

Abstract
This review focuses on the molecular characteristics and development of rare malignant ovarian germ cell tumors (mOGCTs). We provide an overview of the genomic aberrations assessed by ploidy, cytogenetic banding, and comparative genomic hybridization. We summarize and discuss the transcriptome profiles of mRNA and microRNA (miRNA), and biomarkers (DNA methylation, gene mutation, individual protein expression) for each mOGCT histological subtype. Parallels between the origin of mOGCT and their male counterpart testicular GCT (TGCT) are discussed from the perspective of germ cell development, endocrinological influences, and pathogenesis, as is the GCT origin in patients with disorders of sex development. Integrated molecular profiles of the 3 main histological subtypes, dysgerminoma (DG), yolk sac tumor (YST), and immature teratoma (IT), are presented. DGs show genomic aberrations comparable to TGCT. In contrast, the genome profiles of YST and IT are different both from each other and from DG/TGCT. Differences between DG and YST are underlined by their miRNA/mRNA expression patterns, suggesting preferential involvement of the WNT/β-catenin and TGF-β/bone morphogenetic protein signaling pathways among YSTs. Characteristic protein expression patterns are observed in DG, YST and IT. We propose that mOGCT develop through different developmental pathways, including one that is likely shared with TGCT and involves insufficient sexual differentiation of the germ cell niche. The molecular features of the mOGCTs underline their similarity to pluripotent precursor cells (primordial germ cells, PGCs) and other stem cells. This similarity combined with the process of ovary development, explain why mOGCTs present so early in life, and with greater histological complexity, than most somatic solid tumors.
-
Introduction
Classification of GCTs
mOGCT epidemiology, treatment, and hormonal disturbances
Survival and QOL after mOGCT diagnosis
Gonadal and GCT development
Predisposition to GCT in the dysgenetic and phenotypically normal gonad
Methods of the Review
-
Genome Profiling of mOGCT
DNA ploidy: image and flow cytometry analyses
Chromosome G-banding analyses
In situ hybridization: CGH and 12p FISH analyses
Polymorphic marker studies
-
Transcriptome Profiling of mOGCT
mRNA expression
miRNA expression
-
Biomarkers of mOGCT
DNA methylation
Protein expression and gene mutations
Histology-specific molecular features of mOGCT
Comparing Molecular Development of mOGCT and TGCT
Concluding Remarks and Perspectives
I. Introduction
Neoplasms presenting in the ovary can originate from any of the various cell types present. The tumor may be derived from the surface epithelium, the stroma, or the cellular elements of the follicle, where the latter may result in sex cord-stromal tumors (such as granulosa cell tumor or thecoma) or germ cell tumors (GCTs) (1, 2). The most frequently occurring ovarian GCTs are benign, cystic mature teratomas (MTs) that may show highly differentiated tissue and high morphological heterogeneity. This review focuses on the rare gonadal, malignant ovarian GCTs (mOGCTs), which occur predominantly in girls and young women and have not been as well studied as other ovarian tumors. Given the presumed common cell of origin in mOGCTs and their male counterpart, testicular GCTs (TGCTs), parallels between the molecular mechanisms of these 2 tumor types and the tumors of patients with disorders of sex development (DSD) are discussed. By a critical review and summarization of published data, the current knowledge of the molecular basis underlying mOGCT are presented. Specifically, the review summarizes genomic aberrations in mOGCTs as studied by ploidy, cytogenetic banding, comparative genomic hybridization (CGH) and microsatellite loci analysis, and genome-wide mRNA expression and microRNA (miRNA) expression studies as well as expression of single genes and proteins, including relevant mutational studies. Results from these 3 levels of molecular characterization are compared for concurrence and discussed in context of the pathogenesis of mOGCTs.
A. Classification of GCTs
Across both sexes, malignant GCTs most frequently occur in the gonads of young adult males as TGCTs, more rarely in the gonads of females (as OGCT) and infantile boys, and most rarely at extragonadal sites such as the central nervous system, mediastinum, retroperitoneum, and coccyx (3). GCTs are also frequently observed in individuals with DSD (4), underlining the pathogenetic influence of disturbed gonadal development on the malignant transformation of germ cells.
According to the World Health Organization classification system, OGCTs are divided into 3 categories: primitive GCT, biphasic or triphasic teratoma, and monodermal teratoma and somatic-type tumors associated with dermoid cysts (5, 6). The common benign mature cystic teratomas belong to the subgroup of the biphasic and triphasic teratomas, and the remaining OGCTs are malignant. The primitive GCTs are subdivided into dysgerminoma (DG), the ovarian counterpart of the male testicular seminoma, and non-DGs: yolk sac tumor (YST), also known as endodermal sinus tumor, embryonal carcinoma (EC), polyembryoma, nongestational choriocarcinoma (CC), and mixed GCT containing various histologies, including immature teratoma (IT). The most common mOGCT histologies are the DG followed by YST. Pure EC is relatively rare, and this component of mOGCT should be distinguished from stem-cell/EC-like cells occasionally found in association with epithelial ovarian cancer (within the tumor or in malignant ascites) and often defined as a side population of tumor-initiating cells (7, 8). mOGCTs are staged according to the International Federation of Gynaecology and Obstetrics (FIGO) classification (9).
The mOGCTs are believed to be derived from primordial germ cells (PGCs), where the pluripotency traits they retain facilitate the potential to differentiate into the spectrum of histological subtypes listed above. The development of non-DGs is characterized by differentiation of these cells into histologies that mimic embryonic and extraembryonic tissues. The mOGCTs have earlier been subclassified by Oosterhuis and Looijenga (10), based mainly on similarities to TGCTs in clinical parameters, histology, and genome aberrations, but no complete, dedicated molecular survey of the mOGCTs has been published to date. For this review, all published molecular studies on mOGCTs, specifically all primitive OGCTs and ITs, were surveyed to identify molecular characteristics of mOGCT subgroups.
B. mOGCT epidemiology, treatment, and hormonal disturbances
In Western countries, mOGCTs represent approximately 2% to 3% of all ovarian cancers (11). The mOGCT may present already at infancy, but its incidence increases sharply from the age of approximately 5 years and continues with the onset of puberty to a peak incidence rate of 1.2 per 100 000 women aged 15 to 19 (12–14). Among these young girls and women, the histological subtype DG is the most frequent (14). Among the youngest patients, the incidence of ovarian cancer is reported to be 0.1 per 100 000 in girls aged 9 years or younger, as opposed to 1.1 per 100 000 in those aged 10–19 years, with the majority (∼80%) being mOGCT in all age groups (15). A second peak in incidence for OGCT has been reported around the age of 65, after onset of menopause, where benign MTs are the most frequent histological subtype observed (13). Whereas most DGs appear before 40 years of age, with 75% diagnosed between 10 and 30 years, the ITs are commonly diagnosed around the age of 20, and the YSTs and ECs before the patient reaches the age of 20 (16). Interestingly, discrepancies and similarities in the age distribution of the GCTs between the sexes parallel lifetime profiles of gonadotropin levels in each sex (13).
Contradictory reports of incidence trends have been published, most probably due to the rarity of the malignancy. Some reports of a substantial increase over the last decades exist (14, 17), whereas others have reported a slight decrease (18). Based on the Surveillance, Epidemiology, and End Results (SEER) data of gonadal mOGCTs from the United States from 1973 to 2002, the incidence rates, stratified by ethnicity, were found to be highest among the population of other nonwhites (other than blacks, specifically Asian and Hispanic), followed by the white and black population (18). The DG incidence rate was reported to be 2-fold higher among whites and other nonwhites than among blacks (18).
With regard to treatment, cure rates of mOGCT have increased dramatically with the introduction of platinum-based chemotherapy. Most patients can be cured with either surgery alone, preferably fertility-preserving for young patients, or a combination of surgery, radiotherapy, and chemotherapy (19, 20). Stage IA DGs and stage IA grade 1 ITs are treated adequately with surgery alone, and the rate of recurrence in this group of patients is low (15%–25%). DG is reported curable in more than 95% of the cases, even in advanced disease (20). The standard chemotherapy regimen applied for mOGCTs is a combination of bleomycin, etoposide, and cisplatin (20, 21). The exquisite chemosensitivity of GCT, both in women and men, has improved the previously dismal prognosis, and the cure rate of patients with advanced-stage mOGCT is currently about 75%, whereas that of early-stage disease is close to 100% (20–22). There is even 1 report of 100% survival rate among all 40 mOGCTs treated between 1979 and 2008 at 1 particular center, despite various stages and histologies (23). However, analysis from another institution reports that 10-year estimated survival rates are up to 80% and highlights the importance of mOGCT management at specialized centers to optimize future management of this rare disease (24). In the SEER study from 2006, comparing data in 1973 to 1977 with 1998 to 2002, survival was increased by 12.5% for DG (from 83.5% to 96.9%), by 109% for mixed GCT (from 43.4% to 90.7%), and by 29.3% (from 71.6% to 92.6%) for IT. Furthermore, overall survival rates were associated with histology, race, stage of disease, and age at disease (18).
Bilateral tumors have been reported to occur in up to 10% to 15% of mOGCT patients, preferentially in patients with DG (5, 16, 21). The predominant unilaterality of the tumor allows for fertility-sparing surgery in most patients. Even for patients with metastatic disease who have normal ovarian function before treatment, chemotherapy does not adversely affect reproductive function in the long term (21, 25–27). The fact that mOGCTs commonly affect young women, before or during their reproductive years, as well as the aggressiveness of the disease and good response to therapy, underlines the importance of a correct differential diagnosis and optimal therapy. Physicians' awareness of complications associated with chemotherapy is important to maximize efficacy, minimize toxicity, and avoid long-term side effects as well as preserve fertility, all which are critical factors for the patients' quality of life (QOL). This is further discussed in Section I.C.
The various mOGCT subtypes in general produce different serum tumor markers that may be measured in the patients' blood; DGs produce lactate dehydrogenase and in some cases β-human chorionic gonadotropin (βHCG), YSTs produce α-fetoprotein (AFP), whereas ITs may produce AFP in up to one-third of cases (16, 20). ECs may be associated with the markers AFP or βHCG depending on their composition, and CCs produce βHCG (16, 20). Clinicians may overlook this fact and misinterpret tumor-producing factors as hormonal disturbances, as in a recently published case report of a woman of fertile age with a 7-week period of amenorrhea, who consulted for vaginal bleeding. A pelvic mass along with elevated βHCG was misinterpreted for an ectopic pregnancy but proved to be an ovarian DG with nests of syncytiotrophoblastic cells (28). Other atypical presentations of OGCT include cases mimicking pubertas tarda, where in 1 case a pubertal female with primary amenorrhea, short stature, and a vaginal septum proved to have 45,X/46,XY mixed gonadal dysgenesis with DG arising from gonadoblastoma (GB) (29). Hyperandrogenemia mimicking polycystic ovarian syndrome from androgen-secreting ovarian teratomas is another atypical presentation (30). These anecdotal case reports illustrate the difficulties in the diagnosis and management of the patients.
C. Survival and QOL after mOGCT diagnosis
Due to the low incidence of mOGCT, few survivors have been followed long-term, but some studies of mOGCT survivors have been published with focus on both physical and psychological effects (27, 31–38) (reviewed in Ref. 19). Restored regular menstrual cycles and the ability to conceive were seen in 80% or more of the survivors who had fertility-sparing surgery (23, 27, 31, 33, 39). However, regular menstruations were reported in only 38% of young patients, including those who had undergone severe combined chemotherapy and radiation therapy (32). Normal pregnancies and healthy offspring are reported among survivors, and no consistent major congenital birth defect has been associated with chemotherapy (27, 31, 32).
Long-term side effects of cisplatin-based chemotherapy are well-known for TGCT patients (40–42), and associations between long-term side effects and TGCT patient genotypes have also been identified (43, 44). Interview-based QOL studies have shown that mOGCT survivors have more sexual and reproductive concerns, but have in general better dyadic cohesion and appreciation of life than control groups (37, 39). Both interview- and physical examination-based studies find a correlation between cisplatin treatment and mOGCT survivors suffering chronic problems like neurotoxicity, Raynaud's symptoms, and hypertension (35–38). Ototoxicity, or hearing loss, is reported among survivors (32), but in a self-reported, interview-based study, no difference was found in hearing loss and tinnitus between survivors and controls (38). For male GCT patients, there have been reports on the risk of developing secondary malignancies after chemotherapy, especially etoposide with risk estimates of 0.9% or a doubling in relative risk, which can presumably be extrapolated to mOGCT patients. For a thorough discussion of this issue, please see Ref. 19.
D. Gonadal and GCT development
A simplified scheme illustrating the development from zygote via PGC to normal oocyte or to mOGCT and TGCT, including ploidy and the main signaling pathways discussed in this review, is shown in Figure 1.
Figure 1.

A simplified diagram illustrating the development of GCT. The main aberrant signaling pathways and ploidy levels identified in this paper are indicated. Development from zygote via PGC to normal oocyte or to mOGCT and TGCT is also shown. For the description of DSD patient groups with gonadal dysgenesis and increased risk for GCT, see Section I.E. Ter, teratoma.
Most of our knowledge of mammalian ovary development comes from mouse studies. Although there are differences between murine and human ovaries (45), genes identified from mouse studies are often also found to be involved in human ovary development. In the embryo, bipotential gonads develop from the urogenital ridge in close association with the future kidneys and adrenal glands. Studies in mice have found the genes Emx2, Igf1r/Insr/Insrr, Lhx9, Cbx2–001, Sf1, Wt1, and Tcf21 to regulate early development and maintenance of the gonads by preventing apoptosis and promoting cell proliferation between 10.5 and 11.5 days post coitum (46). In humans, WT1, SF1, and NR0B1 are essential genes involved at this developmental stage, and defects are associated with pathology (47). At this initial stage, the undifferentiated gonads do not contain any germ cells, because these migrate to the gonads only at a later stage.
The importance of ensuring the germ cell lineage is evident, because it effects the perpetuation and diversification of genetic information to the following generations in most multicellular organisms. The specification of germ cells, fundamental to organism development, reproduction, and heredity, establishes a group of cells as PGCs. PGC specification is an integration of at least 3 key events: repression of the somatic program, reacquisition of potential pluripotency, and genome-wide epigenetic reprogramming (48). The PGCs arise from the proximal epiblast adjacent to the extraembryonic ectoderm and can be detected in human embryos for the first time around weeks 3 to 4 of gestation (46, 49). In the mouse embryo, the development of epiblast cells into PGCs involves signaling of Bmp4 and Bmp8B from the extraembryonic ectoderm and Bmp2 from the visceral endoderm, generating a gradient that leads to mesoderm induction. However, a few cells escape mesoderm induction and are detectable at embryonic day 7.25 as alkaline phosphatase (ALP)-positive PGCs (reviewed in Ref. 50). PGC specification is marked first by expression of Fragilis followed by Stella in mouse (equivalent to the human genes IFITM1 and DPPA3) (51), which repress homeobox genes and allow escape from somatic cell fate (46). A model and complete summary of the genetic pathways involved in PGC specification in mice has been published (50), showing that specification by Bmp4 and Mothers against decapentaplegic homolog 1/5 is further orchestrated by PR domain zinc finger protein (Prdm)1 and 14. Prdm1, a histone methylator transcriptional repressor, is responsible for repression of the somatic differentiation program, with transcription factor AP-2γ as a likely downstream target of Prdm1 (52, 53). Meta-analysis of genome-scale datasets from murine trophectoderm and human breast cancer cells have identified an overlap of 199 common genes. Among these, Nanos3, Dmrt1, and Dnmt3b were suggested to be potential target genes of TFAP2C with relevance for human germ cell development and GCT (52).
Both PGCs and cells from the inner cell mass of the blastocyst, the starting point for developing embryonal stem cells (ESCs), are pluripotent. Expression of ALPP, NANOG, and POU5F1 mediates pluripotency as well as survival and proliferative mechanisms, where these are expressed in both human fetal germ cells and GCTs (54–60). The PGCs migrate from the yolk sac along the hindgut toward the genital ridges in a process controlled by mast/stem cell growth factor receptor Kit (KIT) and its ligand, Kit ligand (KITLG) (58, 61). Recent studies of human embryos at 4 to 8 weeks indicate that the PGCs migrate along nerve fibers and Schwann cells (62). From the time that the PGCs arrive at the genital ridge, the latter are termed indifferent or bipotential gonads, whereas the PGCs themselves are termed gonocytes in the testis or oogonia in the ovary.
The human bipotential gonads remain morphologically and structurally similar until the initiation of sex-determining region Y protein (SRY)-dependent sex determination, from gestational weeks 6 to 7 onward. At this time, the Sertoli cell differentiating factors SOX9 and FGF9 are expressed, leading to testicular differentiation of the gonad via cascades of downstream genes including AMH, prostaglandins, and steroidogenic genes. The pre-Sertoli cells and gonocytes start to develop cord-like structures (the sex cords) and seminiferous tubules with germs cells in the center and Sertoli cells at the periphery of the tubules (63). Hereafter, the maturation of germ cells is largely dependent on somatic cells that mediate the response to the retinoic acid (RA) produced by the mesonephros in both sexes. In fetal ovaries, RA induces initiation of meiosis, whereas in the developing testes, meiotic entry is prevented by the RA-degrading enzyme cytochrome P450 26B1, which is expressed in Sertoli cells (64, 65) and regulated by an as yet unelucidated complex interaction of several other factors, including nodal homolog and doublesex- and mab-3-related transcription factor 1 (DMRT1) (66, 67). The male germ cells continue dividing mitotically, and gradually migrate to the periphery of the tubules, change morphology, mature to prespermatogonia, and cease to express the gonocyte markers, such as POU5F1, NANOG, TFAP2C, ALPP, and KIT (56, 57, 68, 69). In the fetal ovary, the oogonia arrange in cyst-like structures with supportive pregranulosa cells, rapidly lose POU domain, class 5, transcription factor 1 (POU5F1) expression, enter the meiotic prophase I and become oocytes enclosed in primordial follicles. The ovaries of normal neonates have hardly any POU5F1 expression and there is no expression in the ovaries of 4-mo old infants (63, 70, 71).
Development of the ovary was until recently believed to be the default pathway that occurred in the absence of SRY (72). However, opinion has changed since the identification of FOXL2, WNT4, and RSPO1 as crucial for female gonadal development (73–76). The importance of the WNT/β-catenin pathway in opposing testis determination has been extensively reviewed (77). Recently, the transcription factor FOXL2 was also shown to be important for maintance of ovary differentiation (75, 78) and has been termed the gatekeeper of ovarian identity (79). Somatic mutations of codon 134 in FOXL2 are reported in 94% of adult stromal granulosa cell tumors. Furthermore, its absence in juvenile granulosa cell tumors and in other tumor types suggests this mutation to be pathognomonic for adult granulosa cell tumors (reviewed in Ref. 80). The mutation is not found in stromal granulosa tumors in testis (81), and to our knowledge, mutation of FOXL2 has not been evaluated in mOGCTs.
Expression of the transcription factor DMRT1 has also recently been reported to be important for sex determination and has a labile effect on sexual fate in testis (82). Loss of the Dmrt1 transcription factor in mouse Sertoli cells, even in adult mice, activates Foxl2 and reprograms Sertoli cells into granulosa cells. In this microenvironment, theca cells are formed, estrogen is produced, and germ cells appear feminized (82). Mouse studies also indicate the presence of licensing factors for passing through a gateway to sexual differentiation to either ovary or testis differentiation and, importantly, where germ cells retain expression of pluripotency markers from their primordal state if not licensed (83). We believe that in addition to the genetic alterations, disturbances in the expression of licensing factors in human PGCs could create a time window during which more PGCs are kept in an immature state, postponing their development and maturation and thus increasing the risk of GCT development.
Both regulation of gene expression and communication between cells are essential for the development of the oogonia to the antral follicle. A genome-wide expression analysis has revealed differential expression between human male and female fetal gonads during organ development (84). Gap junction proteins have been described as players in the regulation of folliculogenesis and oogenesis. Connexin 43, encoded by the gene Gja1, is an important protein component of gap junctions, is detected at day 11.5 in somatic cells of the undifferentiated mouse gonad (after migration of germ cells but before sexual differentiation), and is later localized at the interface between somatic and germ cells (85). Involvement in WNT signaling is suggested because connexin 43 has been reported to bind to β-catenin (CTNNB1) and inhibit Wnt signaling in rat cardiomyocytes (86) and has been shown to have a tumor suppressor function and negatively regulate WNT signaling in cancer (87). Bone morphogenetic protein (BMP) signaling in the human fetal ovary is also developmentally regulated, where BMP4 negatively regulates postmigratory PGC numbers by promoting PGC apoptosis (88). Interestingly, the link between hormones and further development of PGC is strengthened by the finding that estrogen inhibits early development of mouse follicles through regulation of Kitlg expression (89).
It has been a longstanding assumption that ovaries, unlike testes, do not continue to produce gametes after birth. However, recently, human ovarian stem cells, which are mitotically active germ cells and capable of producing oocytes in vitro and in vivo, were reported to be isolated from ovaries of reproductive-age women (90). However, this finding remains disputed, and a thorough study failed to identify any oogonia in a series of 23 ovaries from individuals older than 2 years (91).
E. Predisposition to GCT in the dysgenetic and phenotypically normal gonad
In addition to the animal and human fetal studies described above, further insight into normal and pathological human gonadal development can be gained from patients with DSD (earlier termed intersex disorders) (59). The term DSD refers to a heterogeneous group of conditions of incomplete or disordered genital and/or gonadal development that leads to discordance between genetic, gonadal, and phenotypic sex. Recently, an extensive review of published DSD reports formed the basis of a new classification of DSD patients (92), intended to incorporate the patients' genetic background and karyotype and gonadal differentiation as well as risk of tumor development (47, 92, 93).
The earlier the gonadal development program is interrupted by a crucial defect, the less the gonad is able to differentiate into either a testis or ovary. Depending on the genetic defect, the gonadal location and histology will be somewhere in between a well-differentiated testicle and ovary (47). Strikingly, GCTs are the most frequently occurring tumors among DSD patients, but with large variation in the risk of developing such tumors depending on the specific condition. In an analysis of all published patient series, the risk was found to range from no increased risk to an estimated 60% increased risk (47). The different DSD groups, their respective GCT risk, and their associated genetic alterations have been extensively reviewed in several recent papers (4, 47, 59, 93). The main groups of DSD patients with gonadal dysgenesis and increased GCT risk are 1) 46,XY DSD patients with disorders of male gonadal development due to mutations in, for example, SRY, SOX9, SF1, or WT1, or disorders in androgen synthesis or action, including mutations in the AR gene that cause androgen insensitivity syndrome (which in its complete form causes phenotypic male-to-female sex reversal); 2) constitutional sex chromosomal alterations, for example, the karyotypes 45,X/46,XY (mixed gonadal dysgenesis), 47,XXY (Klinefelter syndrome and variants), 45,X (Turner syndrome and variants), and 46,XX/46,XY (chimerism); and 3) 46,XX DSD patients with disorders of female gonadal development due to various genomic aberrations, among others, the presence of SRY, duplication of SOX9, and mutations in RSPO1 (59) (Figure 1). By contrast, the DSD 46,XX patients with androgen excess and hypervirilization do not have increased risk of GCT development compared with the normal population (47, 59, 93).
