

INTRODUCTION: THE CONTRIBUTION OF PLATELETS TO THE HEMOSTATIC RESPONSE
Platelets have evolved to be the primary cellular component of the hemostatic response to injury, replacing the nucleated thrombocytes found in most nonmammalian species. After maturing from megakaryocyte-derived proplatelets, platelets circulate in an essentially quiescent state for 10 days in humans, and about half of that in mice. During that time, the quiescent state is maintained by extrinsic regulators such as nitrogen monoxide (NO) and prostacyclin (PGI2) by the presence of healthy endothelium, which separates platelets from activators within the vessel wall; and, perhaps most critically, by the absence of physiologic platelet activators such as collagen, thrombin, and adenosine diphosphate (ADP). Hemostatic thrombi form when the vessel wall has been breached. Pathologic thrombi form in response to vessel wall disease in the arterial system, stasis and inflammation in the venous system, and the inappropriate presence of platelet activators anywhere in the vascular system. Even in a healthy vascular system, differences in local conditions within arteries and veins affect platelet activation. Because pressures and flow rates are higher, hemostasis in the arterial system requires activated platelets to form the initial obstacle to blood loss, accelerate the production of thrombin, and provide a stable base and protected environment in which fibrin can accumulate. However, platelet activation is not limited to the arterial system; it occurs in the venous system and in capillaries as well and contributes to the hemostatic response in those locations. Patients with marked thrombocytopenia develop petechiae because of small leaks in the microcirculation that are otherwise prevented by platelets.
The primary goal of the hemostatic response is to limit blood loss when penetrating injuries occur. Many of the attributes of platelets that allow them to sense and respond rapidly to vascular injury are the same attributes that contribute to heart attacks and strokes when misdirected. The hemostatic response is sometimes modeled as occurring in 3 overlapping phases: initiation, extension, and stabilization. Initiation occurs when moving platelets become tethered to von Willebrand factor (VWF)/collagen complexes within the injured vessel wall and remain in place long enough to become activated by collagen. Extension occurs when additional platelets adhere to the initial monolayer, extending it in the lateral and luminal directions. Locally generated thrombin plus ADP and thromboxane A2 (TXA2) released by platelets play an important role in this step, activating platelets via G protein–coupled receptors (GPCRs). Subsequent intracellular signaling activates the integrin αIIbβ3 on the platelet surface. Activated platelets stick to each other via bridges formed by the binding of fibrinogen, fibrin or VWF to activated αIIbβ3. Stabilization refers to the events that help to consolidate the platelet plug and prevent premature disaggregation, in part by amplifying signaling within the platelet. The net result of these 3 phases is a hemostatic plug comprising activated platelets and cross-linked fibrin, a structure stable enough to withstand the forces generated by flowing blood in the arterial circulation.
This model for thinking about the hemostatic response can be helpful, because it emphasizes the initial role of collagen and VWF in anchoring platelets to a site of injury and the subsequent roles of thrombin, ADP, and TxA2. Platelets require some form of initial tethering to remain in place long enough to become fully activated. Moving platelets are pushed to the periphery of the blood stream by the flow properties of blood and the presence of red cells at the center of the blood stream. In the first seconds after injury, plasma VWF joins VWF already present in the vascular wall in becoming anchored to collagen. Flow forces working on the anchored VWF expose cryptic binding sites for platelet glycoprotein (GP) Ibα, allowing platelets to become tethered without requiring prior activation. In the absence of functional VWF, platelets can still be activated by collagen, thrombin, ADP, and TxA2, but in vivo, they are unable to remain at the site of injury long enough for the initial layers of the hemostatic thrombus to form. Hence, VWF deficiency or dysfunction results in a bleeding disorder, as do disorders affecting secretion. As platelet activation progresses, additional VWF is released locally from platelet storage granules.1
However, although useful in some respects, a limitation of thinking about platelet activation as successive waves of initiation, extension, and stabilization is that it does not completely describe what has been observed at sites of injury. Recent studies of platelet activation after penetrating injuries in the microcirculation show that platelet activation is not uniform throughout the hemostatic mass.2–8 Instead of a homogeneous mass, there is a core of fully activated, stably adherent platelets overlaid by a shell of less-activated, mostly unstable platelets (Fig. 1). As the response to injury continues, the shell persists even as the stable core expands, suggesting that not all of the platelets associated with the hemostatic response become fully activated. Rather than a homogeneous mass in which all platelets are activated to the same extent and fibrin is dispersed throughout the mass, the hemostatic thrombus retains regional heterogeneity and a hierarchical structure. Fibrin is found primarily in the parts of the thrombus core that are closest to the site of injury. At present, observations such as these are still largely limited to flow chambers and the cremasteric and mesenteric microcirculations in rodent models, but they raise several questions. Do the same events occur when penetrating injuries occur in larger vessels? What normally limits the growth of the hemostatic response and prevents inappropriate vascular occlusion? What goes wrong in pathologic settings such as dyslipidemia or plaque rupture? How is the hemostatic response regulated to produce an optimal outcome?
Fig. 1.
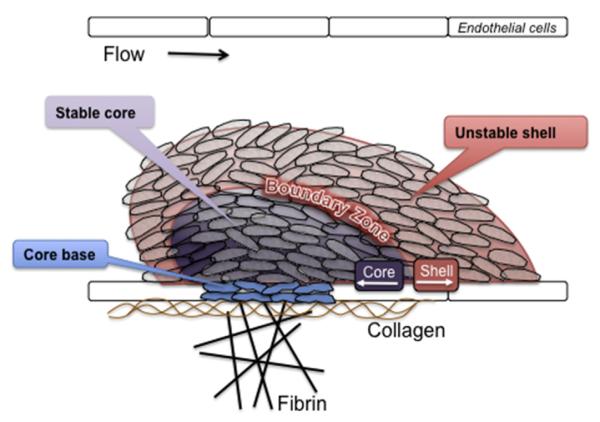
Structural heterogeneity in the hemostatic plug. Recent observations of the platelet response to acute vascular injury in vivo show that there are distinct regions within the hemostatic thrombus where platelets are either more or less activated and where fibrin tends to accumulate. A stable core of closely packed, fully activated platelets is overlaid by an outer shell of less-stable, less-activated platelets, with a boundary zone in between. Fibrin is found within the thrombus core and in a fibrin network formed from fibrinogen that escapes before hemostasis can be achieved.
THE PLATELET SIGNALING NETWORK: THE FOREST AND THE TREES
The molecular mechanisms that underlie the hemostatic response have been the subject of intense scrutiny for many years, reflecting the relevance of this process to clinical medicine and the lives of humans. To the extent that space allows, the remainder of this article summarizes what has been learned. However, before diving into the trees, it is worth considering the forest. Largely for experimental reasons, it has generally been more practical to tease apart platelet activation 1 molecule at a time. Studies with genetically modified mice have helped this process considerably, even although mouse platelets turn out to be different from human platelets in several critical ways. The results make it possible to draw models such as the one in Fig. 2, which summarizes key steps in platelet activation. Knowledge of platelet signaling pathways has aided the development of several antiplatelet agents that have entered clinical practice or are in clinical trials.
Fig. 2.

An overview of some of the major signaling pathways of platelet activation. Targets for antiplatelet agents that are in clinical use or in clinical trials are indicated in blue. Inhibitors are in red. AA, arachidonic acid; IP3, inositol-1,4,5-trisphosphate; PAR, protease-activated receptor; PGI2, prostaglandin I2; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; RGS, regulator of G protein signaling; TXA2, thromboxane A2.
However, Fig. 2 has important limitations. One limitation is that it has little to say about the events that lead to granule secretion in activated platelets. Resting platelets store and activated platelets secrete numerous molecules that support platelet activation and the hemostatic response. Some of these molecules also help to drive events such as wound healing and angiogenesis. Patients lacking storage granules have a clinical bleeding disorder. The presence of both proangiogenic and antiangiogenic factors within platelet storage granules has fueled recent debates about differential selectivity in either the storage or the release of these molecules in vivo.9–12
A second limitation of models such as the one shown in Fig. 2 is that it falls short in answering questions that are critical for understanding platelet activation. Why does platelet activation require such complexity? Why are there so many platelet agonists and so many signaling pathways? If they are merely redundant, why does it seem that genetic or pharmacologic ablation of so many individual molecules has a meaningful effect on platelet function? An answer to some of these questions may come from thinking about signaling networks rather than signaling pathways.
One way of illustrating such networks is shown in Fig. 3. In a network-centric view of platelet activation, instead of responding to a single agonist via a single signaling pathway, platelets accumulating at a site of injury respond to the mixture of inputs (agonists) to which they are exposed. This mixture varies over both time and space, creating agonist concentration gradients. Positive and negative feedback in the form of soluble molecules (some released by platelets, others by damaged cells) plus contact-dependent interactions between adjacent platelets play an important role in modulating platelet reactivity. Information flow within the platelet sums the effects of these inputs and nodal points in the signaling network provide links between them. This view of platelet activation can be tied to the structure shown in Fig. 1 by considering, first, how declining gradients of different platelet agonists originating near the point of injury differentially affect the platelet activation state as the distance from the point of injury grows and, second, how the closer proximity of platelets in the thrombus core facilitates the binding of cell surface ligands to receptors on the surface of adjacent platelets.
Fig. 3.
