

Abstract
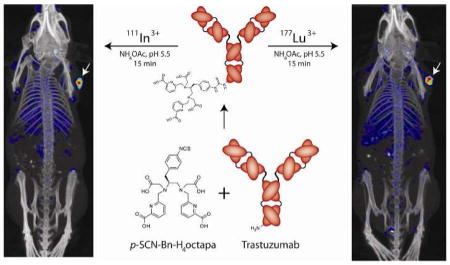
A bifunctional derivative of the versatile acyclic chelator H4octapa, p-SCNBn- H4octapa, has been synthesized for the first time. The chelator was conjugated to the HER2/neu-targeting antibody trastuzumab and labeled in high radiochemical purity and specific activity with the radioisotopes 111In and 177Lu. The in vivo behavior of the resulting radioimmunoconjugates was investigated in mice bearing ovarian cancer xenografts and compared to analogous radioimmunoconjugates employing the ubiquitous chelator DOTA. The H4octapa-trastuzumab conjugates displayed faster radiolabeling kinetics with more reproducible yields under milder conditions (15 min, RT, ~94–95%) than those based on DOTA-trastuzumab (60 min, 37 °C ~50–88%). Further, antibody integrity was better preserved in the 111In- and 177Lu-octapatrastuzumab constructs, with immunoreactive fractions of 0.99 for each compared to 0.93–0.95 for 111In- and 177Lu-DOTA-trastuzumab. These results translated to improved in vivo biodistribution profiles and SPECT imaging results for 111In- and 177Lu-octapa-trastuzumab compared to 111In- and 177Lu-DOTA-trastuzumab, with increased tumor uptake and higher tumor-to-tissue activity ratios.
Introduction
Radiometallated bioconjugates possess an inherent modularity that can prove extremely useful in the design and construction of agents for nuclear imaging and therapy. Indeed, they contain a set of four distinct chemical components that can be swapped systematically to tune the properties and functions of the whole: the radiometal can be exchanged to harness isotopes with different decay characteristics; the chelator can be altered to accommodate different radiometals; the nature of the covalent linkage between the chelator and the biomolecule can be changed to take advantage of different types of conjugation chemistry; and, of course, the biomolecule itself can be replaced in order to change the targeting properties or pharmacokinetics of the construct.1–7 In clinical practice, the use of different isotopes with a single targeting vector becomes especially important in pretherapy imaging and dosimetry studies, procedures in which an agent bearing an imaging radioisotope is used for scouting scans prior to radiotherapy using the same vector labeled with a therapeutic nuclide. Two isotopes that are ideally suited for this purpose are 111In, a cyclotron produced radiometal (111Cd(p, n)111In) for SPECT imaging (t1/2 ~2.8 days), and 177Lu, a reactor produced therapeutic radiometal (176Lu(n, γ)177Lu) that emits β-particles as well as γ-rays (t1/2 ~6.6 days).8
Due to their exceptional specificity and affinity, antibodies have emerged as extremely promising biomolecules for the delivery of radioactive payloads to cancerous tissues.9,10 However, antibodies are not without their limitations as radiopharmaceutical vectors. For example, while they are generally considered robust in vivo, antibodies can become damaged or break down when subjected to the elevated temperatures required by many radiometal chelation reactions.11 As a result, the choice of a chelator for a radioimmunoconjugate becomes particularly critical. In any radiopharmaceutical, the purpose of the chelator is to sequester the radiometal such that the isotope remains attached to the targeting vector at all times, resisting spontaneous demetallation as well as transchelation to serum proteins. Macrocyclic chelators such as 1,4,7,10- tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA; Chart 1) have long been staples of radiochemical research. They are versatile and are generally more kinetically inert than acyclic chelators, primarily due to their constrained geometries and partially pre-organized coordination sites.12–19 One unfortunate and restrictive property of macrocycles like DOTA, however, is that heating (40–90 °C) and extended reaction times (30–180 min) are typically required for quantitative radiolabeling, conditions that can be incompatible with antibodies and other biomolecules.12–26 Acyclic chelators offer improvements in this regard, but not without cost. The acyclic chelator diethylenetriaminetetraacetic acid (DTPA), for example, exhibits much faster reaction kinetics than DOTA and can radiolabel with a variety of radiometals quantitatively in a matter of minutes at ambient temperature. However, it is not nearly as stable in vivo as its macrocyclic counterparts.14,15,27–29
Chart 1. Structures of some selected chelators.

Chelators used or discussed in this work, including two based on the pyridinecarboxylate scaffold that were developed in our laboratory: the promising 67/68Ga chelator H2dedpa and H4octapa, the non-bifunctional variant of the chelator p-SCN-Bn-H4octapa used in this work.
Thus, choosing a radiometal chelator for an antibody conjugate requires a delicate balancing act. An immunoconjugate bearing a macrocyclic chelator such as DOTA can require extended incubations at elevated temperatures —often at the expense of antibody stability or immunoreactivity —in order to achieve suitable radiochemical yields; however, a radioimmunoconjugate bearing an acyclic chelator such as DTPA risks the unintentional release of the radiometal in vivo, an especially pressing problem considering the long pharmacokinetic half-life of many antibodies.8,10,19–22,26–28 It thus becomes clear that a versatile chelator that combines the thermodynamic stability and kinetic inertness of macrocyclic chelators with the facile radiolabeling of acyclic chelators could prove tremendously valuable in the synthesis and development of novel radioimmunoconjugates.
Herein, we report the synthesis, characterization, and in vivo validation of p-SCN-Bn- H4octapa, a bifunctional, versatile, acyclic chelator that can be labeled rapidly at room temperature and exhibits significant thermodynamic stability and kinetic inertness with 111In and 177Lu. This ligand has evolved out of our laboratory’s extensive study of acyclic chelators based on the pyridinecarboxylate scaffold (Chart 1).30–38 Members of the versatile “pa family” of chelators — including H4octapa, H2dedpa, H5decapa, and H2azapa — cover nearly all of the current medicinally relevant radiometals.30–38 H4octapa specifically has demonstrated high in vitro and in vivo stability and rapid radiolabeling kinetics with 111In. Further, comparisons with 111In-DOTA have revealed that H4octapa possesses much more facile radiolabeling kinetics, can be labeled in higher specific activity, undergoes less fluxional isomerization in solution, and exhibits comparable stability in vitro and in vivo.33 A major hurdle during this work, however, has been the cumbersome syntheses of the chelators.30–36 As a result, in the investigation at hand, we report the construction of the bifunctional p-SCN-Bn-H4octapa via a vastly improved synthetic protocol. More importantly, we have employed the HER2/neu-targeted antibody trastuzumab as a model system for the in vitro and in vivo validation of 111In-octapa- and 177Lu-octapa-based radioimmunoconjugates in a murine model of ovarian cancer as well as the comparative evaluation of these radioimmunoconjugates to those employing the more traditional macrocyclic chelator DOTA.
Results and Discussion
Synthesis and Characterization
In order to properly evaluate the potential of H4octapa for use in biomolecular radiopharmaceuticals, we first sought to develop a highly efficient synthesis of a novel bifunctional variant of the versatile acyclic chelator: p-SCN-Bn-H4octapa (Scheme 1), which had never been synthesized before. Like many bifunctional chelators, we have utilized the versatile and enantiopure starting material L-4-nitrophenylalanine.29,30,33 While the N-benzyl-based protecting group chemistry we have previously reported for the synthesis of non-bifunctional H4octapa proved adequate for the construction of the bare chelator, the requisite vigorous hydrogenation conditions resulted in poor yields and were completely incompatible with the synthesis of bifunctional derivatives based on 4- nitrophenylalanine.30,33 Consequently, we revamped the synthesis to utilize the 2- nitrobenzenesulfonamide (nosyl) protecting group, a change that has significantly improved the synthesis of H4octapa (5) and has made the synthesis of p-SCN-Bn-H4octapa (12) feasible.30,33,36,39–41 Specifically, the switch to the nosyl protecting group has dramatically increased the cumulative yield of H4octapa (5) from ~10–12% to ~45–50% over five synthetic steps.33 Further, using this strategy, the bifunctional variant p-SCN-Bn-H4octapa — previously inaccessible using N-benzyl protection chemistry — can now be routinely produced with cumulative yields over 7 steps of ~25–30 %. To our knowledge only a few previous publications have utilized the 2-nitrobenzenesulfonamide (nosyl) amineprotecting group for the synthesis of chelating or imaging agents.39–43
Scheme 1. Improved synthesis of chelators utilizing nosyl protection chemistry.

a, (i) THF, NaHCO3, 2-nitrobenzenesulfonyl chloride (2.2 equiv), RT, 24 h, 87%; (ii) DMF, Na2CO3, methyl-6-bromomethylpicolinate (2.2 equiv), 50 °C, 24 h, 95%; (iii) THF, thiophenol (2.2 equiv), K2CO3, RT, 72 h, 91%; (iv) MeCN, Na2CO3, tert-butylbromoacetate (2.2 equiv), 50 °C, 24 h, 92%; (v) 6 M HCl, reflux, 24 h, 66%. b, (i) THF, NaHCO3, 2-nitrobenzenesulfonyl chloride (2.2 equiv), 50 °C, 24 h, 74%; (ii) DMF, Na2CO3, tert-butylbromoacetate (2.2 equiv), 50 °C, 24 h, 88%; (iii) THF, thiophenol (2.2 equiv), K2CO3, RT, 72 h, 89%; (iv) MeCN, Na2CO3, methyl-6-bromomethylpicolinate (2.2 equiv), 50 °C, 24 h, 96%; (v) 5 mL of (1:1) Glacial acetic acid:3 M HCl, Pd/C (20 wt%), H2 (g) balloon, RT, 1 h; (vi) 6 M HCl, reflux, 24 h; (vii) thiophosgene in DCM (15 equiv), 3 M HCl, RT, 24 h, 50% over steps v-vii.
Compared to the challenging syntheses of bifunctional macrocycles like p-SCN-Bn- DOTA, this streamlined route to p-SCN-Bn-H4octapa should be of significant utility. The bulky nature of the nosyl protecting groups is admittedly atom inefficient; however, their use results in a reduced number of purification steps (compounds 1, 2, and 8 can be purified via crystallization), greater yields, and, most relevantly, the accessibility of p-SCNBn- H4octapa. The final 3 steps in the synthesis of p-SCN-Bn-H4octapa are performed sequentially without purification due to the high polarity and difficult separation of the intermediates, and the final molecule is purified via reverse-phase HPLC chromatography.
Following synthesis, the HCl salt of pure H4octapa was successfully metallated via incubation with indium perchlorate33 [In(ClO4)3] and lutetium chloride [LuCl3] to form quantitatively coordinated complexes. Further, all of the new chelators and their metal complexes were fully chemically characterized using 1H-NMR, 13C-NMR, HR-ESI-MS, and ATR-IR where appropriate (Figures S1–13). Particularly interesting, however, were the 1H NMR spectra of the metallated H4octapa ligand. Variable temperature NMR experiments with In3+ complexes of DOTA have demonstrated fluxional isomerization in solution, despite the macrocyclic nature of the chelator.19,33,44 In this case, however, the 1H-NMR spectra obtained for [In(octapa)]− and [Lu(octapa)]− (6) reveal sharp and distinct coupling patterns, indicating little fluxional isomerization in solution at ambient temperature (Figure S5).33
Thermodynamic Stability and Density Functional Theory Structure Prediction
The thermodynamic stability constants (log KML) for DOTA with Lu3+ have previously been determined by spectrophotometric methods (UV) to be 23.5 and ~25,45,46 and by a competitive potentiometric titration with oxalate to be 29.2 ± 0.2.47 In these studies the only species for which log KML has been documented is for the ML species (no metal hydroxide), and titrations were not done in the presence of sodium, which is known to lower formation constants values (our titrations are done in 0.15 M sodium for physiologically relevant conditions).48 These factors combined with the inconsistencies in the reported log KML values and methods used prompted us to perform competitive potentiometric titration experiments of DOTA with EDTA and Lu3+ in order to provide a formation constant that was determined under identical experimental conditions to our own system. Using potentiometric titrations, we have experimentally determined the thermodynamic stability constant (log KML) for Lu3+ with H4octapa to be 20.08 ± 0.09 (pM = 19.8) and with DOTA to be 21.62 ± 0.10 (pM = 17.1). In addition, the log KML value for Lu3+ with DTPA has been reported in the literature to be 22.4, and we have calculated a pM value of 19.1.17 Although the presence of sodium in our experiments can be expected to lower the stability constant of DOTA with Lu3+ from previously reported values, and the different experimental methods used are expected to vary in their results, the low log KML value we have obtained could be inaccurate as a result of the slow complex formation kinetics of DOTA. During the potentiometric titrations we performed, up to 15 minutes was allowed for stabilization of the electrode reading between each addition of base, which may have been too brief for a proper equilibrium to be established with DOTA. Previous work has waited over 2 weeks for equilibrium to be reached in this system, which highlights the sluggish formation kinetics.45 Because of this uncertainty, our experimentally determined log KML value for DOTA with Lu3+ cannot be relied on solely or be considered to be more accurate than previously determined values, and therefore all reported formation constants must be considered. The thermodynamic stability constants of the In3+ complexes of H4octapa, DOTA, and DTPA have been determined to be 26.6 (pM = 26.5), 23.9 (pM = 18.8), and 29.0 (pM = 25.7), respectively.33 It is important to note that thermodynamic stability is generally regarded as an unreliable predictor of in vivo stability on its own,16 and the kinetic inertness of a radiometal-complex is a much more valuable factor. For example, the [111In(DOTA)]− complex is widely established as being significantly more stable than [111In(DTPA)]2− both in vivo and in vitro, yet the thermodynamic stability constant for In3+ with DTPA is ~5 orders of magnitude higher than with DOTA (29.0 and 23.9, respectively). In fact, pM values — which are calculated from log KML values while taking into account ligand pKa’s and metal ion hydroxide formation under specific and physiologically relevant conditions — are generally considered more useful in predicting in vivo stability than log KML values. H4octapa has been shown to have high pM values with both In3+ (26.5) and Lu3+ (19.8), further substantiating its promising properties with these metals and their radioactive isotopologues.