The existence of a heritable genetic cause or predisposition to GCT is underlined by epidemiological studies of patients with normal gonads and GCT development. TGCT families, mOGCT families, and families with both TGCT and mOGCT patients have all been reported (94–97). Families with mOGCT cases are rare. To date, only 10 families have been reported with both TGCT and mOGCT and 8 families with 2 or more family members with mOGCT (95, 96). In most of these families, histology is discordant among the affected members. Pure DG is the most common GCT among the female cases, followed by teratoma and mixed GCT. Seminoma is the most common TGCT histology among male relatives (96). The mean age at diagnosis of familial cases of DG is reported to be significantly lower than the age of DG diagnosis in the SEER population (96).
The risk of brothers and fathers of TGCT patients is estimated to be as high as 8- to 10-fold and 4- to 6-fold, respectively, compared with the general population (98–100). The genetic contribution to development of TGCT is calculated to be 25%, third highest among all cancer types (101). Linkage analyses have been performed in several TGCT families, identifying Xq27 (102) and a deletion on the Y-chromosome (gr/gr deletion) (103) to be associated with familial TGCT. Genome-wide association studies (GWAS) have also been performed on sporadic cases of TGCT, revealing several predisposing loci (104–106). The predisposing alleles are located in or near several genes, including KITLG, BAK1, SPRY4, TERT, ATF7IP, and DMRT1. These loci have now also been verified in patients with familial and bilateral TGCT (107), and associations to selected loci have recently been reported in a small study of pediatric GCTs from patients of both sexes, including mOGCTs (108), see next paragraph. In addition, a recent study investigating several outcomes linked with testicular dysgenesis syndrome, including TGCT, genital malformations, and male infertility, confirmed the association with KITLG and TGCT (109). At above 2.5, the calculated per-allele odds ratios (ORs) reported for variants in the region of KITLG (104, 105) are the highest reported for any malignancy so far (110, 111), in agreement with epidemiological data that indicate that testicular cancer has higher heritability than most other cancers (101). Collectively, the 8 variants identified by GWAS increase the relative risk of testicular cancer up to 10.5-fold as compared with the general white male population (111). Other reported TGCT risk factors include cryptorchidism, infertility or subfertility, birth weight, and birth order, underlining the association of embryonic development and GCT (111–114).
To the authors' knowledge, there is no information about the mOGCT risk of sisters and mothers of mOGCT cases, probably due to the incidence of this disease being too low to enable an accurate risk assessment. To date, no linkage or GWAS have been published for mOGCT. Allelic distribution of 4 selected single nucleotide polymorphisms in TGCT-associated loci have been evaluated in a group of female GCTs from pediatric patients (age at diagnosis below 21 years) but only borderline significant associations were reported for the KITLG and BAK1 loci (108). However, the group of female GCTs tested in this study consisted of both gonadal and extragonadal tumors, the majority being teratoma and YST. These loci need further evaluation to clarify their relevance in mOGCT.
Hormonal exposure is among the reported risk factors for OGCT. Maternal hormonal factors have been linked with an increased risk of OGCT in the daughters, analogous to the increased risk of TGCT in male offspring and the association with testicular dysgenesis syndrome for which endocrine disruption by environmental chemicals has been proposed to be involved (115, 116). Increased risk for OGCT is linked with maternal use of exogenous hormones (ie, supportive hormones or oral contraceptives after conception) (OR = 3.6), maternal elevated body mass index (OR = 2.7), more rapid achievement of regular menstruation after menarche (OR = 1.8), and age at index pregnancy under 20 years (OR = 2.8) (117). Other studies have also reported association of OGCTs with reproductive risk factors such as parity, oral contraceptive use, ages at the first and last births, and time since last birth (118–120). However, in an epidemiological study of nonepithelial ovarian cancers, including OGCTs, doubling of maternal testosterone, androstenedione, and 17-OH-progesterone concentrations was associated with about 2-fold higher risk of sex cord-stromal tumors but not with OGCT. The analysis was performed in blood samples obtained during the last pregnancy before diagnosis of nonepithelial ovarian cancer (121).
Development of mOGCT may involve the combined effect of genetic alterations or predisposition and disadvantageous environmental factors such as exposure to maternal hormones, external endocrine disruptors, or adverse lifestyle-related factors that disturb the cells' normal biochemistry. The hazardous involvement of environmental compounds in the pathogenesis of OGCT remains a hypothesis but was recently supported by a rat study showing transgenerational epigenetic inheritance and altered phenotypes of pubertal onset and gonadal disease in the third generation after exposure to compounds such as plastics, dioxins, hydrocarbons, and pesticides (122).
The common risk factors identified for ovarian and testicular GCT support the model that the initial step in GCT pathogenesis occurs during embryonic development. Screening mutation analyses of genes known to be affected in other tumor types have so far revealed relatively few GCTs with mutations. However, it is perhaps not surprising that alterations other than mutations of the more classical tumor suppressor genes and oncogenes discovered in cancers of somatic tissue are involved in GCT. Cells of the germ line are set aside during early embryonic development and retain special characteristics in their ability to modulate the genome and transcriptome. One may speculate that the transforming mechanisms are based on the inherent characteristics of the PGCs, which are clearly different from those of somatic cells.
In contrast to the well-accepted precursor carcinoma in situ (CIS) of TGCTs, no precursor lesion has been established for mOGCTs, that is, excluding the gonadal dysgenesis patients (59). The CIS counterpart of the dysgenetic gonads in both phenotypic males and females is known as GB (123), which may originate from surviving POU5F1 positive germ cells in areas of undifferentiated gonadal tissue within the dysgenetic gonad (124). However, a few instances of GB have been reported to present alongside mOGCT in phenotypically normally developed ovaries (125), including in fertile women with 46,XX karyotype (126, 127). The analysis of more mOGCT cases is critical for the identification of the association of mOGCT with GB.
As knowledge of the genetic constitution of more DSD patients increases, so will our understanding of mOGCT in DSD patients as well as in patients with phenotypically normal gonads. We have previously proposed that alterations in even a limited number of cells in the developing gonad may lead to a delay of normal differentiation of oogonia into oocytes in fetal ovaries (55). The subsequent postponement or even failure of the onset of meiotic prophase may increase the chance of neoplastic transformation in the mitotically dividing germ cells (55). Transformation could target cells that are part of the PGC pool, but critical alterations in the supporting (granulosa/theca) cells or in connexin gap junctions, disturbing communication between cells, may also contribute to GCT development. The number of mOGCTs with documented gene mutations remains low, and most mOGCTs in DSD patients include the presence of some Y-chromosome material (59). mOGCTs may also develop against a background of gonadal Y-chromosome mosaicism (128). This also leads to a diagnostic problem for identification of patients with increased risk for bilateral disease. Array CGH analysis in 2 recent studies have found various DNA copy number gains and losses in DSD patients (129, 130). With the recent technological developments, full genome sequencing has become more achievable and will hopefully further extend our knowledge of the genetic backgound of GCT development among these patients.
II. Methods of the Review
This review encompasses mOGCTs of all subtypes and mixed histologies and excludes other gonadal tumors such as sex cord-stromal tumors of the ovary, juvenile or adult granulosa cell tumors, Sertoli or Leydig cell tumors, and fibro-thecomas. All mOGCT histologies were included, without distinguishing between mOGCTs arising in phenotypically normal females and GCTs from phenotypically female DSD patients. Because there is no consistently applied age cutoff separating pediatric and adult mOGCTs, all papers describing gonadal mOGCTs were included, regardless of age classification.
PubMed searches were carried out to thoroughly review all articles detailing DNA copy number and ploidy studies, gene and/or protein expression, mutation, and epigenetic studies in mOGCTs. As of March 2012, the search term “ovarian germ cell tumors” in PubMed returned 8338 articles, which were then filtered for additional terms specific to the relevant molecular studies. A flowchart of the article searches and filtering applied in this review is shown in Supplemental Figure 1 (published on The Endocrine Society's Journals Online web site at http://edrv.endojournals.org). The combination of more general search term usage with additional filtering of articles applied here was necessary to be able to include all relevant articles while distinguishing between epithelial and germ cell ovarian tumors as well as other spuriously returned articles.
Results from cytogenetic banding of mOGCTs were derived from our previous review (131) as well as 3 newer case reports (132–134). DNA ploidy results were summarized from a total of 15 articles (132, 135–148). An overview of the ploidy results from these 15 articles is presented in Figure 2A and Supplemental Table 1. Seven articles described CGH in mOGCTs, with the exclusion of repeatedly published cases and case reports with constitutional alterations. Details of chromosomal imbalances could be extracted from 6 of these 7 articles (131, 132, 149–152) and are presented in Figure 3 and Supplemental Table 2.
Figure 2.
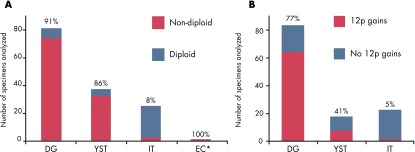
Low-resolution genome instability of mOGCT histological subtypes. A, Results from ploidy analyses from the following 15 articles: Refs. 132 and 135–148. The percentage of nondiploid samples is indicated above each bar. B, DNA copy number gain at chromosome arm 12p, as reported in 8 FISH articles (137, 146, 153, 155–158, 291) and 7 CGH articles (both chromosomal and array-based) (131, 132, 149–152, 340). The percentages of samples with gain at chromosome arm 12p are indicated above each bar. In both plots, the number of cases analyzed is shown on the y-axis. *, EC is represented by only 2 specimens, both nondiploid.
Figure 3.

DNA copy number alterations for histological subtypes of mOGCT as reported by 6 CGH articles (131, 132, 149–152). The percentages of samples with gains and losses are shown in green and red, respectively, on the y-axis. Genomic positions given by the chromosome numbers are indicated along the x-axis. Centromere positions are indicated by vertical, dashed lines, and alterations on the p- and q-arms are mapped to the left and right of the dashed lines, respectively. The different panels summarize the findings per histology for DG (n = 35), YST (n = 14), and IT (n = 16).
DNA copy number gain at chromosome arm 12p could be inferred from 8 fluorescence in situ hybridization (FISH) articles (137, 146, 153–158) and 7 CGH articles (both chromosomal and array-based) (131, 132, 134, 149–152). The results are summarized in Figure 2B and Supplemental Table 3. A total of 6 articles described microsatellite analysis of mOGCTs (150, 159–163).
For gene/protein expression studies, the 8338 OGCT articles were filtered for the word “expression” either in the title or abstract. Among these were 5 articles describing mRNA gene expression profiling (164–168) and 4 articles describing miRNA profiling (166, 168–170). Figure 4 and Supplemental Tables 4 and 5 give an overview of the mOGCT samples included, statistical analyses run, number of genes identified, and platforms used in these mRNA/miRNA expression profiling studies.
Figure 4.

Overlapping results among genome-wide mRNA and among miRNA expression studies of mOGCTs are shown in Venn diagrams where each number indicates the number of common mRNAs or miRNAs identified as overexpressed. A, To the left, mRNAs overexpressed in DG/seminoma vs other GCT types as described, and to the right, mRNAs overexpressed in YST vs other GCT types as described. B, To the left, miRNA overexpressed in DG/seminoma vs YST, and to the right, miRNAs overexpressed in YST vs DG/seminoma. Abbreviations: ova., ovarian; Sem, seminoma (TGCT histological subtype); test., testicular.
A total of 41 articles described expression of proteins by immunohistochemistry (IHC) in 1 or more mOGCT histological subtypes. Most of these articles (n = 35) detailed the number of samples per subtype and how many of these stained positively; results from all of these 35 with references are summarized in Figure 5A. For the remaining 7 articles (164, 171–176), either scoring was reported as a labeling index from which it was not possible to extract the number of positively scoring samples, or TGCT and mOGCT histologies were grouped in such a way that it was not possible to extract the number of positively scoring mOGCT samples. Additionally, 6 articles reported gene/protein expression in single patients only. The single patients' reports are not discussed in this review but are listed in Supplemental Table 6.
Figure 5.

Overview of single-marker results in mOGCT ordered by number of cases for protein expression (A) and gene mutations (B). The number of cases with positive protein staining or gene mutation of the total number analyzed is shown within each circle. The circles are color-coded according to the percentage of positive staining. The total number of tumors studied (n) for each protein is followed by the relevant references (Ref).
Ten articles studied gene mutations, both germline and somatic, in more than 1 patient, and these are included in this review as well as illustrated in Figure 5B (55, 144, 156, 163, 177–182). A further 8 articles reported mutations detected in single patients only and are listed in Supplemental Table 6 as germline or somatic, along with the reported effect of the change, if any. Finally, 8 articles reported epigenetic and DNA methylation studies in mOGCTs (163, 171, 172, 183–187).
Lists of genes with significant differences in expression, as reported by each individual profiling study, were extracted. From these lists, we identified those genes that were recurrently found to be differentially expressed. To ensure concordance across the gene expression profiling studies, the Ensembl gene identifier (ENSG number) for each gene was mapped using the BioMart Central Portal (www.biomart.org) (188) with Ensembl release 59 and the Genome Reference Consortium Human genome build 37. Failing this, 2 other online databases were manually queried for missing ENSG numbers, the HUGO Gene Nomenclature Committee database (www.genenames.org) (189) or GeneCards version 3 (www.genecards.org) (190). The one gene for which an ENSG number could not be identified after these 3 searches, CD24 on chromosome 6q21 (167), was excluded from further analysis.
For all these studies, results were summarized for the gonadal mOGCT samples, excluding tumors from extragonadal sites, relapses, and metastases. For CGH, ploidy, and 12p studies, analyses, and calculations were performed for pure histologies of mOGCT. However, for IHC protein studies, results for both pure histologies and pure components of mixed mOGCT were occasionally reported together, so that these groups were indistinguishable and samples from both subgroups were included in the summary. An exception was made for genome-wide microarray studies, where the nature of the analyses performed made it impossible to exclude female GCTs of extragonadal origin or TGCT samples, when these were grouped together with the mOGCTs. Fisher's exact tests were run on www.statpages.org and SPSS (PASW Statistics version 18.0) to calculate and statistically evaluate the frequencies between the histological subtypes, and a cutoff of P < .02 was set for significance. Statistical comparisons were performed only among samples of pure DG, IT, and YST histologies.
III. Genome Profiling of mOGCT
The genome studies of mOGCTs mirror the developments in technology, starting with ploidy determination, cytogenetic banding analyses, and more recently, application of CGH methods. This section reviews these results as well as FISH and loss of heterozygosity (LOH) studies performed on mOGCTs.
A. DNA ploidy: image and flow cytometry analyses
A total of 181 primary mOGCT specimens in 15 articles have been analyzed for ploidy by image cytometry (ICM) and/or flow cytometry (132, 135–148). Five studies were based on analyses performed on 20 or more cases (135, 138–140, 142). The results from all 15 articles are summarized in Figure 2A and Supplemental Table 1.
The pure DGs and YSTs analyzed were mainly nondiploid (ie, tetraploid, polyploid, or aneuploid), at 91% (74 of 81) and 86% (32 of 37), respectively. Conversely, only 8% (2 of 25) of the ITs reported were identified as nondiploid. The DG nonploidy is in line with early densitometric measurements of DNA content in DGs being about double of control lymphocyte nuclei (191). Whether all published ITs are truly pure ITs or may contain other undetected histological components may be questioned. This is supported by Baker et al (139) who on central review identified YST components in ITs (previously classified by the submitting institutions), 2 of 3 of which were aneuploid. The rare histological subtypes CC and EC are apparently nondiploid, but ploidy analyses have been reported for only a few cases (135, 137, 139, 142, 143, 148), mainly as part of mixed tumors. Interestingly, ICM of 47 mOGCTs found that patients with nondiploid and FIGO stage II-IV mOGCT have poorer survival compared to patients with diploid or FIGO stage I mOGCT (135).
It has been suggested that the pathogenesis of malignant GCT in patients with gonadal dysgenesis differs from GCT in genetically normal females, in that polyploidization is not required (139). However, in a case with bilateral DG and GB, both components were nondiploid (132), and in 3 patients with DG/GB, all 3 DGs were nondiploid (135).
B. Chromosome G-banding analyses
A total of 20 reports describing cytogenetic banding analysis of 50 mOGCTs were published between 1982 and 2000, as previously reviewed (131). Thirteen tumors revealed normal karyotypes, whereas 37 had clonal chromosome aberrations. Among 20 tumors with structural changes, isochromosome 12p or i(12p) was present in 100% of DGs (4 of 4), 50% of YSTs (1 of 2), 40% of mixed mOGCTs (4 of 10), 1 metastasis with MT differentiation from an IT, but not in any of the pure ITs (0 of 3). However, extra copies of chromosome 12 were reported in 2 pure ITs. Trisomy 14 is a recurrent numerical chromosomal abnormality in mOGCT observed in 10 of 37 cases (131). Since 2000, 3 new papers with a total of 6 mOGCTs (1 YST, 2 ITs, 2 mixed mOGCTs, and 1 of unspecified histology) containing karyotypes with numerical and/or structural alterations have been published (133, 158, 192). The YST contained a complex karyotype with several alterations, including extra copies of chromosome 12 and 14 (case 100 in Ref. 158). Gain of chromosome 14 in mOGCT is further supported by the case report of a 16-year-old patient with mixed mOGCT (IT/YST) with both tumor and constitutional karyotype 48,XXX,+mar, resolved by CGH as 14pter→q21 (133). Brassesco et al (192) reported a mixed mOGCT (teratoma/YST) with the tumor karyotype 46,XX t(3;20)(q27;1q13.3)[4]/46,XX,del3q27[3]/46,XX[30]. The 2 ITs presented the karyotypes 47,XX,+mar[2]/46,XX[15] and 45,XX-16[5]/46,XX[15], respectively (158). Due to difficulties related to cell culturing and associated selection of tumor cells, but also advantages of newer technologies including higher resolution, the cytogenetic banding technique has largely been replaced by CGH and other molecular genetic techniques. However, G-banding is still used to identify the constitutional karyotype of patients with GCT, mainly for the evaluation of potential Y-chromosome presence (132–134). The copy number of chromosome 12 among mOGCTs is further discussed in Section III.C. and summarized for the in situ hybridization techniques in Figure 2B and Supplemental Table 3.
C. In situ hybridization: CGH and 12p FISH analyses
DNA copy number alterations as detected by chromosomal (5- to 10-Mb resolution) or array CGH (800-kb resolution) have been published for 79 primary mOGCTs (131, 132, 149–152). The low incidence of mOGCT combined with the histological complexity limit the availability of samples for determining subtype-specific changes. Histograms presenting the copy number changes (gains and losses) identified by CGH in the 3 main histological subgroups DG, YST, and IT are presented in Figure 3. The specific gains and losses in all 79 mOGCTs are listed in Supplemental Table 2.
Gains of parts or the whole of chromosome arm 12p, including isochromosome 12p, are frequently detected among mOGCTs, similarly to TGCT (193). The gain of 12p, as detected by CGH and 12p FISH, is summarized in Figure 2B and Supplemental Table 3. Gain of 12p was significantly associated with DG as compared with both YST and IT (P = 6 × 10−3 and P = 4 × 10−10).
CGH results from a total of 35 DGs, 16 ITs, 14 YSTs, 1 GB component, in a DG and 13 mixed or components of mixed OGCTs, are reported. The average number of chromosome arm gains varies from 7.1 in DG to 4.8 in YST and 1.2 in IT. The average number of chromosome arm losses varies from 2.6 in YST to 2.0 in DG and only 0.06 in IT. The DGs and YSTs show many of the same but also some unique and histology-specific patterns of genomic alterations. One of the IT samples in Supplemental Table 2 (151) contained certain DG-associated gains, ie, gains from 7, 8, 12p, and 21. It could be speculated that this sample may in fact have originated from a tumor of mixed histology but has been misinterpreted as an IT due to a limited amount of tumor tissue available. Finally, in a study of components from 6 mixed mOGCTs, 83% (5 of 6) of the cases had detectable i(12p) in their nonteratomatous components and 66% (4 of 6) in the teratomatous components, as compared with 5 pure MTs and 3 pure ITs, none of which showed evidence of i(12p) or other gains from 12p (155). This supports the idea that teratoma components in mixed mOGCTs have a different pathogenesis than pure teratomas of the ovary (155).
With the caveat that the numbers of YSTs and ITs analyzed are small (n = 14 and 16, respectively), Fisher's exact test was applied to calculate the statistical significance of differences in DNA copy number changes among DG, YST, and IT. All statistically significant differences in copy number changes (P ≤ .02) identified by pair-wise comparisons of the 3 main mOGCT histology subtypes are visualized in Figure 6.
Figure 6.
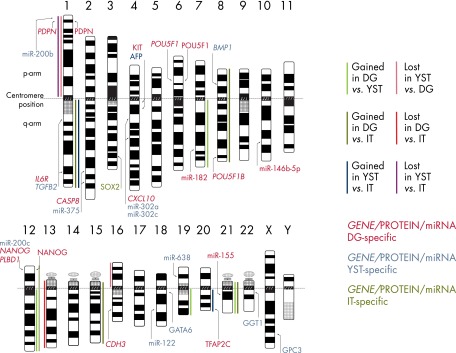
An overview of histology-specific differences in DNA copy number alterations (solid bars), mRNA/miRNA expression (gene names, italics), and protein level (protein names) in mOGCT. Significant differences in DNA copy number gains and losses detected by CGH are presented as colored bars. For DG vs YST, light green bars reflect gains in DG as compared with YST and light red bars reflect losses in YST as compared with DG. For DG vs IT, dark red and dark green bars reflect regions of loss and gain, respectively. The significant gains and losses in YST vs IT are represented by blue and purple bars. Histology-specific gene expression identified from our overlap analysis of microarray results (Figure 4) is indicated to the left of chromosomes. The histology-specific protein expression from IHC studies (Figure 5A) is indicated to the right of chromosomes. The proteins included in the figure are those that were positive in ≥50% of the samples tested and with significantly different expression (P ≤ .02) among histology subtypes. Gene/protein symbols are color-coded according to the histological subtype associated with overexpression.
D. Polymorphic marker studies
To the authors' knowledge, only 6 publications have reported studies on microsatellites in mOGCTs and only a limited part of the genome has been covered. Two studies contain 35 (32 patients) and 36 cases, respectively (159, 160), whereas the last 4 papers contain only from 1 to 6 cases of mOGCT each (150, 161–163). The LOH study by Faulkner and Friedlander (159) analyzed 35 mOGCTs in regions previously associated with TGCT, ie, 62 loci on the chromosome arms 3q, 5q, 9p, 11p, 11q, 12q, 17p, and 18q. LOH was found most frequently at 12q22 (53%), and allelic losses were identified in these regions at similar frequencies as in TGCT (mean 35.5%, range 18%–53%) (159). However, based on the results of the LOH pattern among the subgroups YST (n = 6), DG (n = 6), and IT (n = 16), Faulkner and Friedlander (159) propose that the complete absence of deletions on chromosome 11, 17p13, and 18q21 and the relatively low incidence at 5q31 and 9p22-p21 among DGs suggest that loss of these regions in DG may result in progression to a non-DG phenotype. Radice et al (161) tested only 1 IT and reported LOH at all 24 analyzed loci on 15 chromosome arms. Zahn et al (150) studied 19 loci at chromosome arm 1p and identified LOH in the DGs tested, but not in any of the ITs or the 3 mixed mOGCTs. For analysis of the APC gene among GCTs, 8 loci in the chromosomal region 5q21.1–5q23.1 were evaluated for LOH in 6 mOGCTs (2 DGs, 1 YST, and 3 of mixed histologies). LOH was observed among the YSTs and 2 of the mixed samples, but neither of the DGs showed LOH in this region (163).