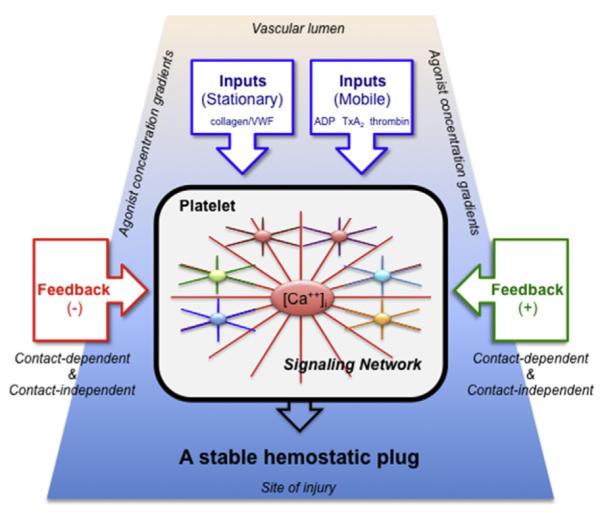
A network-centric view of platelet activation. Signaling in platelets is represented as a balanced network, in which the effects of multiple inputs (mainly agonists) are modulated by positive and negative feedback. Signaling pathways communicate with each other and share nodal points in the network. The extent of activation of any given platelet varies according to the combination and concentration of the agonists to which the platelet is exposed and the sum of the feedback to which it is subjected. Gradients of agonist concentration are expected to be highest at the vessel wall near the site of injury (bottom) and lowest in the thrombus shell (top). Referring to the model in Fig. 1, platelets in the core of the thrombus are most subject to thrombin, collagen, and contact-dependent feedback, whereas those in the shell are affected primarily by soluble mediators such as ADP and TxA2.
NORMAL PLATELET ACTIVATORS IN VIVO
Once vascular injury has occurred, platelets are principally activated by locally exposed collagen, locally generated thrombin, platelet-derived TXA2, and ADP that has either been released from damaged cells or secreted from platelet dense granules. The flow pattern of circulating red cells facilitates platelet adhesion to collagen by pushing platelets closer to the vessel wall, allowing GPIbα on the platelet surface to be snared by the A1 domain of VWF bound to collagen. Human platelets express 15,000 to 25,000 copies of GPIbα in a multiprotein complex with GPIbβ, GPIX, and GPV. Mutations in GPIbα that prevent its expression on the platelet surface or impair its receptor function produce an inherited recessive bleeding disorder (Bernard-Soulier syndrome) because platelet adhesion to the vessel wall is impaired. Macrothrombocytopenia arises in this disorder in part from a failure to form the normal linkages between GPIbα complex and filamin in the platelet membrane cytoskeleton.13
Once platelets are captured at the damaged vessel wall, the primary drivers for platelet activation are collagen, thrombin, ADP, TXA2, and, to a more limited extent, epinephrine (see Fig. 2). With the exception of collagen, each of these agonists signals through 1 or more members of the GPCR superfamily. Properties that are common to this class of receptors make them well suited for their tasks in platelets. Most bind their ligands with high affinity and, because they act as exchange factors, each occupied receptor can theoretically activate multiple G proteins, amplifying the initial signal. Binding studies show that agonist GPCRs are expressed on the platelet surface in low numbers, ranging from a few hundred (ADP and epinephrine) to a few thousand (thrombin) per cell.14–20 The duration of GPCR signaling is subject to receptor internalization, receptor desensitization, and the accelerated inactivation of G proteins by members of the RGS (regulator of G protein signaling) family. This multiplicity of mechanisms allows for tight regulation of platelet activation.
Human platelets express at least 10 heterotrimeric G proteins, including at least 1 Gs, 4 Gi (Gi1, Gi2, Gi3 and Gz), 3 Gq family members (Gq, G11 and G16), and 2 G12 family members (G12 and G13). Much has been learned about the critical role of these G proteins by knocking out the genes encoding their α subunits in mice.21 Less is known about the Gβγ isoforms that are expressed in platelets and the selectivity (if any) of their individual contributions to platelet activation.
Most of the time agonist-initiated platelet activation begins with the activation of a phospholipase C (PLC) isoform, which by hydrolyzing membrane phosphatidylinositol-4,5-bisphosphate (PIP2) produces the second messenger (inositol-1,4,5-trisphosphate [IP3]) needed to increase the cytosolic Ca2+ concentration. This situation leads to integrin activation via an intracellular pathway that includes a Ca2+-dependent exchange factor (CalDAG-GEF), a switch (predominantly Rap1b), an adaptor (RIAM), and proteins that interact directly with the integrin cytosolic domains (kindlin and talin).22 Which isoform of PLC is activated depends on the agonist. Collagen activates PLCγ2 using a mechanism that depends on scaffold molecules and protein tyrosine kinases. Thrombin, ADP, and TxA2 activate PLCβ using Gq as an intermediary. The a subunit of Gq binds directly to the phospholipase, activating it.
It is the binding of bivalent fibrinogen to αIIbβ3 that enables platelets to stick to one another, forming an aggregate. Other proteins that can substitute for fibrinogen include fibrin, VWF, and fibronectin. Average expression levels of αIIbβ3 range from approximately 50,000 per cell on resting platelets to 80,000 on activated platelets. Mutations in αIIbβ3 that prevent its expression or suppress its function produce a bleeding disorder (Glanzmann thrombasthenia) because platelets are unable to form stable aggregates. Antiplatelet agents such as eptifibatide (Integrilin), tirofiban (Aggrastat), and abciximab (ReoPro) take advantage of this situation by blocking αIIbβ3.
Platelet Activation by Collagen
Under static conditions, collagen can activate platelets without the assistance of co-factors, but under arterial flow conditions, VWF plays an essential role. Collagen polymers are better platelet agonists than collagen monomers.23,24 Platelets can adhere to monomeric collagen but require the more complex structure found in fibrillar collagen for optimal activation.25 Four collagen receptors have been identified on human and mouse platelets. Two bind directly to collagen (integrin α2β1 and GPVI); the other 2 (αIIbβ3 and GPIbα) bind to collagen via VWF (Fig. 4). Of these receptors, GPVI is the most potent signaling receptor.26 The structure of the GPVI extracellular domain places it in the immunoglobulin superfamily. Its ability to generate signals rests on its constitutive association with the immunoreceptor tyrosine-based activation domain (ITAM)-containing Fc receptor γ-chain (FcRγ). Platelets from mice that lack either GPVI or FcRγ have impaired responses to collagen, as do platelets in which GPVI has been depleted or blocked.27–30 Loss of FcRγ impairs collagen responses in part because of loss of a necessary signaling element and in part because of its role in helping GPVI reach the platelet surface. α2β1 supports adhesion to collagen and acting as a source of further signaling after engagement.31,32 Human platelets with reduced expression of α2β1 have impaired collagen responses,33,34 as do mouse platelets that lack β1 integrins when the ability of these platelets to bind to collagen is tested at high shear.30,35
Fig. 4.

Platelet activation by collagen. Platelets use several different molecular complexes to support platelet activation by collagen. These complexes include (1) VWF-mediated binding of collagen to the GPIb-IX-V complex and integrin αIIbβ3 and (2) a direct interaction between collagen and both the integrin α2β1 and the GPVI/FcRγ-chain complex. Clustering of GPVI results in the phosphorylation of tyrosine residues in the FcRγ cytoplasmic domain, followed by the activation of the tyrosine kinase, Syk. One consequence of Syk activation is the activation of PLCγ2, leading to phosphoinositide hydrolysis, secretion of ADP, and the production of TXA2. COX-1, cyclooxygenase 1; DAG, diacylglycerol; PG, prostaglandin; PI3K, phosphatidylinositol 3-kinase; PKC, protein kinase C; PLA2, phospholipase A2.
Signaling through GPVI can be studied in isolation with the snake venom protein, convulxin, or with synthetic collagen-related peptides, both of which bind to GPVI, but not to other collagen receptors. Each of these entities causes clustering of GPVI, leading to the phosphorylation of FcRγ by Src family tyrosine kinases constitutively associated with a proline-rich domain in GPVI.36 Phosphorylation creates an ITAM motif recognized by the tandem SH2 domains of the tyrosine kinase Syk. Association of Syk with the GPVI/FcRγ-chain complex activates Syk, produces signaling complexes based on the scaffold proteins LAT and SLP-76, and leads to the activation of PLCγ2. Loss of Syk impairs collagen responses.29 PLCγ2 hydrolyzes PIP2 to form IP3 and diacylglycerol (DAG). IP3 opens Ca2+ channels in the platelet dense tubular system, increasing the cytosolic Ca2+ concentration through passive efflux. Depletion of the Ca2+ stores within the dense tubular system triggers Ca2+ influx across the platelet plasma membrane. The changes in the cytosolic Ca2+ concentration that occur when platelets adhere to collagen under flow can be visualized in real time.37,38 Diacylglycerol activates the more common protein kinase C (PKC) isoforms that are expressed in platelets, allowing the serine/threonine phosphorylation events that are needed for platelet activation.
Collectively, collagen receptors support the capture of fast-moving platelets at sites of injury, cause activation of the captured platelets, and stimulate the cytoskeletal reorganization that allows the previously discoid platelets to flatten out and adhere more closely to the exposed vessel wall. As noted earlier, VWF supports this process. It increases the density of potential binding sites for collagen per platelet, because the number of copies of GPIbα and αIIbβ3 greatly exceeds the number of copies of GPVI and α2β1. Because VWF is highly multimeric, it also increases the number of binding sites per collagen molecule. This situation increases the likelihood that platelets encounter an available binding site and, once bound, are able to increase the number of contacts with collagen by bringing additional receptors into play. GPVI and GPIbα are able to bind collagen and VWF without prior platelet activation, but once activation begins, α2β1 and αIIbβ3 are able to bind their respective ligands as well. Some of the integrin-activating signaling occurs downstream of GPVI, but there is evidence that the GPIb-IX-V complex can signal as well, as can α2β1 and αIIbβ3 once they are engaged.
