijo.13.5.987. [DOI] [PubMed] [Google Scholar]
- 24.Li Q, Yu JJ, Mu C, Yunmbam MK, Slavsky D, Cross CL, Bostick-Bruton F, Reed E. Association between the level of ERCC-1 expression and the repair of cisplatin-induced DNA damage in human ovarian cancer cells. Anticancer Res. 2000;20:645–652. [PubMed] [Google Scholar]
- 25.Wang G, Reed E, Li QQ. Molecular basis of cellular response to cisplatin chemotherapy in non-small cell lung cancer (Review) Oncol Rep. 2004;12:955–965. [PubMed] [Google Scholar]
- 26.Gosland M, Lum B, Schimmelpfennig J, Baker J, Doukas M. Insights into mechanisms of cisplatin resistance and potential for its clinical reversal. Pharmacotherapy. 1996;16:16–39. [PubMed] [Google Scholar]
- 27.Parker RJ, Eastman A, Bostick-Bruton F, Reed E. Acquired cisplatin resistance in human ovarian cancer cells is associated with enhanced DNA repair of cisplatin-DNA lesions and reduced drug accumulation. J Clin Invest. 1991;87:773–777. doi: 10.1172/JCI115080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Godwin A, Meister A, O’Dwyer P, Huang C, Hamilton T, Anderson M. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc Natl Acad Sci USA. 1992;89:3070–3074. doi: 10.1073/pnas.89.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masuda H, Ozols RF, Lai GM, Fojo A, Rothenberg M, Hamilton TC. Increased DNA repair as a mechanism of acquired resistance to cis-diamminedichloroplatinum (II) in human ovarian cancer cell lines. Cancer Res. 1988;48:5713–5716. [PubMed] [Google Scholar]
- 30.Ferry KV, Hamilton TC, Johnson SW. Increased nucleotide excision repair in cisplatin-resistant ovarian cancer cells: role of ERCC1-XPF. Biochem Pharmacol. 2000;60:1305–1313. doi: 10.1016/s0006-2952(00)00441-x. [DOI] [PubMed] [Google Scholar]
- 31.Li Q, Bostick-Bruton F, Reed E. Effect of interleukin-1α and tumor necrosis factor-α on cisplatin-induced ERCC-1 mRNA expression in a human ovarian carcinoma cell line. Anticancer Res. 1998;18:2283–2288. [PubMed] [Google Scholar]
- 32.Li Q, Gardner K, Zhang L, Tsang B, Bostick-Bruton F, Reed E. Cisplatin induction of ERCC-1 mRNA expression in A2780/CP70 human ovarian cancer cells. J Biol Chem. 1998;273:23419–23425. doi: 10.1074/jbc.273.36.23419. [DOI] [PubMed] [Google Scholar]
- 33.Li Q, Tsang B, Bostick-Bruton F, Reed E. Modulation of excision repair cross complementation group 1 (ERCC-1) mRNA expression by pharmacological agents in human ovarian carcinoma cells. Biochem Pharmacol. 1999;57:347–353. doi: 10.1016/s0006-2952(98)00291-3. [DOI] [PubMed] [Google Scholar]
- 34.Li Q, Zhang L, Tsang B, Gardner K, Bostick-Bruton F, Reed E. Phorbol ester exposure activates an AP-1-mediated increase in ERCC-1 messenger RNA expression in human ovarian tumor cells. Cell Mol Life Sci. 1999;55:456–466. doi: 10.1007/s000180050302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li QQ, Ding L, Reed E. Proteasome inhibition suppresses cisplatin-dependent ERCC-1 mRNA expression in human ovarian tumor cells. Res Commun Mol Pathol Pharmacol. 2000;107:387–396. [PubMed] [Google Scholar]
- 36.Li QQ, Yunmbam MK, Zhong X, Yu JJ, Mimnaugh EG, Neckers L, Reed E. Lactacystin enhances cisplatin sensitivity in resistant human ovarian cancer cell lines via inhibition of DNA repair and ERCC-1 expression. Cell Mol Biol. 2001;47:61–72. [PubMed] [Google Scholar]
- 37.Sancar A. Mechanisms of DNA excision repair. Science. 1994;266:1954–1956. doi: 10.1126/science.7801120. [DOI] [PubMed] [Google Scholar]
- 38.Calsou P, Barret JM, Cros S, Salles B. DNA excision repair synthesis is enhanced in a murine leukemia L1210 cell line resistant to cisplatin. Eur J Biochem. 1993;211:403–409. doi: 10.1111/j.1432-1033.1993.tb17563.x. [DOI] [PubMed] [Google Scholar]