Microsatellite instability (also termed replication error, RER) has also been reported. Of the 64 loci analyzed in 36 mOGCTs, RER was reported at 1 or more loci in 33% (12 of 36), including 57% (4 of 7) DGs, 33% (4 of 12) YSTs, and 21% (4 of 19) ITs. Only 3 mOGCTs presented RER at 6 or more loci (160). DGs and YSTs have a higher frequency of RER-positive cases than ITs, reflecting the different origins and/or molecular development of the histological subgroups. The lower frequency of RER observed among IT has been suggested to reflect the fact that this histological subtype may derive from either mitotic or meiotic errors. However, in King et al (162), the 2 mOGCTs analyzed at 2 loci (4q12–13 and 5q11–13) showed microsatellite instability in the IT but not in the DG.The pathogenetic significance of this type of instability in GCT remains uncertain, but the results indicate a more significant role in mOGCT as compared with TGCT (160).
IV. Transcriptome Profiling of mOGCT
In this section, we summarize and discuss the published transcriptome studies of mOGCT. Several methodological issues (sample content, tumor histology, and the mix of male/female GCTs) influence the reported results in these studies. In subsections, the recurrently identified, aberrantly expressed mRNA and miRNA reported among these studies, and their potential biological relevance, are discussed.
To date, only 7 studies have investigated the transcriptome of mOGCTs, reporting mRNA expression data (164–168) and/or miRNA expression data (166, 168–170). Many of the same issues complicate the analysis of microarray gene expression data of mOGCT as for TGCT, the latter discussed in our recent review (194). Not least of these is the availability of tumor tissue because mOGCTs occur rarely and the difficulty in obtaining and analyzing the corresponding normal tissue, the female PGCs, or fetal oogonia. Furthermore, both mOGCTs and TGCTs show a similar range of distinct histologies, which consequently reduces the effective sample sizes. Histology-specific expression patterns have been reported for TGCT (195). Indeed, 6 of the 7 expression profiling studies chose to group testicular seminoma and ovarian DG together for analysis due to previously identified similarities, presumably also to improve power (165–170). YSTs from male and female patients were similarly grouped (166, 167, 170). One of the studies combined seminoma and DG for comparison to spermatocytic seminoma (SS), a TGCT type that is thought to arise from mature germ cells and that occurs in older men (165). In this review, we compared lists of significant genes (mRNAs/miRNAs) published in all these articles and present those that recur between studies in Venn diagrams (see Figure 4). An overview of the expression profiling studies with number of samples, histology, and number of genes listed as significantly altered is presented in Supplemental Table 4. Array platform information on each of the 7 studies is listed in Supplemental Table 5. In addition, an overview of the published case reports of mRNA expression performed on mOGCTs from single patients is presented in Supplemental Table 6.
Combining similar histologies from mOGCT and TGCT undoubtedly influences which genes are identified as significant, as will be discussed in Sections IV.A. and VI.B. No study directly compared mOGCTs with their presumed cell of origin and arguably the most appropriate nonmalignant control, the PGCs, or fetal oogonia. However, a study of fetal ovaries, at stages from 9 to 18 weeks of gestation, has identified genes that are differentially expressed in the fetal ovary during this developmental time window (84). The genes listed herein were examined for recurrence in the published mOGCT studies and are also discussed in Section IV.A.
In addition to the fact that mOGCT samples often contain a proportion of normal/immune cells, mOGCTs may have a varied and complex histology, presenting as admixtures. This underlines the necessity of careful examination of hematoxylin- and eosin-stained tissue sections by an experienced pathologist to ensure which histological subtype is being profiled for gene expression. Because of the substantial correlation observed between histology and expression, ideally, either samples of pure histology or single microdissected components should be analyzed, as in the TGCT study by Skotheim et al (195). Histologies from females and males should also be analyzed separately to detect intersex differences. Additionally, different probe designs and resolutions of the various microarray platforms, and the choice of statistical approach will all influence which genes are ultimately reported as significant. Despite this, recurrent findings are identified across studies, indicating biological relevance and robustness across platforms.
A. mRNA expression
Only 1 paper reported a gene expression profiling study of strictly mOGCTs, comparing 4 DGs and 4 YSTs (164). Interestingly, this study revealed that a subset of 8 WNT/β-catenin signaling components (CTNNB1, DVL1, DVL3, APC, WNT2, WNT5A, WNT8B, and WNT2B) was sufficient to distinguish between the 2 histological subtypes, underlining that there is differential expression in the WNT signaling pathway between DGs and YSTs and the importance of this pathway to pathogenesis in mOGCT. The results were confirmed by RT-PCR and IHC for several of the components, including CTNNB1 (encoding β-catenin) and WNT2B, where β-catenin was found to be expressed in the cytoplasm within all histological subtypes, but with only focal and weak staining for DGs and ECs. β-Catenin nuclear accumulation was observed only in YSTs and teratomas (MT/IT) (164).
In our review analysis, we compared the gene list identified in the study of strictly ovarian DGs and YSTs (164) with the genes identified in the study of the DG/seminoma relative to SS (165). The results showed that 4 of the top 50 genes that differentiated DG/seminoma from SS were also among those reported overexpressed in DG (Figure 4A). The genes were CASP8, CDH3, CXCL10, and IL6R. The biological functions of these genes, also during germ cell (tumor) development, are beginning to be elucidated. Caspase-8 (CASP8) is traditionally thought of as an apoptosis regulator (196), and studies of germ cell proliferation and apoptosis in the developing human ovary between weeks 14 and 19 have indicated that CASP8 contributes to increased apoptosis and increased germ cell loss preceding primordial follicle formation (197). Additionally, significant CASP8 activity has more recently been observed in certain tumor cells to promote cell motility (198) and adhesion (199). Similarly, the β-catenin–interacting cadherin-3 (CDH3) is known to regulate motility and invasion in prostate cancer cells (200) and has been suggested to be a marker of poor prognosis in breast cancer (201). CDH3 could also be related to the PGC origin of the GCTs, because these cells have been shown to express CDH3 both during and after migration to the genital ridges in mice (202).
The 2 other genes found to be recurrently overexpressed among DGs, IL6R and CXCL10, are known to be involved in cytokine signaling and part of the immune response. The proinflammatory cytokine receptor interleukin-6 receptor subunit α (IL6R) is a central component in autocrine IL-6 signaling, leading to the production of growth and survival factors in both breast and lung cancer patients (203). Expression of IL6R in human fetal ovary was also found to be developmentally regulated, significantly increasing with gestation from 8 to 16 weeks, and specific expression in germ cells of the fetal ovary indicates these to be the target of IL-6 signaling (204). Interestingly, based on analogous in vitro studies of mouse PGCs (205), it is suggested that the up-regulation of receptor expression prevents premature entry into meiosis and maintains an immature germ cell population in the human fetal ovary (204). Studies of TGCTs (n = 21) have revealed that C-X-C motif chemokine 10 (CXCL10) is constantly expressed among seminomas and mixed TGCTs (seminoma/nonseminoma), and that TGCT-infiltrating T lymphocytes express the CXCL10 receptor CXCR3 (206). This could in turn lead to recruitment of T lymphocytes to the tumor site (206), in agreement with the observation of lymphocyte infiltration in seminomas as well as DGs (207). However, the effect of tumor-infiltrating lymphocytes depends on their diversity and composition and may positively or negatively affect both tumor growth and patient clinical outcomes (reviewed in Ref. 208). Together with other chemokines, CXCL10 may exert an antimalignancy effect by inducing leukocyte infiltration to tumors (209), and it has also been indicated that CXCL10 can repress angiogenesis (210). These genes appear to have implications both for male and female GCTs, even though the functions or pathways in which they are involved need not be common to both sexes.
Recurrent overexpression of 5 genes was revealed when comparing the 2 studies of DG/seminoma, one comparing the DG/seminoma group with SS (165) and the other comparing DG/seminoma with ovarian and testicular YSTs (167) (Figure 4A). These 5 genes were the pluripotency genes NANOG, POU5F1, POU5F1B, PLBD1, and PDPN. The recurrence of NANOG, POU5F1, and possibly POU5F1B underline the PGC-like traits and origin of DG and seminoma. Interestingly, POU5F1B encodes a putative POU5F1P1 protein localized in the nucleus that has been suggested to act as a transcriptional activator and regulate expression in a similar way to the POU5F1 isoform 1 (211). PLBD1 and PDPN are both also specifically reported to be markers of the TGCT precursor CIS (194) and thus support a degree of common expression between DG and TGCT. Little is known about PLBD1, apart from that it contains a phospholipase B domain, whereas PDPN encodes for podoplanin (PDPN), a small mucin-like protein that has been known to mediate cell migration and invasion both in vitro and in vivo (212).
The fact that there are overexpressed genes identified in common for seminoma and DG may reflect either common causal factors involved in tumor development supporting a shared developmental pathway for a subgroup of mOGCT and TGCT or remnant traits of the state of their mutual precursor, the PGC. However, recurrence of these genes could also be due to dominance of these genes in either DG or testicular seminoma, camouflaged by the mix of TGCT and mOGCT. Functional and gene expression studies with larger sample sizes of TGCTs and mOGCTs for comparative analysis are required to resolve this. No genes were found to overlap between all 4 gene expression profiling studies that included mOGCT samples (Figure 4A).
The cell signaling genes BMP1 and TGFB2 were both found to be overexpressed in YSTs in 2 of the 3 studies identifying genes up-regulated among YSTs as compared with seminomas and/or DGs (Figure 4A). One study was based strictly on ovarian YSTs and DGs (164), whereas the other study compared YSTs from both ovary and testis with DGs, including 1 germinoma of the central nervous system in a female patient (166). The latter article specifically focused on the TGF-β/BMP signaling pathway, profiling 84 genes encoding components of this pathway, and found 29 of these to be significantly more highly expressed in YST compared with DG (166). This pathway regulates, among others, embryonic development (213). Its biological relevance is underlined by the fact that mutation of the BMP receptor alk6b impairs germ cell differentiation and causes GCT in zebrafish (214).
Overexpression of certain pathway components may reflect the distinct levels of differentiation between these 2 tumor types and the developmental stage of their cell of origin. However, as previously described, the confounding effect of combining male and female YSTs for comparison with the purely female DGs should not be disregarded, especially because 5 of 7 YSTs were from males compared with female DGs (here 3 DGs and 1 germinoma). Nevertheless, the studies report that the histological subtypes generally cluster together, indicating that the expression is similar across gender for DG and seminoma and ovarian and testicular YST, both on the mRNA (165–167) and miRNA (166, 168–170) level.
Finally, it was interesting to compare the gene lists from the 3 mOGCT microarray studies with those identified by transcriptome analysis of the developing gonad. This study compared normal fetal female gonads at 9 to 11 weeks with those at 12 to 18 weeks of gestation (84), earlier described in Section I.D. One gene, HERC5, which was up-regulated in fetal ovary during normal development, was also overexpressed in DG compared with YST (167). Three genes, BMP7, IGFBP5, and TNC, were down-regulated both during development in normal fetal ovary and in DG compared with YST (84, 164, 166).
B. miRNA expression
A total of 4 papers published miRNA expression profiles for mOGCTs (166, 168–170). In 2 papers, the mOGCTs were included in a larger collection of both male and female GCTs, including cell lines and various nonmalignant tissue controls (168, 169). The other 2 papers (166, 170) compared the global miRNAs profiles of the main mOGCT histological subtypes DG and YST to identify repeatedly identified, characteristic miRNAs (Figure 4B). However, both articles also included expression profiles of testicular YSTs and seminomas in their analyses. Only one article profiled miRNA expression in IT (n = 3), however no IT-specific findings were reported (168).
All 4 papers identified 2 miRNA clusters, mir-302–367 and mir-371–373, as overexpressed in malignant GCTs when compared with nonmalignant tissue controls (166, 168–170). Although not all of these papers specifically aimed to derive histology-specific miRNA signatures, both of these miRNA clusters were found to be responsible for separating the subtypes seminoma from EC, EC from YST, and EC from teratomas in a principal components analysis (169). Knowledge of the transcriptional regulators of the identified clusters mir-302–367 and mir-371–373 is scarce, although expression of these clusters was associated with overexpression of the transcription factors TEAD4 and SOX17 (168). This coordinate overexpression seems specific for malignant GCTs, with no similar findings for other malignancies or diseases to date (168). Gene Ontology (GO) analysis has shown that the down-regulated mRNA targets for mir-302–367 and mir-371–373 mediate cellular processes important in oncogenesis and malignant progression, supporting the functional significance of these miRNA clusters in the biology of malignant GCTs (168). Interestingly, these miRNA clusters have also been found enriched in mouse and human ESCs, and recent studies have revealed insight into their involvement in regulation of the cell cycle, pluripotency, and early development and in PGC migration (as reviewed in Ref. 215). A regulatory feedback loop involving β-catenin/lymphoid enhancer-binding factor 1, mir-371–373, and dickkopf-related protein 1, a well-known inhibitor of WNT/β-catenin signaling, has been identified and may have a critical role in regulating the activity of WNT/β-catenin signaling in human cancer cells (216). Furthermore, RECK protein, essential for mammalian development and a tumor suppressor, is a target of miR-372/373, and miR-372/373 mediate hypoxia-induced down-regulation of RECK through the hypoxia-inducible factor 1-α/twist-related protein 1 pathway (217).
The overall most significantly overexpressed miRNA among YSTs was the same in both studies, miR-375 (166, 170). Dysregulation of miR-375 has been observed for various tumor types, including head and neck and esophageal squamous cell carcinoma, and lung and gastric cancer (218–221). This may have clinical relevance because changes in circulating levels of miR-375 have been associated with prostate cancer metastasis (222), and a combined analysis of miR-375 and miR-142-5p has been suggested to predict recurrence risk for gastric cancer patients (219). Signaling pathway analyses of miR-375–regulated genes have indicated involvement of cell cycle regulation, focal adhesion, MAPK, TGF-β, WNT, vascular endothelial growth factor (VEGF), and apoptosis pathways (219).
In comparing the global miRNA profiles of the histological subtypes DG/seminoma and YST (Figure 4B), ovarian DGs and testicular seminomas were invariably reported to cluster separately from YSTs of both male and female patients (166, 168–170). Two of these studies included multiple histological subtypes from both sexes and demonstrated up-regulation of mir-302–367 and mir-371–373 clusters to be the most significant, regardless of histological subtype (YST and DG/seminoma), tumor site (ovary, testis, or extragonadal), and patient age (168, 169). Recently, miRNAs from the mir-302–367 and mir-371–373 have been suggested as potential serum biomarkers of malignant GCT (223).
Interestingly, both the malignant GCT clusters mir-302–367 and mir-371–373 showed even higher overexpression in YST as compared with DG/seminoma in 2 independent studies (166, 170). The mRNA profiles were used to identify transcription factors with corresponding and associated overexpression in YST compared with DG/seminoma, where the transcription factors GATA6, GATA3, TCF7L2, and MAF were found to have predicted binding sites in the mir-302–367 cluster (170). The mRNAs likely down-regulated in YST due to miR-302 overexpression included mediators of tumor suppressors and apoptosis regulators (170), and miR-302 has also been shown to modulate the BMP response (224).
Three miRNAs were recurrently identified with higher expression among DGs as compared with YSTs: miR-146b-5p, miR-155, and miR-182 (Figure 4B) (166, 170). The specific functions of these miRNAs in GCTs and DGs are unknown, but overexpression of these has also been reported in other cancers as breast, lung, cervix, and colon cancer (225–227), and interactions with BRCA1 have been reported for all 3 (225, 228, 229). Furthermore, miR-155 has also been shown to down-regulate DNA mismatch repair genes and induce mutator activity (230, 231). A mutator phenotype among DGs was also identified in a microsatellite analysis (Section III.D.) (160). We recently reported that induced expression of miR-182 in a colon cancer cell line was linked to the down-regulation of the forkhead transcription factor FOXF2 (232). Interestingly, from mouse studies, Foxf2 mutation is shown to influence the expression of Bmp4 and Wnt5a (233), thus linking miR-182 to the BMP and WNT signaling pathways.
Eight miRNAs were recurrently overexpressed in YST as compared with DG: miRNA-122, miR-200b, miR-200c, miR-302a, miR-302c, miR-302c*, miR-375 (discussed above), and miR-638 (Figure 4B) (166, 170). In mouse testis, miR-122 is shown to bind and reduce the expression of Tnp2 (transition protein 2) mRNA, 1 of the key transition proteins that replaces histones during the early phase of chromatin condensation that accompanies spermiogenesis (234), and is among the miRNAs with the greatest fold enrichment in prepubertal mouse testis (235). Furthermore, recent studies indicate miR-122 to be involved in translational regulation of TP53 mRNA and cellular senescence (236).
Members of the mir-302–367 cluster, also found overexpressed in YST by both studies, have been found to specifically modulate the TGF-β/BMP signaling pathway (166). This is especially interesting in light of the corresponding alterations in mRNA levels of genes encoding components of the TGF-β/BMP signaling pathway identified in the same study. The miR-200 family has been shown to prevent pluripotency and, along with the transcription factors TGFβ and zinc finger E-box-binding homeobox (ZEB), is involved in the epithelial to mesenchymal transitions (237, 238). An analysis of ovarian carcinomas has suggested tumor proteins 63 and 73 (TP63 and TP73) as activators of miR-200 miRNA family transcription (239). Taken together, this underlines the association of TGF-β/BMP signaling and the differentiation state/histology of GCTs, highly active among YSTs, but mostly absent in DGs.
Decreased levels of miR-638 are reported in gastric cancer (240), at the invasion front of colorectal metastases (241) and in non–small-cell lung cancers (242). Interestingly, a recent report links increased miR-638 to the benzo(a)pyrene-induced carcinogenesis and shows that miR-638 targets BRCA1 mRNA, leading to reduced protein expression and may contribute to inhibition of DNA repair (242).
V. Biomarkers of mOGCT
From the genome profiling section, it is clear that 12p gains are not pathognomonic for mOGCT in contrast to what is seen for TGCT, where the presence of additional 12p copies can be used to classify an extragonadal tumor of uncertain cell origin (193, 243). In the transcriptome profiling section, the mir-371–373 and mir-302–367 clusters in mOGCT were suggested as potential serum biomarkers of malignant GCT for both genders (223). The following section reviews single-marker studies, including the epigenetic and mutation events and protein expression among mOGCTs and their potential as biomarkers. Section V.C. is dedicated to an overview of the histology-specific features identified among mOGCTs.
A. DNA methylation
Epigenetic studies of mOGCT have been limited to 2 main groups of genes. The first group includes H19, IGF2, and SNRPN, where the DNA methylation status has been investigated in the tumor because it is thought to reflect the developmental status of the cell of origin, the PGC (183–186). A second group of genes include CDKN1B, CDKN2A, and APC, where the DNA methylation and subsequent expression may have clinical and pathological consequences for tumorigenesis (171, 172). Finally, 1 article has assessed global methylation of mOGCT (187).
During normal development of the gonocyte/oocyte from a PGC, imprinting erasure occurs during migration and maturation, where it is hypothesized that the tumor cells' imprinting status may reflect the stage of development of the PGC when malignant transformation occurs. The reciprocally imprinted genes IGF2 and H19 (which are maternally and paternally imprinted, respectively) are the most extensively studied imprinted genes to date, explaining the interest in their DNA methylation status in male and female GCTs. For mOGCT, increasingly sophisticated methods were applied to study DNA methylation of these genes as they became available. Initially, allele-specific expression of IGF2 and H19 was investigated by RT-PCR (184, 185). Both these studies reported a variable pattern of IGF2 and H19 imprinting for the mOGCTs analyzed, including tumors of DG, YST, and IT histology, but with no reported association to histology. More recently, the technique of methylation-sensitive single nucleotide primer extension was applied to study the promoter methylation status of these genes, and all 8 mOGCTs were found to be hypomethylated at the imprinting control region of IGF2/H19 (183).
All these findings are consistent with the fact that the methylation status of IGF2/H19 in mOGCT may reflect preservation of the imprinting erasure in the PGC. However, it cannot be ruled out that the methylation status and subsequent allele-specific expression of H19 and IGF2 may in fact be a result of abnormalities acquired during tumorigenesis. As such, a new maternally imprinted gene, SNRPN, was proposed and has been studied by bisulfite modification, PCR, and restriction analysis (184) as well as by differential restriction and Southern blot analysis (186). The 5′-flanking region of SNRPN was found to be unmethylated in 15 of 17 mOGCTs by the first method, which included 8 DGs and 2 YSTs; conversely, the 2 ITs studied were found to be methylated (184). By differential restriction and Southern blot analysis, 14 of 16 mOGCTs were found to have a nonsomatic methylation pattern, with no histology-specific differences (186). Both studies support the findings on the imprinting of IGF2/H19, namely, that although demethylation of imprinted genes and alterations in their expression may be implicated in GCT oncogenesis, it may still be a reflection of the imprinting status of the PGC at transformation. Compared with TGCTs, mOGCTs were found to have a more variable pattern of imprinting. This may be due to the fact that ovarian germ cells undergo early arrest in meiosis, whereas those of the testis are subjected to mitotic arrest until puberty and have not entered meiosis. The variable pattern in mOGCTs may reflect the different extent of imprinting erasure and reestablishment during gametogenesis (185).
Promoter methylation of CDKN2A and CDKN1B, which encode the cyclin kinase inhibitor proteins p16 and p27, respectively, and the expression levels of these 2 proteins as well as that of the cyclins D and E were assessed as a proxy for cell cycle regulation and cell proliferation in a large series of mOGCTs (171, 172). Methylation at the 5′-CpG island of CDKN2A was observed in 15 of 37 (41%) across all mOGCT histologies (172), whereas only 1 DG and 1 YST were methylated at the CDKN1B gene promoter CpG island (171). Methylation of the CDKN2A promoter was significantly associated with lower or absent protein expression, as well as higher proliferation activity, in line with active cyclin D1 (172). None of the DGs (0 of 9), YSTs (0 of 3), or IT (0 of 1) showed promoter methylation or mutations at APC in the tumor. By contrast, the other mainly extragonadal samples of unspecified gender showed pronounced hypermethylation in both teratomas and YSTs (163). Despite this, APC expression did not vary significantly between the different histological subgroups, indicating that APC is unlikely to be the main driver behind WNT-controlled pathogenesis of mOGCT in particular and GCT in general.
Only 1 study has reported on global methylation of mOGCTs (187) and analyzed the methylation status of both LINE-1 repetitive elements dispersed throughout the genome as well as that of gene regulatory regions of 807 individual genes previously associated with methylation and/or cancer by the use of methylation arrays. Both DGs and YSTs displayed significant global hypomethylation of LINE-1 elements compared with control samples. However, whereas the DGs and control samples did not differ in methylation of gene regulatory regions, YSTs were found to have significantly increased methylation compared with DGs at 131 of 807 (16%) of these genes, many of which were potential tumor suppressor genes. This YST methylator phenotype coincided with silencing of genes associated with CASP8-dependent apoptosis as well as increased expression of the de novo methyltransferase DNMT3B and is suggested to be a possible explanation for the more aggressive progression and relative therapy resistance of YST (187).