Platelet Activation by Thrombin
Thrombin is able to activate platelets at concentrations as low as 0.1 nM. Although other platelet agonists can also cause phosphoinositide hydrolysis, none seems to be as efficiently coupled to PLC as thrombin. Within seconds of the addition of thrombin, the cytosolic Ca2+ concentration increases 10-fold, triggering downstream Ca2+-dependent events, including the activation of phospholipase A2 and integrin activation via the Rap1b-mediated pathway. All of these responses, but not shape change, are abolished in platelets from mice lacking Gqα.39 Thrombin also activates Rho in platelets, leading to rearrangement of the actin cytoskeleton and shape change, responses that are greatly reduced or absent in mouse platelets that lack G13α.40 Thrombin is able to inhibit adenylyl cyclase activity in human platelets, either directly (via a Gi family member) or indirectly (via released ADP).41,42
Platelet responses to thrombin are mediated by members of the protease-activated receptor (PAR) family of GPCRs. There are 4 members of this family, 3 of which (PAR1, PAR3, and PAR4) can be activated by thrombin. PAR1 and PAR4 are expressed on human platelets; mouse platelets express PAR3 and PAR4. Receptor activation occurs when thrombin cleaves the extended N terminus of each of these receptors, exposing a new N terminus that serves as a tethered ligand.43 Synthetic peptides based on the sequence of the tethered ligand domain of PAR1 and PAR4 are able to activate the receptors, mimicking at least some of the actions of thrombin. PAR3 was originally identified after gene ablation studies showed that platelets from mice lacking PAR1 were still fully responsive to thrombin.44 Approximately half of PAR1−/− mice die in utero, but this seems to be caused by loss of receptor expression in the vasculature, rather than in platelets.45 Whereas human PAR3 has been shown to signal in response to thrombin, PAR3 on mouse platelets primarily serves to facilitate PAR4 cleavage.46 Activation of PAR4 requires higher concentrations of thrombin than are required for activation of PAR1, apparently because it lacks the hirudinlike sequences that can interact with the anion-binding exosite of thrombin and facilitate receptor cleavage.46–49 Kinetic studies in human platelets suggest that thrombin signals first through PAR1 and subsequently through PAR4.50,51
Considerable evidence shows that PAR family members are sufficient to activate platelets. Peptide agonists for either PAR1 or PAR4 cause platelet aggregation and secretion.43,48 Conversely, simultaneous inhibition of human PAR1 and PAR4 abolishes responses to thrombin,52 as does deletion of the gene encoding PAR4 in mice.53 However, there are differences between mice and humans: optimal thrombin responses in human platelets require both PAR1 and PAR4, whereas those in mouse platelets are mediated by PAR3-facilitated cleavage of PAR4. PAR1 and PAR4 are coupled to at least Gq and G13. There is conflicting evidence about whether Gi-dependent signaling in thrombin-activated platelets is entirely mediated by secreted ADP or occurs in part by a direct interaction between PARs and Gi2.54–56 However, given the ability of PAR1 to directly couple to Gi2 in cells other than platelets, it is likely that both occur. This hypothesis is consistent with a recent observation that expression of an RGS-resistant variant of Gi2α enhances thrombin responses in mouse platelets, even when the contribution of secreted ADP is blocked.57 On the other hand, a requirement for PAR family members does not preclude the involvement of other participants in platelet responses to thrombin. GPIbα has a high affinity thrombin binding site within residues 268 to 287.58,59 Deletion or blockade of this site reduces platelet responses to thrombin, particularly at low thrombin concentrations,60–64 and impairs PAR1 cleavage on human platelets.63 PAR1 antagonists have been developed and are undergoing clinical trials.65 The early results show that blocking PAR1 may have some clinical usefulness, but can also increase bleeding risk in patients receiving other antiplatelet agents.66–68
Platelet Activation by ADP
ADP is stored in platelet dense granules and released on platelet activation. It is also released from damaged cells at sites of vascular injury, serving as a stimulus for activating tethered platelets and stabilizing the hemostatic plug. Aggregation studies show that all of the other platelet agonists are dependent to some extent on released ADP to elicit maximal platelet aggregation, although this dependence varies with the agonist and is dose related. Drugs that block platelet ADP P2Y12 receptors have proved to be effective antiplatelet agents despite the fact that ADP, when added alone, is a less potent platelet agonist than thrombin.
When added to platelets in vitro, ADP causes an increase in cytosolic Ca2+, TXA2 formation, protein phosphorylation, shape change, aggregation, and secretion. It also inhibits cyclic adenosine monophosphate (cAMP) formation. These responses are half-maximal at approximately 1 μM ADP. However, even at high concentrations, ADP is a comparatively weak activator of PLC. Instead, its usefulness as a platelet agonist rests more on its ability to activate other pathways.69,70 Human and mouse platelets express 2 distinct receptors for ADP, denoted P2Y1 and P2Y12. Both receptors are members of the purinergic class of GPCRs.17–19,71,72 P2Y1 receptors couple to Gq. P2Y12 receptors couple to Gi family members. Optimal activation of platelets by ADP alone requires activation of both receptors, but the potentiation by ADP of platelet responses to other agonists seems to be mainly caused by P2Y12. Knockouts of P2Y1 and P2Y12 in mice produce effects consistent with those predicted by pharmacologic studies on human platelets.17,54 A third purinergic receptor on platelets, P2X1, is an adenosine trisphosphate (ATP)-gated Ca2+ channel.73–76 Platelet dense granules contain ATP as well as ADP, and studies suggest that there are conditions in which P2X1 activity is essential for platelet activation.77–79 P2X1 is also functional on megakaryocytes.80
When P2Y1 is blocked or deleted, ADP is still able to inhibit cAMP formation, but its ability to cause an increase in cytosolic Ca2+, shape change, and aggregation is greatly impaired, as it is in platelets from mice that lack Gqα.39 P2Y1−/− mice have a minimal increase in bleeding time and show some resistance to thromboembolic mortality after injection of ADP, but no predisposition to spontaneous hemorrhage. Primary responses to platelet agonists other than ADP are unaffected and when combined with serotonin, which is a weak stimulus for PLC in platelets, ADP can still cause aggregation of P2Y1−/− platelets. Taken together, these results show that platelet P2Y1 receptors are coupled to Gqα and responsible for activation of PLC. P2Y1 receptors can also activate Rac and the Rac effector, p21-activated kinase (PAK),81 but do not seem to be coupled to Gi family members.
P2Y12 was independently identified by 2 groups.19,72 As had been predicted by inhibitor studies and by the phenotype of a patient lacking functional P2Y12,82 platelets from P2Y12−/− mice do not aggregate normally in response to ADP.83 P2Y12−/− platelets retain P2Y1-associated responses, including shape change and PLC activation, but lack the ability to inhibit cAMP formation in response to ADP. The Gi family member associated with P2Y12 seems to be primarily Gi2, because platelets from Gi2α−/− mice have an impaired response to ADP,84,85 whereas those lacking Gi3α or Gzα do not.85,86 Conversely, expression of a Gi2α variant that is resistant to the inhibitory effects of RGS proteins produces a gain of function in mouse platelets stimulated with ADP.57 Absence of P2Y12 produces a hemorrhagic phenotype in humans, albeit a mild one.19,82,87 Deletion of either P2Y1 or P2Y12 in mice prolongs the bleeding time and impairs platelet responses not only to ADP but also to thrombin and TXA2, particularly at low concentrations.83,88,89 Because the receptors for thrombin and TXA2 can cause robust activation of PLC, the contribution of ADP when thrombin or TXA2 are present seems to be largely because of its ability to activate Gi2.90 Downstream effectors for Gi2α include Src family members91 as well as PI3 kinase and Rap1b.
Platelet Activation by TXA2
TXA2 is produced from arachidonate in platelets by the aspirin-sensitive cyclooxygenase 1 (COX-1) pathway. When added to platelets in vitro, stable TXA2 analogues such as U46619 cause shape change, aggregation, secretion, phosphoinositide hydrolysis, protein phosphorylation, and an increase in cytosolic Ca2+ and have little, if any, direct effect on cAMP formation. Similar responses are seen when platelets are incubated with exogenous arachidonate.92 TXA2 can diffuse across the plasma membrane and activate nearby platelets.93 Like secreted ADP, release of TXA2 amplifies the initial stimulus for platelet activation and helps to recruit additional platelets.94 This process is effective locally, but is limited by the brief (~30 second) half-life of TXA2 in solution, helping to confine the spread of platelet activation to the original area of injury.
Only 1 gene encodes TXA2 receptors, but 2 splice variants are produced that differ in their cytoplasmic tails. Human platelets express both.95 Loss of Gqα abolishes U46619-induced IP3 formation, but does not prevent shape change,39 which can be blocked by knocking out G13α.40 Although in cells other than platelets, TXA2 receptors have been shown to couple to Gi family members,96 in platelets the inhibitory effects of U46619 on cAMP formation seem to be largely mediated by secreted ADP. These observations have previously been interpreted to mean that platelet TXA2 receptors are coupled to Gq and G12/13, but not to Gi family members. However, the enhanced response observed in mouse platelets carrying an RGS protein-resistant Gi2α variant suggests that this is still an open issue.57
The biological relevance of TXA2 to normal platelet function is supported by genetic and pharmacologic evidence. TP−/− mice have a prolonged bleeding time. Their platelets are unable to aggregate in response to TXA2 agonists and show delayed aggregation with collagen, presumably reflecting the role of TXA2 in platelet responses to collagen.97 A group of Japanese patients with impaired platelet responses to TXA2 analogues have proved to be either homozygous or heterozygous for an R60L mutation in the first cytoplasmic loop of TP.98 However, the most compelling case for the contribution of TXA2 signaling in human platelets comes from the successful use of aspirin as an antiplatelet agent. When added to platelets in vitro, aspirin abolishes TXA2 generation. Aspirin also blocks platelet activation by arachidonate and impairs responses to thrombin and ADP. The defect in thrombin responses appears as a shift in the dose/response curve, indicating that TXA2 generation is supportive of platelet activation by thrombin, but not essential.
Platelet Activation by Epinephrine
Compared with thrombin, epinephrine is a weak activator of human platelets when added on its own. Nonetheless, there are reports of human families in which a mild bleeding disorder is associated with impaired epinephrine-induced aggregation and reduced numbers of catecholamine receptors.99,100 Epinephrine responses in platelets are mediated by α2A-adrenergic receptors.20,101,102 In both mice and humans, epinephrine is able to potentiate the effects of other agonists so that the combination is a stronger stimulus for platelet aggregation than either agonist alone. Potentiation is often attributed to the ability of epinephrine to inhibit cAMP formation, but as is discussed later, there are clearly other effects as well. In contrast to other platelet agonists, epinephrine has no detectable direct effect on PLC and does not cause shape change, although it can trigger phosphoinositide hydrolysis indirectly by stimulating TXA2 formation.103 Taken together, these results suggest that platelet α2A-adrenergic receptors are coupled to Gi, but not Gq or G12 family members. Human and mouse platelets express 4 members of the Gi family: Gi1, Gi2, Gi3, and Gz.104 Of these members, Gzα has the slowest rate of intrinsic guanosine triphosphate (GTP) hydrolysis and is the only one that is not a substrate for pertussis toxin. Knockout studies show that epinephrine responses in platelets are abolished when Gzα is deleted. Loss of Gi2α or Gi3α has no effect.85,86 Gz also seems to be responsible for the ability of epinephrine to activate Rap1b.85,105 Therefore, it seems that in mouse platelets, α2A-adrenergic receptors couple to Gz, but not Gi2 or Gi3.