The coordination geometry of both the 8- and 9-coordinate [Lu(octapa)]− and [Lu(octapa)(H2O)]− complexes were estimated in silico using density functional theory (DFT) calculations, and MEP polar surface area maps were superimposed on the structure (Figure 1). Both Lu3+ complexes were found to be highly symmetrical and very similar to one another; indeed, that the addition of an H2O ligand results in very little change in metalligand bond lengths and angles suggests that water binding bears little influence on the coordination of the ligand. Importantly, the [Lu(octapa)]− and [Lu(octapa)(H2O)]− DFT structures shown here are quite similar to the DFT structure of the 8-coordinate [In(octapa)]− complex.33 Taken together, the high log KML and pM values, the absence of fluxional behavior in solution, and the structural similarity between the [Lu(octapa)]− and [In(octapa)]− complexes strongly suggest that these two complexes should exhibit similar behavior in vitro and in vivo. This is especially important when considering 111In as a SPECT imaging surrogate isotope for 177Lu-based radiotherapies.
Figure 1. In silico DFT structure predictions.

a, 8-coordinate structure of [Lu(octapa)]−. b, 9-coordinate structure of [Lu(octapa)(H2O)]−, as well as the MEP polar-surface area maps (bottom) predicting the charge distribution over the solvent-exposed surface of the metal complexes (red = negative, blue = positive, representing a maximum potential of 0.254 au and a minimum of −0.254 au, mapped onto electron density isosurfaces of 0.002 Å−3), demonstrating very little difference between the 8- and 9-coordinate solution structures in terms of both geometry and charge distribution, suggesting little change in physical properties between the 8- and 9-coordinate geometries.
Bioconjugation and In Vitro Characterization
Synthesis and characterization data aside, the true value of any radiometal chelator ultimately lies in its behavior in vitro and in vivo. As a result, the next step in our investigation was to assess the in vitro and in vivo performance of H4octapa in a model system based on the HER2/neu-targeting antibody trastuzumab and HER2-expressing SKOV-3 ovarian cancer cells. In order to provide a basis for comparison, radioimmunoconjugates bearing the ubiquitous chelator DOTA were also constructed and employed in parallel in all in vitro and in vivo experiments.
To begin, purified trastuzumab was incubated under slightly basic conditions (pH 8.5–9.0) with 4 equivalents of p-SCN-Bn-H4octapa or p-SCN-Bn-DOTA and purified via size exclusion chromatography (PD-10, GE Healthcare, UK). Subsequent radiometric isotopic dilution experiments indicated that these modifications yielded 3.03 ± 0.1 chelates per antibody in the case of p-SCN-Bn-H4octapa and 3.40 ± 0.1 chelates per antibody in the case of p-SCN-Bn-DOTA. H4octapa-trastuzumab was then radiolabeled with either 111In or 177Lu in NH4OAc buffer (pH 5.5, 200 mM) for 15 min at room temperature, rapidly producing quantitatively labeled radioimmunoconjugates (>95% radiochemical yield) in high radiochemical purity (>99% in each case) and specific activity (4.0 ± 0.3 and 3.5 ± 0.4 mCi/mg, respectively) (Figures S14–15). It is important to note that these labile kinetics offer a significant improvement over DOTA-trastuzumab, which required the less favorable incubation conditions of 1 h at 37 °C to achieve highly variable radiochemical yields of ~50- 88% and to produce 111In- and 177Lu-labeled radioimmunoconjugates with specific activities of 2.0 ± 0.2 and 3.4 ± 0.3 mCi/mg, respectively (Table 1). These results are of practical significance because antibodies and radiometals are very expensive, and so the inconsistent yields provided when radiolabeling DOTA-trastuzumab could waste as much as 50% of the antibody construct and radiometal, greatly increasing cost and making translation to a hospital radiopharmacy setting more challenging.
Table 1.
Chemical and in vitro biological characterization data for 111In- and 177Lu-octapa-trastuzumab and 111In- and 177Lu-DOTA-trastuzumab radioimmunoconjugates.
| Immunoconjugate | Isotope | Radiolabeling conditions and yield | Chelates/mAba | Specific activity (mCi/mg) | Immunoreactive fraction (%)b | Serum stability 120 h (%)c |
|---|---|---|---|---|---|---|
| H4octapa-trastuzumab | 111In | 15 min, 25 °C, 94% | 3.0 ± 0.1 | 4.0 ± 0.3 | 99.9 ± 0.02 | 94.9 ± 0.6 |
| 177Lu | 15 min, 25 °C, 95% | 3.0 ± 0.1 | 3.5 ± 0.4 | 99.2 ± 0.8 | 92.4 ± 0.6 | |
| DOTA-trastuzumab | 111In | 60 min, 37 °C, 50-85% | 3.4 ± 0.1 | 2.0 ± 0.2 | 93.2 ± 0.5 | 91.1 ± 0.6 |
| 177Lu | 60 min, 37 °C, 50-88% | 3.4 ± 0.1 | 3.4 ± 0.3 | 95.2 ± 0.2 | 98.6± 0.6 |
Isotopic dilution assays, n = 3.
Determined immediately prior to in vivo experimentation, n = 6.
Calculated for incubation in human serum at 37 °C for 120 h, n = 3.
The mild radiolabeling conditions afforded by H4octapa-trastuzumab likely contributed to the extremely high immunoreactivity of the immunoconjugates as determined by in vitro cellular assays using SKOV-3 cancer cells: 99.9 ± 0.02% for 111Inoctapa- trastuzumab and 98.7 ± 0.8% for 177Lu-octapa-trastuzumab. These values compared favorably to the slightly lower but statistically different values determined for 111In- and 177Lu-DOTA-trastuzumab: 93.2 ± 0.5 % and 95.2 ± 0.2 %, respectively (p-values of 0.003 and 0.02, respectively) (Table 1). In order to assay the stability of the radioimmunoconjugates under biological conditions, all four constructs were incubated in human serum at 37 °C for a period of 5 days. Over the course of the experiment, the stability of the 111In-octapa- and 111In-DOTA-trastuzumab was determined to be 94.9 ± 0.6% and 91.1 ± 0.6%, respectively, and that of the 177Lu-octapa- and 177Lu-DOTA-based conjugates was found to be 92.4 ± 0.6% and 98.6 ± 0.6%, respectively (Table 1). In the future, acid dissociation experiments, and studies probing the ability of H4octapa to radiolabel with 111In and 177Lu in the presence of an excess of metal ions such as Ca2+, Mg2+, Cu2+, Zn2+, and Fe3+ are of interest.49,50
Acute Biodistribution Studies
Biodistribution experiments were performed in order to directly compare the in vivo behavior and pharmacokinetics of the 111In- and 177Lu-based trastuzumab radioimmunoconjugates. To this end, each of the four radiolabeled antibodies — 111In-octapa-trastuzumab, 177Lu-octapa-trastuzumab, 111In-DOTA-trastuzumab, and 177Lu-DOTA-trastuzumab — were injected via tail vein into female nude athymic mice bearing subcutaneous SKOV-3 ovarian cancer xenografts in the right shoulder (~30 μCi, ~8– 15 μg, in 200 μL of sterile saline; tumor volume ~ 100–150 mm3). After 24, 48, 72, 96, or 120 h (n = 4 per time point) the mice were euthanized via CO2 (g) asphyxiation, and 13 organs, including the SKOV-3 tumors, were removed, weighed, and assayed for radioactivity on a γ-counter.
In all four cases, the biodistribution results showed all the hallmarks of tumortargeted radioimmunoconjugates, specifically high levels of uptake in the blood pool at early time points giving way over time to increasing levels of uptake in the tumor and, ultimately, very high tumor-to-background organ activity ratios. In all four cases, the level of uptake in non-target organs is roughly similar, with the highest levels of background uptake in the liver, spleen, and kidneys (Tables S1–3). However, past 24 h, the amount of activity in nontarget organs rarely exceeds 5–10 %ID/g; for example, at 96 h post-injection, 177Lu-octapatrastuzumab displays uptakes of 9.8 ± 1.7 %ID/g, 9.2 ± 1.1 %ID/g, and 3.8 ± 1.8 %ID/g in the liver, spleen, and kidneys, respectively. A close inspection of Table 2 reveals slightly higher uptake of 111In/177Lu-octapa-trastuzumab in the bone when compared to the DOTAtrastuzumab conjugates, but also slightly lower uptake of 111In/177Lu-octapa-trastuzumab in the kidneys.
Table 2.
Biodistribution data of 111In/177Lu-H4octapa-trastuzumab and 111In/177Lu-DOTA-trastuzumab. Performed over a 5 day period in mice bearing SKOV-3 ovarian cancer xenografts, tumor volume ~ 100–150 mm3 (n = 4 for each time point).