B. Protein expression and gene mutations
Results from all protein expression studies on primary mOGCTs by IHC for which we could extract staining frequencies for the individual histological subtypes are summarized in Figure 5A. Importantly, a range of frequencies have been reported across different studies for a single protein, even for the same histological subtype. This may reflect biological variation in expression but could also be due to variation in methods for protein staining as well as for interpretation and scoring. For example, expression of TFAP2C was variously reported at 33% (55), 83% (244), and 100% (245) of the DGs studied. Results from all samples of the same histology across the various studies have been tallied and are reported in Figure 5A. An overview of the published case reports of protein expression and germline or somatic gene mutation analyses performed on mOGCT patients/mOGCT is presented in Supplemental Table 6 for completion.
The proteins whose expression in mOGCTs has been studied to date fall into 2 main functional categories: pluripotency and developmental factors and histology-specific markers. Expression of some proteins, such as protein arginine deiminase type-4 (246) and the claudins 1, 4, 5, and 7 (247), have been studied to aid in differential diagnosis between mOGCT and other ovarian malignancies. Other studies have profiled the expression of some proteins in mOGCTs for descriptive and mechanistic purposes, such as for the estrogen receptors 1 and 2 and their coregulators SNRPN upstream reading frame protein (248), GDNF family receptor α-1 (249), and cold shock domain protein A and Y-box-binding protein 2 (250). However, samples sizes were very small, at between 1 and 3 samples per subtype, and are included in Figure 5A for the sake of completion. In particular, proteins for which histology-specific differences have been identified are discussed in relation to possible implications for mOGCT initiation and development.
Given the PGC origin of mOGCT, it is unsurprising that expression of pluripotency-determining transcription factors is retained during malignant transformation and is sustained by some histological subtypes during tumor development and progression. The extent of expression of the various pluripotency factors is significantly different between the histological subtypes (Figure 5A). Protein lin-28 homolog A is essential for the proper PGC development in mouse (251) and was uniformly expressed across all the DGs, YSTs, ITs, and ECs studied (252). However, both POU5F1 and homeobox protein NANOG (NANOG) are significantly more often expressed in the poorly differentiated DG than in YST (P = 2 × 10−15 and P = 5 × 10−5 for POU5F1 and NANOG, respectively) and in IT (P = 1 × 10−15 and P = 1 × 10−8 for POU5F1 and NANOG, respectively), supporting their application as biomarkers for these subtypes (55, 244, 253–255). Eighteen of the ITs in Figure 5A were also evaluated for grade-specific POU5F1 staining. POU5F1 was detected in the immature neuroepithelium of all (7 of 7) grade-3 ITs but only 40% (2 of 5) grade-2 and none (0 of 5) of grade-1 ITs studied, leading to its proposal as a biomarker for highly malignant IT (256).
By contrast, the pluripotency factor transcription factor SOX-2 (SOX2) was found to be significantly more frequently expressed in IT compared with both DG (P = 1 × 10−5) and YST (P = 4 × 10−4) (Figure 5A) (255) and was the only IT-specific protein in our analysis. Despite the fact that SOX2 acts in concert with POU5F1 and NANOG to maintain pluripotency in ESCs (257), PGCs are not reported to express SOX2 yet remain capable of proliferation (258). The absence of SOX2 expression in DGs underlines their strong resemblance to PGCs.
Dysregulation of genes controlling early development of the embryo, including gonadal differentiation, has been implicated in mOGCT pathogenesis. The transcription factor TFAP2C (also known as AP-2γ) has been reported as expressed in DG in 3 studies (55, 244, 245), at frequencies ranging from 33% to 100%. This underlines the importance of validation in larger sample series, to assess the true level of protein expression, essential for the implementation of a protein as a diagnostic biomarker. In summary, DGs were found to express TFAP2C significantly more frequently than both YSTs (P = 2 × 10−5) and ITs (P = 1 × 10−6) (Figure 5A). TFAP2C has been suggested to be a target gene of the transcription factor PRDM1, and together they act to specify and maintain PGC characteristics and suppress differentiation (52, 259). This may explain the expression of TFAP2C in the less differentiated tumor type DG and its absence in the more differentiated mOGCT subtypes.
TFAP2C is also a homolog of the melanoma tumor suppressor TFAP2, whose loss is associated with down-regulation of KIT (260). Aberrant expression of the tyrosine kinase receptor KIT has been implicated in the development of various cancer types, in particular gastrointestinal stromal tumor (GIST) (261) and acute myeloid leukemia (262) as well as GCT (263). Among mOGCTs, KIT is more commonly expressed in DG than YST (P = 9 × 10−14) and IT (P = 7 × 10−7) (Figure 5A) (55, 156, 264–268), explaining KIT's utility as a differential diagnostic biomarker for DG and YST.
In normal germ cell and gonadal development, KIT and its ligand, KITLG, not only influence germ cell proliferation and survival in the human gonad but also regulate PGC migration (61). Activating mutations in KIT result in increased KIT signaling, which in turn promotes cell proliferation and survival (269). In surveys of somatic mutations in KIT among mOGCTs, mutations have been detected exclusively in DG at frequencies of 27% (6 of 22) and 24% (4 of 17) (55, 156), whereas no mutations have been reported in tumors of patients with pure or mixed histologies of IT and YST (n = 25) (55). All KIT mutations reported in DG, also in case reports (178, 270), were located in exon 17 (D816V, D816H, and D816Y), mirroring other cancer types, including certain leukemias and GIST (271). The success of KIT-targeting imatinib mesylate in the treatment of GIST (272) and chronic myelogenous leukemia (273) may have relevance for DG, given their high frequency of KIT expression and the negative effects of radiotherapy and chemotherapy on fertility (274). There are case reports of complete remission of chemoresistant and/or disseminated TGCT after treatment with imatinib mesylate (275, 276). However, a small trial administering imatinib mesylate to patients with KIT-positive metastatic TGCT (n = 6) did not result in remission for any of them (277).
In addition to POU5F1, PDPN has also been proposed as a DG diagnostic marker, and in our summarization, DGs expressed PDPN more commonly than YSTs (P = 3 × 10−6) and ITs (P = 1 × 10−7) (Figure 5A) (255, 278, 279). By contrast, good molecular markers for YST are limited. The YSTs often display complex and varied histological appearance, making correct differential diagnosis difficult, in particular between YST and clear-cell carcinoma of the ovary. Mixed tumors present an additional level of complexity, where small islands of YST may grow closely with other subtypes such as IT. Misdiagnosis by overlooking the presence of these subtypes may have serious prognostic and clinical management implications (280).
AFP was proposed as early as 1978 as a marker for identification of YST (281) and is in our review, 1 of 5 proteins to be significantly more frequently expressed in YSTs compared with DGs (P = 1 × 10−7 for AFP) (Figure 5A) (55, 264, 281, 282). However, the diagnostic use of AFP IHC is hampered by poor sensitivity and specificity (283). Emerging knowledge of the role of transcription factors GATA4 and GATA6, involved in regulation of early development of visceral and yolk sac endoderm, has led to studies of these factors among mOGCTs. So far, only a few cases (n ≤ 8) of each subtype have been analyzed (Figure 5A), but YSTs were found to express GATA6 more frequently than DGs (P = 2 × 10−4) (284, 285). DGs, YSTs, and ITs all express GATA4, although at different frequencies (282, 285). This pattern has proposed the use of both GATA4 and GATA6 as differential markers between the malignant endodermal cells of YST and DG (285).
Additional proteins have also been tested with limited success as biomarkers for YST, including keratin type II cytoskeletal 7 (used to differentiate between primary and metastatic ovarian adenocarcinomas) and epithelial membrane antigen (previously tested in TGCT) (264). Most recently, however, glypican-3 (GPC3), has emerged as a contender (283). Our summary revealed GPC3 to be more often expressed in YSTs compared with DGs, albeit small sample sizes (P = 3 × 10−3) (Figure 5A) (283, 286). In studies including both ovarian and testicular YSTs as well as other GCT subtypes, GPC3 is shown to have higher sensitivity than AFP (286, 287). However, the specificity for YST is still disputed, because GPC3 is also reported to stain, albeit mostly focally, other GCT types, including EC, teratoma, SS, and syncytiotrophoblastic cells (288). Interestingly, in identification of hepatocellular carcinoma, higher sensitivity and specificity has been shown for serum GPC3 than for AFP (289), which raises the possibility of a similar noninvasive test for YST, of particular interest for monitoring patients for relapse.
The 2 other proteins more frequently expressed in YSTs than in DGs, again with the caveat of small sample numbers, were γ-glutamyl transpeptidase 1 (GGT1) (P = 1 × 10−4) (290) and the epithelial staining antibody mixture AE1/AE3, which recognizes most acidic and all basic cytokeratins (P = 3 × 10−5) (Figure 5A) (264, 291). GGT1 was studied to ascertain the extent of expression in GCT and clinical response, because it has been found to be essential for the nephrotoxicity of cisplatin in animal models; however, its activity was not required for therapeutic effect (290). Both AE1/AE3 studies included broader panels of cytokeratin antibodies, with the purpose of differentiating YST from endometrioid and clear-cell carcinomas in one (264) and characterizing cytokeratin expression in DGs in the other (291). However, the results showed that differences in cytokeratin staining are subtle and nonexclusive, limiting their application for differential diagnosis.
Somatic mutation of TP53 and subsequent accumulation of the TP53 protein have been detected in over 50% of human tumors (292). However, mutations in TP53 are rare among TGCTs (293–297), although high abundance of TP53 is common (176, 295–298). Four studies have analyzed TP53 expression and nuclear accumulation among mOGCTs (144, 176, 179, 299). However, 1 study was excluded because the results did not separate the TGCTs from mOGCTs (176). Among the 3 remaining studies, one reported nuclear staining of TP53 in 100% (7 of 7) of DGs whereas a second showed TP53 accumulation in 2 of 5 YSTs and in the only IT analyzed, however, only in <10% of the tumor cells (179). The final study reported no staining in any of the 2 DGs or 2 ITs studied (144). Overall, no significant histology-specific pattern of expression was observed (Figure 5A). In our summarization, mutation of TP53 was found in only 1 DG (1 of 5) and in none of the other subtypes, IT (0 of 4), YST (0 of 3), CC (0 of 1), and EC (0 of 1), studied (Figure 5B) (144, 179, 180). TP53 overexpression in the tumor is not always due to TP53 mutations. Other mechanisms for regulation as well as methodological considerations such as the proportion of positive tumor cells identified and the sensitivity of the mutation detection can also contribute to the variation reported. For example, in 1 article, no mutations were found in the frequently mutated region of TP53 (exons 4 to 9), but the 3 analyzed cases (2 YSTs and 1 IT) contained less than 10% TP53-positive cells (179). The IHC results in this article have been disputed, with technical issues suggested to be the reason for lack of staining among the DGs here (300).
The TP53 protein family, consisting of the 3 transcription factors, TP53, TP63, and TP73, affects a range of cellular functions including cancer and development and represents one of the most powerful gene families. Observations from both mouse studies as well as human mutation and single-nucleotide polymorphism studies have revealed the importance of the TP53 family members as guardians of human maternal reproduction (301, 302). TP63 is responsible for quality control and survival of the germ cell pool and protection of the female germline during meiotic arrest, TP73 for guaranteeing that the dividing early blastocyst undergoes normal mitotic entry, and TP53 for leukemia inhibitory factor transcription and implantation of the fertilized egg (301–303). Additonally, TP63 has been suggested to potentially bind over 5800 target sites, and whether tumorigenic pathways are also involved, and to what extent, is a subject of both promise and controversy that remains to be resolved (304). Furthermore, mutations in TP63 are associated with defects in genitourinary development (305). Recently, by an integrative genomic approach for identifying transcription factors responsible for altered miRNA expression in ovarian carcinoma, the TP53/TP63/TP73 family members were suggested as candidate drivers for miRNA overexpression (239). Furthermore, analysis of an independent set of ovarian carcinomas revealed TP73 and TP63 as activators of miR-200 family transcription (239). This is interesting in view of the identification of miR-200s as repeatedly overexpressed in certain mOGCT subtypes (see Section IV.B.).
Finally, mutations in DICER1, a gene encoding an RNAIII endoribonuclease that is essential for processing miRNA, were screened for in a survey of 102 nonepithelial ovarian tumors, including 21 mOGCTs (181). Somatic mutations were found in 26 of 43 (60%) of the Sertoli-Leydig cell tumors, of which 4 had additional germline DICER1 mutations. Somatic mutations were also found in 2 of 15 (13%) of YSTs in the region encoding the ribonuclease (RNase) IIIb domain of DICER1, but neither of these had additional germline mutations. No mutations were identified among the DGs and ITs tested (0 of 1 DG and 0 of 5 ITs). A different study analyzed the constitutional DNA of 185 individuals with GCT for DICER1 mutations (306). The gender of the patients was not reported, but of all these, only 1 case, a seminoma, was found to be mutated. In vitro analysis of the mutant endoribonuclease Dicer 1 (DICER1) proteins found in mOGCTs indicated that these proteins have impaired RNase-IIIb activity but retain RNase-IIIa activity. Thus, DICER1 mutations affecting the RNase-IIIb region may result in altered DICER1 function and miRNA processing in specific cell types, a novel mechanism through which perturbation of miRNA processing may be oncogenic (181).
C. Histology-specific molecular features of mOGCT
An overview of the changes in DNA copy number significantly associated with the histological subtypes DG, YST, and IT are mapped to the genome in Figure 6. The complete spectrum of copy number changes found in the respective histologies is presented in Section III.C., Figure 3, and Supplemental Table 2. Similarities and differences between the histology-specific genomic alterations are visualized in Figure 6. Both DG and YST have significantly more gains at chromosome arm 1q than IT (Figure 6). Gains at chromosome 12 and 21 were associated with DG compared with both YST and IT. Losses at chromosome arm 1p were significantly associated with YST as compared with both DG and IT. Further histology specific alterations for DG were gains at 7q and 19q as compared to YST, and gains at 8 and 15 and losses at 13 as compared to IT. YST also typically shows more gains at 20q and more losses at 16p as compared to IT and DG, respectively.
An overview of the recurrent and specific changes in gene and protein expression of the histological subtypes DG, YST, and IT are also mapped to the genome in Figure 6. The histology-associated genes and proteins were identified and discussed in Sections IV and V, Figures 4 and 5, and Supplemental Table 4. The histology-specific mRNA and miRNAs were defined as recurrent findings, identified in 2 or more independent studies. From 2 and up to 9 mRNA and miRNAs were recurrently identified for a specific histology, despite the fact that the studies included in this review published relatively short lists of genes as significantly altered, on average, 24 mRNAs (range, 5–80) and 20 miRNAs (range, 10–37) per study. For DG, 9 mRNAs (CASP8, CDH3, CXCL10, IL6R, NANOG, PDPN, PLBD1, POU5F1, and POU5F1B) and 3 miRNAs (miR-146b-5p, miR-155, and miR-182) were recurrently identified. For YST, 2 mRNAs (BMP1 and TGFB2) and 8 miRNAs (miR-122, miR-200b, miR-200c, miR-302a, miR-302c, mir-302*, miR-375, and miR-638) were recurrently found among the studies.
From the overview of mRNAs overexpressed in DGs as compared with YSTs, we identified 9 recurrently reported mRNAs (Figure 4A) involved in various pathways as discussed in Section IV.A.: apoptosis regulation (CASP8), cell motility, adhesion, and invasion (CASP8 and CDH3), cytokine signaling and immune response (IL6R and CXCL10), possibly in angiogenesis inhibition (CXCL10), pluripotency (NANOG, POU5F1, and POU5F1B), and potentially meiosis regulation (IL6R). The miRNAs recurrently identified with higher expression among DGs as compared with YSTs were miR-146b-5p, miR-155, and miR-182 (Figure 4B). These miRNAs were earlier described in Section IV.B. Briefly, overexpression and interactions of these with BRCA1 are reported for several types of cancer (breast, lung, cervix, and colon). miR-155 is involved in down-regulation of DNA mismatch repair genes and mutator activity. Interestingly, a mutator phenotype among DGs has also been identified in a microsatellite study (Section III.D.) (160). Furthermore, miR-182, acting through FOXF2, may have a role in regulation of BMP and WNT signaling (Section IV.B.).
Two mRNAs were recurrently overexpressed among YSTs as compared with DGs: BMP1 and TGFB2. Both are components of the TGF-β/BMP pathway, involved in embryonic development, and associated with GCT development (see Section IV.A.). The only gene expression study strictly limited to mOGCT (164) reports that mRNA expression of the WNT pathway components (CTNNB1, DVL1, DVL3, APC, WNT2, WNT5A, WNT8B, and WNT2B) discriminates YST from DG, adding that only YST and IT/MT showed nuclear β-catenin staining (see Section IV.A.). The recurrent 8 miRNAs overexpressed in YSTs as compared with DGs were miRNA-122, miR-200b, miR-200c, miR-302a, miR-302c, miR-302c*, miR-375, and miR-638 (Figure 4B). Interestingly, miR-200, miR-302, and miR-375 regulate genes shown to be involved in the TGF-β/BMP signaling pathway (166, 219, 224). Furthermore, analysis of ovarian carcinomas has revealed TP73 and TP63 to be activators of miR-200 miRNA family transcription (239). Mediators of tumor suppressors and apoptosis regulators are among the mRNAs down-regulated due to overexpression of the mir-302–367 cluster in GCT (170). Altogether, this underlines earlier studies (166) showing that TGF-β/BMP signaling correlates with the differentiation state/histology of GCT and is highly active among YST but mostly absent in DG.
To date, only 3 ovarian ITs have been included in an miRNA analysis, and to the best of our knowledge, no IT-specific results have been published.
The functions and potential relevance of the identified histology-specific proteins by IHC are discussed in Section V.B. and presented in Figure 6. Briefly, DG was associated with expression of POU5F1, NANOG, TFAP2C, KIT, and PDPN as compared with both YST and IT. Expression of AFP, GATA6, GPC3, GGT1, and cytokeratins recognized by the antibody AE1/AE3 were significantly associated with YST as compared with DG, with the caveat of small numbers tested (Section V.B.).
There is some concurrence between the differentially gained chromosomal regions identified and the location of the differentially up-regulated genes or proteins. One example is the differential gain of chromosome arm 12p in DG, in agreement with the overexpression of NANOG for DG, both on the gene and protein level. The level of amplification has been shown to influence overexpression of a gene at that particular locus (307). However, not all overexpressed genes and proteins are associated with gain of chromosomal material, such as for POU5F1 and CDH3, on chromosome arms 6p and 16q, respectively. Given the complexity of regulation from gene to active protein as well as the epigenetic and gene expression changes in the PGC genome occurring during normal development, many different and complex mechanisms other than copy number may be responsible for the overexpression of genes and/or proteins in GCT tissue. An example of such a contradiction is DG-specific overexpression of PDPN both on the mRNA and protein level, whereas the gene maps to chromosome arm 1p, reported to be lost in about 10% of DGs as evaluated by CGH (Figure 3A).
Finally, technical limitations should not be discounted when comparing results from different types of analysis. CGH analysis, gene expression profiling, and mutation analysis are all methods that average the signal of all input cells and are therefore by their very nature limited by the fact that most tumor samples are genetically heterogeneous, and may contain variable proportions of normal cells. By contrast, IHC analysis morphologically distinguishes tumor cells from normal cells and specifically evaluates staining intensity and amount of tumor cells in selected sections. Up to now, most genome studies on GCT have had low resolution, and small gains and deletions affecting gene expression may have gone undetected.
VI. Comparing Molecular Development of mOGCT and TGCT
The mOGCTs and TGCTs are believed to have a common cellular origin deriving from PGC, and in contrast to most other cancer types both present relatively early in life with peak incidences around 15 to 20 and 20 to 35 years of age, respectively (14, 18). Both the mOGCTs and TGCTs present with a spectrum of histologies, reflecting these tumors' capability to differentiate, presumably due to retention of pluripotent characteristics of the PGC. The pluripotency traits may also explain why GCT development in both sexes can result in similar histological differentiation, despite the fact that the male and female gonads have distinct gene expression profiles during their normal development from the bipotential gonad (84).
Common molecular factors for mOGCTs and TGCTs are of special interest, because these represent crucial, sex-independent components that may underlie tumor development. Identification of families with both mOGCT and TGCT cases (96) underlines the role of heritability and common predisposing factors, genetic and/or environmental, for GCT in both sexes. The fact that the observed cure rate of mOGCT by use of platinum-derived chemotherapy is high and similar to that of TGCT patients, in contrast to other cancer types, further implies that there are common mechanisms and pathways involved in male and female GCT pathogenesis. However, a certain degree of gender-specific molecular characteristics may also be expected, due to gender-specific differences in the normal development of PGC to gonocytes or oocytes.
Although the range of histological subtypes that may develop among TGCTs and OGCTs is comparable, age at presentation of the different subtypes is dissimilar. DGs are associated with young females, whereas nonseminoma is more frequent among younger males (14). TGCTs are believed to derive from the precursor CIS and may develop and differentiate into the histological subtypes seminoma, EC, IT, MT, CC, and YST, each with characteristic cell morphologies, as well as mixtures of these subtypes, mixed TGCT. By contrast, the presumed precursor lesion of mOGCT, GB, is with few exceptions seen only in dysgenetic gonads, and very few reports exist of GB detected in GCT from otherwise normal, fertile females (125–127). The most common OGCT subtype is the usually benign MT (dermoid cysts). The characterization as benign is again in contrast to what is seen for TGCT patients, where the adult testicular MTs are regarded as malignant, especially because they usually occur as a component of nonseminoma, which contains more rapidly growing and disseminating histological components (308). The most common mOGCT histological subtype is DG, followed by YST, IT, CC, and EC, as well as mixed mOGCT.
An early genetic event in the genesis of TGCT is polyploidization of the PGC, resulting in a near-tetraploid CIS, followed by nonrandom losses and gains of chromosomal regions into either a seminoma and/or a nonseminoma subtype, typically with hyper- and hypo-triploid lesions, respectively. About 90% of DGs and YSTs have nondiploid cell populations and are mostly triploid to tetraploid (Figure 2A and Supplemental Table 1). In contrast, most of the ovarian ITs studied were diploid, although aneuploidy has been detected in some grade-3 IT lesions and in IT with areas of YST differentiation (136, 139). A profound difference in DNA content as measured by flow cytometry and ICM exists between ovarian IT and the other mOGCTs.
Genomic studies with chromosomal band resolution, or CGH, identified recurrent DNA copy number alterations among DGs similar to those identified among TGCT (10, 309). Specifically, these alterations were gains from chromosomes 7, 8, 12, and 21 and loss from chromosome 13 (Figures 3A and 7). The DNA copy number profile identified for YSTs has come unique features, but also some changes in common with DG/TGCT (Figure 3).
Figure 7.
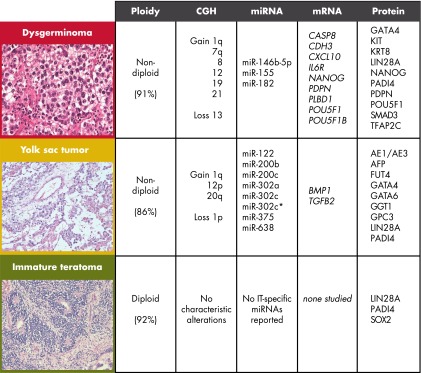
Molecular characteristics of the main histological subtypes of mOGCT. Hematoxylin- and eosin-stained sections of the subtypes DG, YST, and IT are illustrated to the left. The typical diploid or nondiploid stem line is shown, followed by copy number alterations reported in ≥30% of the subtypes (Figure 3). Also listed are recurrent differentially expressed mRNAs and miRNAs, reported in at least 2 papers (Figure 4), and proteins with positive staining in ≥50% of at least 6 samples per subtype (Figure 5A).