CRITICAL EVENTS
Thus far, this review has focused on platelet activation mechanisms from the perspective of individual agonists, many of which trigger a common set of pathways. In this section, some of those pathways are considered.
Phosphoinositide Hydrolysis
With the exception of epinephrine, platelet activation begins with the activation of PLC which, by hydrolyzing membrane phosphatidylinositol-4,5-bisphosphate (PIP2), produces IP3 and (DAG), the second messengers needed to increase the cytosolic Ca2+ concentration and activate some of the PKC isoforms found in platelets. As already noted, how PLC is activated varies with the agonist. Collagen activates PLCγ2 using adaptor molecules and tyrosine kinases. Thrombin, ADP, and TXA2 activate PLCβ isoforms using Gqα and (less efficiently if at all) Gβγ derived from Gi. Regardless of which PLC isoform is activated, the subsequent increase in the Ca2+ concentration triggers downstream events, including integrin activation and TXA2 formation. Thrombin provides a robust stimulus for phosphoinositide hydrolysis and causes the largest and fastest increase in cytosolic Ca2+. Collagen and ADP are more dependent on the synthesis and release of TXA2 to achieve a maximal response.
Cytosolic Ca2+ and Integrin Activation
Resting platelets maintain their cytosolic free Ca2+ concentration at approximately 0.1 μM by (1) limiting Ca2+ influx and (2) pumping Ca2+ out of the cytosol across the plasma membrane or into the dense tubular system (smooth endoplasmic reticulum). This action consumes ATP. In activated platelets, [Ca2+]i can spike 10-fold to greater than 1 μM with potent agonists like thrombin. Measurements made of large populations of platelets suggest that this increase occurs rapidly and uniformly in response to potent agonists such as thrombin and less potently with agonists such as ADP and collagen. However, observations made with single platelets suggest that the Ca2+ response is heterogeneous, occurring to a different extent in different platelets, with some showing spiking behavior rather than a sustained increase and some not responding at all.106–108 The molecular basis of this heterogeneity is not fully understood, although simulation studies suggest that platelet size is a factor.108
The increase in cytosolic Ca2+ that occurs during platelet activation derives from 2 sources. The first part is caused by the IP3-mediated release of Ca2+ from the platelet dense tubular system. The second part occurs when depletion of the dense tubular system Ca2+ pool triggers store operated Ca2+ entry through a conformational change in STIM1, a protein in the dense tubular system membrane, binding of STIM1 to Orai1 in the plasma membrane, allowing an influx of plasma Ca2+.109 These events can be separated by chelating extracellular Ca2+, thereby precluding Ca2+ influx. The increase in [Ca2+]i activates the GTP-binding protein, Rap1b, via the Ca2+-dependent guanine nucleotide exchange factor (GEF), CalDAG-GEF.110 Activated Rap1b has been shown to bind to RIAM, bringing it to the plasma membrane, where it can bind talin. This situation allows talin to bind to the cytoplasmic domain of αIIbβ3, triggering integrin activation and exposing a binding site for fibrinogen.22
TXA2 Production
The initial events of platelet activation lead to the activation of phospholipase A2, which cleaves phosphatidylcholine and other membrane phospholipids, liberating arachidonate from the C2 position of the glycerol backbone. Arachidonate can be transformed into many bioactive compounds, but in platelets, TXA2 is the key product. TXA2 synthesis begins with COX-1, which forms prostaglandin (PG) G2 and PGH2 from arachidonate in a 2-step process (see Fig. 4). The PGH2 is then metabolized to TXA2 by thromboxane synthetase.111 Evidence suggests that phospholipase A2 activation in platelets can occur in more than 1 way. It can clearly happen in response to an increase in cytosolic Ca2+, because the addition of a Ca2+ ionophore is sufficient to cause phospholipase A2 activation.112 There is also evidence that mitogen-activated protein kinase pathway signaling activates phospholipase A2, although not necessarily by direct phosphorylation of phospholipase A2.113,114 TXA2 formation is promoted by platelet aggregation and may, therefore, also occur as a consequence of integrin signaling, although this has not been tested directly.115 TXA2 can diffuse out of the platelet, activating nearby receptors in an autocrine or paracrine fashion before it is hydrolyzed to inactive TXB2. Even although TXA2 is a potent platelet activator, its half-life in aqueous solution is short (about 30 seconds), which has implications for both its duration of action and its impact downstream from a growing thrombus. This situation may be especially relevant for the model of the hemostatic response shown in Fig. 1, which postulates that declining gradients of soluble platelet agonists originating in the thrombus core contribute to the heterogeneity in platelet activation that we and others have observed within the hemostatic mass.
Shape Change and Rearrangement of the Actin Cytoskeleton
Activated platelets lose their characteristic discoid shape, either assuming a globular shape with filopodial extensions if they are activated in suspension or flattening out and developing lamellopodia if they are activated on a suitable surface. Shape change reflects loss of the circumferential microtubule ring and rearrangement of the actin cytoskeleton of the platelet, a process mediated by monomeric G proteins in the Rho and Rac families. When platelets in suspension are activated by soluble agonists, shape change precedes platelet aggregation. The soluble agonists (thrombin, ADP, and TXA2) that trigger shape change typically act through receptors that are coupled to members of the Gq and G12 families. Epinephrine is unable to cause shape change, because its receptors are coupled solely to Gz.
At least 2 pathways are involved in the reorganization of the actin cytoskeleton: Ca2+-dependent activation of myosin light-chain kinase downstream of Gq family members and activation of Rho family members downstream of G13.21,116 Several proteins having both Gα-interacting domains and GEF domains can link G12 family members to Rho family members, including p115RhoGEF.117 With the exception of ADP, shape change persists in platelets from mice that lack Gqα39 but is lost when G13α expression is suppressed, alone or in combination with G12α.40 A combination of inhibitor and genetic approaches suggests that G13-dependent Rho activation leads to shape change via pathways that include the Rho-activated kinase (p160ROCK) and LIM kinase.116,118,119 Activation of these kinases results in phosphorylation of myosin light-chain kinase and cofilin, helping to regulate both actin filament formation and myosin. ADP, on the other hand, depends more heavily on Gq-dependent activation of PLC to produce shape change and is able to activate G13 only as a consequence of TXA2 generation; hence, the loss of ADP-induced shape change when Gq signaling is suppressed.
PI3 Kinase Activation
An additional category of critical events needed for robust platelet activation involves activation of the PI3-kinase (PI3K) isoforms expressed in platelets, either by Gi family members (PI3Kγ) or by phosphotyrosine-dependent signaling pathways downstream of collagen receptors (PI3Kαβδ). PI3-kinases phosphorylate PI-4-P and PI-4,5-P2 to produce PI-3,4-P2 and PI-3,4,5-P3, respectively. Among the best-described consequence of PI3K activation in platelets is the activation of the protein kinase, Akt. Knockout and inhibitor studies show that all 3 Akt isoforms are necessary for normal platelet activation.120–122 Knockout and inhibitor studies also show a Gi/PI3K-dependent mechanism for activating Rap1b,85,105 which, given the clinical usefulness of P2Y12 antagonists as antiplatelet agents, seems to be a mechanism that is important for stabilizing platelet aggregates. However, aside from Rap1b and GSK3β, a kinase the activity of which is negatively regulated by Akt, little is known about Akt-dependent signaling pathways in platelets. To our knowledge, the Akt-dependent GEF for Rap1b has yet to be identified. PI3Kβ has been proposed as a target for antiplatelet agents.123
CONTACT-DEPENDENT AND CONTACT-FACILITATED SIGNALING
As platelet activation proceeds, platelets that were previously mobile come into increasingly stable contact with each other, eventually with sufficient proximity that molecules on the surface of 1 platelet can interact directly with molecules on the surface of adjacent platelets.124 Although in theory this situation could occur anywhere within the growing hemostatic thrombus, it is likely to occur most readily in the thrombus core, where platelets are most densely packed (see Fig. 1).8 Stable cohesive contacts between platelets require engagement of αIIbβ3 with 1 of its ligands, after which inward-directed (ie, outside-in) signaling occurs through the integrin. Inward-directed signaling also occurs through other surface molecules, which can engage their counterparts on the surface of adjoining platelets. Some of these molecules have been shown to affect platelet activation and thrombus stability, either positively or negatively. Others serve primarily to help form contacts between platelets and create a protected space in which soluble molecules, including agonists, can accumulate. In the next section, several examples are considered in which contact-dependent signaling has been studied in platelets and shown to have a meaningful impact.