| 111In-octapa-trastuzumab | 111In-DOTA-trastuzumab | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Organ | 24 h | 48 h | 72 h | 96 h | 120 h | Organ | 24 h | 48 h | 72 h | 96 h | 120 h |
| Blood | 6.9 ± 5.1 | 7.6 ± 6.5 | 10.5 ± 7.1 | 6.9 ± 6.6 | 11.4 ± 6 | Blood | 18 ± 1.7 | 17.3 ± 2.6 | 16.8 ± 2.2 | 15 ± 3.6 | 14.3 ± 1.6 |
| Tumor | 57.4 ± 22 | 58.3 ± 17.2 | 57.8 ± 14.6 | 68.7 ± 20.5 | 72.4 ± 21.3 | Tumor | 22.3 ± 6.4 | 28.4 ± 5.2 | 30.6 ± 9.1 | 32.8 ± 7.3 | 31.5 ± 3.8 |
| Heart | 2.6 ± 0.5 | 2.5 ± 1.5 | 2.7 ± 1.4 | 1.9 ± 1.4 | 3.6 ± 1.6 | Heart | 6.1 ± 0.9 | 6.6 ± 2 | 5.6 ± 1.3 | 5.2 ± 1.8 | 5.2 ± 0.9 |
| Lungs | 4.4 ± 2.1 | 3.9 ± 2.3 | 5 ± 2.8 | 3.8 ± 3.3 | 6.3 ± 3 | Lungs | 7.2 ± 1.6 | 6.8 ± 3.3 | 5.2 ± 1.4 | 6.3 ± 1.2 | 5.7 ± 2.2 |
| Liver | 12 ± 2.4 | 10.6 ± 1.2 | 9.1 ± 0.8 | 8.5 ± 2 | 6.6 ± 0.6 | Liver | 6.2 ± 0.4 | 5.5 ± 1.4 | 4.8 ± 1.3 | 4.4 ± 1.7 | 5.2 ± 1.2 |
| Spleen | 8.9 ± 2.4 | 7.6 ± 1 | 8.1 ± 0.8 | 7.4 ± 1.1 | 6.5 ± 1.6 | Spleen | 3.6 ± 0.8 | 3.7 ± 1.6 | 3 ± 0.7 | 2.7 ± 0.7 | 3.4 ± 1.4 |
| Stomach | 1.4 ± 0.2 | 0.8 ± 0.2 | 1.2 ± 0.1 | 0.7 ± 0.3 | 0.7 ± 0.2 | Stomach | 0.7 ± 0.1 | 0.7 ± 0.4 | 0.8 ± 0.2 | 0.3 ± 0.2 | 0.6 ± 0.3 |
| Lg Intestine | 3.7 ± 0.4 | 2.5 ± 0.7 | 2 ± 0.9 | 1.7 ± 0.4 | 1.2 ± 0.1 | Lg Intestine | 0.5 ± 0.1 | 0.6 ± 0.1 | 0.7 ± 0.1 | 0.5 ± 0.1 | 0.5 ± 0.1 |
| Sm Intestine | 3.8 ± 0.7 | 2.1 ± 0.4 | 2.5 ± 0.6 | 2.1 ± 0.4 | 1.5 ± 0.1 | Sm Intestine | 1.9 ± 0.1 | 1.6 ± 0.5 | 1.7 ± 0.1 | 1.1 ± 0.2 | 1.3 ± 0.1 |
| Kidney | 3.6 ± 1.2 | 4.2 ± 2.1 | 4.3 ± 2.1 | 3.5 ± 2 | 4.3 ± 1.6 | Kidney | 5.3 ± 0.5 | 6.3 ± 0.9 | 5.1 ± 1.7 | 5.8 ± 1.7 | 4.9 ± 0.4 |
| Muscle | 1.3 ± 0.5 | 0.9 ± 0.5 | 1 ± 0.5 | 0.8 ± 0.5 | 0.9 ± 0.5 | Muscle | 0.7 ± 0.2 | 0.8 ± 0.3 | 0.5 ± 0.1 | 0.4 ± 0.1 | 0.3 ± 0.1 |
| Bone | 4.1 ± 2 | 3.5 ± 0.4 | 2.8 ± 1 | 3.3 ± 0.5 | 1.4 ± 0.3 | Bone | 1 ± 0.3 | 0.9 ± 0.1 | 1.1 ± 0.4 | 0.9 ± 0.4 | 0.6 ± 0.2 |
| Skin | 10.8 ± 1.4 | 9.3 ± 1.4 | 7.2 ± 2.1 | 5.4 ± 1.2 | 3 ± 1.1 | Skin | 1.8 ± 0.4 | 2.1 ± 0.7 | 2.6 ± 0.1 | 1.7 ± 0.8 | 2.2 ± 0.8 |
|
| |||||||||||
| 177Lu-octapa-trastuzumab | 177Lu-DOTA-trastuzumab | ||||||||||
| Organ | 24 h | 48 h | 72 h | 96 h | 120 h | Organ | 24 h | 48 h | 72 h | 96 h | 120 h |
|
| |||||||||||
| Blood | 19.1 ± 1.5 | 9.1 ± 9.5 | 9.7 ± 5.2 | 8.9 ± 6.9 | 7.6 ± 5.7 | Blood | 20.4 ± 1.7 | 17.4 ± 1 | 18.1 ± 2.8 | 15 ± 0.8 | 14.7 ± 0.7 |
| Tumor | 50 ± 5.4 | 63.5 ± 15 | 62.4 ± 20 | 70.4 ± 25.8 | 56.6 ± 12.7 | Tumor | 18.3 ± 4.8 | 31.6 ± 2.9 | 32.1 ± 7.4 | 37 ± 9.5 | 37.4 ± 10.3 |
| Heart | 4.2 ± 0.7 | 3.2 ± 1.9 | 2.5 ± 0.9 | 2.3 ± 0.7 | 2.6 ± 1.4 | Heart | 7.2 ± 1 | 4.9 ± 3.3 | 5.8 ± 1.6 | 4.6 ± 1.1 | 5.4 ± 0.6 |
| Lung | 7.8 ± 3.6 | 3.9 ± 3.1 | 5 ± 2.3 | 4.4 ± 2.5 | 2.7 ± 1.8 | Lungs | 10 ± 1.5 | 6.7 ± 4.6 | 7.1 ± 2.3 | 6.3 ± 0.6 | 6 ± 2.1 |
| Liver | 10.1 ± 1.3 | 13.2 ± 4 | 9.5 ± 3.5 | 9.8 ± 1.7 | 8 ± 2.6 | Liver | 4.5 ± 2 | 5.7 ± 3.6 | 5.7 ± 2.5 | 2.8 ± 0.9 | 3.7 ± 1.9 |
| Spleen | 8.8 ± 2.3 | 11.5 ± 2.5 | 7.4 ± 2.8 | 9.2 ± 1.2 | 6.7 ± 2.4 | Spleen | 3.8 ± 0.5 | 3.1 ± 2.9 | 3.2 ± 1 | 3.5 ± 0.4 | 3.8 ± 1 |
| Stomach | 1.4 ± 0.3 | 1 ± 0.5 | 0.9 ± 0.4 | 1.1 ± 0.3 | 0.6 ± 0.2 | Stomach | 0.7 ± 0.2 | 1.3 ± 1.9 | 0.9 ± 0.2 | 0.8 ± 0.2 | 0.7 ± 0.2 |
| Lg Intestine | 1.3 ± 0.3 | 1 ± 0.3 | 1.1 ± 0.5 | 1.1 ± 0.2 | 0.8 ± 0.2 | Lg Intestine | 0.6 ± 0.1 | 0.5 ± 0.3 | 0.8 ± 0.2 | 0.5 ± 0.1 | 0.6 ± 0.2 |
| Sm Intestine | 1.9 ± 0.5 | 2.5 ± 1.1 | 2 ± 0.9 | 2.3 ± 0.4 | 1.6 ± 0.4 | Sm Intestine | 1.7 ± 0.2 | 1.5 ± 1 | 1.5 ± 0.4 | 1.4 ± 0.3 | 1.2 ± 0.1 |
| Kidney | 4.9 ± 0.9 | 3.8 ± 2 | 3.7 ± 1.2 | 3.8 ± 1.8 | 2.8 ± 1.2 | Kidney | 5.5 ± 1.7 | 7.3 ± 0.7 | 5.1 ± 0.8 | 4.4 ± 0.2 | 7.1 ± 5 |
| Muscle | 0.9 ± 0.2 | 0.7 ± 0.5 | 0.8 ± 0.5 | 0.5 ± 0.2 | 0.4 ± 0.2 | Muscle | 0.7 ± 0.2 | 0.5 ± 0.4 | 0.4 ± 0.1 | 0.5 ± 0.2 | 0.5 ± 0.1 |
| Bone | 3.2 ± 0.5 | 4 ± 0.6 | 3.1 ± 0.7 | 4.2 ± 1.5 | 3.6 ± 1.7 | Bone | 1.2 ± 0 | 1.3 ± 0.8 | 1.3 ± 0.3 | 0.9 ± 0.2 | 1.4 ± 0.3 |
| Skin | 8.4 ± 2 | 6.8 ± 2.9 | 6.9 ± 0.5 | 5.6 ± 1.2 | 4.1 ± 2.2 | Skin | 1.9 ± 0.9 | 2.4 ± 1.7 | 2.1 ± 0.2 | 2.1 ± 0.7 | 3.1 ± 1.3 |
Far more interesting are the levels of tumor uptake, which are markedly higher with the 111In-octapa- and 177Lu-octapa-based variants compared to their 111In-DOTA- and 177Lu- DOTA-based analogs. For example, at 96 h post-injection the tumor uptake for 111In-octapatrastuzumab and 177Lu-octapa-trastuzumab was found to be 68.7 ± 20.5 %ID/g and 70.4 ± 25.8 %ID/g, respectively, compared to 32.8 ± 7.3 %ID/g and 37.0 ± 9.5 %ID/g for the 111In- DOTA-trastuzumab and 177Lu-DOTA-trastuzumab constructs. Notably, despite the large range in errors, the p-values for these differences are 0.04 for the 111In data sets and 0.05 for 177Lu data sets, indicating statistically significant differences (two-tailed students t-test, Tables S1–3). Statistical analysis of tumor uptake by one-way ANOVA provided p-values of 0.02 for the 111In data sets and 0.05 for 177Lu data sets. In imaging, tumor-to-tissue activity ratios are arguably even more important measures than absolute uptake values, as these ratios provide quantitative information regarding the contrast that will be observed during imaging. As a result of the excellent tumor uptake of the H4octapa-based radioimmunoconjugates, the tumor-to-background activity ratios were higher than the corresponding DOTA-based agents for almost all organs. For example, the tumor-to-blood ratios for 111In-octapa- and 111In-DOTA-trastuzumab at 120 h were found to be 6.4 ± 3.8 and 2.2 ± 0.4, respectively, while for 177Lu-octapa- and 177Lu-DOTA-trastuzumab they were 7.5 ± 5.8 and 2.5 ± 0.7, respectively (Table S1).
The origin of the elevated tumor uptake values observed for the H4octapatrastuzumab agents could lie in the enhanced immunoreactivity conferred by the fast and mild radiolabeling conditions. However, the difference in immunoreactivity was only about 5%, a discrepancy that would not be expected to cause such a change in vivo. Alternatively, it could be postulated that while the antigen-binding region of the antibody was only slightly adversely affected by temperature in the DOTA-trastuzumab conjugate, a separate region of the immunoglobulin may have suffered bond cleavage or protein structure disruption during radiolabeling, which in turn affected tumor uptake and retention. Finally, the differences in pharmacokinetic properties from attaching three to four relatively small chelating moieties to a 150 kD antibody is almost certainly insignificant, but it is not inconceivable that this modification could somehow affect the distribution of the radioimmunoconjugates in vivo. Experiments are currently underway to probe the origin of this elevated tumor uptake. Ultimately, regardless of the causation, the superior tumor uptake and tumor-to-tissue activity ratios plainly show that 111In-octapa-trastuzumab and 177Lu-octapa-trastuzumab are highly effective and versatile radioimmunoconjugates and, more generally, that H4octapa is a very promising ligand for the construction of biomolecular radiopharmaceuticals.
Small Animal SPECT/CT Imaging and Cerenkov Luminescence Imaging
Single photon emission computed tomography (SPECT) was used in conjunction with standard helical Xray CT for in vivo imaging of all 111In and 177Lu labeled immunoconjugates over 5 days, visually demonstrating high uptake and clear delineation of the SKOV-3 ovarian cancer xenografts by 111In- and 177Lu-octapa-trastuzumab as well as 111In- and 177Lu-DOTAtrastuzumab (Figure 2). Images taken at early time points show residual activity in the blood pool at 24 hours; however, at later time points, the background activity in the blood clears and is accompanied by a concomitant increase in the amount of activity in the tumor, culminating at a point at which the tumor is by far the most prominent feature in the image (Figures 2, S17–20). Overall, the image quality, tumor uptake, and tumor contrast observed are very similar between the H4octapa- and DOTA-based constructs. In all four cases, high levels of tumor uptake and excellent tumor-to-background contrast are observed at later time points, data which correlates well with the high tumor uptake and tumor-to-tissue ratios revealed in the biodistribution experiments (Tables 2, S1–3). It is important to note that while the 111In- and 177Lu-octapa-trastuzumab images do not appear qualitatively superior to those produced by the 111In-DOTA- and 177Lu-DOTA variants, the enhanced tumor-to-background activity ratios of the 111In- and 177Lu-octapa-based constructs could potentially be of value in identifying small lesions or metastases in which absolute uptake of radioactivity is limited and for which contrast is crucial. The use of Cerenkov radiation (CR) for Cerenkov luminescence imaging (CLI) has been emerging as a new imaging modality with applications such as intraoperative radionuclide-guided surgery for the resection of cancers.51 In order to probe the potential of our 177Lu-labeled immunoconjugate, we have successfully imaged 177Lu-octapa-trastuzumab via external optical imaging through the skin 24 hours post injection via CLI (Figure 3). To our knowledge, we report the first published example of in vivo CLI imaging of 177Lu (Figure 3).
Figure 2. SPECT/CT imaging of the 111In/177Lu-octapa-trastuzumab and 111In/177Lu- DOTA-trastuzumab immunoconjugates.