The genome profiles of ovarian IT indicate that they appear to develop through a different pathway from the DG and the TGCT, because the ITs are typically diploid (Figure 2A), rarely present gain of chromosome arm 12p (5%, Figure 2B), and rarely show any DNA copy number alterations (Figure 3C). For the testicular teratomas, data support separate pathogeneses for prepubertal and postpubertal teratomas (10, 308). The ovarian ITs have previously been suggested to have a different pathogenesis from DG and TGCT (131, 149, 310). The fact that ovarian YST and IT are components often found in the same tumor lends to support the theory that ITs and YSTs may develop through mechanisms different from the DGs and TGCTs. Altogether, the similarities between DNA copy alterations of DG and TGCT, as well as the differences between ovarian DG, YST, and IT indicate the presence of distinct mOGCT development and associated histological differentiation. The gain of material from chromosome arm 12p is a central feature for TGCT (309) and is also common among DGs (Figure 2B). In up to 80% of TGCTs, 12p gain occurs through the generation of an i(12p), such that this aberration is pathognomonic, and the remaining 20% of cases gain 12p through other rearrangements, including by amplifications of smaller regions (243). In contrast, a gain of 12p is found in 77% and 41% of DGs and YSTs, respectively (Figure 2B).
Genes located on chromosome arm 12p are among the most extensively studied candidates for their involvement in the pathogenesis of TGCT. These include KRAS and CCND2, both associated with malignant transformation and proliferation (311, 312). A single article has profiled 9 OGCTs for somatic mutations in KRAS and found 1 teratoma (1 of 7) (not further specified as mature or immature) but no DGs (0 of 2) to be mutated at codon 12 of exon 2 (Figure 5B) (182). However, a cluster of genes associated with stemness and the maintenance of pluripotency, including DPPA3, NANOG, and GDF3, are also located on 12p. Other candidate genes have also been identified through various genome-scale studies (Section IV and Figure 4). In general, each histological subtype reflects their gene expression and protein expression profile, independent of gender, ie, TGCT or OGCT. However, differences in gene and protein expression are identified among GCT histologies. For example, the less differentiated tumor types represented by the DG among mOGCT, the precursor CIS and seminoma among TGCT, and EC among both mOGCTs and TGCTs all express the pluripotency factors POU5F1 and NANOG at significantly higher levels than the more differentiated subtypes YST, CC, and teratomas in both mOGCT and TGCT.
In our overview of mOGCT results, we found that DGs gained 12q more frequently than YSTs (Section III.C. and Figure 3A). Interestingly, an association between genotypes at KITLG at 12q22 and an increased risk of developing TGCT has been found in 2 independent GWAS (104, 105) and has been confirmed in familial and bilateral TGCT (107). Association with a KITLG variant has been reported in a small study of 20 pediatric GCTs but was not significant for pediatric ovarian GCT (P = .08) (108). Larger studies are needed to evaluate whether the variants associated with TGCT constitute a risk factor for ovarian GCT or specific subgroups therein.
Differences also exist between the ovarian DG and testicular seminoma. For example, the DGs express GATA4 but not GATA6 (285), whereas seminoma express both (313). Another example is KIT mutation, reported in around 30% of both DG and seminoma analyzed (55, 156, 177). However, whereas KIT mutations have been detected only in unilateral DG (55, 156), reports have suggested KIT mutations to be more common in bilateral seminoma (314, 315), with some contradictions (316, 317). This suggests that KIT mutations in TGCT may predispose for bilateral cancer, whereas no such evidence exists in mOGCT. Based on the identical mutations found for both tumors of bilateral TGCT patients, it is logical that KIT mutation occurs before or during the migration of PGC to the gonadal ridges. In OGCT, there is no evidence of mutation occurring in bilateral tumors, although the number of bilateral mOGCTs analyzed is still low. Strikingly, all tumors with KIT mutations were from patients with unilateral DG (5 of 11), but no mutations were found in the tumors from 4 patients with bilateral DG disease (55). This suggests that KIT mutations in mOGCT appear after the migration of PGC to the gonadal ridges. Activating KIT mutations in early oogonia may be a factor in the stimulation of proliferation and delay of meiotic entry during fetal life. This may increase the pool of immature germ cells and potentially induce a delay and time window for transformation to occur. In general, differences observed in expression of specific genes and proteins in DG and seminoma may be remnants of the PGCs settling and maturation in disparate gonadal environments and/or the fact that gender-specific PGCs are primed for differentiation into 2 different entities, the oocyte/follicle in females and spermatozoa in males.
The possibilities afforded by microarray technology to simultaneously measure the expression levels of thousands of genes in a single sample have greatly enhanced the study of gene expression. The gene expression profiles identified by genome-wide expression studies of mOGCT and TGCT provide much insight into histology and gender specificity. The one study that profiled strictly mOGCTs returned significant genes up-regulated in DG compared with YST and in YST compared with DG as well as genes highly expressed in both DG and YST (164). These lists of candidate mOGCT genes were compared with corresponding lists from 5 TGCT studies with lists of genes overexpressed in seminoma as compared with normal testis (318–322). From this comparison, we identified 3 genes overexpressed in DG and in 2 or more independent TGCT studies: CDH3, IL6R, and MYCN. Five genes were overexpressed both in the DG and 1 TGCT study: CASP8, CDK5, CXCL10, MSH2, and PRODH. MYCN overexpression among DGs and TGCTs is of interest due to the function of the MYCN oncogene as a positive regulator of LIN28B (323), whereas deregulation of MYCN, LIN28B, and LET7 is associated with a molecular subtype of high-grade serous ovarian cancer (324). The specific relevance or function of CDK5, MSH2, and PRODH in mOGCT cells and development is so far unknown. However, among the interesting functions identified in other cell types are the phosphorylation of serine/threonine-protein kinase PAK 1, influence on astral microtubules and the fidelity of the mitosis by CDK5 (325, 326), participation in DNA mismatch repair by MSH2, and both up- and down-regulation of PRODH relevant to proline metabolism and cancer development (327).
Interestingly, when comparing genes overexpressed in ovarian YST with testicular YST, no overlap was found: none of the genes overexpressed in the ovarian YST had previously been reported as YST-specific genes in TGCT transcriptomic studies (195, 328–330), indicating divergence in molecular profiles between the ovarian and testicular YST.
Finally, for a complete mOGCT with TGCT comparison, a combined list of genes up-regulated in both DG and ovarian YST (164) were compared with a combined list of all genes published in 7 studies as overexpressed among TGCT compared with normal testis (319, 330–335). Only 2 genes, CD9 and RAC1, were up-regulated in GCT regardless of histology, namely in ovarian DG and YST as well as in TGCT. Both genes are candidates for further studies as predicted common biomarkers for GC pathogenesis in male and female GCT. CD9 is located on chromosome arm 12p, is highly expressed in TGCT, and has been suggested as a biomarker for neoplastic germ cells (333, 334). RAC1 encodes a Rho-family protein and is known to contribute to transformation and cancer (336). Studies in zebrafish have shown that polarization of PGC, important for PGC migration to the gonads, are regulated by Rac1 (337). Interestingly, CD9 and RAC1 were identified by our group in 2006 as potential proto-oncogenes in GCT, based on results from the very first genome-wide integrated array-CGH and gene expression profiling of TGCT (333).
VII. Concluding Remarks and Perspectives
Development of the ovaries is a critical process requiring careful timing and simultaneous regulation of gonadal development and PGC migration and maturation. The steps before final colocalization of PGC with the gonad involve mitosis, epigenetic erasure by DNA demethylation, cell migration, cell communication, and cell differentiation. The microenvironment of the female PGC, consisting of the granulosa and theca cells, is critical for their maturation to oocytes and development into antral follicles. Both the timing and level of expression of the genes controlling these processes are crucial. A delay in the action of these licensing factors could potentially set development on hold, including postponement of meiotic prophase, and thereby provide a prolonged window for tumor initiation.
For dysgenetic ovaries, a step-wise progression from GB to DG is proposed. The similarities between GB and the precursor of TGCT, CIS (338), indicate that GB is a precursor stage of at least a subset of ovarian GCT. For most cases of mOGCT in DSD gonads, etiology is associated with phenotypic females with male karyotype or at least parts of the Y-chromosome present in their genome. More extensive studies are required to improve understanding of genetic variation and the associated GCT risk, both among DSD patients as well as within the spectrum of phenotypically normal females with low risk. For normal female gonads with no Y-chromosome material, alterations of autosomal genes or genes on the X-chromosome may be tumor promoting. These may be genes that are directly involved in the normal ovary/tumor development or indirectly involved by influencing gene expression and/or regulation. An example of the latter is miRNA alteration, which may have large implications for global gene expression profiles due to their multiple downstream targets (339).
The alterations in critical factors may either be inherited or somatically acquired in the PGCs and/or gonadal cells of the GCT patient. DSD patients with Y-chromosome material may be considered to be one extreme of the constitutional carriers. The possibilities afforded by new technologies like deep sequencing for revealing the genetic constitution of otherwise normal female OGCT patients are intriguing. Such studies should preferably include both blood and gonadal tumor samples, because these patients may also have gonadal mosaicism, such as that reported for mOGCT patients with GB (127). Polymorphic sites in crucial genes could also influence mOGCT development, as already identified for TGCT patients. Genetic alterations or polymorphisms could affect the functions of genes previously known from general somatic tumor development as oncogenes or tumor suppressor genes, genes relevant for normal ovarian development or the internal endocrine system controlling hormone production.
We believe that the complex development of the gonad, involving critical timing and regulation of genes, in addition to the pluripotent traits of the PGC, render the developing PGC extremely sensitive to tumor development. The fact that GCTs rarely contain mutations in well-known oncogenes or tumor suppressor genes shows that the pathogenesis of GCT differs from somatic tumor development, possibly by exploiting the retained pluripotency and the primed demethylation process of the PGC. The difference in incidence of male and female GCT may be related either to the absolute number of susceptible cells in males compared with females (14) or to the phenotype of the male as opposed to the female PGCs and their respective microenvironments. In that sense, the male PGC may, relative to the female PGC, be more likely to have fewer barriers against developing the tumorigenic phenotype.
Each of the histological subtypes DG, YST, and IT shows recurrent molecular characteristics of ploidy indices, DNA copy number changes, and specific expression patterns of mRNA, miRNA, and proteins. Seemingly, there is preferential involvement of the TGFβ/BMP and WNT pathways in YST compared to DG.
Future research on both constitutional and somatic alterations in all GCT patients across the spectrum of gender phenotypes are needed to fully understand the pathogenesis of mOGCT. This includes evaluation of the contributions of epigenetic, genetic, and (micro)environmental alterations, both in the male and female gonads. Whole-genome and transcriptome sequencing will shed light on the range of constitutional as well as somatic alterations among mOGCT and TGCT patients. Due to the low incidence of mOGCT, there is a need for multicenter collaborations for the molecular, clinical and epidemiologic studies to reach critical sample sizes.
Note added in proof: After the completion of the literature search for the current review, additional relevant papers have been published. We consider the following papers and topics to be of particular interest: karyopherin suggested a prognostic marker in mOGCT patients by He et al (341); a study of KIT mutations among GBs and DGs by Hersmus et al (342); molecular profiling of ovarian EC cells based on pluripotency markers by Malecki et al (343); and a report of GCT incidence during the last 40 years in Finland by Pauniaho et al (344).
Supplementary Material
Acknowledgments
This work was supported by the Health Region South-East Norway, the Research Council of Norway, the Danish Cancer Society, the Lundbeck Foundation, and the Childhood Cancer Foundation.
Disclosure Summary: There are no conflicts of interest to declare.
Footnotes
- AFP
- α-fetoprotein
- βHCG
- β human chorionic gonadotropin
- BMP
- bone morphogenetic protein
- CASP8
- caspase-8
- CC
- choriocarcinoma
- CDH3
- cadherin-3
- CGH
- comparative genomic hybridization
- CIS
- carcinoma in situ
- CXCL10
- C-X-C motif chemokine 10
- DG
- dysgerminoma
- DMRT1
- doublesex- and mab-3-related transcription factor 1
- DICER1
- endoribonuclease Dicer
- DSD
- disorders of sex development
- EC
- embryonal carcinoma
- ESC
- embryonal stem cell
- FIGO
- the International Federation of Gynaecology and Obstetrics
- FISH
- fluorescence in situ hybridization
- GB
- gonadoblastoma
- GCT
- germ cell tumor
- GGT-1
- γ-glutamyl transpeptidase 1
- GIST
- gastrointestinal stromal tumor
- GPC3
- glypican-3
- GWAS
- genome wide association studies
- ICM
- image cytometry
- IHC
- immunohistochemistry
- IL6R
- IL-6 receptor subunit α
- IT
- immature teratoma
- KIT
- mast/stem cell growth factor receptor Kit
- KITLG
- Kit ligand
- LOH
- loss of heterozygosity
- miRNA
- microRNA
- mOGCT
- malignant ovarian GCT
- MT
- mature teratoma
- NANOG
- homeobox protein NANOG
- OR
- odds ratio
- PDPN
- podoplanin
- PGC
- primordial germ cell
- POU5F1
- POU domain, class 5, transcription factor 1
- Prdm
- PR domain zinc finger protein
- QOL
- quality of life
- RA
- retinoic acid
- RER
- replication error
- RNase
- ribonuclease
- SEER
- Surveillance, Epidemiology, and End Results
- SOX2
- transcription factor SOX-2
- SRY
- sex-determining region Y protein
- SS
- spermatocytic seminoma
- TGCT
- testicular GCT
- TP
- tumor protein
- YST
- yolk sac tumor
- ZEB
- zinc finger E-box-binding homeobox.
References
- 1. Clement PB, Young RH. Ovarian surface epithelial-stromal tumors. In: Mills SE, ed. Sternberg's Diagnostic Surgical Pathology. 5th ed Philadelphia, PA: Lippincott Williams, Wilkins; 2009;2278–2308. [Google Scholar]
- 2. Young RH, Clement PB. Sex cord-stromal, steroid cell, and germ cell tumors of the ovary. In: Mills SE, ed. Sternberg's Diagnostic Surgical Pathology. 5th ed Philadelphia, PA: Lippincott Williams, Wilkins; 2009;2309–2341. [Google Scholar]
- 3. Palmer RD, Nicholson JC, Hale JP. Management of germ cell tumours in childhood. Curr Paediatr. 2003;13:213–220. [Google Scholar]
- 4. Pleskacova J, Hersmus R, Oosterhuis JW, et al. Tumor risk in disorders of sex development. Sex Dev. 2010;4:259–269. [DOI] [PubMed] [Google Scholar]
- 5. Nogales F, Talerman A, Kubik-Huch RA, Tavassoli FA, Devouassoux-Shisheboran M. Germ cell tumours. In: Tavassoli FA, Deville P, eds. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of the Breast and Female Genital Organs. Lyon, France: IARC Press; 2003:163–175. [Google Scholar]
- 6. Roth LM, Talerman A. Recent advances in the pathology and classification of ovarian germ cell tumors. Int J Gynecol Pathol. 2006;25:305–320. [DOI] [PubMed] [Google Scholar]
- 7. Hu L, McArthur C, Jaffe RB. Ovarian cancer stem-like side-population cells are tumourigenic and chemoresistant. Br J Cancer. 2010;102:1276–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peng S, Maihle NJ, Huang Y. Pluripotency factors Lin28 and Oct4 identify a sub-population of stem cell-like cells in ovarian cancer. Oncogene. 2010;29:2153–2159. [DOI] [PubMed] [Google Scholar]
- 9. Scully RE. Ovarian tumors. A review. Am J Pathol. 1977;87:686–720. [PMC free article] [PubMed] [Google Scholar]
- 10. Oosterhuis JW, Looijenga LH. Testicular germ-cell tumours in a broader perspective. Nat Rev Cancer. 2005;5:210–222. [DOI] [PubMed] [Google Scholar]
- 11. Quirk JT, Natarajan N. Ovarian cancer incidence in the United States, 1992–1999. Gynecol Oncol. 2005;97:519–523. [DOI] [PubMed] [Google Scholar]
- 12. Quirk JT, Natarajan N, Mettlin CJ. Age-specific ovarian cancer incidence rate patterns in the United States. Gynecol Oncol. 2005;99:248–250. [DOI] [PubMed] [Google Scholar]
- 13. dos Santos Silva I, Swerdlow AJ. Ovarian germ cell malignancies in England: epidemiological parallels with testicular cancer. Br J Cancer. 1991;63:814–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Møller H, Evans H. Epidemiology of gonadal germ cell cancer in males and females. APMIS. 2003;111:43–48. [DOI] [PubMed] [Google Scholar]
- 15. Brookfield KF, Cheung MC, Koniaris LG, Sola JE, Fischer AC. A population-based analysis of 1037 malignant ovarian tumors in the pediatric population. J Surg Res. 2009;156:45–49. [DOI] [PubMed] [Google Scholar]
- 16. Guillem V, Poveda A. Germ cell tumours of the ovary. Clin Transl Oncol. 2007;9:237–243. [DOI] [PubMed] [Google Scholar]
- 17. Walker AH, Ross RK, Pike MC, Henderson BE. A possible rising incidence of malignant germ cell tumours in young women. Br J Cancer. 1984;49:669–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith HO, Berwick M, Verschraegen CF, et al. Incidence and survival rates for female malignant germ cell tumors. Obstet Gynecol. 2006;107:1075–1085. [DOI] [PubMed] [Google Scholar]
- 19. Pectasides D, Pectasides E, Kassanos D. Germ cell tumors of the ovary. Cancer Treat Rev. 2008;34:427–441. [DOI] [PubMed] [Google Scholar]
- 20. Parkinson CA, Hatcher HM, Ajithkumar TV. Management of malignant ovarian germ cell tumors. Obstet Gynecol Surv. 2011;66:507–514. [DOI] [PubMed] [Google Scholar]
- 21. Gershenson DM. Management of ovarian germ cell tumors. J Clin Oncol. 2007;25:2938–2943. [DOI] [PubMed] [Google Scholar]
- 22. Murugaesu N, Schmid P, Dancey G, et al. Malignant ovarian germ cell tumors: Identification of novel prognostic markers and long-term outcome after multimodality treatment. J Clin Oncol. 2006;24:4862–4866. [DOI] [PubMed] [Google Scholar]
- 23. Weinberg LE, Lurain JR, Singh DK, Schink JC. Survival and reproductive outcomes in women treated for malignant ovarian germ cell tumors. Gynecol Oncol. 2011;121:285–289. [DOI] [PubMed] [Google Scholar]
- 24. Luis IV, Coleman R. Ovarian germ cell malignancy: a heterogeneous tumour requiring supra-regional management. Eur J Gynaecol Oncol. 2011;32:387–392. [PubMed] [Google Scholar]
- 25. Low JJ, Perrin LC, Crandon AJ, Hacker NF. Conservative surgery to preserve ovarian function in patients with malignant ovarian germ cell tumors. A review of 74 cases. Cancer. 2000;89:391–398. [PubMed] [Google Scholar]
- 26. Low JJ, Ilancheran A, Ng JS. Malignant ovarian germ-cell tumours. Best Pract Res Clin Obstet Gynaecol. 2012;26:347–355. [DOI] [PubMed] [Google Scholar]
- 27. Zanetta G, Bonazzi C, Cantù MG, Locatelli A, Bratina G, Mangioni C. Survival and reproductive function after treatment of malignant germ cell ovarian tumors. J Clin Oncol. 2001;19:1015–1020. [DOI] [PubMed] [Google Scholar]
- 28. Rozenholc A, Abdulcadir J, Pelte MF, Petignat P. A pelvic mass on ultrasonography and high human chorionic gonadotropin level: not always an ectopic pregnancy. BMJ Case Rep. 2012; doi: 10.1136/bcr.01.2012.5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McKnight KK, Bates J. Atypical presentation and management dilemma of mixed gonadal dysgenesis. Fertil Steril. 2011;95:291.e7–291.e9. [DOI] [PubMed] [Google Scholar]
- 30. Dennedy MC, Smith D, O'Shea D, McKenna TJ. Investigation of patients with atypical or severe hyperandrogenaemia including androgen-secreting ovarian teratoma. Eur J Endocrinol. 2010;162:213–220. [DOI] [PubMed] [Google Scholar]
- 31. Gershenson DM. Menstrual and reproductive function after treatment with combination chemotherapy for malignant ovarian germ cell tumors. J Clin Oncol. 1988;6:270–275. [DOI] [PubMed] [Google Scholar]
- 32. Hale GA, Marina NM, Jones-Wallace D, Greenwald CA, Jenkins JJ, Rao BN, Luo X, Hudson MM. Late effects of treatment for germ cell tumors during childhood and adolescence. J Pediatr Hematol Oncol. 1999;21:115–122. [DOI] [PubMed] [Google Scholar]
- 33. Brewer M, Gershenson DM, Herzog CE, Mitchell MF, Silva EG, Wharton JT. Outcome and reproductive function after chemotherapy for ovarian dysgerminoma. J Clin Oncol. 1999;17:2670–2675. [DOI] [PubMed] [Google Scholar]