Outside-In Signaling by Integrins
Activated αIIbβ3 bound to fibrinogen, fibrin, or VWF provides the dominant cohesive force that holds platelet aggregates together. It also contributes a further impetus for sustained platelet activation by serving as a scaffold for the assembly of signaling molecules.125–127 The term outside-in signaling refers to the effects of these molecules.128 Integrin signaling depends in large part on the formation of protein complexes that link to the integrin cytoplasmic domain. Some of the protein-protein interactions that involve the cytoplasmic domains of αIIbβ3 help regulate integrin activation; others participate in outside-in signaling and clot retraction. Proteins that are reportedly capable of binding directly to the cytoplasmic domains of αIIbβ3 include β3-endonexin, CIB1, talin, kindlin, myosin, Shc, and the tyrosine kinases, Src, Fyn, and Syk. As noted earlier, the binding of talin and kindlin is believed to be one of the final events in the allosteric regulation of integrin activation.129–132 Some other interactions require the phosphorylation of tyrosine residues Y773 and Y785 (Y747 and Y759 in mice) in the β3 cytoplasmic domain by Src family members. Substitution of Y747 and Y759 with phenylalanine produces mice with platelets that tend to disaggregate and that show impaired clot retraction and a tendency to rebleed from tail bleeding time sites.133 Fibrinogen binding to the extracellular domain of activated αIIbβ3 stimulates an increase in the activity of Src family kinases and Syk.133,134 Studies of platelets from mice lacking these kinases suggest that these events are required for the initiation of outside-in signaling and for full platelet spreading, irreversible aggregation, and clot retraction.135–138 There is evidence that the ITAM-containing receptor, FcRγIIA, is the link between Src family kinase and Syk activation in human platelets activated by αIIbβ3, although if so, a different receptor must perform this role in mouse platelets, which do not express FcRγIIA.139
Contact-Dependent Ligand-Receptor Interactions
The proximity of 1 platelet to another can also permit the direct binding of cell surface ligands to cell surface receptors on adjacent platelets. Two examples that we have studied are the interactions of ephrinB1 and sema4D with their cognate receptors. Ephrins are cell surface molecules attached by either a GPI anchor (ephrin [Eph] A family members) or a single transmembrane domain (ephrin B family members) that serve as ligands for cell surface receptor tyrosine kinases in the Eph A and B families. Eph kinases, like Eph B family members, have a single transmembrane domain that includes the catalytic domain and protein/protein interaction motifs. Human platelets express at least 2 Eph kinases, EphA4 and EphB1, and their ligand, ephrinB1.140 Forced clustering of either EphA4 or ephrinB1 results in cytoskeletal changes leading to platelet spreading, as well as increased adhesion to fibrinogen, Rap1b activation, and granule secretion.140,141 EphA4 associates with αIIbβ3 and Eph/ephrin interactions promote phosphorylation of the β3 cytoplasmic domain. Conversely, blockade of Eph/ephrin interactions impairs clot retraction and causes platelet disaggregation at low agonist concentrations, resulting in impaired thrombus growth.140,142 It also inhibits platelet accumulation on collagen under flow.140,142 These observations suggest that close contacts between platelets during the early stages of thrombus formation allow the binding of ephinB1 to EphA4 and EphB1 in trans to provide signaling for sustained integrin activation.
Semaphorin 4D (Sema4D, CD100) provides another example of a ligand that is involved in contact-dependent signaling in platelets. Like the ephrins, semaphorins are best known for their role in the developing nervous system,143 but they have also been implicated in organogenesis, vascularization, and immune cell regulation.144 The 25 members of the semaphorin family are defined by a large sema domain145 and divided into 8 classes differing in part by how (and whether) they are anchored to the surface of the cell that expresses them.146 Sema4D, which has a transmembrane domain, is expressed on the surface of both mouse and human platelets. Platelet activation causes a rapid increase in surface expression, after which there is a gradual decline below the original expression level as the sema4D exodomain is cleaved and shed by the metalloprotease, ADAM17.147 Inhibiting or deleting ADAM17 blocks this process. Sema4D−/− mouse platelets have a defect in their responses to collagen and convulxin in vitro, and a reduced response to vascular injury in vivo.147 Responses to thrombin, ADP, and TXA2 mimetics are normal.147,148 The collagen defect has been mapped to a failure to maximally activate Syk downstream of the collagen receptor, GPVI. Events in the pathway upstream of Syk occur normally in sema4D−/− platelets.147,148 These defects are observed only when platelets come in contact with each other and can be reversed by adding soluble recombinant sema4D. Thus, the evidence suggests that sema4D provides a contact-dependent boost in collagen signaling. The platelets that are most likely to be activated via GPVI are those in contact with collagen. If so, then the sema4D signaling is likely most relevant for the platelets closest to the vessel wall at the site of injury. Human platelets express at least 2 receptors for sema4D: CD72 and a member of the plexin B family.147 Mouse platelets express the plexin, but not CD72. However, although the presence of these Sema4D receptors has been established, the extent to which they mediate Sema4D signaling in platelets remains to be determined.
OBTAINING AN OPTIMAL RESPONSE TO INJURY
The molecular mechanisms that drive platelet activation reflect an evolutionary compromise. This compromise can be thought of as establishing a threshold for platelet activation. If the threshold is too high, then platelets become useless for hemostasis. If too low, then opportunities for unwarranted platelet activation increase (Fig. 5). An optimal platelet response to injury can be defined as one in which blood loss is restrained and hemostasis is achieved without the penalty of further tissue damage caused by unwarranted vascular occlusion. The set point is established by balancing the intracellular signaling mechanisms that drive platelet activation in response to injury with regulatory mechanisms that either dampen those responses or prevent their initiation in the first place. The normal environment of the platelet includes the other blood cells, the soluble molecules found in plasma and, most critically, the vascular wall and the endothelial cells that line it, all of which are subject to change in the face of injury, disease, circadian rhythms, and aging. Many of these entities serve as extrinsic regulators of platelet activation. A healthy endothelium provides a physical barrier that limits platelet activation. It also produces inhibitors of platelet activation, including NO,149 PGI2,150–152 and the ecto-ADPase, CD39, which hydrolyzes plasma ADP that would otherwise sensitize platelets to activation by other agonists.153 In addition to these extrinsic regulators, several intrinsic regulators of platelet activation have been identified. A few of them are considered here.
Fig. 5.
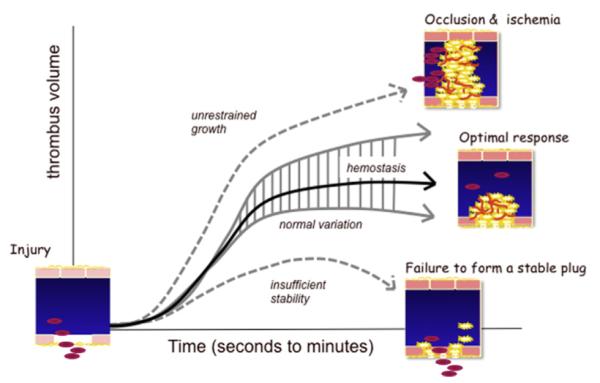
Formation of an optimal platelet plug. Vascular injury produces a hemostatic response that can be too aggressive (leading to occlusion and ischemia), inadequate (leading to further bleeding), or optimal. This model suggests that an optimal response varies in detail (hence, a range of normal), but is best viewed as a response that results in hemostasis with a minimum of blood loss and an avoidance of unwarranted vascular occlusion. In the setting of a vascular wall disease such as atherosclerosis, the rapid accumulation of platelets on top of a ruptured plaque represents an escape from normal restraints.
Regulation of G Protein-Dependent Signaling
As noted earlier, most of the platelet agonists act via GPCRs. Because mechanisms exist that can limit the activation of GPCRs, platelet activation can be tightly regulated. The role of RGS proteins in platelets is just beginning to be explored. RGS proteins limit signaling intensity and duration by accelerating the intrinsic hydrolysis of GTP by activated G protein α subunits.154 As many as 10 RGS proteins have been identified in platelets at the RNA level, but to our knowledge, only RGS10 and RGS18 have been confirmed at the protein level.155–160 The evidence that RGS proteins are biologically relevant in platelets comes from studies on mice in which glycine 184 in the α subunit of Gi2 has been replaced with serine, rendering it unable to interact with RGS proteins without impairing the ability of Gi2 to interact with either receptors or downstream effectors. This substitution produces the predicted gain of platelet function in vitro and in vivo, even in the heterozygous state.57 At the molecular level, the Gi2α(G184S) allele causes an attenuated increase in cAMP levels in response to PGI2 and a substantial increase in basal Akt activation, 2 events that occur downstream of Gi2 in platelets. In contrast, agonist-stimulated increases in [Ca2+]i and Rap1 activation are unaffected, indicating no crossover into Gqα-dependent signaling pathways.
These results show that removing the restraining hand of RGS proteins on Gi2 in platelets is sufficient to produce a pronounced gain of function, arguing that the normal role of the RGS proteins is to limit platelet activation. If so, then the timing of the interaction of RGS proteins with activated G proteins becomes critical as well. If signaling is constrained too early, then platelet activation might never occur, whereas if signaling is shut down too late, more platelets might accumulate than are needed. Two recent studies show that RGS proteins interact with other molecules the role of which may be to regulate RGS protein availability. The first of these proteins is the scaffold protein, spinophilin, which binds RGS10 and RGS18 in resting platelets, and then releases them on platelet activation. Knocking out spinophilin in mice produces a loss of function in platelets, as would be expected if RGS10 and RGS18 interact prematurely with activated G proteins when spinophilin is not present.161 A second group of RGS protein-interacting proteins in platelets includes members of the 14-3-3 scaffold protein family. 14-3-3 family members can bind to phosphorylated serine residues in RGS18, an interaction that is displaced when cAMP-dependent protein kinase A phosphorylates RGS18.162 Taken together, these recent observations suggest that spinophilin serves as a critical node within the platelet signaling network.