Imaging studies in female nude athymic mice with subcutaneous SKOV-3 xenografts (identified by arrow at right shoulder, tumor volume ~ 100–150 mm3), showing transverse (top) and coronal (bottom) planar images bisecting the tumor, imaged at 24, 48, 72, 96, and 120 h post injection, see Figures S17–20 for 48 and 96 h time points.
Figure 3. Cerenkov luminescence image of 177Lu-octapa-trastuzumab.
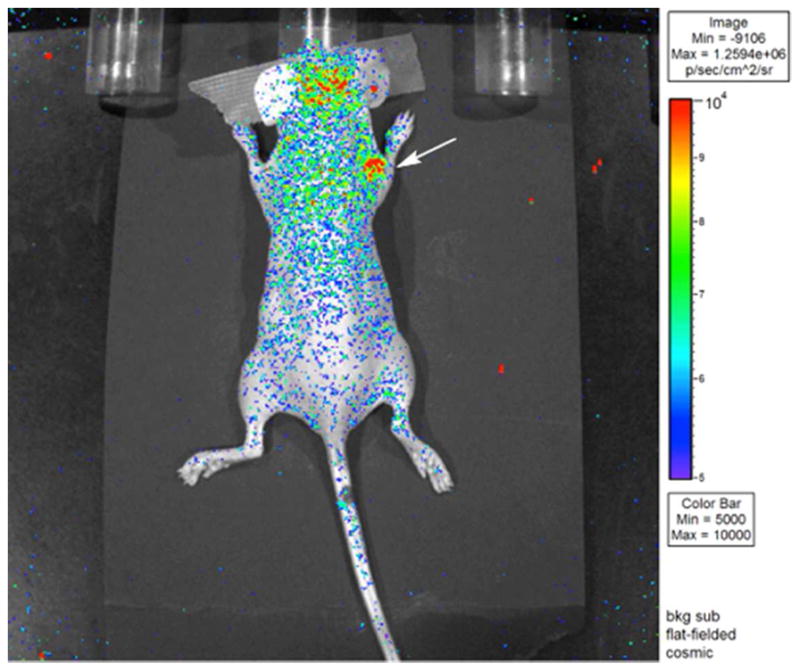
CLI obtained 24 h post-injection of a female nude athymic mouse bearing an SKOV-3 ovarian cancer xenograft on the right shoulder, indicated by white arrow (2 minute acquisition time). The bright spots on the head of the mouse are the result of fluorescent molecules in the mouse food.
Conclusions
Ultimately, the major benefit of the acyclic H4octapa chelator system over macrocyclic chelators such as DOTA lies in the ability to radiolabel with rapid kinetics at room temperature, a trait which has the potential to shorten radiolabeling times (< 15 min), streamline radiopharmaceutical production, and aid in the retention of antibody integrity and immunoreactivity during radiolabeling. Trastuzumab is a robust antibody and can generally withstand the radiolabeling conditions required by DOTA, and so although H4octapa provides significant and tangible benefits over DOTA as outlined in this manuscript, its use is not strictly required; however, when working with antibodies that are more sensitive than trastuzumab and are less forgiving of elevated temperatures and extended reaction times, a chelator with more facile reaction kinetics like p-SCN-Bn- H4octapa may become crucially important. The high and reproducible radiochemical yields that H4octapa provides (>95%) is additionally important because antibodies are the most expensive component of these radiolabeled immunoconjugate systems by a large margin. The inconsistent yields obtained with DOTA (50–90%) can waste as much as 50% of these expensive compounds and radiometals, and could make it difficult to reliably produce specific doses in a hospital radiopharmacy setting. Yet the advantages provided by H4octapa do not end with facile radiolabeling protocols. Potentiometric titrations, spectroscopic measurements, and prolonged serum stability assays indicate that this broadly versatile chelator forms highly thermodynamically stable and kinetically inert coordination complexes with isotopes of both Lu3+ and In3+. Finally, and most importantly, acute biodistribution studies have shown that 111In- and 177Lu-octapa-trastuzumab specifically and selectively accumulate in SKOV-3 ovarian cancer xenografts in vivo to a degree unmatched by analogous 111In-DOTA- and 177Lu-DOTA-trastuzumab radioimmunoconjugates. Further, SPECT/CT imaging reveals that both 111In- and 177Luoctapa- trastuzumab are capable of delineating HER2-positive tumors in vivo with excellent contrast and high tumor-to-background activity ratios, producing images that are comparable to those created using 111In- and 177Lu-DOTA-trastuzumab. Taken together, these data clearly indicate that p-SCN-Bn-H4octapa is a versatile and exceedingly promising bifunctional chelator for the construction of highly potent and effective 111In- and 177Lulabeled radiopharmaceuticals.
Experimental Section
Materials and Methods
All solvents and reagents were purchased from commercial suppliers (Sigma Aldrich, St. Louis, MO; TCI America, Portland, OR; Fisher Scientific, Waltham, MA) and were used as received unless otherwise indicated. DMSO used for chelator stock solutions was of molecular biology grade (>99.9%: Sigma, D8418). 1,4,7,10- Tetraazacyclododecane-1,4,7,10-tetraacetic acid para-benzylisothiocyanate (p-SCN-Bn- DOTA) was purchased from Macrocyclics, Inc. (Dallas, TX). Methyl-6- bromomethylpicolinate was synthesized according to a literature protocol.33 Water used was ultra-pure (18.2 MΩ cm−1 at 25 °C, Milli-Q, Millipore, Billerica, MA). The analytical thinlayer chromatography (TLC) plates were aluminum-backed ultrapure silica gel (Siliaplate™, 60 Å pore size, 250 μM plate thickness, Silicycle, Quebec, QC). Flash column silica gel was provided by Silicycle (Siliaflash® Irregular Silica Gels F60, 60 Å pore size, 40–63 mm particle size, Silicycle, Quebec, QC). Automated column chromatography was performed using a Teledyne Isco (Lincoln, NE) CombiFlash® Rf automated system with solid load cartridges packed with flash column silica gel and RediSep Rf Gold® reusable normal-phase silica columns and neutral alumina columns (Teledyne Isco, Lincoln, NE). 1H and 13C NMR spectra were recorded on Bruker AV300, AV400, or AV600 instruments; all spectra were internally referenced to residual solvent peaks except for 13C NMR spectra in D2O, which were externally referenced to a sample of CH3OH/D2O. Low-resolution mass spectrometry was performed using a Waters liquid chromatography-mass spectrometer (LC-MS) consisting of a Waters ZQ quadrupole spectrometer equipped with an ESCI electrospray/chemical ionization ion source and a Waters 2695 HPLC system (Waters, Milford, MA). Highresolution electrospray-ionization mass spectrometry (ESI-MS) was performed on a Waters Micromass LCT time of flight instrument. Microanalyses for C, H, and N were performed on a Carlo Erba EA 1108 elemental analyzer. The HPLC system used for purification of nonradioactive compounds consisted of a semi-preparative reverse phase C18 Phenomenex synergi hydro-RP (80 Å pore size, 250 × 21.2 mm, Phenomenex, Torrance, CA) column connected to a Waters 600 controller, a Waters 2487 dual wavelength absorbance detector, and a Waters delta 600 pump. 177Lu(chelate) analysis was performed using an HPLC system comprised of a Shimadzu SPD-20A prominence UV/Vis, LC-20AB prominence LC, a Bioscan flow-count radiation detector, and a C18 reverse phase column (Phenomenex Luna Analytical 250 × 4.6 mm). UV/Vis measurements for determining antibody stock solution concentrations were taken on a Thermo Scientific Nanodrop 2000 spectrophotometer (Wilmington, DE).
111In was cyclotron produced (Advanced Cyclotron Systems, Model TR30) by proton bombardment through the reactions 111Cd(p,n)111In and was provided by Nordion as 111InCl3 in 0.05 M HCl. 177Lu was procured from Perkin Elmer (Perkin Elmer Life and Analytical Sciences, Wellesley, MA, effective specific activity of 29.27 Ci/mg) as 177LuCl3 in 0.05 M HCl. Labeling reactions were monitored using silica-gel impregnated glass-microfiber instant thin layer chromatography paper (iTLC-SG, Varian, Lake Forest, CA) and analyzed on a Bioscan AR-2000 radio-TLC plate reader using Winscan Radio-TLC software (Bioscan Inc., Washington, DC). All radiolabeling chemistry was performed with ultrapure water (>18.2 MΩ cm−1 at 25 °C, Milli-Q, Millipore, Billerica, MA) that had been passed through a 10 cm column of Chelex resin (BioRad Laboratories, Hercules, CA). Human blood serum (Sigma, Sera, human, aseptically filled, S7023–100 mL) competition solutions were agitated at 550 rpm and held at 37 °C using an Eppendorf Thermomixer and then 177Lu(chelate) mixtures were analyzed using GE Healthcare Life Sciences PD-10 desalting columns (GE Healthcare, United Kingdom, MW < 5000 Da filter) that were conditioned by elution of 25 mL phosphate-buffered saline (PBS) before use. 177Lu/111In-immunoconugates were analyzed using iTLC as described above, and purified using PD-10 desalting columns and Corning 50k MW Amicon® Ultra centrifugation filters. Radioactivity in samples was measured using a Capintec CRC-15R dose calibrator (Capintec, Ramsey, NJ), and for biodistribution studies a Perkin-Elmer (Waltham, MA) Automated Wizard Gamma Counter was used for counting organ activities and creating calibration curves. SPECT/CT imaging was performed using a four-headed NanoSPECT/CT®PLUS camera (Bioscan Inc., Washington DC, USA) with a multi-pinhole focused collimator and a temperature controlled animal bed (Minerve equipment veterinaire). Imaging of Cerenkov radiation (CR) was performed using a IVIS 200 (Caliper Life Sciences) optical imager. Human breast cancer cell line SKOV-3 was obtained from the American Type Culture Collection (ATCC, Manassas, VA) and was grown by serial passage. All animals used were female nude athymic mice (Taconic Farms, Inc., Hudson, NY).
N,N′-(2-Nitrobenzenesulfonamide)-1,2-diaminoethane (1)
Ethylenediamine (548 μL, 8.2 mmol) was dissolved in THF (10 mL) and placed in an ice bath, then sodium bicarbonate (~2 g) was added, followed by slow addition of 2-nitrobenzenesulfonyl chloride (4.00 g, 18.1 mmol). The reaction mixture was allowed to warm to ambient temperature and stirred overnight. The yellow mixture was filtered to remove sodium bicarbonate, rotovapped to red oil, and then dissolved in a minimum volume of dichloromethane and placed in the freezer overnight. Product precipitated quickly and was filtered and then washed with cold dichloromethane (3 × 10 mL), and this process was repeated with the filtrate once more to recover more material. The faint yellow powder (1) was dried in vacuo for a yield of 87% (~3.07 g). 1H NMR (300 MHz, DMSO-d6, 25 °C) δ: 8.16 (br s, 2H), 8.00–7.94 (m, 4H), 7.90– 7.83 (m, 4H), 3.00 (s, 4H). 13C NMR (75 MHz, DMSO-d6, 25 °C) δ: 147.62, 134.18, 132.76, 132.41, 129.45, 124.54, 42.39. HR-ESI-MS calcd. for [C14H14N4O8S2+Na]+: 453.0151; found: 453.0154 [M+Na]+, PPM = 0.7.
N,N′-(2-Nitrobenzenesulfonamide)-N,N′-[6-(methoxycarbonyl)pyridin-2-yl]methyl]-1,2-diaminoethane (2)
To a solution of 1 (1.17 g, 2.72 mmol) in dimethylformamide (10 mL, dried over molecular sieves 4 Å) was added methyl-6-bromomethyl picolinate33 (1.38 g, 5.98 mmol) and sodium carbonate (~2 g). The faint yellow reaction mixture was stirred at 50 °C overnight, filtered to remove sodium carbonate, and concentrated in vacuo. The crude product was purified by silica chromatography (CombiFlash Rf automated column system; 40 g HP silica; A: dichloromethane, B: methanol, 100% A to 20% B gradient) to yield the product 2 as white fluffy solid (95%, ~1.88 g) (Rf: 0.9, TLC in dichloromethane). 1H NMR (300 MHz, CDCl3, 25 °C) δ: 8.10–8.08 (m, 2H), 7.93 (dd, J = 7.8, 0.9 Hz, 2H), 7.75 (t, J = 7.8 Hz, 2H), 7.66–7.62 (m, 4H), 7.60–7.57 (m, 2H), 7.51 (dd, J = 7.8, 0.9 Hz, 2H), 4.71 (s, 4H), 3.91 (s, 6H), 3.51 (s, 4H). 13C NMR (75 MHz, CDCl3, 25 °C) δ: 165.07, 156.21, 147.83, 147.41, 137.89, 133.65, 132.34, 131.93, 131.22, 125.64, 124.09, 124.04, 53.52, 52.65, 46.69. HR-ESI-MS calcd. for [C30H28N6O12S2+H]+: 729.1285; found: 729.1263, [M+H]+, PPM = −3.0.