- 34. Swenson MM, MacLeod JS, Williams SD, Miller AM, Champion VL. Quality of life after among ovarian germ cell cancer survivors: a narrative analysis. Oncol Nurs Forum. 2003;30:E48–E54. [DOI] [PubMed] [Google Scholar]
- 35. de Vos FY, Nuver J, Willemse PH, et al. Long-term survivors of ovarian malignancies after cisplatin-based chemotherapy: cardiovascular risk factors and signs of vascular damage. Eur J Cancer. 2004;40:696–700. [DOI] [PubMed] [Google Scholar]
- 36. Champion V, Williams SD, Miller A, et al. Quality of life in long-term survivors of ovarian germ cell tumors: a gynecologic oncology group study. Gynecol Oncol. 2007;105:687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Monahan PO, Champion VL, Zhao Q, et al. Case-control comparison of quality of life in long-term ovarian germ cell tumor survivors: a gynecologic oncology group study. J Psychosoc Oncol. 2008;26:19–42. [DOI] [PubMed] [Google Scholar]
- 38. Matei D, Miller AM, Monahan P, et al. Chronic physical effects and health care utilization in long-term ovarian germ cell tumor survivors: a gynecologic oncology group study. J Clin Oncol. 2009;27:4142–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gershenson DM, Miller AM, Champion VL, et al. Reproductive and sexual function after platinum-based chemotherapy in long-term ovarian germ cell tumor survivors: a gynecologic oncology group study. J Clin Oncol. 2007;25:2792–2797. [DOI] [PubMed] [Google Scholar]
- 40. Fung C, Vaughn DJ. Complications associated with chemotherapy in testicular cancer management. Nat Rev Urol. 2011;8:213–222. [DOI] [PubMed] [Google Scholar]
- 41. Winter C, Albers P. Testicular germ cell tumors: pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2011;7:43–53. [DOI] [PubMed] [Google Scholar]
- 42. Fosså SD, Oldenburg J, Dahl AA. Short- and long-term morbidity after treatment for testicular cancer. BJU Int. 2009;104:1418–1422. [DOI] [PubMed] [Google Scholar]
- 43. Oldenburg J, Kraggerud SM, Brydoy M, Cvancarova M, Lothe RA, Fosså SD. Association between long-term neuro-toxicities in testicular cancer survivors and polymorphisms in glutathione-s-transferase-P1 and -M1, a retrospective cross sectional study. J Transl Med. 2007;5:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oldenburg J, Kraggerud SM, Cvancarova M, Lothe RA, Fossa SD. Cisplatin-induced long-term hearing impairment is associated with specific glutathione S-transferase genotypes in testicular cancer survivors. J Clin Oncol. 2007;25:708–714. [DOI] [PubMed] [Google Scholar]
- 45. Jiménez R. Ovarian organogenesis in mammals: mice cannot tell us everything. Sex Dev. 2009;3:291–301. [DOI] [PubMed] [Google Scholar]
- 46. Oktem O, Oktay K. The ovary. Ann N Y Acad Sci. 2008;1127:1–9. [DOI] [PubMed] [Google Scholar]
- 47. Cools M, Looijenga LHJ, Wolffenbuttel K, Drop S. Disorders of sex development: update on the genetic background, terminology and risk for the development of germ cell tumors. World J Pediatr. 2009;5:93–102. [DOI] [PubMed] [Google Scholar]
- 48. Saitou M. Germ cell specification in mice. Curr Opin Genet Dev. 2009;19:386–395. [DOI] [PubMed] [Google Scholar]
- 49. Oktem O, Oktay K. Stem cells. Ann N Y Acad Sci. 2008;1127:20–26. [DOI] [PubMed] [Google Scholar]
- 50. Saitou M, Yamaji M. Germ cell specification in mice: signaling, transcription regulation, and epigenetic consequences. Reproduction. 2010;139:931–942. [DOI] [PubMed] [Google Scholar]
- 51. Tres LL, Rosselot C, Kierszenbaum AL. Primordial germ cells: what does it take to be alive? Mol Reprod Dev. 2004;68:1–4. [DOI] [PubMed] [Google Scholar]
- 52. Schäfer S, Anschlag J, Nettersheim D, Haas N, Pawig L, Schorle H. The role of BLIMP1 and its putative downstream target TFAP2C in germ cell development and germ cell tumours. Int J Androl. 2011;34:e152–e159. [DOI] [PubMed] [Google Scholar]
- 53. Kurimoto K, Yamaji M, Seki Y, Saitou M. Specification of the germ cell lineage in mice: a process orchestrated by the PR-domain proteins, Blimp1 and Prdm14. Cell Cycle. 2008;7:3514–3518. [DOI] [PubMed] [Google Scholar]
- 54. Hart AH, Hartley L, Parker K, et al. The pluripotency homeobox gene NANOG is expressed in human germ cell tumors. Cancer. 2005;104:2092–2098. [DOI] [PubMed] [Google Scholar]
- 55. Hoei-Hansen CE, Kraggerud SM, Abeler VM, Kaern J, Rajpert-De Meyts E, Lothe RA. Ovarian dysgerminomas are characterised by frequent KIT mutations and abundant expression of pluripotency markers. Mol Cancer. 2007;6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hoei-Hansen CE, Nielsen JE, Almstrup K, et al. Transcription factor AP-2g is a developmentally regulated marker of testicular carcinoma in situ and germ cell tumors. Clin Cancer Res. 2004;10:8521–8530. [DOI] [PubMed] [Google Scholar]
- 57. Hoei-Hansen CE, Almstrup K, Nielsen JE, et al. Stem cell pluripotency factor NANOG is expressed in human fetal gonocytes, testicular carcinoma in situ and germ cell tumours. Histopathology. 2005;47:48–56. [DOI] [PubMed] [Google Scholar]
- 58. Hersmus R, de Leeuw BH, Wolffenbuttel KP, et al. New insights into type II germ cell tumor pathogenesis based on studies of patients with various forms of disorders of sex development (DSD). Mol Cell Endocrinol. 2008;291:1–10. [DOI] [PubMed] [Google Scholar]
- 59. Looijenga LH, Hersmus R, de Leeuw BH, et al. Gonadal tumours and DSD. Best Pract Res Clin Endocrinol. 2010;24:291–310. [DOI] [PubMed] [Google Scholar]
- 60. Cheng L, Sung MT, Cossu-Rocca P, et al. OCT4: biological functions and clinical applications as a marker of germ cell neoplasia. J Pathol. 2007;211:1–9. [DOI] [PubMed] [Google Scholar]
- 61. Molyneaux K, Wylie C. Primordial germ cell migration. Int J Dev Biol. 2004;48:537–544. [DOI] [PubMed] [Google Scholar]
- 62. Møllgård K, Jespersen A, Lutterodt MC, Yding Andersen C, Høyer PE, Byskov AG. Human primordial germ cells migrate along nerve fibers and Schwann cells from the dorsal hind gut mesentery to the gonadal ridge. Mol Hum Reprod. 2010;16:621–631. [DOI] [PubMed] [Google Scholar]
- 63. Wilhelm D, Palmer S, Koopman P. Sex determination and gonadal development in mammals. Physiol Rev. 2007;87:1–28. [DOI] [PubMed] [Google Scholar]
- 64. Bowles J, Knight D, Smith C, et al. Retinoid signaling determines germ cell fate in mice. Science. 2006;312:596–600. [DOI] [PubMed] [Google Scholar]
- 65. Koubova J, Menke DB, Zhou Q, Capel B, Griswold MD, Page DC. Retinoic acid regulates sex-specific timing of meiotic initiation in mice. Proc Natl Acad Sci U S A. 2006;103:2474–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Souquet B, Tourpin S, Messiaen S, Moison D, Habert R, Livera G. Nodal signaling regulates the entry into meiosis in fetal germ cells. Endocrinology. 2012;153:2466–2473. [DOI] [PubMed] [Google Scholar]
- 67. Jørgensen A, Nielsen JE, Blomberg Jensen M, Graem N, Rajpert-De Meyts E. Analysis of meiosis regulators in human gonads: a sexually dimorphic spatio-temporal expression pattern suggests involvement of DMRT1 in meiotic entry. Mol Hum Reprod. 2012;18:523–534. [DOI] [PubMed] [Google Scholar]
- 68. Honecker F, Stoop H, de Krijger RR, Lau YF, Bokemeyer C, Looijenga LH. Pathobiological implications of the expression of markers of testicular carcinoma in situ by fetal germ cells. J Pathol. 2004;203:849–857. [DOI] [PubMed] [Google Scholar]
- 69. Pauls K, Schorle H, Jeske W, et al. Spatial expression of germ cell markers during maturation of human fetal male gonads: an immunohistochemical study. Hum Reprod. 2006;21:397–404. [DOI] [PubMed] [Google Scholar]
- 70. Rajpert-De Meyts E, Hanstein R, Jørgensen N, Græm N, Vogt PH, Skakkebæk NE. Developmental expression of POU5F1 (OCT-3/4) in normal and dysgenetic human gonads. Hum Reprod. 2004;19:1338–1344. [DOI] [PubMed] [Google Scholar]
- 71. Stoop H, Honecker F, Cools M, de Krijger R, Bokemeyer C, Looijenga LH. Differentiation and development of human female germ cells during prenatal gonadogenesis: an immunohistochemical study. Hum Reprod. 2005;20:1466–1476. [DOI] [PubMed] [Google Scholar]
- 72. Brennan J, Capel B. One tissue, two fates: molecular genetic events that underlie testis versus ovary development. Nat Rev Genet. 2004;5:509–521. [DOI] [PubMed] [Google Scholar]
- 73. Parma P, Radi O, Vidal V, et al. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat Genet. 2006;38:1304–1309. [DOI] [PubMed] [Google Scholar]
- 74. Mandel H, Shemer R, Borochowitz ZU, et al. SERKAL syndrome: an autosomal-recessive disorder caused by a loss-of-function mutation in WNT4. Am J Hum Genet. 2008;82:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Uhlenhaut NH, Jakob S, Anlag K, et al. Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell. 2009;139:1130–1142. [DOI] [PubMed] [Google Scholar]
- 76. Tomaselli S, Megiorni F, Lin L, et al. Human RSPO1/R-spondin1 is expressed during early ovary development and augments β-catenin signaling. PLoS One. 2011;6:e16366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tevosian SG, Manuylov NL. To β or not to β: canonical β-catenin signaling pathway and ovarian development. Dev Dynam. 2008;237:3672–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hersmus R, Kalfa N, de Leeuw B, et al. FOXL2 and SOX9 as parameters of female and male gonadal differentiation in patients with various forms of disorders of sex development (DSD). J Pathol. 2008;215:31–38. [DOI] [PubMed] [Google Scholar]
- 79. Pannetier M, Pailhoux E. FOXL2, le gardien de l'identité ovarienne. Med Sci (Paris). 2010;26:470–473. [DOI] [PubMed] [Google Scholar]
- 80. Jamieson S, Fuller PJ. Molecular pathogenesis of granulosa cell tumors of the ovary. Endocr Rev. 2012;33:109–144. [DOI] [PubMed] [Google Scholar]
- 81. Hes O, Vanecek T, Petersson F, et al. Mutational analysis (c. 402C>G) of the FOXL2 gene and immunohistochemical expression of the FOXL2 protein in testicular adult type granulosa cell tumors and incompletely differentiated sex cord stromal tumors. Appl Immunohistochem Mol Morphol. 2011;19:347–351. [DOI] [PubMed] [Google Scholar]
- 82. Matson CK, Murphy MW, Sarver AL, Griswold MD, Bardwell VJ, Zarkower D. DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature. 2011;476:101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gill ME, Hu YC, Lin Y, Page DC. Licensing of gametogenesis, dependent on RNA binding protein DAZL, as a gateway to sexual differentiation of fetal germ cells. Proc Natl Acad Sci U S A. 2011;108:7443–7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Houmard B, Small C, Yang L, et al. Global gene expression in the human fetal testis and ovary. Biol Reprod. 2009;81:438–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pérez-Armendariz EM, Sáez JC, Bravo-Moreno JF, López-Olmos V, Enders GC, Villalpando I. Connexin43 is expressed in mouse fetal ovary. Anat Rec. 2003;271A:360–367. [DOI] [PubMed] [Google Scholar]
- 86. Ai Z, Fischer A, Spray DC, Brown AM, Fishman GI. Wnt-1 regulation of connexin43 in cardiac myocytes. J Clin Invest. 2000;105:161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sirnes S, Bruun J, Kolberg M, et al. Connexin43 acts as a colorectal cancer tumor suppressor and predicts disease outcome. Int J Cancer. 2011;131:570–581. [DOI] [PubMed] [Google Scholar]
- 88. Childs AJ, Kinnell HL, Collins CS, et al. BMP signaling in the human fetal ovary is developmentally regulated and promotes primordial germ cell apoptosis. Stem Cells. 2010;28:1368–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Huansheng D, Qingjie P, Hanqiong Z, Lianjun Z, Bo C, Wenbin Y. Estrogen inhibits the early development of mouse follicles through regulating the expression of Kit ligand. Biochem Biophys Res Commun. 2011;410:659–664. [DOI] [PubMed] [Google Scholar]
- 90. White YA, Woods DC, Takai Y, Ishihara O, Seki H, Tilly JL. Oocyte formation by mitotically active germ cells purified from ovaries of reproductive-age women. Nat Med. 2012;18:413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Byskov AG, Høyer PE, Yding Andersen C, Kristensen SG, Jespersen A, Möllgård K. No evidence for the presence of oogonia in the human ovary after their final clearance during the first two years of life. Hum Reprod. 2011;26:2129–2139. [DOI] [PubMed] [Google Scholar]
- 92. Lee PA, Houk CP, Ahmed SF, Hughes IA; International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. Consensus statement on management of intersex disorders. Pediatrics. 2006;118:e488–e500. [DOI] [PubMed] [Google Scholar]
- 93. Cools M, Drop SL, Wolffenbuttel KP, Oosterhuis JW, Looijenga LH. Germ cell tumors in the intersex gonad: old paths, new directions, moving frontiers. Endocr Rev. 2006;27:468–484. [DOI] [PubMed] [Google Scholar]
- 94. Stettner AR, Hartenbach EM, Schink JC, et al. Familial ovarian germ cell cancer: Report and review. Am J Med Genet. 1999;84:43–46. [DOI] [PubMed] [Google Scholar]
- 95. Cyriac S, Rajendranath R, Robert LA, Sagar TG. Familial germ cell tumor. Indian J Hum Genet. 2012;18:119–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Giambartolomei C, Mueller CM, Greene MH, Korde LA. A mini-review of familial ovarian germ cell tumors: an additional manifestation of the familial testicular germ cell tumor syndrome. Cancer Epidemiol. 2009;33:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kratz CP, Mai PL, Greene MH. Familial testicular germ cell tumours. Best Pract Res Clin Endocrinol. 2010;24:503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Heimdal K, Olsson H, Tretli S, Flodgren P, Borresen AL, Fosså SD. Familial testicular cancer in Norway and southern Sweden. Br J Cancer. 1996;73:964–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dong C, Hemminki K. Modification of cancer risks in offspring by sibling and parental cancers from 2,112,616 nuclear families. Int J Cancer. 2001;92:144–150. [PubMed] [Google Scholar]
- 100. Dong C, Lönnstedt I, Hemminki K. Familial testicular cancer and second primary cancers in testicular cancer patients by histological type. Eur J Cancer. 2001;37:1878–1885. [DOI] [PubMed] [Google Scholar]
- 101. Czene K, Lichtenstein P, Hemminki K. Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish family-cancer database. Int J Cancer. 2002;99:260–266. [DOI] [PubMed] [Google Scholar]
- 102. Rapley EA, Crockford GP, Teare D, et al. Localization to Xq27 of a susceptibility gene for testicular germ-cell tumours. Nat Genet. 2000;24:197–200. [DOI] [PubMed] [Google Scholar]
- 103. Nathanson KL, Kanetsky PA, Hawes R, et al. The Y deletion gr/gr and susceptibility to testicular germ cell tumor. Am J Hum Genet. 2005;77:1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Rapley EA, Turnbull C, Al Olama AA, et al. A genome-wide association study of testicular germ cell tumor. Nat Genet. 2009;41:807–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kanetsky PA, Mitra N, Vardhanabhuti S, et al. Common variation in KITLG and at 5q31.3 predisposes to testicular germ cell cancer. Nat Genet. 2009;41:811–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Turnbull C, Rapley EA, Seal S, et al. Variants near DMRT1, TERT and ATF7IP are associated with testicular germ cell cancer. Nat Genet. 2010;42:604–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kratz CP, Han SS, Rosenberg PS, et al. Variants in or near KITLG, BAK1, DMRT1, and TERT-CLPTM1L predispose to familial testicular germ cell tumour. J Med Genet. 2011;48:473–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Poynter JN, Hooten AJ, Lindsay Frazier A, Ross JA. Associations between variants in KITLG, SPRY4, BAK1, and DMRT1 and pediatric germ cell tumors. Genes Chromosome Cancer. 2012;51:266–271. [DOI] [PubMed] [Google Scholar]
- 109. Dalgaard MD, Weinhold N, Edsgärd D, et al. A genome-wide association study of men with symptoms of testicular dysgenesis syndrome and its network biology interpretation. J Med Genet. 2012;49:58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Chanock S. High marks for GWAS. Nat Genet. 2009;41:765–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kratz CP, Greene MH, Bratslavsky G, Shi J. A stratified genetic risk assessment for testicular cancer. Int J Androl. 2011;34:e98–e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Stephansson O, Wahnström C, Pettersson A, et al. Perinatal risk factors for childhood testicular germ-cell cancer: a Nordic population-based study. Cancer Epidemiol. 2011;35:e100–e104. [DOI] [PubMed] [Google Scholar]
- 113. Cook MB, Graubard BI, Rubertone MV, Erickson RL, McGlynn KA. Perinatal factors and the risk of testicular germ cell tumors. Int J Cancer. 2008;122:2600–2606. [DOI] [PubMed] [Google Scholar]
- 114. McGlynn KA, Cook MB. Etiologic factors in testicular germ-cell tumors. Future Oncol. 2009;5:1389–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Sharpe RM, Skakkebaek NE. Testicular dysgenesis syndrome: mechanistic insights and potential new downstream effects. Fertil Steril. 2008;89:e33–e38. [DOI] [PubMed] [Google Scholar]
- 116. Skakkebæk NE, Rajpert-De Meyts E, Main KM. Testicular dysgenesis syndrome: an increasingly common developmental disorder with environmental aspects: opinion. Hum Reprod. 2001;16:972–978. [DOI] [PubMed] [Google Scholar]
- 117. Walker AH, Ross RK, Haile RW, Henderson BE. Hormonal factors and risk of ovarian germ cell cancer in young women. Br J Cancer. 1988;57:418–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Horn-Ross PL, Whittemore AS, Harris R, Itnyre J. Characteristics relating to ovarian cancer risk: Collaborative analysis of 12 U.S. case-control studies. VI. Nonepithelial cancers among adults. Collaborative Ovarian Cancer Group. Epidemiology. 1992;3:490–495. [DOI] [PubMed] [Google Scholar]
- 119. Adami HO, Lambe M, Persson I, et al. Parity, age at first childbirth, and risk of ovarian cancer. Lancet. 1994;344:1250–1254. [DOI] [PubMed] [Google Scholar]
- 120. Albrektsen G, Heuch I, Kvåle G. Full-term pregnancies and incidence of ovarian cancer of stromal and germ cell origin: a Norwegian prospective study. Br J Cancer. 1997;75:767–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Chen T, Surcel HM, Lundin E, et al. Circulating sex steroids during pregnancy and maternal risk of non-epithelial ovarian cancer. Cancer Epidemiol Biomarkers Prev. 2011;20:324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Manikkam M, Guerrero-Bosagna C, Tracey R, Haque MM, Skinner MK. Transgenerational actions of environmental compounds on reproductive disease and identification of epigenetic biomarkers of ancestral exposures. PLoS One. 2012;7:e31901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Scully RE. Gonadoblastoma. A review of 74 cases. Cancer. 1970;25:1340–1356. [DOI] [PubMed] [Google Scholar]
- 124. Cools M, Stoop H, Kersemaekers AM, et al. Gonadoblastoma arising in undifferentiated gonadal tissue within dysgenetic gonads. J Clin Endocrinol Metab. 2006;91:2404–2413. [DOI] [PubMed] [Google Scholar]
- 125. Talerman A, Vang R. Germ cell tumors of the ovary. In: Kurman RJ, Ellenson LH, Ronnett BM, eds. Blaustein's Pathology of the Female Genital Tract. New York, NY: Springer; 2011:847–907. [Google Scholar]
- 126. Koo YJ, Chun YK, Kwon YS, et al. Ovarian gonadoblastoma with dysgerminoma in a woman with 46XX karyotype. Pathol Int. 2011;61:171–173. [DOI] [PubMed] [Google Scholar]
- 127. Zhao S, Kato N, Endoh Y, Jin Z, Ajioka Y, Motoyama T. Ovarian gonadoblastoma with mixed germ cell tumor in a woman with 46,XX karyotype and successful pregnancies. Pathol Int. 2000;50:332–335. [DOI] [PubMed] [Google Scholar]
- 128. Shahsiah R, Jahanbin B, Rabiei R, Ardalan FA, Sarhadi B, Izadi-Mood N. Malignant ovarian germ cell tumours in gonadal Y chromosome mosaicism. J Clin Pathol. 2011;64:973–976. [DOI] [PubMed] [Google Scholar]
- 129. Ledig S, Hiort O, Scherer G, et al. Array-CGH analysis in patients with syndromic and non-syndromic XY gonadal dysgenesis: Evaluation of array CGH as diagnostic tool and search for new candidate loci. Hum Reprod. 2010;25:2637–2646. [DOI] [PubMed] [Google Scholar]
- 130. Bashamboo A, Ledig S, Wieacker P, Achermann JC, McElreavey K. New technologies for the identification of novel genetic markers of disorders of sex development (DSD). Sex Dev. 2010;4:213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Kraggerud SM, Szymanska J, Abeler VM, et al. DNA copy number changes in malignant ovarian germ cell tumors. Cancer Res. 2000;60:3025–3030. [PubMed] [Google Scholar]
- 132. Kildal W, Kraggerud SM, Abeler VM, et al. Genome profiles of bilateral dysgerminomas, a unilateral gonadoblastoma, and a metastasis from a 46,XY phenotypic female. Hum Pathol. 2003;34:946–949. [DOI] [PubMed] [Google Scholar]
- 133. Lee-Jones L, Williams T, Little E, Sampson J. Trisomy 14pter→q21: a case with associated ovarian germ cell tumor and review of the literature. Am J Med Genet. 2004;128A:78–84. [DOI] [PubMed] [Google Scholar]
- 134. Gimelli S, Beri S, Drabkin HA, et al. The tumor suppressor gene TRC8/RNF139 is disrupted by a constitutional balanced translocation t(8;22)(q24.13;q11.21) in a young girl with dysgerminoma. Mol Cancer. 2009;8:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Kildal W, Kærn J, Kraggerud SM, et al. Evaluation of genomic changes in a large series of malignant ovarian germ cell tumors-relation to clinicopathologic variables. Cancer Genet Cytogenet. 2004;155:25–32. [DOI] [PubMed] [Google Scholar]
- 136. Silver SA, Wiley JM, Perlman EJ. DNA ploidy analysis of pediatric germ cell tumors. Mod Pathol. 1994;7:951–956. [PubMed] [Google Scholar]
- 137. Gibas Z, Talerman A. Analysis of chromosome aneuploidy in ovarian dysgerminoma by flow cytometry and fluorescence in situ hybridization. Diagn Mol Pathol. 1993;2:50–56. [PubMed] [Google Scholar]
- 138. Palmquist MB, Webb MJ, Lieber MM, Gaffey TA, Nativ O. DNA ploidy of ovarian dysgerminomas: correlation with clinical outcome. Gynecol Oncol. 1992;44:13–16. [DOI] [PubMed] [Google Scholar]