cAMP and Protein Kinase A
Rising cAMP levels turn off signaling in platelets. Regulatory molecules released from endothelial cells cause Gsα-mediated increases in adenylyl cyclase activity (PGI2) and inhibit the hydrolysis of cAMP by phosphodiesterases (NO).163 When added to platelets in vitro, PGI2 can cause a greater than 10-fold increase in the platelet cAMP concentration, but even small increases in cAMP levels (2-fold or less) impair platelet activation.164 Deletion of either Gi2α or Gzα causes an increase in the basal cAMP concentration in mouse platelets.85 cAMP phosphodiesterase inhibitors such as dipyridamole act as antiplatelet agents by raising cAMP levels. Conversely, loss of PGI2 receptor (IP) expression causes a decrease in basal cAMP levels, an enhanced response to agonists, and a predisposition to thrombosis in murine arterial injury models.85,165 Despite ample evidence that cAMP inhibits platelet activation, the mechanism by which it does this is not fully defined. cAMP-dependent protein kinase A is believed to be involved, but other mechanisms may be as well. Substrates for the kinase in platelets include serine and threonine residues in some GPCRs, IP3 receptors, GPIbβ, vasodilator-stimulated phosphoprotein, RGS18, and Rap1b, but it remains unclear whether a single substrate or small group of substrates accounts for most of the effect or whether it is the accumulated effect of phosphorylation of many substrates.162,166–174
Adhesion/Junction Receptors Contribute to Contact-Dependent Signaling
In addition to amplifying platelet activation, contact-dependent signaling can also help to limit thrombus growth and stability. Examples of this phenomenon include platelet/endothelial cell adhesion molecule-1 (PECAM-1), carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) and at least 2 members of the CTX family of adhesion molecules, ESAM and JAM-A. PECAM-1 is a type 1 transmembrane protein with 6 extracellular domains, the most distal of which can form homophilic interactions in trans.175 The cytoplasmic domain contains 2 immunoreceptor tyrosine inhibitory motifs (ITIMs) that can bind the tyrosine phosphatase, SHP-2.176 PECAM-1-deficient platelets show enhanced responses to collagen in vitro and in vivo.177–179 CEACAM1 is a second ITIM family member expressed on the platelet surface that can form homophilic and heterophilic interactions with other CEACAM superfamily members. In contrast to PECAM-1, the CEACAM1 ITIMs prefer SHP-1 over SHP-2, although either can become bound.180,181 The CEACAM1 knockout, like the PECAM-1 knockout, produces an enhanced platelet response, showing increased platelet activation in vitro in response to collagen and increased thrombus formation in a FeCl3 injury model.182 ESAM and JAM-A have 2 extracellular immunoglobulin domains, a single transmembrane region and a cytoplasmic tail of varying length that terminates in a binding site for PDZ domain-containing proteins. ESAM is associated with a-granules in resting platelets, but localizes to junctions during platelet activation.183 ESAM−/− platelets show increased aggregation in response to low doses of GPCR agonists and are more resistant to disaggregation compared with wild-type platelets. In vivo, ESAM−/− mice form larger and more stable thrombi compared with their wild-type counterparts.183 Collectively, these studies suggest that ESAM negatively regulates platelet function through contact-dependent homophilic interactions, although the mechanistic basis for these observations remains to be identified. JAM-A−/− mice also show a gain of function phenotype.184
SUMMARY
Platelets are part of the initial response to vascular injury intended to resist further blood loss by producing a hemostatic plug or thrombus. Conventional models portray such thrombi as relatively uniform masses of fully activated platelets intermixed with fibrin. Recent observational studies suggest that reality is more complex in at least the microcirculation, with fibrin deposition concentrated close to the vessel wall at the site of injury and the extent of platelet activation varying inversely with distance from the injury such that a hierarchical structure is formed. Underlying platelet activation is flexible signaling network within the platelets that sums the response to multiple platelet agonists, including collagen, thrombin, ADP, TxA2, and epinephrine. Critical events in this network include the activation of PLC, an increase in cytosolic Ca2+, reorganization of the platelet cytoskeleton, granule secretion, and the steps that link Ca2+ to activation of the platelet fibrinogen receptor, αIIbβ3, so that platelet aggregates can form. Extrinsic regulators provided by a healthy endothelium dampen platelet responsiveness and resist unwarranted platelet activation, whereas intrinsic regulators such as RGS proteins limit signaling downstream of platelet agonists once it has been initiated. As activated platelets come into increasingly stable contact with each other, molecules on their surface are able to interact directly, generating a wave of contact-dependent signaling events. Such events can amplify platelet activation, but can also help to mold the growing thrombus by limiting the extent of platelet activation.
KEY POINTS.
Circulating platelets possess the ability to undergo rapid activation at sites of vascular injury and resist unwarranted activation, which can lead to heart attacks and strokes.
The signaling mechanisms underlying the regulation of platelet activation have been approached as a collection of individual pathways unique to agonist.
This review takes a different approach, casting platelet activation as the product of a signaling network, in which activating and restraining mechanisms interact in a flexible network that regulates platelet adhesiveness, cohesion between platelets, granule secretion, and the formation of a stable hemostatic thrombus.
Footnotes
Disclosure: The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Kanaji S, Fahs SA, Shi Q, et al. Contribution of platelet versus endothelial VWF to platelet adhesion and hemostasis. J Thromb Haemost. 2012;10(8):1646–52. doi: 10.1111/j.1538-7836.2012.04797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reininger AJ, Heijnen HF, Schumann H, et al. Mechanism of platelet adhesion to von Willebrand factor and microparticle formation under high shear stress. Blood. 2006;107:3537–45. doi: 10.1182/blood-2005-02-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruggeri ZM, Orje JN, Habermann R, et al. Activation-independent platelet adhesion and aggregation under elevated shear stress. Blood. 2006;108:1903–10. doi: 10.1182/blood-2006-04-011551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munnix IC, Cosemans JM, Auger JM, et al. Platelet response heterogeneity in thrombus formation. Thromb Haemost. 2009;102:1149–56. doi: 10.1160/TH09-05-0289. [DOI] [PubMed] [Google Scholar]
- 5.Nesbitt WS, Westein E, Tovar-Lopez FJ, et al. A shear gradient-dependent platelet aggregation mechanism drives thrombus formation. Nat Med. 2009;15:665–73. doi: 10.1038/nm.1955. [DOI] [PubMed] [Google Scholar]
- 6.Bellido-Martin L, Chen V, Jasuja R, et al. Imaging fibrin formation and platelet and endothelial cell activation in vivo. Thromb Haemost. 2011;105:776–82. doi: 10.1160/TH10-12-0771. [DOI] [PubMed] [Google Scholar]
- 7.Nishimura S, Manabe I, Nagasaki M, et al. In vivo imaging visualizes discoid platelet aggregations without endothelium disruption and implicates contribution of inflammatory cytokine and integrin signaling. Blood. 2012;119:e45–56. doi: 10.1182/blood-2011-09-381400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brass LF, Wannemacher KM, Ma P, et al. Regulating thrombus growth and stability to achieve an optimal response to injury. J Thromb Haemost. 2011;9(Suppl 1):66–75. doi: 10.1111/j.1538-7836.2011.04364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Italiano JE, Jr, Richardson JL, Patel-Hett S, et al. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood. 2008;111:1227–33. doi: 10.1182/blood-2007-09-113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peters CG, Michelson AD, Flaumenhaft R. Granule exocytosis is required for platelet spreading: differential sorting of alpha-granules expressing VAMP-7. Blood. 2012;120:199–206. doi: 10.1182/blood-2011-10-389247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Battinelli EM, Markens BA, Italiano JE., Jr Release of angiogenesis regulatory proteins from platelet alpha granules: modulation of physiologic and pathologic angiogenesis. Blood. 2011;118:1359–69. doi: 10.1182/blood-2011-02-334524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamykowski J, Carlton P, Sehgal S, et al. Quantitative immunofluorescence mapping reveals little functional coclustering of proteins within platelet alpha-granules. Blood. 2011;118:1370–3. doi: 10.1182/blood-2011-01-330910. [DOI] [PubMed] [Google Scholar]
- 13.Kato K, Martinez C, Russell S, et al. Genetic deletion of mouse platelet glycoprotein Ibbeta produces a Bernard-Soulier phenotype with increased alpha-granule size. Blood. 2004;104:2339–44. doi: 10.1182/blood-2004-03-1127. [DOI] [PubMed] [Google Scholar]
- 14.Brass LF, Vassallo RR, Jr, Belmonte E, et al. Structure and function of the human platelet thrombin receptor: studies using monoclonal antibodies against a defined epitope within the receptor N-terminus. J Biol Chem. 1992;267:13795–8. [PubMed] [Google Scholar]
- 15.Mills DC. ADP receptors on platelets. Thromb Haemost. 1996;76:835–56. [PubMed] [Google Scholar]
- 16.Gachet C, Hechler B, Léon C, et al. Activation of ADP receptors and platelet function. Thromb Haemost. 1997;78:271–5. [PubMed] [Google Scholar]