N,N′-[6-(Methoxycarbonyl)pyridin-2-yl]methyl-1,2-diaminoethane (3)
To a solution of 2 (1.48 g, 2.03 mmol) in tetrahydrofuran (10 mL) was added thiophenol (457 μL, 4.47 mmol) and potassium carbonate (excess, ~1 g). The reaction mixture was stirred at ambient temperature for 72 hours, where a slow color change from colorless to dark yellow occurred. The reaction mixture was split into two 20 mL falcon tubes, diluted with additional tetrahydrofuran, centrifuged for 5 minutes at 4000 rpm, and then the solvent was decanted. The potassium carbonate in the falcon tubes was rinsed/centrifuged/decanted with tetrahydrofuran 3 times, pooled, and the organic fractions were concentrated to dryness in vacuo. Please note that the potassium carbonate can be very sticky (varies between supplier) and can clog gravity and vacuum filters, and so to prevent product loss the method of centrifugation is recommended if a sticky texture is observed after reaction. The resulting crude yellow oil was purified by silica chromatography (CombiFlash Rf automated column system; 24 g neutral alumina; A: dichloromethane, B: methanol, 100% A to 30% B gradient) to yield 3 as clear colorless oil (91%, ~0.66 g). Compound 3 was purified using neutral alumina, as it demonstrates an abnormally high affinity for silica and requires the use of ammonium hydroxide and >20% methanol to be eluted, resulting in partial methyl-ester deprotection and dissolving of some silica. 1H NMR (300 MHz, MeOD, 25 °C) δ: 8.02 (d, J = 7.5 Hz, 2H), 7.94 (t, J = 7.6 Hz, 2H), 7.66 (d, J = 7.5 Hz, 2H), 3.96 (s, 6H), 2.79 (s, 4H). 13C NMR (75 MHz, MeOD, 25 °C) δ: 167.04, 161.45, 148.48, 139.51, 127.61, 124.84, 54.92, 53.37, 50.00, 49.27. HR-ESI-MS calcd. for [C18H22N4O4+H]+: 359.1719; found: 359.1720, [M+H]+, PPM = 0.2.
N,N′-[(tert-Butoxycarbonyl)methyl-N,N′-[6-(methoxycarbonyl)pyridin-2-yl]methyl]-1,2-diaminoethane (4)
To a solution of 3 (212 mg, 0.593 mmol) in dry acetonitrile (10 mL, distilled over CaH2) was added tert-butylbromoacetate (202 μL, 1.36 mmol) and sodium carbonate (~300 mg). The reaction was stirred at 60 °C overnight. Sodium carbonate was removed by filtration and the crude reaction mixture was concentrated in vacuo. The crude oil was purified by column chromatography (CombiFlash Rf automated column system; 24 g HP silica; A: dichloromethane, B: methanol, 100% A to 20% B gradient) to afford the product 4 as light yellow oil (92%, ~0.32 g). 1H NMR (300 MHz, CDCl3, 25 °C) δ: 7.95 (dd, J = 7.2, 1.4 Hz, 2H), 7.80–7.70 (m, 4H), 3.97 (s, 4H), 3.95 (s, 6H), 3.28 (s, 4H), 2.78 (s, 4H), 1.40 (s, 18H). 13C NMR (75 MHz, CDCl3, 25 °C) δ: 170.36, 165.77, 160.79, 147.09, 137.29, 126.02, 123.48, 80.91, 60.51, 56.27, 52.75, 52.39, 28.05. HR-ESI-MS calcd. for [C30H42N4O8+H]+: 587.3081; found: 587.3085, [M+H]+, PPM = 0.7.
H4octapa, N,N′-(6-carboxy-2-pyridylmethyl)-N,N′-diacetic acid-1,2-diaminoethane (5)
Compound 4 (138 mg, 0.235 mmol) was dissolved in HCl (10 mL, 6 M) and refluxed for 12 hours at 140 °C. The reaction mixture was concentrated in vacuo and then purified via semiprep reverse-phase HPLC (10 mL/min, gradient: A: 0.1% TFA (trifluoroacetic acid) in deionized water, B: 0.1% TFA in CH3CN. 0 to 70% B linear gradient 30 min. tR = 12.6 min, broad). Product fractions were pooled, concentrated in vacuo, dissolved in 6M HCl, and then concentrated in vacuo again to remove trifluoroacetic acid. The HCl salt H4octapa-4HCl- 2H2O (5) was obtained as a white solid (66% yield, ~0.097 g, using the molecular weight of the HCl salt as determined by elemental analysis). 1H NMR (300 MHz, D2O) δ: 8.11–8.01 (m, 4H, pyr-H), 7.72–7.69 (m, 2H, pyr-H), 4.53 (s, 4H, Pyr-CH2-N), 3.97 (s, 4H, HOOC-CH2-N), 3.57 (s, 4H, ethylene-H). 13C NMR (75 MHz, D2O) δ: 170.33, 165.27, 151.19, 145.09, 142.64, 128.60, 126.12, 57.81, 54.97, 51.22. IR (neat, ATR-IR): = 1721 cm−1 (C=O), 1636/1618 cm−1 (C=C py). HR-ESI-MS calcd. for [C20H22N4O8 + H]+: 447.1515; found [M + H]+: 447.1526, PPM = 0.7. Elemental analysis: calcd % for H4octapa•4HCl•2H2O (C20H22N4O8•4HCl•2H2O = 628.2839): C 38.23, H 4.81, N 8.92; found: C 37.94, H 4.66, N 8.59.
[Na][Lu(octapa)] (6). H4octapa•4HCl•2H2O (5) (9.8 mg, 0.013 mmol) was suspended in 0.1 M HCl (1.5 mL) and LuCl3•6H2O (6.1 mg, 0.015 mmol) was added. The pH was adjusted to 4–5 using 0.1 M NaOH and then the solution was stirred at room temperature. After 1 hour the product was confirmed via mass spectrometry and the solvent was removed in vacuo to yield [Lu(octapa)][Na] (6). 1H NMR (600 MHz, D2O, 25 °C) δ: 8.28–8.12 (m, 3H), 8.06 (d, J = 8.2 Hz, 2H), 7.81–7.67 (m, 2H), 4.56–4.48 (m, 1.5H), 4.33–4.05 (m, 2H), 3.81–3.74 (m, 1H), 3.51–3.43 (m, 2H) 3.26–3.16 (m, 4H), 3.00–2.98 (m, 0.5H), 2.57–2.55 (m, 0.5H), 2.12 (br s, 0.5H). 13C NMR (150 MHz, D2O, 25 °C) δ: 180.75, 177.07, 173.29, 172.78, 171.72, 158.17, 157.23, 155.65, 154.34, 150.65, 149.89, 149.55, 143.81, 142.96, 142.54, 141.42, 126.78, 126.43, 124.64, 124.50, 123.97, 65.71, 64.25, 62.27, 61.01, 60.51, 59.11, 57.63, 55.96. HR-ESI-MS calcd. for [C20H18175LuN4O8 + 2Na]+: 663.0328; found: 663.0318, [M + 2Na]+, PPM = −1.5.
1-(p-Nitrobenzyl)ethylenediamine (7)
Compound 7 was prepared according to a literature preparation.52 Product was purified with a modified procedure using column chromatography (CombiFlash Rf automated column system; 40 g HP silica;, A: 95% dichloromethane 5% ammonium hydroxide, B: 95% methanol 5% ammonium hydroxide, 100% A to 30% B gradient) to afford 7 as brown/amber oil in a cumulative yield of 40% over 3 steps (~3 g). 1H NMR (300 MHz, CDCl3, 25 °C) δ: 7.91 (d, J = 8.9 Hz, 2H), 7.18 (d, J = 8.5 Hz, 2H), 2.80–2.57 (m, 3H), 2.45–2.31 (m, 2H). 13C NMR (75 MHz, CDCl3, 25 °C) δ: 147.13, 147.71, 129.39, 122.83, 54.26, 47.52, 41.36. HR-ESI-MS calcd. for [C9H13N3O2+H]+: 196.1086; found: 196.1084 [M + H], PPM = −1.0.
N,N′-(2-Nitrobenzenesulfonamide)-1-(p-nitrobenzyl)-1,2-diaminoethane (8)
Compound 7 (578 mg, 2.96 mmol) was dissolved in THF (10 mL) and placed in an ice bath, then sodium bicarbonate (~2 g) was added, followed by slow addition of 2- nitrobenzenesulfonyl chloride (1.58 g, 7.1 mmol). The reaction mixture was heated to 50 °C and stirred overnight. The yellow/orange mixture was filtered to remove sodium bicarbonate, rotovapped to orange oil, and then dissolved in a minimum volume of dichloromethane and placed in the freezer. Product precipitated quickly and was filtered, then washed with cold dichloromethane (3 × 10 mL), and this process was repeated with the filtrate twice more to recover more product. The faint yellow powder (8) was dried in vacuo for a yield of 74% (~1.24 g) (Rf: 0.90, TLC in 10% methanol in dichloromethane). 1H NMR (300 MHz, acetone-d6, 25 °C) δ: 8.18–8.15 (m, 1H), 7.99–7.94 (m, 2H), 7.79–7.57 (m, 5H), 7.33 (d, J = 8.5 Hz, 2H), 7.04–6.98 (m, 2H), 4.01 (br s, 1H), 3.41–3.38 (m, 2H), 3.26 (dd, J = 3.4, 13.7 Hz, 1H), 2.98 (m, 1H). 13C NMR (75 MHz, acetone-d6, 25 °C) δ: 206.33, 149.17, 148.14, 147.58, 146.74, 135.23, 134.79, 134.26, 133.89, 133.77, 133.57, 131.67, 131.38, 130.94, 126.00, 125.67, 123.90, 57.67, 49.35, 38.20. HR-ESI-MS calcd. for [C21H19N5O10S2+Na]+: 588.0471; found: 588.0465 [M+Na]+, PPM = −1.0.
N,N′-(2-Nitrobenzenesulfonamide)-N,N′-[(tert-butoxycarbonyl)methyl]-1-(pnitrobenzyl)- 1,2-diaminoethane (9)
To a solution of 8 (224 mg, 0.390 mmol) in dimethylformamide (5 mL, dried over molecular sieves 4 Å) was added tertbutylbromoacetate (169.7 mg, 0.87 mmol) and sodium carbonate (~400 mg). The yellow reaction mixture was stirred at 50 °C overnight, filtered to remove sodium carbonate, and concentrated in vacuo. The crude product was purified by silica chromatography (CombiFlash Rf automated column system; 40 g HP silica; A: ethyl acetate, B: petroleum ether, 100% A to 100% B gradient) to yield 9 as white fluffy solid (88%, ~0.27 g). 1H NMR (300 MHz, CDCl3, 25 °C) δ: 7.98 (d, J = Hz, 2H), 7.84–7.39 (m, 8H), 7.25 (d, J = Hz, 2H), 4.46 (Br s, 1H), 4.33–3.85 (m, 6 H), 3.53 (m, 1H), 3.39 (m, 1H), 2.91 (m, 1H), 1.40 (s, 18H). 13C NMR (75 MHz, CDCl3, 25 °C) δ: 168.35, 167.64, 148.00, 147.09, 146.31, 144.83, 133.91, 133.61, 132.53, 131.81, 131.72, 131.60, 131.51, 130.65, 129.85, 124.01, 123.01, 82.71, 82.53, 58.48, 52.47, 50.70, 45.37, 35.04, 27.69. HR-ESI-MS calcd. for [C33H39N5O14S2+Na]+: 816.1833; found: 816.1838, [M+Na]+, PPM = 0.6.