- 139. Baker BA, Frickey L, Yu IT, Hawkins EP, Cushing B, Perlman EJ. DNA content of ovarian immature teratomas and malignant germ cell tumors. Gynecol Oncol. 1998;71:14–18. [DOI] [PubMed] [Google Scholar]
- 140. Oud PS, Soeters RP, Pahlplatz MM, et al. DNA cytometry of pure dysgerminomas of the ovary. Int J Gynecol Pathol. 1988;7:258–267. [DOI] [PubMed] [Google Scholar]
- 141. Kelley JL, Naus GJ, Christopherson WA. Endodermal sinus tumor of the ovary: a case series with flow cytometric DNA content analysis. Gynecol Oncol. 1991;42:34–38. [DOI] [PubMed] [Google Scholar]
- 142. Kommoss F, Bibbo M, Talerman A. Nuclear deoxyribonucleic acid content (ploidy) of endodermal sinus (yolk sac) tumor. Lab Invest. 1990;62:223–231. [PubMed] [Google Scholar]
- 143. Atkin NB. Modal DNA value and chromosome number in ovarian neoplasia. A clinical and histopathologic assessment. Cancer. 1971;27:1064–1073. [DOI] [PubMed] [Google Scholar]
- 144. Kihana T, Tsuda H, Teshima S, Okada S, Matsuura S, Hirohashi S. High incidence of p53 gene mutation in human ovarian cancer and its association with nuclear accumulation of p53 protein and tumor DNA aneuploidy. Cancer Sci. 1992;83:978–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Feichter GE, Kühn W, Czernobilsky B, et al. DNA flow cytometry of ovarian tumors with correlation to histopathology. Int J Gynecol Pathol. 1985;4:336–345. [DOI] [PubMed] [Google Scholar]
- 146. Jenderny J, Koster E, Meyer A, et al. Detection of chromosome aberrations in paraffin sections of seven gonadal yolk sac tumors of childhood. Hum Genet. 1995;96:644–650. [DOI] [PubMed] [Google Scholar]
- 147. Kühn W, Kaufmann M, Feichter GE, Rummel HH, Schmid H, Heberling D. DNA flow cytometry, clinical and morphological parameters as prognostic factors for advanced malignant and borderline ovarian tumors. Gynecol Oncol. 1989;33:360–367. [DOI] [PubMed] [Google Scholar]
- 148. Stock C, Ambros IM, Lion T, et al. Detection of numerical and structural chromosome abnormalities in pediatric germ cell tumors by means of interphase cytogenetics. Genes Chromosomes Cancer. 1994;11:40–50. [DOI] [PubMed] [Google Scholar]
- 149. Riopel MA, Spellerberg A, Griffin CA, Perlman EJ. Genetic analysis of ovarian germ cell tumors by comparative genomic hybridization. Cancer Res. 1998;58:3105–3110. [PubMed] [Google Scholar]
- 150. Zahn S, Sievers S, Alemazkour K, et al. Imbalances of chromosome arm 1p in pediatric and adult germ cell tumors are caused by true allelic loss: a combined comparative genomic hybridization and microsatellite analysis. Genes Chromosomes Cancer. 2006;45:995–1006. [DOI] [PubMed] [Google Scholar]
- 151. Veltman I, Veltman J, Janssen I, et al. Identification of recurrent chromosomal aberrations in germ cell tumors of neonates and infants using genomewide array-based comparative genomic hybridization. Genes Chromosomes Cancer. 2005;43:367–376. [DOI] [PubMed] [Google Scholar]
- 152. Palmer RD, Foster NA, Vowler SL, et al. Malignant germ cell tumours of childhood: new associations of genomic imbalance. Br J Cancer. 2007;96:667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Cheng L, Zhang S, Talerman A, Roth LM. Morphologic, immunohistochemical, and fluorescence in situ hybridization study of ovarian embryonal carcinoma with comparison to solid variant of yolk sac tumor and immature teratoma. Hum Pathol. 2010;41:716–723. [DOI] [PubMed] [Google Scholar]
- 154. Cossu-Rocca P, Zhang S, Roth LM, et al. Chromosome 12p abnormalities in dysgerminoma of the ovary: a FISH analysis. Mod Pathol. 2006;19:611–615. [DOI] [PubMed] [Google Scholar]
- 155. Poulos C, Cheng L, Zhang S, Gersell DJ, Ulbright TM. Analysis of ovarian teratomas for isochromosome 12p: evidence supporting a dual histogenetic pathway for teratomatous elements. Mod Pathol. 2006;19:766–771. [DOI] [PubMed] [Google Scholar]
- 156. Cheng L, Roth LM, Zhang S, et al. KIT gene mutation and amplification in dysgerminoma of the ovary. Cancer. 2011;117:2096–2103. [DOI] [PubMed] [Google Scholar]
- 157. Wehle D, Yonescu R, Long PP, Gala N, Epstein J, Griffin CA. Fluorescence in situ hybridization of 12p in germ cell tumors using a bacterial artificial chromosome clone 12p probe on paraffin-embedded tissue: clinical test validation. Cancer Genet Cytogenet. 2008;183:99–104. [DOI] [PubMed] [Google Scholar]
- 158. Bussey KJ, Lawce HJ, Himoe E, et al. Chromosomes 1 and 12 abnormalities in pediatric germ cell tumors by interphase fluorescence in situ hybridization. Cancer Genet Cytogenet. 2001;125:112–118. [DOI] [PubMed] [Google Scholar]
- 159. Faulkner SW, Friedlander ML. Molecular genetic analysis of malignant ovarian germ cell tumors. Gynecol Oncol. 2000;77:283–288. [DOI] [PubMed] [Google Scholar]
- 160. Faulkner SW, Friedlander ML. Microsatellite instability in germ cell tumors of the testis and ovary. Gynecol Oncol. 2000;79:38–43. [DOI] [PubMed] [Google Scholar]
- 161. Radice P, Pierotti MA, Lacerenza S, et al. Loss of heterozygosity in human germinal tumors. Cytogenet Cell Genet. 1989;52:72–76. [DOI] [PubMed] [Google Scholar]
- 162. King BL, Carcangiu ML, Carter D, et al. Microsatellite instability in ovarian neoplasms. Br J Cancer. 1995;72:376–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Okpanyi V, Schneider DT, Zahn S, et al. Analysis of the adenomatous polyposis coli (APC) gene in childhood and adolescent germ cell tumors. Pediatr Blood Cancer. 2011;56:384–391. [DOI] [PubMed] [Google Scholar]
- 164. Fritsch MK, Schneider DT, Schuster AE, Murdoch FE, Perlman EJ. Activation of Wnt/β-catenin signaling in distinct histologic subtypes of human germ cell tumors. Pediatr Dev Pathol. 2006;9:115–131. [DOI] [PubMed] [Google Scholar]
- 165. Looijenga LH, Hersmus R, Gillis AJ, et al. Genomic and expression profiling of human spermatocytic seminomas: primary spermatocyte as tumorigenic precursor and DMRT1 as candidate chromosome 9 gene. Cancer Res. 2006;66:290–302. [DOI] [PubMed] [Google Scholar]
- 166. Fustino N, Rakheja D, Ateek CS, Neumann JC, Amatruda JF. Bone morphogenetic protein signalling activity distinguishes histological subsets of paediatric germ cell tumours. Int J Androl. 2011;34:e218–e233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Palmer RD, Barbosa-Morais NL, Gooding EL, et al. ; Children's Cancer and Leukaemia Group. Pediatric malignant germ cell tumors show characteristic transcriptome profiles. Cancer Res. 2008;68:4239–4247. [DOI] [PubMed] [Google Scholar]
- 168. Palmer RD, Murray MJ, Saini HK, et al. ; Children's Cancer and Leukaemia Group. Malignant germ cell tumors display common microRNA profiles resulting in global changes in expression of messenger RNA targets. Cancer Res. 2010;70:2911–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Gillis AJ, Stoop HJ, Hersmus R, et al. High-throughput microRNAome analysis in human germ cell tumours. J Pathol. 2007;213:319–328. [DOI] [PubMed] [Google Scholar]
- 170. Murray M, Saini H, van Dongen S, et al. The two most common histological subtypes of malignant germ cell tumour are distinguished by global microRNA profiles, associated with differential transcription factor expression. Mol Cancer. 2010;9:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Kawauchi S, Yamamoto Y, Uchida K, et al. Significance of cyclin E and p27 expression in malignant ovarian germ cell tumors: Correlation with the cell proliferation activity and clinicopathologic features. Oncol Rep. 2006;16:1029–1033. [PubMed] [Google Scholar]
- 172. Kawauchi S, Liu XP, Kawasaki K, et al. Significance of β-catenin and pRB pathway components in malignant ovarian germ cell tumours: INK4A promoter CpG island methylation is associated with cell proliferation. J Pathol. 2004;204:268–276. [DOI] [PubMed] [Google Scholar]
- 173. Cobellis L, Cataldi P, Reis FM, et al. Gonadal malignant germ cell tumors express immunoreactive inhibin/activin subunits. Eur J Endocrinol. 2001;145:779–784. [DOI] [PubMed] [Google Scholar]
- 174. Tanoguchi K, Sasano H, Yabuki N, et al. Immunohistochemical and two-parameter flow cytometric studies of DNA topoisomerase II alpha in human epithelial ovarian carcinoma and germ cell tumor. Mod Pathol. 1998;11:186–193. [PubMed] [Google Scholar]
- 175. Jin JS, Yao CW, Loh SH, Cheng MF, Hsieh DS, Bai CY. Increasing expression of extracellular matrix metalloprotease inducer in ovary tumors: tissue microarray analysis of immunostaining score with clinicopathological parameters. Int J Gynecol Pathol. 2006;25:140–146. [DOI] [PubMed] [Google Scholar]
- 176. Soini Y, Pääkkö P. Extent of apoptosis in relation to p53 and bcl-2 expression in germ cell tumors. Hum Pathol. 1996;27:1221–1226. [DOI] [PubMed] [Google Scholar]
- 177. Kemmer K, Corless CL, Fletcher JA, et al. KIT mutations are common in testicular seminomas. Am J Pathol. 2004;164:305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Tian Q, Frierson HF, Krystal GW, Moskaluk CA. Activating c-kit gene mutations in human germ cell tumors. Am J Pathol. 1999;154:1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Liu FS, Ho ES, Chen JT, Shih RT, Yang CH, Shih A. Overexpression or mutation of the p53 tumor suppressor gene does not occur in malignant ovarian germ cell tumors. Cancer. 1995;76:291–295. [DOI] [PubMed] [Google Scholar]
- 180. Okamoto A, Sameshima Y, Yokoyama S, et al. Frequent allelic losses and mutations of the p53 gene in human ovarian cancer. Cancer Res. 1991;51:5171–5176. [PubMed] [Google Scholar]
- 181. Heravi-Moussavi A, Anglesio MS, Cheng SW, et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med. 2011;366:234–242. [DOI] [PubMed] [Google Scholar]
- 182. Ichikawa Y, Yoshida S, Koyama Y, et al. Inactivation of p16/CDKN2 and p15/MTS2 genes in different histological types and clinical stages of primary ovarian tumors. Int J Cancer. 1996;69:466–470. [DOI] [PubMed] [Google Scholar]
- 183. Sievers S, Alemazkour K, Zahn S, et al. IGF2/H19 imprinting analysis of human germ cell tumors (GCTs) using the methylation-sensitive single-nucleotide primer extension method reflects the origin of GCTs in different stages of primordial germ cell development. Genes Chromosomes Cancer. 2005;44:256–264. [DOI] [PubMed] [Google Scholar]
- 184. Schneider DT, Schuster AE, Fritsch MK, et al. ; Pediatric Oncology Group, German Pediatric Germ Cell Tumor Study Group. Multipoint imprinting analysis indicates a common precursor cell for gonadal and nongonadal pediatric germ cell tumors. Cancer Res. 2001;61:7268–7276. [PubMed] [Google Scholar]
- 185. Ross JA, Schmidt PT, Perentesis JP, Davies SM. Genomic imprinting of H19 and insulin-like growth factor-2 in pediatric germ cell tumors. Cancer. 1999;85:1389–1394. [PubMed] [Google Scholar]
- 186. Bussey KJ, Lawce HJ, Himoe E, et al. SNRPN methylation patterns in germ cell tumors as a reflection of primordial germ cell development. Genes Chromosomes Cancer. 2001;32:342–352. [DOI] [PubMed] [Google Scholar]
- 187. Jeyapalan JN, Noor DA, Lee SH, et al. Methylator phenotype of malignant germ cell tumours in children identifies strong candidates for chemotherapy resistance. Br J Cancer. 2011;105:575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Haider S, Ballester B, Smedley D, Zhang J, Rice P, Kasprzyk A. BioMart Central Portal: unified access to biological data. Nucleic Acids Res. 2009;37:W23–W27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Bruford EA, Lush MJ, Wright MW, Sneddon TP, Povey S, Birney E. The HGNC Database in 2008: a resource for the human genome. Nucleic Acids Res. 2008;36:D445–D448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Safran M, Dalah I, Alexander J, et al. GeneCards Version 3: the human gene integrator. Database. 2010;2010:baq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Asadourian LA, Taylor HB. Dysgerminoma. An analysis of 105 cases. Obstet Gynecol. 1969;33:370–379. [PubMed] [Google Scholar]
- 192. Brassesco MS, Castro-Gamero AM, Valera ET, Neder L, Elias J, Tone LG. 3q27 aberrations in a childhood ovary teratoma with associated malignant germ cell component. Pediatr Blood Cancer. 2009;52:398–401. [DOI] [PubMed] [Google Scholar]
- 193. Atkin NB, Baker MC. i(12p): specific chromosomal marker in seminoma and malignant teratoma of the testis? Cancer Genet Cytogenet. 1983;10:199–204. [DOI] [PubMed] [Google Scholar]
- 194. Alagaratnam S, Lind GE, Kraggerud SM, Lothe RA, Skotheim RI. The testicular germ cell tumour transcriptome. Int J Androl. 2011;34:e133–e151. [DOI] [PubMed] [Google Scholar]
- 195. Skotheim RI, Lind GE, Monni O, et al. Differentiation of human embryonal carcinomas in vitro and in vivo reveals expression profiles relevant to normal development. Cancer Res. 2005;65:5588–5598. [DOI] [PubMed] [Google Scholar]
- 196. Fulda S. Caspase-8 in cancer biology and therapy. Cancer Lett. 2009;281:128–133. [DOI] [PubMed] [Google Scholar]
- 197. Fulton N, Martins da Silva SJ, Bayne RA, Anderson RA. Germ cell proliferation and apoptosis in the developing human ovary. J Clin Endocrinol Metab. 2005;90:4664–4670. [DOI] [PubMed] [Google Scholar]
- 198. Helfer B, Boswell BC, Finlay D, et al. Caspase-8 promotes cell motility and calpain activity under nonapoptotic conditions. Cancer Res. 2006;66:4273–4278. [DOI] [PubMed] [Google Scholar]
- 199. Finlay D, Vuori K. Novel noncatalytic role for caspase-8 in promoting Src-mediated adhesion and Erk signaling in neuroblastoma cells. Cancer Res. 2007;67:11704–11711. [DOI] [PubMed] [Google Scholar]
- 200. Kumper S, Ridley AJ. p120ctn and P-cadherin but not E-cadherin regulate cell motility and invasion of DU145 prostate cancer cells. PLoS One. 2010;5:e11801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Turashvili G, McKinney SE, Goktepe O, et al. P-cadherin expression as a prognostic biomarker in a 3992 case tissue microarray series of breast cancer. Mod Pathol. 2011;24:64–81. [DOI] [PubMed] [Google Scholar]
- 202. Bendel-Stenzel MR, Gomperts M, Anderson R, Heasman J, Wylie C. The role of cadherins during primordial germ cell migration and early gonad formation in the mouse. Mech Dev. 2000;91:143–152. [DOI] [PubMed] [Google Scholar]
- 203. Grivennikov S, Karin M. Autocrine IL-6 signaling: a key event in tumorigenesis? Cancer Cell. 2008;13:7–9. [DOI] [PubMed] [Google Scholar]
- 204. Eddie SL, Childs AJ, Jabbour HN, Anderson RA. Developmentally regulated IL6-type cytokines signal to germ cells in the human fetal ovary. Mol Hum Reprod. 2012;18:88–95. [DOI] [PubMed] [Google Scholar]
- 205. Chuma S, Nakatsuji N. Autonomous transition into meiosis of mouse fetal germ cells in vitro and its inhibition by gp130-mediated signaling. Dev Biol. 2001;229:468–479. [DOI] [PubMed] [Google Scholar]
- 206. Schweyer S, Soruri A, Baumhoer D, et al. Expression of CXC chemokine IP-10 in testicular germ cell tumours. J Pathol. 2002;197:89–97. [DOI] [PubMed] [Google Scholar]
- 207. Dietl J, Horny HP, Ruck P, Kaiserling E. Dysgerminoma of the ovary. An immunohistochemical study of tumor-infiltrating lymphoreticular cells and tumor cells. Cancer. 1993;71:2562–2568. [DOI] [PubMed] [Google Scholar]
- 208. Sasada T, Suekane S. Variation of tumor-infiltrating lymphocytes in human cancers: controversy on clinical significance. Immunotherapy. 2011;3:1235–1251. [DOI] [PubMed] [Google Scholar]
- 209. Ben-Baruch A. The multifaceted roles of chemokines in malignancy. Cancer Metastasis Rev. 2006;25:357–371. [DOI] [PubMed] [Google Scholar]
- 210. Belperio JA, Keane MP, Arenberg DA, et al. CXC chemokines in angiogenesis. J Leukocyte Biol. 2000;68:1–8. [PubMed] [Google Scholar]
- 211. Panagopoulos I, Moller E, Collin A, Mertens F. The POU5F1P1 pseudogene encodes a putative protein similar to POU5F1 isoform 1. Oncol Rep. 2008;20:1029–1033. [PubMed] [Google Scholar]
- 212. Wicki A, Lehembre F, Wick N, Hantusch B, Kerjaschki D, Christofori G. Tumor invasion in the absence of epithelial-mesenchymal transition: podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell. 2006;9:261–272. [DOI] [PubMed] [Google Scholar]
- 213. Rider CC, Mulloy B. Bone morphogenetic protein and growth differentiation factor cytokine families and their protein antagonists. Biochem J. 2010;429:1–12. [DOI] [PubMed] [Google Scholar]
- 214. Neumann JC, Chandler GL, Damoulis VA, et al. Mutation in the type IB bone morphogenetic protein receptor alk6b impairs germ-cell differentiation and causes germ-cell tumors in zebrafish. Proc Natl Acad Sci U S A. 2011;108:13153–13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Alves Vidigal J, Ventura A. Embryonic stem cell miRNAs and their roles in development and disease. Semin Cancer Biol. 2012;22:428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Zhou AD, Diao LT, Xu H, et al. β-Catenin/LEF1 transactivates the microRNA-371–373 cluster that modulates the Wnt/β-catenin-signaling pathway. Oncogene. 2011;31:2968–2978. [DOI] [PubMed] [Google Scholar]
- 217. Loayza-Puch F, Yoshida Y, Matsuzaki T, Takahashi C, Kitayama H, Noda M. Hypoxia and RAS-signaling pathways converge on, and cooperatively downregulate, the RECK tumor-suppressor protein through microRNAs. Oncogene. 2010;29:2638–2648. [DOI] [PubMed] [Google Scholar]
- 218. Harris T, Jimenez L, Kawachi N, et al. Low-level expression of miR-375 correlates with poor outcome and metastasis while altering the invasive properties of head and neck squamous cell carcinomas. Am J Pathol. 2012;180:917–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Zhang X, Yan Z, Zhang J, et al. Combination of hsa-miR-375 and hsa-miR-142–5p as a predictor for recurrence risk in gastric cancer patients following surgical resection. Ann Oncol. 2011;22:2257–2266. [DOI] [PubMed] [Google Scholar]
- 220. Kong KL, Kwong DL, Chan TH, et al. MicroRNA-375 inhibits tumour growth and metastasis in oesophageal squamous cell carcinoma through repressing insulin-like growth factor 1 receptor. Gut. 2012;61:33–42. [DOI] [PubMed] [Google Scholar]
- 221. Yu L, Todd NW, Xing L, et al. Early detection of lung adenocarcinoma in sputum by a panel of microRNA markers. Int J Cancer. 2010;127:2870–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Bryant RJ, Pawlowski T, Catto JW, et al. Changes in circulating microRNA levels associated with prostate cancer. Br J Cancer. 2012;106:768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Murray MJ, Halsall DJ, Hook CE, Williams DM, Nicholson JC, Coleman N. Identification of microRNAs from the miR-371∼373 and miR-302 clusters as potential serum biomarkers of malignant germ cell tumors. Am J Clin Pathol. 2011;135:119–125. [DOI] [PubMed] [Google Scholar]
- 224. Lipchina I, Elkabetz Y, Hafner M, et al. Genome-wide identification of microRNA targets in human ES cells reveals a role for miR-302 in modulating BMP response. Gene Dev. 2011;25:2173–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Garcia AI, Buisson M, Bertrand P, et al. Down-regulation of BRCA1 expression by miR-146a and miR-146b–5p in triple negative sporadic breast cancers. EMBO Mol Med. 2011;3:279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Faraoni I, Antonetti FR, Cardone J, Bonmassar E. miR-155 gene: a typical multifunctional microRNA. Biochim Biophys Acta. 2009;1792:497–505. [DOI] [PubMed] [Google Scholar]
- 227. Sarver A, French A, Borralho P, et al. Human colon cancer profiles show differential microRNA expression depending on mismatch repair status and are characteristic of undifferentiated proliferative states. BMC Cancer. 2009;9:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Chang S, Wang RH, Akagi K, et al. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nat Med. 2011;17:1275–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Moskwa P, Buffa FM, Pan Y, et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol Cell. 2011;41:210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Valeri N, Gasparini P, Fabbri M, et al. Modulation of mismatch repair and genomic stability by miR-155. Proc Natl Acad Sci U S A. 2010;107:6982–6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Tili E, Michaille JJ, Wernicke D, et al. Mutator activity induced by microRNA-155 (miR-155) links inflammation and cancer. Proc Natl Acad Sci U S A. 2011;108:4908–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Cekaite L, Rantala J, Bruun J, et al. MicroRNAs miR-9, -31, and -182 are implicated in cell proliferation and apoptosis in colon cancer. Neoplasia. 2012;14:838–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Ormestad M, Astorga J, Landgren H, et al. Foxf1 and Foxf2 control murine gut development by limiting mesenchymal Wnt signaling and promoting extracellular matrix production. Development. 2006;133:833–843. [DOI] [PubMed] [Google Scholar]
- 234. Yu Z, Raabe T, Hecht NB. MicroRNA Mirn122a reduces expression of the posttranscriptionally regulated germ cell transition protein 2 (Tnp2) messenger RNA (mRNA) by mRNA cleavage. Biol Reprod. 2005;73:427–433. [DOI] [PubMed] [Google Scholar]
- 235. Buchold GM, Coarfa C, Kim J, Milosavljevic A, Gunaratne PH, Matzuk MM. Analysis of microRNA expression in the prepubertal testis. PLoS One. 2010;5:e15317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Burns DM, D'Ambrogio A, Nottrott S, Richter JD. CPEB and two poly(A) polymerases control miR-122 stability and p53 mRNA translation. Nature. 2011;473:105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Gregory PA, Bracken CP, Smith E, et al. An autocrine TGF-β/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell. 2011;22:1686–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Peter ME. Let-7 and miR-200 microRNAs: guardians against pluripotency and cancer progression. Cell Cycle. 2009;8:843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Knouf EC, Garg K, Arroyo JD, et al. An integrative genomic approach identifies p73 and p63 as activators of miR-200 microRNA family transcription. Nucleic Acids Res. 2012;40:499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Yao Y, Suo AL, Li ZF, et al. MicroRNA profiling of human gastric cancer. Mol Med Report. 2009;2:963–970. [DOI] [PubMed] [Google Scholar]