- 17.Daniel JL, Dangelmaier C, Jin JG, et al. Molecular basis for ADP-induced platelet activation I. Evidence for three distinct ADP receptors on human platelets. J Biol Chem. 1998;273:2024–9. doi: 10.1074/jbc.273.4.2024. [DOI] [PubMed] [Google Scholar]
- 18.Jin JG, Daniel JL, Kunapuli SP. Molecular basis for ADP-induced platelet activation II. The P2Y1 receptor mediates ADP-induced intracellular calcium mobilization and shape change in platelets. J Biol Chem. 1998;273:2030–4. doi: 10.1074/jbc.273.4.2030. [DOI] [PubMed] [Google Scholar]
- 19.Hollopeter G, Jantzen HM, Vincent D, et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. 2001;409:202–7. doi: 10.1038/35051599. [DOI] [PubMed] [Google Scholar]
- 20.Motulsky HJ, Insel PA. [3H]Dihydroergocryptine binding to alpha-adrenergic receptors of human platelets. A reassessment using the selective radioligands [3H]prazosin, [3H]yohimbine, and [3H]rauwolscine. Biochem Pharmacol. 1982;31:2591–7. doi: 10.1016/0006-2952(82)90705-5. [DOI] [PubMed] [Google Scholar]
- 21.Offermanns S. In vivo functions of heterotrimeric G-proteins: studies in Galpha-deficient mice. Oncogene. 2001;20:1635–42. doi: 10.1038/sj.onc.1204189. [DOI] [PubMed] [Google Scholar]
- 22.Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brass LF, Bensusan HB. The role of collagen quaternary structure in the platelet: collagen interaction. J Clin Invest. 1974;54:1480–7. doi: 10.1172/JCI107896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santoro SA. Identification of a 160,000 dalton platelet membrane protein that mediates the initial divalent cation-dependent adhesion of platelets to collagen. Cell. 1986;46:913–20. doi: 10.1016/0092-8674(86)90073-5. [DOI] [PubMed] [Google Scholar]
- 25.Brass LF, Faile D, Bensusan HB. Direct measurement of the platelet: collagen interaction by affinity chromatography on collagen/Sepharose. J Lab Clin Med. 1976;87:525–34. [PubMed] [Google Scholar]
- 26.Clemetson JM, Polgar J, Magnenat E, et al. The platelet collagen receptor glycoprotein VI is a member of the immunoglobulin superfamily closely related to FcalphaR and the natural killer receptors. J Biol Chem. 1999;274:29019–24. doi: 10.1074/jbc.274.41.29019. [DOI] [PubMed] [Google Scholar]
- 27.Massberg S, Gawaz M, Gruner S, et al. A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J Exp Med. 2003;197:41–9. doi: 10.1084/jem.20020945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato K, Kanaji T, Russell S, et al. The contribution of glycoprotein VI to stable platelet adhesion and thrombus formation illustrated by targeted gene deletion. Blood. 2003;102:1701–7. doi: 10.1182/blood-2003-03-0717. [DOI] [PubMed] [Google Scholar]
- 29.Poole A, Gibbins JM, Turner M, et al. The Fc receptor gamma-chain and the tyrosine kinase Syk are essential for activation of mouse platelets by collagen. EMBO J. 1997;16:2333–41. doi: 10.1093/emboj/16.9.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nieswandt B, Brakebusch C, Bergmeier W, et al. Glycoprotein VI but not alpha2-beta1 integrin is essential for platelet interaction with collagen. EMBO J. 2001;20:2120–30. doi: 10.1093/emboj/20.9.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keely PJ, Parise LV. The alpha2beta1 integrin is a necessary co-receptor for collagen-induced activation of Syk and the subsequent phosphorylation of phospholipase Cgamma2 in platelets. J Biol Chem. 1996;271:26668–76. [PubMed] [Google Scholar]
- 32.Consonni A, Cipolla L, Guidetti G, et al. Role and regulation of phosphatidylinositol 3-kinase beta in platelet integrin alpha2beta1 signaling. Blood. 2012;119:847–56. doi: 10.1182/blood-2011-07-364992. [DOI] [PubMed] [Google Scholar]
- 33.Nieuwenhuis HK, Akkerman JW, Houdijk WP, et al. Human blood platelets showing no response to collagen fail to express glycoprotein Ia. Nature. 1985;318:470–2. doi: 10.1038/318470a0. [DOI] [PubMed] [Google Scholar]
- 34.Sixma JJ, Van Zanten GH, Huizinga EG, et al. Platelet adhesion to collagen: an update. Thromb Haemost. 1997;78:434–8. [PubMed] [Google Scholar]
- 35.Kuijpers MJ, Schulte V, Bergmeier W, et al. Complementary roles of glycoprotein VI and alpha2beta1 integrin in collagen-induced thrombus formation in flowing whole blood ex vivo. FASEB J. 2003;17:685–7. doi: 10.1096/fj.02-0381fje. [DOI] [PubMed] [Google Scholar]
- 36.Schmaier AA, Zou Z, Kazlauskas A, et al. Molecular priming of Lyn by GPVI enables an immune receptor to adopt a hemostatic role. Proc Natl Acad Sci U S A. 2009;106:21167–72. doi: 10.1073/pnas.0906436106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nesbitt WS, Giuliano S, Kulkarni S, et al. Intercellular calcium communication regulates platelet aggregation and thrombus growth. J Cell Biol. 2003;160:1151–61. doi: 10.1083/jcb.200207119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kulkarni S, Nesbitt WS, Dopheide SM, et al. Techniques to examine platelet adhesive interactions under flow. Methods Mol Biol. 2004;272:165–86. doi: 10.1385/1-59259-782-3:165. [DOI] [PubMed] [Google Scholar]
- 39.Offermanns S, Toombs CF, Hu YH, et al. Defective platelet activation in Galphaq-deficient mice. Nature. 1997;389:183–6. doi: 10.1038/38284. [DOI] [PubMed] [Google Scholar]
- 40.Moers A, Nieswandt B, Massberg S, et al. G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat Med. 2003;9:1418–22. doi: 10.1038/nm943. [DOI] [PubMed] [Google Scholar]
- 41.Barr AJ, Brass LF, Manning DR. Reconstitution of receptors and GTP-binding regulatory proteins (G proteins) in Sf9 cells: a direct evaluation of selectivity in receptor-G protein coupling. J Biol Chem. 1997;272:2223–9. doi: 10.1074/jbc.272.4.2223. [DOI] [PubMed] [Google Scholar]
- 42.Kim S, Foster C, Lecchi A, et al. Protease-activated receptor 1 and 4 do not stimulate G(i) signaling pathways in the absence of secreted ADP and cause human platelet aggregation independently of G(i) signaling. Blood. 2002;99:3629. doi: 10.1182/blood.v99.10.3629. [DOI] [PubMed] [Google Scholar]
- 43.Vu TK, Hung DT, Wheaton VI, et al. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–68. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- 44.Connolly AJ, Ishihara H, Kahn ML, et al. Role of the thrombin receptor in development and evidence for a second receptor. Nature. 1996;381:516–9. doi: 10.1038/381516a0. [DOI] [PubMed] [Google Scholar]
- 45.Griffin CT, Srinivasan Y, Zheng YW, et al. A role of thrombin receptor signaling in endothelial cells during embryonic development. Science. 2001;293:1666–70. doi: 10.1126/science.1061259. [DOI] [PubMed] [Google Scholar]
- 46.Nakanishi-Matsui M, Zheng YW, Sulciner DJ, et al. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. 2000;404:609–10. doi: 10.1038/35007085. [DOI] [PubMed] [Google Scholar]
- 47.Xu WF, Andersen H, Whitmore TE, et al. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci U S A. 1998;95:6642–6. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kahn ML, Zheng YW, Huang W, et al. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690–4. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- 49.Ishii K, Gerszten R, Zheng YW, et al. Determinants of thrombin receptor cleavage. Receptor domains involved, specificity, and role of the P3 aspartate. J Biol Chem. 1995;270:16435–40. doi: 10.1074/jbc.270.27.16435. [DOI] [PubMed] [Google Scholar]
- 50.Covic L, Gresser AL, Kuliopulos A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry. 2000;39:5458–67. doi: 10.1021/bi9927078. [DOI] [PubMed] [Google Scholar]
- 51.Shapiro MJ, Weiss EJ, Faruqi TR, et al. Protease-activated receptors 1 and 4 are shut off with distinct kinetics after activation by thrombin. J Biol Chem. 2000;275:25216–21. doi: 10.1074/jbc.M004589200. [DOI] [PubMed] [Google Scholar]
- 52.Kahn ML, Nakanishi-Matsui M, Shapiro MJ, et al. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest. 1999;103:879–87. doi: 10.1172/JCI6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sambrano GR, Weiss EJ, Zheng Y-W, et al. Role of thrombin signaling in platelets in hemostasis and thrombosis. Nature. 2001;413:74–8. doi: 10.1038/35092573. [DOI] [PubMed] [Google Scholar]
- 54.Jin JG, Kunapuli SP. Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Proc Natl Acad Sci U S A. 1998;95:8070–4. doi: 10.1073/pnas.95.14.8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brass LF, Woolkalis MJ. Dual regulation of cAMP formation by thrombin in HEL cell, a leukemic cell line with megakaryocytic properties. Biochem J. 1992;281:73–80. doi: 10.1042/bj2810073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brass LF, Woolkalis MJ, Manning DR. Interactions in platelets between G proteins and the agonists that stimulate phospholipase C and inhibit adenylyl cyclase. J Biol Chem. 1988;263:5348–55. [PubMed] [Google Scholar]
- 57.Signarvic RS, Cierniewska A, Stalker TJ, et al. RGS/Gi2alpha interactions modulate platelet accumulation and thrombus formation at sites of vascular injury. Blood. 2010;116:6092–100. doi: 10.1182/blood-2010-05-283846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.De Cristofaro R, De Candia E, Rutella S, et al. The Asp272-Glu282 region of platelet glycoprotein Ibalpha interacts with the heparin-binding site of alpha-thrombin and protects the enzyme from the heparin-catalyzed inhibition by anti-thrombin III. J Biol Chem. 2000;275:3887–95. doi: 10.1074/jbc.275.6.3887. [DOI] [PubMed] [Google Scholar]
- 59.Celikel R, McClintock RA, Roberts JR, et al. Modulation of alpha-thrombin function by distinct interactions with platelet glycoprotein Ibalpha. Science. 2003;301:218–21. doi: 10.1126/science.1084183. [DOI] [PubMed] [Google Scholar]
- 60.De Marco L, Mazzucato M, Masotti A, et al. Function of glycoprotein Ibalpha in platelet activation induced by alpha-thrombin. J Biol Chem. 1991;266:23776–83. [PubMed] [Google Scholar]