N,N′-[(tert-Butoxycarbonyl)methyl]-1-(p-nitrobenzyl)-1,2-diaminoethane (10)
To a solution of 9 (151 mg, 0.189 mmol) in tetrahydrofuran (5 mL) was added thiophenol (41 μL, 0.398 mmol) and potassium carbonate. The reaction mixture was stirred at ambient temperature for 72 hours, where a slow color change from colorless to dark yellow occurred. The reaction mixture was split into two 20 mL falcon tubes, diluted with additional tetrahydrofuran, centrifuged for 5 minutes at 4000 rpm, and then the solvent was decanted. The potassium carbonate in the falcon tubes was rinsed with tetrahydrofuran and centrifuged 3 times, pooled, and then concentrated to dryness in vacuo. The resulting crude yellow oil was purified by silica chromatography (CombiFlash Rf automated column system; 24 g HP silica; A: dichloromethane, B: methanol, 100% A to 20% B gradient) to yield 10 as clear colorless oil (89%, ~0.071 g). 1H NMR (300 MHz, CDCl3, 25 °C) δ: 8.12 (d, J = 8.2 Hz, 2H), 7.35 (d, J = 8.2 Hz, 2H), 3.30 (s, 2H), 3.23–3.22 (m, 2H), 2.87–2.84 (m, 3H), 2.62 (m, 1H), 2.40 (m, 1H), 2.12 (Br s, 2H, -NH), 1.41 (s, 18H). 13C NMR (75 MHz, CDCl3, 25 °C) δ: 171.68, 171.53, 147.07, 146.51, 130.07, 123.46, 81.17, 81.04, 58.28, 51.57, 49.34, 39.16, 28.01. HR-ESI-MS calcd. for [C21H33N3O6+H]+: 424.2448; found: 424.2451, [M+H]+, PPM = 0.7.
N,N′-[(tert-Butoxycarbonyl)methyl]-N,N′-[(6-methoxycarbonyl)pyridin-2-yl)methyl]-1-(p-nitrobenzyl)-1,2-diaminoethane (11)
To a solution of 10 (71.7 mg, 0.169 mmol) in dry acetonitrile (5 mL, distilled over CaH2) was added methyl 6-(bromomethyl)picolinate33 (85.7 mg, 0.372 mmol) and sodium carbonate (~200 mg). The reaction was stirred at 60 °C overnight. Sodium carbonate was removed by filtration and the crude reaction mixture was concentrated in vacuo. The crude oil was purified by column chromatography (CombiFlash Rf automated column system; 24 g HP silica; A: dichloromethane, B: methanol, 100% A to 20% B gradient) to afford the product 11 as light yellow oil (96%, ~0.117 g). 1H NMR (300 MHz, MeOD, 25 °C) δ: 8.18 (d, J = 8.7 Hz, 2H), 8.11–8.02 (m, 3H), 7.95 (t, J = 7.8 Hz, 1H), 7.73- 7.72 (m, 1H), 7.48–7.45 (m, 3H), 4.05 (s, 3H, methyl ester), 3.98 (s, 3H, methyl ester), 3.76 (m, 2H), 3.51 (d, J = 17.4 Hz, 1H), 3.38–3.31 (m, 2H), 3.25–3.20 (m, 2H), 3.16–3.01 (m, 2H), 2.89–2.82 (m, 2H), 2.71–2.64 (m, 1H), 2.55–2.51 (m, 1H), 1.39 (s, 18H). 13C NMR (75 MHz, MeOD, 25 °C) δ: 174.44, 173.45, 167.07, 166.81, 160.17, 160.06, 149.25, 148.67, 148.51, 148.09, 140.34, 140.13, 131.60, 129.00, 128.57, 125.14, 125.04, 124.84, 83.39, 83.26, 63.82, 61.82, 58.41, 57.05, 53.78, 53.61, 34.10, 28.38. HR-ESI-MS calcd. for [C36H48N5O10+H]+: 722.3401; found: 722.3390, [M+H]+, PPM = −1.5.
p-SCN-Bn-H4octapa, N,N′-[(Carboxylato)methyl]-N,N′-[(6-carboxylato)pyridin-2- yl)methyl]-1-(p-benzyl-isothiocyanato)-1,2-diaminoethane (12)
Compound 11 (118 mg, 0.16 mmol) was dissolved in glacial acetic acid (2.5 mL) with hydrochloric acid (2.5 mL, 3 M), palladium on carbon (20 wt%), and hydrogen gas (balloon). The reaction was stirred vigorously at RT for 1 hour, then filtered to remove Pd/C and washed ad libitum with methanol and hydrochloric acid (3 M). The crude reaction mixture was concentrated in vacuo, dissolved in 15 mL of hydrochloric acid (6 M), and refluxed overnight to facilitate deprotection of the methyl and tert-butyl esters. HR-ESI-MS calcd. for [C27H29N5O8+H]+: 552.2094; found: 552.2098, [M+H]+, PPM = 0.7. Without purification, the crude reaction mixture was dissolved in hydrochloric acid (1 mL, 3 M) and then reacted with thiophosgene (purchased suspended in chloroform) in ~0.2 mL of additional chloroform (15 eq, 2.45 mmol) overnight at ambient temperature with vigorous stirring. The reaction mixture was washed with chloroform (5 × 1 mL) by vigorous biphasic stirring followed by decanting of the organic phase with a pipette to remove excess thiophosgene, diluted to a volume of 4.5 mL with deionized water, and injected directly onto a semi-prep HPLC column for purification (A: 0.1% TFA in deionized water, B: 0.1% TFA in CH3CN, 100% A to 60% B gradient over 40 minutes). p-SCN-Bn-H4octapa (12) was found in the largest peak at Rt = 34 minutes (broad, Figure S22), lyophilized overnight, and was isolated as a fluffy white solid (50% over 3 steps from 11, ~0.047 g). 1H NMR (400 MHz, MeOD, 25 °C) δ: 8.00–7.87 (m, 4H), 7.59–7.54 (m, 2H), 7.14 (dd, J = 7.7 Hz, 35.4 Hz, 4H), 5.03 (Br m, 1H), 4.66 (Br m, 2H), 4.49–4.45 (Br m, 1H), 4.11 (Br m, 1H), 3.92 (Br m, 2H), 3.66–3.59 (m, 2H), 3.35–3.18 (Br m, 3H), 2.66–2.60 (m, 1H). 13C NMR (100 MHz, MeOD, 25 °C) δ: 173.10, 167.95, 166.02, 165.82, 161.34, 160.97, 159.40, 151.40, 147.74, 147.21, 138.63, 138.54, 137.60, 135.72, 130.30, 129.75, 127.51, 126.61, 125.65, 124.72, 123.94, 60.31, 59.74, 54.20, 54.14, 33.35. HR-ESI-MS calcd. for [C28H27N5O8S+H]+: 594.1659; found: 594.1661, [M+H]+, PPM = 0.3.
Solution Thermodynamics
The experimental procedures and details of the apparatus closely followed those of a previous study for H2dedpa with Ga3+ and H4octapa for In3+.30,33 As a result of the strength of the binding of the Lu3+ complexes, [177Lu(octapa)]−and [Lu(DOTA)], the complex formation constants with these ligands could not be determined directly and the ligand-ligand competition method using the known competitor Na2H2EDTA was used. Potentiometric titrations were performed using a Metrohm Titrando 809 equipped with a Ross combination pH electrode and a Metrohm Dosino 800. Data were collected in triplicate using PC Control (Version 6.0.91, Metrohm). The titration apparatus consisted of a water-jacketed glass vessel maintained at 25.0 (± 0.1 °C, Julabo water bath). Prior to and during the course of the titration, a blanket of nitrogen, passed through 10% NaOH to exclude any CO2, was maintained over the sample solution. Lutetium ion solutions were prepared by dilution of the appropriate atomic absorption standard (AAS) solution. The exact amount of acid present in the lutetium standard was determined by titration of an equimolar solution of Lu3+ and Na2H2EDTA. The amount of acid present was determined by Gran’s method.53 Calibration of the electrode was performed prior to each measurement by titrating a known amount of HCl with 0.1 M NaOH. Calibration data were analyzed by standard computer treatment provided within the program MacCalib54 to obtain the calibration parameters E0. Equilibration times for titrations were 10 minutes pKa titrations and 15 minutes for metal complex titrations. Ligand and metal concentrations were in the range of 0.75–1.0 mM for potentiometric titrations. The data were treated by the program Hyperquad2008.55 The proton dissociation constant corresponding to hydrolysis of Lu3+(aq) ion included in the calculations were taken from Baes and Mesmer.56 The acid dissociation constants for DOTA and the KML value for the lutetium-edta complex were taken from Martell (REF). pM values were calculated at physiologically relevant conditions of pH 7.4, 100μM ligand, and 10 μM metal. All values and errors represent the average of at least three independent experiments.
Molecular Modelling
Calculations were performed using the Gaussian 0957 and GaussView packages. Molecular geometries and electron densities were obtained from density functional theory calculations, with the B3LYP functional employing the 6–31+G(d,p) basis set for 1st and 2nd row elements and the Stuttgart–Dresden effective core potential, SDD for lutetium.58–60 Solvent (water) effects were described through a continuum approach by means of the IEF PCM as implemented in G09. The electrostatic potential was mapped onto the calculated electron density surface
[177Lu(chelate)] Radiolabeling
For 177Lu experiments, the chelators H4octapa, DTPA, and DOTA were used. Aliquots of each chelator stock solution (1 mg/mL) were transferred to Corning® 2.0 mL self-standing micro-centrifuge tubes containing ~1.2 mCi of 177Lu to a ligand concentration of ~180 μM, and made up to 1 mL with NaOAc buffer (10 mM, pH 5.0). H4octapa and DTPA were allowed to radiolabel at ambient temperature for 10 minutes and DOTA was radiolabeled for 1 hour at 90 °C. Radiometal complexes were then evaluated using radio-HPLC (linear gradient A: 0.1% TFA in H2O, B: CH3CN, 0 – 80% B over 30 minutes): [177Lu(octapa)]− tR = 5 min, >99%; [177Lu(DOTA)]− tR = 6.8 min, >99%, [177Lu(DTPA)]2− tR = 5.2 min, >99%. Radiolabeled chelators were then used for blood serum stability assays.
Trastuzumab Antibody Modification/Thiourea Bioconjugation
Trastuzumab (purchased commercially as Herceptin, Genentech, San Francisco, CA) was purified using centrifugal filter units with a 50000 molecular weight cutoff (Amicon® ultra centrifuge filters, Ultracel®-50: regenerated cellulose, Millipore Corp., Billerica, MA) and phosphate buffered saline (PBS, pH 7.4) to remove α-;-trehalose dihydrate, L-histidine, and polysorbate 20 additives. After purification, the antibody was taken up in PBS pH 7.4. Subsequently, 300 μL of antibody solution (150–250 μM) were combined with 100 μL PBS (pH 8.0), the pH of the resulting solution was adjusted to 8.8–9.0 with 0.1 M Na2CO3, and 4 equiv of the p-SCN-Bn-H4octapa or p-SCN-Bn-DOTA were added in 10 μL DMSO. The reactions were incubated at 37 °C for 1 h, followed by centrifugal filtration to purify the resultant antibody conjugate. The final modified antibody stock solutions were stored in PBS (pH 7.4) at 4 °C.
111In- and 177Lu-octapa/DOTA-trastuzumab Radiolabeling
Aliquots of H4octapa/DOTAtrastuzumab immunoconjugates were transferred to 2 mL microcentrifuge tubes and made up to 1 mL of ammonium acetate buffer (pH 5.5, 200 mM), and then aliquots of 177Lu or 111In were added (~2–3 mCi). The H4octapa-trastuzumab mixtures were allowed to radiolabel at room temperature for 15 minutes and then analyzed via iTLC with an eluent of 50 mM EDTA (pH 5) and confirmed reproducible values >95% RCY. The DOTA-trastuzumab mixtures were heated to 37 ± 0.1 °C for 1 hour and then analyzed via iTLC with RCYs ranging from ~50–90%. 30 μL of EDTA solution (50 mM, pH 5) was then added to the reaction mixture, the resultant radiolabeled immunoconjugates were then purified using size-exclusion chromatography (Sephadex G-25 M, PD-10 column, 30 kDa, GE Healthcare; dead volume = 2.5 mL, eluted with 1 mL fractions of PBS, pH 7.4) and centrifugal column filtration (Amicon® ultra 50k). The radiochemical purity of the final radiolabeled bioconjugate was assayed by radio-iTLC and was found to be >99% in all preparations. In the iTLC experiments, 111In- and 177Lu-octapa/DOTA-trastuzumab remained at the baseline, while 177Lu3+/111In3+ ions complexed as [111In/177Lu]-EDTA eluted with or near the solvent front.