- 39.Hill BT, Scanlon KJ, Hansson J. Deficient repair of cisplatin-DNA adducts identified in human testicular teratoma cell lines established from tumours from untreated patients. Eur J Cancer. 1994;30:832–837. doi: 10.1016/0959-8049(94)90301-8. [DOI] [PubMed] [Google Scholar]
- 40.Pooter CMD, Oosterom ATV, Scalliet PG, Maes RA, Brujin EAD. Correlation of the response to cisplatin of human ovarian cancer cell lines, originating from one tumor but with different sensitivity, with the recovery of DNA adducts. Biochem Pharmacol. 1996;51:629–634. doi: 10.1016/s0006-2952(95)02229-5. [DOI] [PubMed] [Google Scholar]
- 41.Zhen W, Link CJ, O’Connor PM, Reed E, Parker RJ, Howell SB, Bohr VA. Increased gene-specific repair of cisplatin interstrand cross-links in cisplatin-resistant human ovarian cancer cell lines. Mol Cell Biol. 1992;12:3689–3698. doi: 10.1128/mcb.12.9.3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dabholkar M, Vionnet J, Bostick-Bruton F, Yu JJ, Reed E. Messenger RNA levels of XPA and ERCC1 in ovarian cancer tissue correlate with response to platinum-based chemotherapy. J Clin Invest. 1994;94:703–708. doi: 10.1172/JCI117388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dabholkar M, Bostick-Bruton F, Weber C, Bohr V, Egwuagu C, Reed E. ERCC1 and ERCC2 expression in malignant tissues from ovarian cancer patients. J Natl Cancer Inst. 1992;84:1512–1517. doi: 10.1093/jnci/84.19.1512. [DOI] [PubMed] [Google Scholar]
- 44.Potapova O, Haghighi A, Bost F, Liu C, Birrer MJ, Gjerset R, Mercola D. The Jun kinase/stress-activated protein kinase pathway functions to regulate DNA repair and inhibition of the pathway sensitizes tumor cells to cisplatin. J Biol Chem. 1997;272:14041–14044. doi: 10.1074/jbc.272.22.14041. [DOI] [PubMed] [Google Scholar]
- 45.Potapova O, Gorospe M, Bost F, Dean NM, Gaarde WA, Mercola D, Holbrook NJ. c-Jun N-terminal kinase is essential for growth of human T98G glioblastoma cells. J Biol Chem. 2000;275:24767–24775. doi: 10.1074/jbc.M904591199. [DOI] [PubMed] [Google Scholar]
- 46.Zhong X, Thornton K, Reed E. Computer based analyses of the 5′-flanking regions of selected genes involved in the nucleotide excision repair complex. Int J Oncol. 2000;17:375–380. doi: 10.3892/ijo.17.2.375. [DOI] [PubMed] [Google Scholar]
- 47.Kudo K, Gavin E, Das S, Amable L, Shevde LA, Reed E. Inhibition of Gli1 results in altered c-Jun activation, inhibition of cisplatin-induced upregulation of ERCC1, XPD and XRCC1, and inhibition of platinum-DNA adduct repair. Oncogene. 2012;31:4718–4724. doi: 10.1038/onc.2011.610. [DOI] [PubMed] [Google Scholar]
- 48.Britten RA, Liu D, Tessier A, Hutchison MJ, Murray D. ERCC-1 expression as a molecular marker of cisplatin resistance in human cervical tumor cells. Int J Cancer. 2000;89:453–457. [PubMed] [Google Scholar]
- 49.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 50.Liu ZG, Lea-Chou ET, Wood LD, Chen Y, Karin M, Wang JY. Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature. 1996;384:273–276. doi: 10.1038/384273a0. [DOI] [PubMed] [Google Scholar]
- 51.Carpenter CL, Cantley LC. Phosphoinositide 3-kinase and the regulation of cell growth. Biochim Biophys Acta. 1996;1288:M11–M16. doi: 10.1016/0304-419x(96)00018-2. [DOI] [PubMed] [Google Scholar]
- 52.Burgering BMT, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol 3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- 53.Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3 phosphoinositide-regulated kinase: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem. 1999;68:965–1014. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- 54.Romashkova JA, Makarov SS. NF-κB is a target of Akt in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 55.Del Peso L, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 56.Klippel A, Reinhard C, Apell G, Escobedo MA, Williams LT. Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways. Mol Cell Biol. 1996;16:4117–4127. doi: 10.1128/mcb.16.8.4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eastman A. The mechanism of action of cisplatin: from adducts to apoptosis. In: Lippert E, editor. Cisplatin, Chemistry and Biochemistry of a Leading Anticancer Drug. Wiley-VCH; Basel: 1999. pp. 111–134. [Google Scholar]