- 241. Kahlert C, Klupp F, Brand K, et al. Invasion front-specific expression and prognostic significance of microRNA in colorectal liver metastases. Cancer Sci. 2011;102:1799–1807. [DOI] [PubMed] [Google Scholar]
- 242. Li D, Wang Q, Liu C, et al. Aberrant expression of miR-638 contributes to benzo(a)pyrene-induced human cell transformation. Toxicol Sci. 2012;125:382–391. [DOI] [PubMed] [Google Scholar]
- 243. Gilbert D, Rapley E, Shipley J. Testicular germ cell tumours: predisposition genes and the male germ cell niche. Nat Rev Cancer. 2011;11:278–288. [DOI] [PubMed] [Google Scholar]
- 244. Salonen J, Leminen A, Stenman UH, Bützow R, Heikinheimo M, Heikinheimo O. Tissue AP-2g and Oct-3/4, and serum CA 125 as diagnostic and prognostic markers of malignant ovarian germ cell tumors. Tumor Biol. 2008;29:50–56. [DOI] [PubMed] [Google Scholar]
- 245. Pauls K, Jäger R, Weber S, et al. Transcription factor AP-2g, a novel marker of gonocytes and seminomatous germ cell tumors. Int J Cancer. 2005;115:470–477. [DOI] [PubMed] [Google Scholar]
- 246. Wang L, Chang X, Yuan G, Zhao Y, Wang P. Expression of peptidylarginine deiminase type 4 in ovarian tumors. Int J Biol Sci. 2010;6:454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Soini Y, Talvensaari-Mattila A. Expression of claudins 1, 4, 5, and 7 in ovarian tumors of diverse types. Int J Gynecol Pathol. 2006;25:330–335. [DOI] [PubMed] [Google Scholar]
- 248. Salonen J, Butzow R, Palvimo JJ, Heikinheimo M, Heikinheimo O. Oestrogen receptors and small nuclear ring finger protein 4 (RNF4) in malignant ovarian germ cell tumours. Mol Cell Endocrinol. 2009;307:205–210. [DOI] [PubMed] [Google Scholar]
- 249. Bing Z, Pasha TL, Lal P, Tomaszewski JE. Expression of glial cell line-derived neurotropic factor receptor a-1 in immature teratomas. Am J Clin Pathol. 2008;130:892–896. [DOI] [PubMed] [Google Scholar]
- 250. Kohno Y, Matsuki Y, Tanimoto A, et al. Expression of Y-box-binding protein dbpC/contrin, a potentially new cancer/testis antigen. Br J Cancer. 2006;94:710–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. West JA, Viswanathan SR, Yabuuchi A, et al. A role for Lin28 in primordial germ-cell development and germ-cell malignancy. Nature. 2009;460:909–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Xue D, Peng Y, Wang F, Allan RW, Cao D. RNA-binding protein LIN28 is a sensitive marker of ovarian primitive germ cell tumours. Histopathology. 2011;59:452–459. [DOI] [PubMed] [Google Scholar]
- 253. Palma I, Peña RY, Contreras A, et al. Participation of OCT3/4 and β-catenin during dysgenetic gonadal malignant transformation. Cancer Lett. 2008;263:204–211. [DOI] [PubMed] [Google Scholar]
- 254. Cheng L, Thomas A, Roth LM, Zheng W, Michael H, Karim FW. OCT4: a novel biomarker for dysgerminoma of the ovary. Am J Surg Pathol. 2004;28:1341–1346. [DOI] [PubMed] [Google Scholar]
- 255. Chang MC, Vargas SO, Hornick JL, Hirsch MS, Crum CP, Nucci MR. Embryonic stem cell transcription factors and D2–40 (podoplanin) as diagnostic immunohistochemical markers in ovarian germ cell tumors. Int J Gynecol Pathol. 2009;28:347–355. [DOI] [PubMed] [Google Scholar]
- 256. Abiko K, Mandai M, Hamanishi J, et al. Oct4 expression in immature teratoma of the ovary: relevance to histologic grade and degree of differentiation. Am J Surg Pathol. 2010;34:1842–1848. [DOI] [PubMed] [Google Scholar]
- 257. Chambers I, Tomlinson SR. The transcriptional foundation of pluripotency. Development. 2009;136:2311–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Perrett RM, Turnpenny L, Eckert JJ, et al. The early human germ cell lineage does not express SOX2 during in vivo development or upon in vitro culture. Biol Reprod. 2008;78:852–858. [DOI] [PubMed] [Google Scholar]
- 259. Weber S, Eckert D, Nettersheim D, et al. Critical function of AP-2γ/TCFAP2C in mouse embryonic germ cell maintenance. Biol Reprod. 2010;82:214–223. [DOI] [PubMed] [Google Scholar]
- 260. Huang S, Jean D, Luca M, Tainsky MA, Bar-Eli M. Loss of AP-2 results in downregulation of c-KIT and enhancement of melanoma tumorigenicity and metastasis. EMBO J. 1998;17:4358–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Sartini S, Dario B, Morelli M, Da SF, La Motta C. Receptor tyrosine kinase kit and gastrointestinal stromal tumours: an overview. Curr Med Chem. 2011;18:2893–2903. [DOI] [PubMed] [Google Scholar]
- 262. Renneville A, Roumier C, Biggio V, et al. Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia. 2008;22:915–931. [DOI] [PubMed] [Google Scholar]
- 263. Rajpert-De Meyts E, Skakkebaek NE. Expression of the c-kit protein product in carcinoma-in-situ and invasive testicular germ cell tumours. Int J Androl. 1994;17:85–92. [DOI] [PubMed] [Google Scholar]
- 264. Ramalingam P, Malpica A, Silva EG, Gershenson DM, Liu JL, Deavers MT. The use of cytokeratin 7 and EMA in differentiating ovarian yolk sac tumors from endometrioid and clear cell carcinomas. Am J Surg Pathol. 2004;28:499–505. [DOI] [PubMed] [Google Scholar]
- 265. Inoue M, Kyo S, Fujita M, Enomoto T, Kondoh G. Coexpression of the c-kit receptor and the stem cell factor in gynecological tumors. Cancer Res. 1994;54:3049–3053. [PubMed] [Google Scholar]
- 266. Tsuura Y, Hiraki H, Watanabe K, et al. Preferential localization of c-kit product in tissue mast cells, basal cells of skin, epithelial cells of breast, small cell lung carcinoma and seminoma/dysgerminoma in human: immunohistochemical study on formalin-fixed, paraffin-embedded tissues. Virchows Arch. 1994;424:135–141. [DOI] [PubMed] [Google Scholar]
- 267. Went PT, Dirnhofer S, Bundi M, et al. Prevalence of KIT expression in human tumors. J Clin Oncol. 2004;22:4514–4522. [DOI] [PubMed] [Google Scholar]
- 268. Sever M, Jones TD, Roth LM, et al. Expression of CD117 (c-kit) receptor in dysgerminoma of the ovary: diagnostic and therapeutic implications. Mod Pathol. 2005;18:1411–1416. [DOI] [PubMed] [Google Scholar]
- 269. Masson K, Rönnstrand L. Oncogenic signaling from the hematopoietic growth factor receptors c-Kit and Flt3. Cell Signal. 2009;21:1717–1726. [DOI] [PubMed] [Google Scholar]
- 270. Pauls K, Wardelmann E, Merkelbach-Bruse S, Büttner R, Zhou H. c-KIT codon 816 mutation in a recurrent and metastatic dysgerminoma of a 14-year-old girl: Case study. Virchows Arch. 2004;445:651–654. [DOI] [PubMed] [Google Scholar]
- 271. Heinrich MC, Blanke CD, Druker BJ, Corless CL. Inhibition of KIT tyrosine kinase activity: a novel molecular approach to the treatment of KIT-positive malignancies. J Clin Oncol. 2002;20:1692–1703. [DOI] [PubMed] [Google Scholar]
- 272. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11:865–878. [DOI] [PubMed] [Google Scholar]
- 273. Eiring A, Khorashad J, Morley K, Deininger M. Advances in the treatment of chronic myeloid leukemia. BMC Med. 2011;9:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Fleischer RT, Vollenhoven BJ, Weston GC. The effects of chemotherapy and radiotherapy on fertility in premenopausal women. Obstet Gynecol Surv. 2011;66:248–254. [DOI] [PubMed] [Google Scholar]
- 275. Pedersini R, Vattemi E, Mazzoleni G, Graiff C. Complete response after treatment with imatinib in pretreated disseminated testicular seminoma with overexpression of c-KIT. Lancet Oncol. 2007;8:1039–1040. [DOI] [PubMed] [Google Scholar]
- 276. Pectasides D, Nikolaou M, Pectasides E, Koumarianou A, Valavanis C, Economopoulos T. Complete response after imatinib mesylate administration in a patient with chemoresistant stage IV seminoma. Anticancer Res. 2008;28:2317–2320. [PubMed] [Google Scholar]
- 277. Einhorn LH, Brames MJ, Heinrich MC, Corless CL, Madani A. Phase II study of imatinib mesylate in chemotherapy refractory germ cell tumors expressing KIT. Am J Clin Oncol. 2006;29:12–13. [DOI] [PubMed] [Google Scholar]
- 278. Schacht V, Dadras SS, Johnson LA, Jackson DG, Hong YK, Detmar M. Up-regulation of the lymphatic marker podoplanin, a mucin-type transmembrane glycoprotein, in human squamous cell carcinomas and germ cell tumors. Am J Pathol. 2005;166:913–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Sonne S, Herlihy A, Hoei-Hansen CE, et al. Identity of M2A (D2–40) antigen and gp36 (Aggrus, T1A-2, podoplanin) in human developing testis, testicular carcinoma in situ and germ-cell tumours. Virchows Arch. 2006;449:200–206. [DOI] [PubMed] [Google Scholar]
- 280. Stenning SP, Parkinson MC, Fisher C, et al. Postchemotherapy residual masses in germ cell tumor patients. Cancer. 1998;83:1409–1419. [PubMed] [Google Scholar]
- 281. Palmer PE, Wolfe HJ. Immunocytochemical localization of oncodevelopmental proteins in human germ cell and hepatic tumors. J Histochem Cytochem. 1978;26:523–531. [DOI] [PubMed] [Google Scholar]
- 282. Siltanen S, Anttonen M, Heikkilä P, et al. Transcription factor GATA-4 is expressed in pediatric yolk sac tumors. Am J Pathol. 1999;155:1823–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Esheba GE, Pate LL, Longacre TA. Oncofetal protein glypican-3 distinguishes yolk sac tumor from clear cell carcinoma of the ovary. Am J Surg Pathol. 2008;32:600–607. [DOI] [PubMed] [Google Scholar]
- 284. Siltanen S, Heikkilä P, Bielinska M, Wilson DB, Heikinheimo M. Transcription factor GATA-6 is expressed in malignant endoderm of pediatric yolk sac tumors and in teratomas. Pediatr Res. 2003;54:542–546. [DOI] [PubMed] [Google Scholar]
- 285. Mannisto S, Butzow R, Salonen J, Leminen A, Heikinheimo O, Heikinheimo M. Transcription factors GATA-4 and GATA-6, and their potential downstream effectors in ovarian germ cell tumors. Tumor Biol. 2005;26:265–273. [DOI] [PubMed] [Google Scholar]
- 286. Zynger DL, Everton MJ, Dimov ND, Chou PM, Yang XJ. Expression of glypican 3 in ovarian and extragonadal germ cell tumors. Am J Clin Pathol. 2008;130:224–230. [DOI] [PubMed] [Google Scholar]
- 287. Zynger DL, McCallum JC, Luan C, Chou PM, Yang XJ. Glypican 3 has a higher sensitivity than α-fetoprotein for testicular and ovarian yolk sac tumour: immunohistochemical investigation with analysis of histological growth patterns. Histopathology. 2010;56:750–757. [DOI] [PubMed] [Google Scholar]
- 288. Preda O, Nicolae A, Aneiros-Fernández J, Borda A, Nogales FF. Glypican 3 is a sensitive, but not a specific, marker for the diagnosis of yolk sac tumours. Histopathology. 2011;58:312–314. [DOI] [PubMed] [Google Scholar]
- 289. Hippo Y, Watanabe K, Watanabe A, et al. Identification of soluble NH2-terminal fragment of glypican-3 as a serological marker for early-stage hepatocellular carcinoma. Cancer Res. 2004;64:2418–2423. [DOI] [PubMed] [Google Scholar]
- 290. Hanigan MH, Frierson HF, Jr, Abeler VM, Kaern J, Taylor PT., Jr Human germ cell tumours: Expression of γ-glutamyl transpeptidase and sensitivity to cisplatin. Br J Cancer. 1999;81:75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Cossu-Rocca P, Jones TD, Roth LM, et al. Cytokeratin and CD30 expression in dysgerminoma. Hum Pathol. 2006;37:1015–1021. [DOI] [PubMed] [Google Scholar]
- 292. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. [DOI] [PubMed] [Google Scholar]
- 293. Ye DW, Zheng J, Qian SX, et al. p53 gene mutations in Chinese human testicular seminoma. J Urol. 1993;150:884–886. [PubMed] [Google Scholar]
- 294. Fleischhacker M, Strohmeyer T, Imai Y, Slamon DJ, Koeffler HP. Mutations of the p53 gene are not detectable in human testicular tumors. Mod Pathol. 1994;7:435–439. [PubMed] [Google Scholar]
- 295. Schenkman NS, Sesterhenn IA, WA L, et al. Increased p53 protein does not correlate to p53 gene mutations in microdissected human testicular germ cell tumors. J Urol. 1995;154:617–621. [DOI] [PubMed] [Google Scholar]
- 296. Lothe RA, Peltomaki P, Tommerup N, et al. Molecular genetic changes in human male germ cell tumors. Lab Invest. 1995;73:606–614. [PubMed] [Google Scholar]
- 297. Guillou L, Estreicher A, Chaubert P, et al. Germ cell tumors of the testis overexpress wild-type p53. Am J Pathol. 1996;149:1221–1228. [PMC free article] [PubMed] [Google Scholar]
- 298. Ehteshami M, Ro J, Shin H, Lee J, Ordonez N, Ayala A. p53 expression and tumor proliferative activity in testicular germ-cell tumors. Int J Oncol. 1996;9:787–793. [PubMed] [Google Scholar]
- 299. Dietl J, Horny HP, Kaiserling E. Frequent overexpression of p53 in dysgerminoma of the ovary. Gynecol Obstet Invest. 1994;37:141–142. [DOI] [PubMed] [Google Scholar]
- 300. Marx L, Dietl J, Horny HP. Overexpression or mutation of the p53 tumor suppressor gene does not occur in malignant ovarian germ cell tumors. Cancer. 1996;78:179–180. [DOI] [PubMed] [Google Scholar]
- 301. Hu W, Zheng T, Wang J. Regulation of fertility by the p53 family members. Genes Cancer. 2011;2:420–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G. The p53 family: Guardians of maternal reproduction. Nat Rev Mol Cell Biol. 2011;12:259–265. [DOI] [PubMed] [Google Scholar]
- 303. Suh EK, Yang A, Kettenbach A, et al. p63 protects the female germ line during meiotic arrest. Nature. 2006;444:624–628. [DOI] [PubMed] [Google Scholar]
- 304. Crum CP, McKeon FD. p63 in epithelial survival, germ cell surveillance, and neoplasia. Annu Rev Pathol Mech Dis. 2010;5:349–371. [DOI] [PubMed] [Google Scholar]
- 305. Ince TA, Cviko AP, Quade BJ, et al. p63 coordinates anogenital modeling and epithelial cell differentiation in the developing female urogenital tract. Am J Pathol. 2002;161:1111–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Slade I, Bacchelli C, Davies H, et al. DICER1 syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J Med Genet. 2011;48:273–278. [DOI] [PubMed] [Google Scholar]
- 307. Menezes R, Boetzer M, Sieswerda M, van Ommen GJ, Boer J. Integrated analysis of DNA copy number and gene expression microarray data using gene sets. BMC Bioinformatics. 2009;10:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Ulbright TM. Germ cell tumors of the gonads: a selective review emphasizing problems in differential diagnosis, newly appreciated, and controversial issues. Mod Pathol. 2005;18:S61–S79. [DOI] [PubMed] [Google Scholar]
- 309. Kraggerud SM, Skotheim RI, Szymanska J, et al. Genome profiles of familial/bilateral and sporadic testicular germ cell tumors. Genes Chromosomes Cancer. 2002;34:168–174. [DOI] [PubMed] [Google Scholar]
- 310. Ihara T, Ohama K, Satoh H, Fujii T, Nomura K, Fujiwara A. Histologic grade and karyotype of immature teratoma of the ovary. Cancer. 1984;54:2988–2994. [DOI] [PubMed] [Google Scholar]
- 311. Vakiani E, Solit DB. KRAS and BRAF: drug targets and predictive biomarkers. J Pathol. 2011;223:220–230. [DOI] [PubMed] [Google Scholar]
- 312. Katoh Y, Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med. 2009;9:873–886. [DOI] [PubMed] [Google Scholar]
- 313. Salonen J, Rajpert-De Meyts E, Mannisto S, et al. Differential developmental expression of transcription factors GATA-4 and GATA-6, their cofactor FOG-2 and downstream target genes in testicular carcinoma in situ and germ cell tumors. Eur J Endocrinol. 2010;162:625–631. [DOI] [PubMed] [Google Scholar]
- 314. Biermann K, Göke F, Nettersheim D, et al. c-KIT is frequently mutated in bilateral germ cell tumours and down-regulated during progression from intratubular germ cell neoplasia to seminoma. J Pathol. 2007;213:311–318. [DOI] [PubMed] [Google Scholar]
- 315. Looijenga LH, de Leeuw H, van Oorschot M, et al. Stem cell factor receptor (c-KIT) codon 816 mutations predict development of bilateral testicular germ-cell tumors. Cancer Res. 2003;63:7674–7678. [PubMed] [Google Scholar]
- 316. Sakuma Y, Matsukuma S, Yoshihara M, et al. Mutations of c-kit gene in bilateral testicular germ cell tumours in Japan. Cancer Lett. 2008;259:119–126. [DOI] [PubMed] [Google Scholar]
- 317. Coffey J, Linger R, Pugh J, et al. Somatic KIT mutations occur predominantly in seminoma germ cell tumors and are not predictive of bilateral disease: Report of 220 tumors and review of literature. Genes Chromosomes Cancer. 2008;47:34–42. [DOI] [PubMed] [Google Scholar]
- 318. Okada K, Katagiri T, Tsunoda T, et al. Analysis of gene-expression profiles in testicular seminomas using a genome-wide cDNA microarray. Int J Oncol. 2003;23:1615–1635. [PubMed] [Google Scholar]
- 319. Skotheim RI, Monni O, Mousses S, et al. New insights into testicular germ cell tumorigenesis from gene expression profiling. Cancer Res. 2002;62:2359–2364. [PubMed] [Google Scholar]
- 320. Yamada S, Kohu K, Ishii T, et al. Gene expression profiling identifies a set of transcripts that are up-regulated in human testicular seminoma. DNA Res. 2004;11:335–344. [DOI] [PubMed] [Google Scholar]
- 321. Gashaw I, Grümmer R, Klein-Hitpass L, et al. Gene signatures of testicular seminoma with emphasis on expression of ets variant gene 4. Cell Mol Life Sci. 2005;62:2359–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Port M, Schmelz HU, Stockinger M, et al. Gene expression profiling in seminoma and nonseminoma. J Clin Oncol. 2005;23:58–69. [DOI] [PubMed] [Google Scholar]
- 323. Cotterman R, Knoepfler PS. N-Myc regulates expression of pluripotency genes in neuroblastoma including lif, klf2, klf4, and lin28b. PLoS One. 2009;4:e5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Helland Å, Anglesio MS, George J, et al. Deregulation of MYCN, LIN28B and LET7 in a molecular subtype of aggressive high-grade serous ovarian cancers. PLoS One. 2011;6:e18064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Kumar R, Gururaj AE, Barnes CJ. p21-activated kinases in cancer. Nat Rev Cancer. 2006;6:459–471. [DOI] [PubMed] [Google Scholar]
- 326. Banerjee M, Worth D, Prowse DM, Nikolic M. Pak1 phosphorylation on T212 affects microtubules in cells undergoing mitosis. Curr Biol. 2002;12:1233–1239. [DOI] [PubMed] [Google Scholar]
- 327. Phang JM, Liu W, Zabirnyk O. Proline metabolism and microenvironmental stress. Annu Rev Nutr. 2010;30:441–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Juric D, Sale S, Hromas RA, et al. Gene expression profiling differentiates germ cell tumors from other cancers and defines subtype-specific signatures. Proc Natl Acad Sci U S A. 2005;102:17763–17768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Korkola JE, Houldsworth J, Dobrzynski D, et al. Gene expression-based classification of nonseminomatous male germ cell tumors. Oncogene. 2005;24:5101–5107. [DOI] [PubMed] [Google Scholar]
- 330. Korkola JE, Heck S, Olshen AB, et al. In vivo differentiation and genomic evolution in adult male germ cell tumors. Genes Chromosomes Cancer. 2008;47:43–55. [DOI] [PubMed] [Google Scholar]
- 331. Bourdon V, Naef F, Rao PH, et al. Genomic and expression analysis of the 12p11–p12 amplicon using EST arrays identifies two novel amplified and overexpressed genes. Cancer Res. 2002;62:6218–6223. [PubMed] [Google Scholar]
- 332. Rodriguez S, Jafer O, Goker H, et al. Expression profile of genes from 12p in testicular germ cell tumors of adolescents and adults associated with i(12p) and amplification at 12p11.2-p12.1. Oncogene. 2003;22:1880–1891. [DOI] [PubMed] [Google Scholar]
- 333. Skotheim RI, Autio R, Lind GE, et al. Novel genomic aberrations in testicular germ cell tumors by array-CGH, and associated gene expression changes. Cell Oncol. 2006;28:315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Biermann K, Heukamp LC, Steger K, et al. Gene expression profiling identifies new biological markers of neoplastic germ cells. Anticancer Res. 2007;27:3091–3100. [PubMed] [Google Scholar]
- 335. McIntyre A, Summersgill B, Lu YJ, et al. Genomic copy number and expression patterns in testicular germ cell tumours. Br J Cancer. 2007;97:1707–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Malliri A, Collard JG. Role of Rho-family proteins in cell adhesion and cancer. Curr Opin Cell Biol. 2003;15:583–589. [DOI] [PubMed] [Google Scholar]
- 337. Xu H, Kardash E, Chen S, Raz E, Lin F. Gbg signaling controls the polarization of zebrafish primordial germ cells by regulating Rac activity. Development. 2012;139:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Jørgensen N, Müller J, Jaubert F, Clausen OP, Skakkebaek NE. Heterogeneity of gonadoblastoma germ cells: similarities with immature germ cells, spermatogonia and testicular carcinoma in situ cells. Histopathology. 1997;30:177–186. [DOI] [PubMed] [Google Scholar]
- 339. Kasinski AL, Slack FJ. MicroRNAs en route to the clinic: Progress in validating and targeting microRNAs for cancer therapy. Nat Rev Cancer. 2011;11:849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Schneider DT, Schuster AE, Fritsch MK, et al. Genetic analysis of childhood germ cell tumors with comparative genomic hybridization. Klin Padiatr. 2001;213:204–211. [DOI] [PubMed] [Google Scholar]
- 341. He L, Ding H, Wang JH, et al. Overexpression of karyopherin 2 in human ovarian malignant germ cell tumor correlates with poor prognosis. PLoS One. 2012;7:e42992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Hersmus R, Stoop H, van de Geijn GJ, et al. Prevalence of c-KIT mutations in gonadoblastoma and dysgerminomas of patients with disorders of sex development (DSD) and ovarian dysgerminomas. PLoS One. 2012;7:e43952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Malecki M, Anderson M, Beauchaine M, Seo S, Tambokan X. TRA-1-60(+), SSEA-4(+), Oct4A(+), Nanog(+) clones of pluripotent stem cells in the embryonal carcinomas of the ovaries. J Stem Cell Res Ther. 2012;2:130. [PMC free article] [PubMed] [Google Scholar]
- 344. Pauniaho SL, Salonen J, Helminen M, Vettenranta K, Heikinheimo M, Heikinheimo O. The incidences of malignant gonadal and extragonadal germ cell tumors in males and females: a population-based study covering over 40 years in Finland. Cancer Causes Control. 2012; 23:1921–1927. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.