Chelate Number Isotopic Dilution Assay
The number of accessible H4octapa and DOTA chelates conjugated to trastuzumab was measured by radiometric isotopic dilution assays following methods similar to those described by Anderson et al. and Holland et al.9,61–63 All experiments were performed in triplicate.
Immunoreactivity – Lindmo Cellular Assay
The immunoreactivity of the111In/177Luoctapa- trastuzumab and 111In/177Lu-DOTA-trastuzumab bioconjugates was determined using specific radioactive cellular-binding assays following procedures derived from Lindmo et al.64,65 To this end, SKOV-3 cells were suspended in microcentrifuge tubes at concentrations of 5.0, 4.0, 3.0, 2.5, 2.0, 1.5, and 1.0 × 106 cells/mL in 500 μL PBS (pH 7.4). Aliquots of either 111In/177Lu-octapa-trastuzumab or 111In/177Lu-DOTA-trastuzumab (50 μL of a stock solution of 10 μCi in 10 mL of 1% bovine serum albumin in PBS pH 7.4) were added to each tube (n = 6; final volume: 550 μL), and the samples were incubated on a mixer for 60 min at room temperature. The treated cells were then pelleted via centrifugation (3000 rpm for 5 min), resuspended, and washed twice with cold PBS before removing the supernatant and counting the activity associated with the cell pellet. The activity data were background-corrected and compared with the total number of counts in appropriate control samples. Immunoreactive fractions were determined by linear regression analysis of a plot of (total/bound) activity against (1/[normalized cell concentration]). No weighting was applied to the data, and data were obtained as n = 6.
[177Lu(chelate)] Blood Serum Competition Experiments
Frozen human blood serum was thawed for 30 minutes, and 750 μL aliquots were transferred to 2.0 mL Corning centrifuge vials. 300 μL of each 177Lu(chelator) was transferred to 750 μL of blood serum, along with 450 μL of PBS to a total volume of 1.5 mL (n = 3 for each chelator). The final 177Lu(chelate) concentration present in serum was ~36 μM. Serum competition samples were then incubated at 37 ± 0.1 °C with constant agitation (550 rpm) and analyzed via PD- 10 size-exclusion column elution (filters MW < 5000 Da) at 1.5 hr and 24 hr time points and counted using a Capintec CRC-15R dose calibrator. 500 μL of each serum/177Lu(chelator) competition solution (n = 3) was removed from the competition vial, diluted to 2.5 mL with PBS, and counted. The diluted aliquot of serum competition mixture was loaded onto a conditioned PD-10 column. The loading volume (2.5 mL) was eluted into radioactive waste, and then an additional 3.5 mL of PBS was loaded, collected, and counted in the dose calibrator as the serum-bound 177Lu (non-chelate bound). Percent stability was reported as a percentage of 177Lu still chelate-bound and not associated with serum proteins (MW < 5000 Da).
111In- and 177Lu-octapa/DOTA-trastuzumab Blood Serum Competition Experiments
Frozen human blood serum was thawed for 30 minutes, and 300 μL aliquots were transferred to 2.0 mL Corning centrifuge vials. A portion of radiolabeled immunoconjugate (~300 μCi) was transferred to the blood serum (n = 3 for each chelator). Serum competition samples were then incubated at 37 ± 0.1 °C with gentle agitation (300 rpm) and analyzed via iTLC with an EDTA eluent (50 mM, pH 5.0) (Bioscan AR-2000) at time points of 0, 24, 48, 72, 96, and 120 hours.
Cell Culture
Human ovarian cancer cell line SKOV-3 was obtained from the American Tissue Culture Collection (ATCC, Bethesda, MD) and maintained in a 1:1 mixture of Dulbecco’s Modified Eagle medium: F-12 medium, supplemented with 10% heatinactivated fetal calf serum (Omega Scientific, Tarzana, Ca), 2.0 mM glutamine, nonessential amino acids, 100 units/mL penicillin, and 100 units/mL streptomycin in a 37 °C environment containing 5% CO2. Cell lines were harvested and passaged weekly using a formulation of 0.25% trypsin/0.53 mM EDTA in Hank’s Buffered Salt Solution without calcium and magnesium.
SKOV-3 Xenograft Mouse Models
All experiments were performed under an Institutional Animal Care and Use Committee-approved protocol, and the experiments followed institutional guidelines for the proper and humane use of animals in research. Six- to eightweek- old athymic nu/nu female mice (NCRNU-M) were obtained from Taconic Farms Incorporated (Hudson, NY). Animals were housed in ventilated cages, were given food and water ad libitum, and were allowed to acclimatize for approximately 1 week prior to treatment. After several days, SKOV-3 tumors were induced on the right shoulder by a subcutaneous injection of 1.0 × 106 cells in a 100 μL cell suspension of a 1:1 mixture of fresh media/BD Matrigel (BD Biosciences, Bedford, Ma).
111In- and 177Lu-octapa/DOTA-trastuzumab Biodistribution Studies
Biodistribution studies were used to evaluate the 5-day stability and tumor uptake of the 111In- and 177Luoctapa- trastuzumab preparations and were compared to the same system using the “gold standard” 111In- and 177Lu-DOTA-Trastuzumab. DOTA- and H4octapa-trastuzumab constructs were radiolabeled with 111In- and 177Lu under the same conditions as described above and prepared for biodistribution and SPECT/CT imaging studies in nude athymic mice (female, 6–8 weeks old) bearing subcutaneous SKOV-3 ovarian cancer xenografts (HER2-positive, small with tumor volume ~ 100–150 mm3). The radiolabeled immunoconjugates were purified via PD-10 size exclusion columns, followed by purification and concentrated via centrifuge filtration (Amicon® Ultra centrifuge filters, Ultracel®-50: regenerated cellulose, Millipore Corp., Billerica, MA). The purified immunoconjugates were suspended in sterile saline (0.9% NaCl) to a concentration and dose of ~30 μCi (1.1 MBq) in 200 μL per mouse. The specific activity of 111In-octapa-trastuzumab was ~3.7 μCi/μg, and for 177Lu-octapa-trastuzumab it was 3.3 μCi/μg. The specific activity of 111In-DOTAtrastuzumab was ~ 2.0 μCi/μg, and for 177Lu-DOTA-trastuzumab it was 3.4 μCi/μg. The amount of immunoconjugate injected per mouse was ~ 10–15 μg. Each mouse was intravenously injected through the tail vein and euthanized by CO2 (g) asphyxiation at time points of 24, 48, 72, 96, and 120 hours (n = 4 per time point). Organs collected after sacrifice included blood, tumor, heart, lungs, liver, spleen, kidneys, large intestine, small intestine, muscle, bone (femur), and skin (ear). All organs were rinsed in water after removal and airdried for 5 minutes. Tissues were weighed and counted (calibrated for 111In or 177Lu), the counts were background- and decay-corrected from the time of injection and then converted to the percentage of injected dose (%ID) per gram of organ tissue (%ID/g). The radioactivity counts measured in each organ were converted to activity (μCi) using a calibration curve created from known standards of 111In or 177Lu.
111In- and 177Lu-octapa/DOTA-trastuzumab SPECT/CT Imaging Studies
Mice with SKOV-3 ovarian cancer xenografts were imaged with 111In (n = 2) and 177Lu (n = 2) labeled immunoconjugates, using a four-headed NanoSPECT/CT®PLUS camera (Bioscan Inc., Washington DC, USA) with a multi-pinhole focused collimator and a temperature control animal bed unit (Minerve equipment veterinaire). Nine pinhole apertures with a diameter of 2.5 mm were used on each head, with a field of view (FOV) of 24 mm. Settings of the 111In energy peaks were 245 and 171 keV, and the 177Lu peaks were 208, 112, and 56 keV. A CT at 45 kVp was acquired (180 projections, pitch of 1). Based on the helical CT topogram, SPECT images were obtained over a range of 85 mm. For the octapa-based radioimmunoconjugates, the mice were administered with either ~550 μCi of 111In-octapatrasuzumab (specific activity ~3.7 μCi/μg) or ~450 μCi of 177Lu-octapa-trastuzumab (3.3 μCi/μg) in 200 μL of sterile saline (0.9% NaCl) via intravenous tail vein injection. For the DOTA-based radioimmunoconjugates, the mice were administered with either ~400 μCi of 111In-DOTA-trasuzumab (specific activity ~2.0 μCi/μg) or ~550 μCi of 177Lu-octapatrastuzumab (3.4 μCi/μg) in 200 μL of sterile saline (0.9% NaCl) via intravenous tail vein injection. The amount of immunoconjugate injected per mouse was ~ 150–200 μg. Each Approximately 5 minutes prior to SPECT/CT image acquisition, mice were anesthetized via inhalation of 2% isoflurane/oxygen gas mixture (Baxter Healthcare, Deerfield, IL) and placed on the scanner bed. Anesthesia was maintained during imaging using a reduced 1.5% isoflurane/oxygen mixture. Animals were imaged at 24, 48, 72, 96, and 120 h p.i. Mice injected with 177Lu-labeled immunoconjugates were imaged for 1.5–2 hours each, and mice injected with 111In-labeled immunoconjugates were imaged for 40 min (24 and 48 h time points), 50 min (72 h), and 60 min (96 and 120 h). Image collection was performed using Nucline software (V1.02 build 009), and images were processed and reconstructed using InVivoScope (2.00 patch3, 64 bit) and HiSPECT (v 1.4.3049) software. To derive the isotopespecific calibration factors, a mouse-sized phantom filled with 1.5–2 mCi of 111InCl3 or 177LuCl3 was scanned.
Cerenkov Luminescence Imaging (CLI)
Optical images of Cerenkov radiation (CR) were obtained using an IVIS 200 (Caliper Life Sciences) optical imaging machine. This system uses a cryo-cooled charge-coupled device for high-sensitivity detection of low-intensity sources. Two-minute exposures were obtained of isoflurane anesthetized mice (n = 2) 24 hours post injection of ~450 μCi of 177Lu-octapa-trastuzumab (3.3 μCi/μg) in 200 μL of sterile saline (0.9% NaCl) via intravenous tail vein injection.
Statistics
All data used was obtained in at least triplicate (n = 3), and all values were expressed as mean ± S.D. A student’s two-tailed t-test was used to compare all biodistribution data sets. A p-value ≤ 0.05 was considered statistically significant. A oneway ANOVA analysis was also performed on select data points, a p-value ≤ 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work was presented at the 2013 Society of Nuclear Medicine and Molecular Imaging (SNMMI) annual meeting, and the abstract was awarded the Berson-Yalow award. The authors thank Blesida Punzalan for skillful tail-vein injections, Dr. Pat Zanzonico and Valerie A. Longo for aid with SPECT/CT calibration, Dr. Daniel L. J. Thorek for help with Cerenkov optical imaging, and Michael Doran for cell culture work. We thank Dr. Cara L. Ferreira and Dr. Dennis W. Wester (Nordion) for radiochemistry mentoring and collaboration throughout the project, and Gwendolyn A. Bailey for early synthesis help. We acknowledge Nordion (Canada) and the Natural Sciences and Engineering Research Council (NSERC) of Canada for grant support (CR&D), NSERC for CGS-M/CGS-D fellowships (E.W.P., C.F.R), and the University of British Columbia for 4YF fellowships (E.W.P., C.F.R). C.O. acknowledges the Canada Council for the Arts for a Killam Research Fellowship (2011–2013) and the University of Canterbury for a Visiting Erskine Fellowship (2013). Services provided by the MSKCC Small-Animal Imaging Core Facility were supported in part by NIH grants R24 CA83084 and P30 CA08748. The authors also thank the NIH (Award 1F32CA1440138–01, BMZ) and the DOE (Award DE-SC0002184, JSL) for their generous funding.
Footnotes
Notes
The authors declare no competing financial interests.
Supporting information. Cerenkov imaging of 177Lu-octapa-trastuzumab, additional SPECT/CT images, selected 1H/13C NMR spectra, potentiometric titration data, and select FT-ATR-IR spectra of final synthesized compounds are available in the supporting information. This information is available free of charge via the internet at http://pubs.acs.org.
Contributor Information
Jason S. Lewis, Email: lewisj2@mskcc.org.
Michael J. Adam, Email: adam@triumf.ca.
Chris Orvig, Email: orvig@chem.ubc.ca.