- 58.Johnstone R, Ruefli A, Lowe S. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 59.Wang X, Martindale JL, Holbrook NJ. Requirement for ERK activation in cisplatin-induced apoptosis. J Biol Chem. 2000;275:39435–39443. doi: 10.1074/jbc.M004583200. [DOI] [PubMed] [Google Scholar]
- 60.Makin G, Corfe B, Griffiths G, Thistlethwaite A, Hickman J, Dive C. Damage-induced Bax N-terminal change, translocation to mitochondria and formation of Bax dimers/complexes occur regardless of cell fate. EMBO J. 2001;20:6306–6315. doi: 10.1093/emboj/20.22.6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang G, Reed E, Li QQ. Apoptosis in prostate cancer: progressive and therapeutic implications (Review) Int J Mol Med. 2004;14:23–34. [PubMed] [Google Scholar]
- 62.Cryns V, Yuan J. Proteases to die for. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- 63.Okouoyo S, Herzer K, Ucur E, Mattern J, Krammer P, Debatin K, Herr I. Rescue of death receptor and mitochondrial apoptosis signaling in resistant human NSCLC in vivo. Int J Cancer. 2004;108:580–587. doi: 10.1002/ijc.11585. [DOI] [PubMed] [Google Scholar]
- 64.Fulda S, Los M, Friesen C, Debatin K. Chemosensitivity of solid tumor cells in vitro is related to activation of the CD95 system. Int J Cancer. 1998;76:105–114. doi: 10.1002/(sici)1097-0215(19980330)76:1<105::aid-ijc17>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 65.Spierings D, de Vries E, Vellenga E, de Jong S. Loss of drug-induced activation of the CD95 apoptotic pathway in a cisplatin-resistant testicular germ cell tumor cell line. Cell Death Differ. 2003;10:808–822. doi: 10.1038/sj.cdd.4401248. [DOI] [PubMed] [Google Scholar]
- 66.Cory S, Huang D, Adams J. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–8607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 67.Gross A, McDonnell J, Korsmeyer S. Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 68.Hopkins-Donaldson S, Cathomas R, Simoes-Wust A, Kurtz S, Belyanskaya L, Stahel R, Zangemeister-Wittke U. Induction of apoptosis and chemosensitization of mesothelioma cells by Bcl-2 and Bcl-xL antisense treatment. Int J Cancer. 2003;106:160–166. doi: 10.1002/ijc.11209. [DOI] [PubMed] [Google Scholar]
- 69.Kojima H, Endo K, Moriyama H, Tanaka Y, Alnemri E, Slapak C, Teicher B, Kufe D, Datta R. Abrogation of mitochondrial cytochrome c release and caspase-3 activation in acquired multidrug resistance. J Biol Chem. 1998;273:16647–16650. doi: 10.1074/jbc.273.27.16647. [DOI] [PubMed] [Google Scholar]
- 70.Del Bello B, Valentini M, Zunino F, Comporti M, Maellaro E. Cleavage of Bcl-2 in oxidant- and cisplatin-induced apoptosis of human melanoma cells. Oncogene. 2001;20:4591–4595. doi: 10.1038/sj.onc.1204618. [DOI] [PubMed] [Google Scholar]
- 71.Mansouri A, Ridgway LD, Zhang Q, Tian L, Wang Y, Claret FX. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J Biol Chem. 2003;278:19245–19256. doi: 10.1074/jbc.M208134200. [DOI] [PubMed] [Google Scholar]
- 72.Li J, Feng Q, Kim J, Schneiderman D, Liston P, Li M, Vanderhyden B, Faught W, Fung M, Senterman M, Korneluk R, Tsang B. Human ovarian cancer and cisplatin resistance: possible role of inhibitor of apoptosis proteins. Endocrinol. 2001;142:370–380. doi: 10.1210/endo.142.1.7897. [DOI] [PubMed] [Google Scholar]
- 73.Mansouri A, Zhang Q, Ridgway LD, Tian L, Claret FX. Cisplatin resistance in an ovarian carcinoma is associated with a defect in programmed cell death control through XIAP regulation. Oncol Res. 2003;13:399–404. doi: 10.3727/096504003108748410. [DOI] [PMC free article] [PubMed] [Google Scholar]