References
- 1.Rösch F, Baum RP. Dalton Trans. 2011;40:6104–6111. doi: 10.1039/c0dt01504k. [DOI] [PubMed] [Google Scholar]
- 2.Zeglis BM, Lewis JS. Dalton Trans. 2011;40:6168–6195. doi: 10.1039/c0dt01595d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wadas TJ, Wong EH, Weisman GR, Anderson CJ. Chem Rev. 2010;110:2858–2902. doi: 10.1021/cr900325h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deri MA, Zeglis BM, Francesconi LC, Lewis JS. Nucl Med Biol. 2013;40:3–14. doi: 10.1016/j.nucmedbio.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeglis BM, Mohindra P, Weissmann GI, Divilov V, Hilderbrand SA, Weissleder R, Lewis JS. Bioconjugate Chem. 2011;22:2048–2059. doi: 10.1021/bc200288d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartholoma MD, Louie AS, Valliant JF, Zubieta J. Chem Rev. 2010;110:2903–2920. doi: 10.1021/cr1000755. [DOI] [PubMed] [Google Scholar]
- 7.Ramogida CF, Orvig C. Chem Commun. 2013;49:4720–4739. doi: 10.1039/c3cc41554f. [DOI] [PubMed] [Google Scholar]
- 8.Holland JP, Williamson MJ, Lewis JS. Mol Imaging. 2010;9:1–20. [PMC free article] [PubMed] [Google Scholar]
- 9.Holland JP, Divilov V, Bander NH, Smith-Jones PM, Larson SM, Lewis JS. J Nucl Med. 2010;51:1293–1300. doi: 10.2967/jnumed.110.076174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boswell CA, Brechbiel MW. Nucl Med Biol. 2007;34:757–778. doi: 10.1016/j.nucmedbio.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vermeer AWP, Norde W. Biophys J. 2000;78:394–404. doi: 10.1016/S0006-3495(00)76602-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stimmel JB, Kull FC., Jr Nucl Med Biol. 1998;25:117–125. doi: 10.1016/s0969-8051(97)00151-0. [DOI] [PubMed] [Google Scholar]
- 13.Stimmel JB, Stockstill ME, Kull FC. Bioconjugate Chem. 1995;6:219–225. doi: 10.1021/bc00032a010. [DOI] [PubMed] [Google Scholar]
- 14.Camera L, Kinuya S, Garmestani K, Wu C, Brechbiel MW, Pai LH, McMurry TJ, Gansow OA, Pastan I, Paik CH, Carrasquillo JA. J Nucl Med. 1994;35:882–889. [PubMed] [Google Scholar]
- 15.Harrison A, Walker CA, Parker D, Jankowski KJ, Cox JPL, Craig AS, Sansom JM, Beeley NRA, Boyce RA, Chaplin L, Eaton MAW, Farnsworth APH, Millar K, Millican AT, Randall AM, Rhind SK, Secher DS, Turner A. Int J Rad Appl Instrum [B] 1991;18:469–476. doi: 10.1016/0883-2897(91)90107-v. [DOI] [PubMed] [Google Scholar]
- 16.Cooper MS, Ma MT, Sunassee K, Shaw KP, Williams JD, Paul RL, Donnelly PS, Blower PJ. Bioconjugate Chem. 2012;23:1029–1039. doi: 10.1021/bc300037w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martell AE, Smith RM. Critical Stability Constants. 1–6. Plenum Press; New York: 1974–1989. [Google Scholar]
- 18.Hancock RD. J Chem Educ. 1992;69:615–621. [Google Scholar]
- 19.Liu S, Pietryka J, Ellars CE, Edwards DS. Bioconjugate Chem. 2002;13:902–913. doi: 10.1021/bc010134h. [DOI] [PubMed] [Google Scholar]
- 20.Liu S, Edwards DS. Bioconjugate Chem. 2000;12:7–34. doi: 10.1021/bc000070v. [DOI] [PubMed] [Google Scholar]
- 21.Kukis DL, DeNardo SJ, DeNardo GL, O’Donnell RT, Meares CF. J Nucl Med. 1998;39:2105–2110. [PubMed] [Google Scholar]
- 22.Jang YH, Blanco M, Dasgupta S, Keire DA, Shively JE, Goddard WA. J Am Chem Soc. 1999;121:6142–6151. [Google Scholar]
- 23.Milenic DE, Garmestani K, Chappell LL, Dadachova E, Yordanov A, Ma D, Schlom J, Brechbiel MW. Nucl Med Biol. 2002;29:431–442. doi: 10.1016/s0969-8051(02)00294-9. [DOI] [PubMed] [Google Scholar]
- 24.Kang CS, Sun X, Jia F, Song HA, Chen Y, Lewis M, Chong HS. Bioconjugate Chem. 2012;23:1775–1782. doi: 10.1021/bc200696b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chappell LL, Ma D, Milenic DE, Garmestani K, Venditto V, Beitzel MP, Brechbiel MW. Nucl Med Biol. 2003;30:581–595. doi: 10.1016/s0969-8051(03)00033-7. [DOI] [PubMed] [Google Scholar]
- 26.Rasaneh S, Rajabi H, Babaei MH, Johari Daha F. J Label Compd Radiopharm. 2010;53:575–579. [Google Scholar]
- 27.Förster GJ, Engelbach MJ, Brockmann JJ, Reber HJ, Buchholz HG, Mäcke HR, Rösch FR, Herzog HR, Bartenstein PR. Eur J Nucl Med. 2001;28:1743–1750. doi: 10.1007/s002590100628. [DOI] [PubMed] [Google Scholar]
- 28.Wu C, Kobayashi H, Sun B, Yoo TM, Paik CH, Gansow OA, Carrasquillo JA, Pastan I, Brechbiel MW. Bioorg Med Chem. 1997;5:1925–1934. doi: 10.1016/s0968-0896(97)00130-2. [DOI] [PubMed] [Google Scholar]
- 29.Brechbiel MW, Gansow OA, Atcher RW, Schlom J, Esteban J, Simpson D, Colcher D. Inorg Chem. 1986;25:2772–2781. [Google Scholar]
- 30.Boros E, Ferreira CL, Cawthray JF, Price EW, Patrick BO, Wester DW, Adam MJ, Orvig C. J Am Chem Soc. 2010;132:15726–15733. doi: 10.1021/ja106399h. [DOI] [PubMed] [Google Scholar]
- 31.Boros E, Ferreira CL, Yapp DTT, Gill RK, Price EW, Adam MJ, Orvig C. Nucl Med Biol. 2012;39:785–794. doi: 10.1016/j.nucmedbio.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Boros E, Cawthray JF, Ferreira CL, Patrick BO, Adam MJ, Orvig C. Inorg Chem. 2012;51:6279–6284. doi: 10.1021/ic300482x. [DOI] [PubMed] [Google Scholar]
- 33.Price EW, Cawthray JF, Bailey GA, Ferreira CL, Boros E, Adam MJ, Orvig C. J Am Chem Soc. 2012;134:8670–8683. doi: 10.1021/ja3024725. [DOI] [PubMed] [Google Scholar]
- 34.Boros E, Lin YHS, Ferreira CL, Patrick BO, Hafeli UO, Adam MJ, Orvig C. Dalton Trans. 2011;40:6253–6259. doi: 10.1039/c0dt01458c. [DOI] [PubMed] [Google Scholar]
- 35.Boros E, Ferreira CL, Patrick BO, Adam MJ, Orvig C. Nucl Med Biol. 2011;38:1165–1174. doi: 10.1016/j.nucmedbio.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 36.Bailey GA, Price EW, Zeglis BM, Ferreira CL, Boros E, Lacasse MJ, Patrick BO, Lewis JS, Adam MJ, Orvig C. Inorg Chem. 2012;51 doi: 10.1021/ic302225z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Platas-Iglesias C, Mato-Iglesias M, Djanashvili K, Muller RN, Elst LV, Peters JA, de Blas A, Rodríguez-Blas T. Chem Eur J. 2004;10:3579–3590. doi: 10.1002/chem.200306031. [DOI] [PubMed] [Google Scholar]
- 38.Ferreiros-Martinez R, Esteban-Gomez D, Platas-Iglesias C, Blas Ad, Rodriguez-Blas T. Dalton Trans. 2008:5754–5765. doi: 10.1039/b808631a. [DOI] [PubMed] [Google Scholar]
- 39.Masuda R, Oishi S, Ohno H, Kimura H, Saji H, Fujii N. Bioorg Med Chem. 2011;19:3216–3220. doi: 10.1016/j.bmc.2011.03.059. [DOI] [PubMed] [Google Scholar]
- 40.Dioury F, Guéné E, Scala-Roulleau AD, Ferroud C, Guy A, Port M. Tetrahedron Lett. 2005;46:611–613. [Google Scholar]
- 41.Favre-Réguillon A, Segat-Dioury F, Nait-Bouda L, Cosma C, Siaugue JM, Foos J, Guy A. Synlett. 2000;2000:0868–0871. [Google Scholar]
- 42.Fukuyama T, Jow CK, Cheung M. Tetrahedron Lett. 1995;36:6373–6374. [Google Scholar]
- 43.Salvatore RN, Yoon CH, Jung KW. Tetrahedron. 2001;57:7785–7811. [Google Scholar]
- 44.Viola NA, Rarig RS, Jr, Ouellette W, Doyle RP. Polyhedron. 2006;25:3457–3462. [Google Scholar]
- 45.Tóth Ė, Brücher E. Inorg Chim Acta. 1994;221:165–167. [Google Scholar]
- 46.Cacheris WP, Nickle SK, Sherry AD. Inorg Chem. 1987;26:958–960. [Google Scholar]
- 47.Loncin MF, Desreux JF, Merciny E. Inorg Chem. 1986;25:2646–2648. [Google Scholar]
- 48.Anderegg G, Arnaud-Neu F, Delgado R, Felcman J, Popov K. Pure Appl Chem. 2005;77:1445–1495. [Google Scholar]
- 49.Šimeček J, Schulz M, Notni J, Plutnar J, Kubíček V, Havlíčková J, Hermann P. Inorg Chem. 2011;51:577–590. doi: 10.1021/ic202103v. [DOI] [PubMed] [Google Scholar]
- 50.Šimeček J, Hermann P, Wester HJ, Notni J. ChemMedChem. 2013;8:95–103. doi: 10.1002/cmdc.201200471. [DOI] [PubMed] [Google Scholar]
- 51.Thorek DLJ, Abou DS, Beattie BJ, Bartlett RM, Huang R, Zanzonico PB, Grimm J. J Nucl Med. 2012;53:1438–1445. doi: 10.2967/jnumed.112.104349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumar Mishra A, Panwar P, Chopra M, Kumar Sharma R, Chatal J-F. New J Chem. 2003;27:1054–1058. [Google Scholar]
- 53.Gran G. Analyst. 1952;77:661–671. [Google Scholar]
- 54.Duckworth P. Private communication.
- 55.Gans P, Sabatini A, Vacca A. Talanta. 1996;43:1739–1753. doi: 10.1016/0039-9140(96)01958-3. [DOI] [PubMed] [Google Scholar]
- 56.Baes CF, Jr, Mesmer RE. Wiley-Interscience; New York: 1976. [Google Scholar]
- 57.Frisch MJ, GWT, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 58.Lee C, Yang W, Parr RG. Physical Review B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 59.Becke AD. The Journal of Chemical Physics. 1993;98:5648–5652. [Google Scholar]
- 60.Cao X, Dolg M. The Journal of Chemical Physics. 2001;115:7348–7355. [Google Scholar]
- 61.Holland JP, Caldas-Lopes E, Divilov V, Longo VA, Taldone T, Zatorska D, Chiosis G, Lewis JS. PLoS One. 2010;5:e8859. doi: 10.1371/journal.pone.0008859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anderson CJ, Connett JM, Schwarz SW, Rocque PA, Guo LW, Philpott GW, Zinn KR, Meares CF, Welch MJ. J Nucl Med. 1992;33:1685–1691. [PubMed] [Google Scholar]
- 63.Anderson CJ, Schwarz SW, Connett JM, Cutler PD, Guo LW, Germain CJ, Philpott GW, Zinn KR, Greiner DP, Meares CF, Welch MJ. J Nucl Med. 1995;36:850–858. [PubMed] [Google Scholar]
- 64.Lindmo T, Boven E, Cuttitta F, Fedorko J, Bunn PA., Jr J Immunol Methods. 1984;72:77–89. doi: 10.1016/0022-1759(84)90435-6. [DOI] [PubMed] [Google Scholar]
- 65.Lindmo T, Bunn PA., Jr Methods Enzymol. 1986;121:678–691. doi: 10.1016/0076-6879(86)21067-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.