- 61.Harmon JT, Jamieson GA. Platelet activation by thrombin in the absence of the high affinity thrombin receptor. Biochemistry. 1988;27:2151–7. doi: 10.1021/bi00406a050. [DOI] [PubMed] [Google Scholar]
- 62.Mazzucato M, De Marco L, Masotti A, et al. Characterization of the initial alpha-thrombin interaction with glycoprotein Ibalpha in relation to platelet activation. J Biol Chem. 1998;273:1880–7. doi: 10.1074/jbc.273.4.1880. [DOI] [PubMed] [Google Scholar]
- 63.De Candia E, Hall SW, Rutella S, et al. Binding of thrombin to glycoprotein Ib accelerates hydrolysis of PAR1 on intact platelets. J Biol Chem. 2001;276:4692–8. doi: 10.1074/jbc.M008160200. [DOI] [PubMed] [Google Scholar]
- 64.Dörmann D, Clemetson KJ, Kehrel BE. The GPIb thrombin-binding site is essential for thrombin-induced platelet procoagulant activity. Blood. 2000;96:2469–78. [PubMed] [Google Scholar]
- 65.Tello-Montoliu A, Tomasello SD, Ueno M, et al. Antiplatelet therapy: thrombin receptor antagonists. Br J Clin Pharmacol. 2011;72:658–71. doi: 10.1111/j.1365-2125.2010.03884.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morrow DA, Braunwald E, Bonaca MP, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366:1404–13. doi: 10.1056/NEJMoa1200933. [DOI] [PubMed] [Google Scholar]
- 67.Tricoci P, Huang Z, Held C, et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med. 2012;366:20–33. doi: 10.1056/NEJMoa1109719. [DOI] [PubMed] [Google Scholar]
- 68.Scirica BM, Bonaca MP, Braunwald E, et al. Vorapaxar for secondary prevention of thrombotic events for patients with previous myocardial infarction: a prespecified subgroup analysis of the TRA 2 degrees P-TIMI 50 trial. Lancet. 2012;380(9850):1317–24. doi: 10.1016/S0140-6736(12)61269-0. [DOI] [PubMed] [Google Scholar]
- 69.Fisher GJ, Bakshian S, Baldassare JJ. Activation of human platelets by ADP causes a rapid rise in cytosolic free calcium without hydrolysis of phosphatidylinositol-4,5-bisphosphate. Biochem Biophys Res Commun. 1985;129:958–64. doi: 10.1016/0006-291x(85)91984-9. [DOI] [PubMed] [Google Scholar]
- 70.Daniel JL, Dangelmaier CA, Selak M, et al. ADP stimulates IP3 formation in human platelets. FEBS Lett. 1986;206:299–303. doi: 10.1016/0014-5793(86)81000-6. [DOI] [PubMed] [Google Scholar]
- 71.Léon C, Hechler B, Vial C, et al. The P2Y1 receptor is an ADP receptor antagonized by ATP and expressed in platelets and megakaryoblastic cells. FEBS Lett. 1997;403:26–30. doi: 10.1016/s0014-5793(97)00022-7. [DOI] [PubMed] [Google Scholar]
- 72.Zhang FL, Luo L, Gustafson E, et al. ADP is the cognate ligand for the orphan G protein-coupled receptor SP1999. J Biol Chem. 2001;276:8608–15. doi: 10.1074/jbc.M009718200. [DOI] [PubMed] [Google Scholar]
- 73.McKenzie AB, Mahout-Smith MP, Sage SO. Activation of receptor-operated channels via P2X1 not P2T purinoreceptors in human platelets. J Biol Chem. 1996;271:2879–81. doi: 10.1074/jbc.271.6.2879. [DOI] [PubMed] [Google Scholar]
- 74.Vial C, Hechler B, Léon C, et al. Presence of P2X1 purinoceptors in human platelets and megakaryoblastic cell lines. Thromb Haemost. 1997;78:1500–4. [PubMed] [Google Scholar]
- 75.Sun B, Li J, Okahara K, et al. P2X1 purinoceptor in human platelets–molecular cloning and functional characterization after heterologous expression. J Biol Chem. 1998;273:11544–7. doi: 10.1074/jbc.273.19.11544. [DOI] [PubMed] [Google Scholar]
- 76.Mahaut-Smith MP, Ennion SJ, Rolf MG, et al. ADP is not an agonist at P2X1 receptors: evidence for separate receptors stimulated by ATP and ADP on human platelets. Br J Psychol. 2000;131:108–14. doi: 10.1038/sj.bjp.0703517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fung CY, Brearley CA, Farndale RW, et al. A major role for P2X1 receptors in the early collagen-evoked intracellular Ca21 responses of human platelets. Thromb Haemost. 2005;94:37–40. doi: 10.1160/TH04-11-0732. [DOI] [PubMed] [Google Scholar]
- 78.Mahaut-Smith MP, Tolhurst G, Evans RJ. Emerging roles for P2X1 receptors in platelet activation. Platelets. 2004;15:131–44. doi: 10.1080/09537100410001682788. [DOI] [PubMed] [Google Scholar]
- 79.Hechler B, Lenain N, Marchese P, et al. A role of the fast ATP-gated P2X1 cation channel in thrombosis of small arteries in vivo. J Exp Med. 2003;198:661–7. doi: 10.1084/jem.20030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tolhurst G, Vial C, Leon C, et al. Interplay between P2Y(1), P2Y(12), and P2X(1) receptors in the activation of megakaryocyte cation influx currents by ADP: evidence that the primary megakaryocyte represents a fully functional model of platelet P2 receptor signaling. Blood. 2005;106:1644–51. doi: 10.1182/blood-2005-02-0725. [DOI] [PubMed] [Google Scholar]
- 81.Soulet C, Hechler B, Gratacap MP, et al. A differential role of the platelet ADP receptors P2Y and P2Y in Rac activation. J Thromb Haemost. 2005;3:2296–306. doi: 10.1111/j.1538-7836.2005.01588.x. [DOI] [PubMed] [Google Scholar]
- 82.Nurden P, Savi P, Heilmann E, et al. An inherited bleeding disorder linked to a defective interaction between ADP and its receptor on platelets. Its influence on glycoprotein IIb-IIIa complex function. J Clin Invest. 1995;95:1612–22. doi: 10.1172/JCI117835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Foster CJ. Molecular identification and characterization of the platelet ADP receptor targeted by thienopyridine drugs using P2Yac-null mice. J Clin Invest. 2001;107:1591–8. doi: 10.1172/JCI12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jantzen HM, Milstone DS, Gousset L, et al. Impaired activation of murine platelets lacking Galphai2. J Clin Invest. 2001;108:477–83. doi: 10.1172/JCI12818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang J, Wu J, Jiang H, et al. Signaling through Gi family members in platelets–redundancy and specificity in the regulation of adenylyl cyclase and other effectors. J Biol Chem. 2002;277:46035–42. doi: 10.1074/jbc.M208519200. [DOI] [PubMed] [Google Scholar]
- 86.Yang J, Wu J, Kowalska MA, et al. Loss of signaling through the G protein, Gz, results in abnormal platelet activation and altered responses to psychoactive drugs. Proc Natl Acad Sci U S A. 2000;97:9984–9. doi: 10.1073/pnas.180194597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cattaneo M, Gachet C. ADP receptors and clinical bleeding disorders. Arterioscler Thromb Vasc Biol. 1999;19:2281–5. doi: 10.1161/01.atv.19.10.2281. [DOI] [PubMed] [Google Scholar]
- 88.Fabre JE, Nguyen MT, Latour A, et al. Decreased platelet aggregation, increased bleeding time and resistance to thromboembolism in P2Y1-deficient mice. Nat Med. 1999;5:1199–202. doi: 10.1038/13522. [DOI] [PubMed] [Google Scholar]
- 89.Léon C, Hechler B, Freund M, et al. Defective platelet aggregation and increased resistance to thrombosis in purinergic P2Y1 receptor-null mice. J Clin Invest. 1999;104:1731–7. doi: 10.1172/JCI8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Paul BZ, Jin JG, Kunapuli SP. Molecular mechanism of thromboxane A2-induced platelet aggregation–essential role for P2TAC and alpha2A receptors. J Biol Chem. 1999;274:29108–14. doi: 10.1074/jbc.274.41.29108. [DOI] [PubMed] [Google Scholar]
- 91.Nash CA, Severin S, Dawood BB, et al. Src family kinases are essential for primary aggregation by G(i)-coupled receptors. J Thromb Haemost. 2010;8:2273–82. doi: 10.1111/j.1538-7836.2010.03992.x. [DOI] [PubMed] [Google Scholar]
- 92.Gerrard JM, Carroll RC. Stimulation of protein phosphorylation by arachidonic acid and endoperoxide analog. Prostaglandins. 1981;22:81–94. doi: 10.1016/0090-6980(81)90055-1. [DOI] [PubMed] [Google Scholar]
- 93.FitzGerald GA. Mechanisms of platelet activation: thromboxane A2 as an amplifying signal for other agonists. Am J Correct. 1991;68:11B–5B. doi: 10.1016/0002-9149(91)90379-y. [DOI] [PubMed] [Google Scholar]
- 94.Brass LF, Shaller CC, Belmonte EJ. Inositol 1,4,5-triphosphate-induced granule secretion in platelets. Evidence that the activation of phospholipase C mediated by platelet thromboxane receptors involves a guanine nucleotide binding protein-dependent mechanism distinct from that of thrombin. J Clin Invest. 1987;79:1269–75. doi: 10.1172/JCI112947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hirata T, Ushikubi F, Kakizuka A, et al. Two thromboxane A2 receptor isoforms in human platelets–opposite coupling to adenylyl cyclase with different sensitivity to Arg60 to Leu mutation. J Clin Invest. 1996;97:949–56. doi: 10.1172/JCI118518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gao Y, Tang S, Zhou S, et al. The thromboxane A2 receptor activates mitogen-activated protein kinase via protein kinase C-dependent Gi coupling and Src-dependent phosphorylation of the epidermal growth factor receptor. J Pharmacol Exp Ther. 2001;296:426–33. [PubMed] [Google Scholar]
- 97.Thomas DW, Mannon RB, Mannon PJ, et al. Coagulation defects and altered hemodynamic responses in mice lacking receptors for thromboxane A2. J Clin Invest. 1998;102:1994–2001. doi: 10.1172/JCI5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Higuchi W, Fuse I, Hattori A, et al. Mutations of the platelet thromboxane A2 (TXA2) receptor in patients characterized by the absence of TXA2-induced platelet aggregation despite normal TXA2 binding activity. Thromb Haemost. 1999;82:1528–31. [PubMed] [Google Scholar]
- 99.Rao AK, Willis J, Kowalska MA, et al. Differential requirements for platelet aggregation and inhibition of adenylate cyclase by epinephrine. Studies of a familial platelpha2-adrenergic receptor defect. Blood. 1988;71:494–501. [PubMed] [Google Scholar]
- 100.Tamponi G, Pannocchia A, Arduino C, et al. Congenital deficiency of alpha2-adrenoreceptors on human platelets: description of two cases. Thromb Haemost. 1987;58:1012–6. [PubMed] [Google Scholar]
- 101.Newman KD, Williams LT, Bishopric NH, et al. Identification of alpha-adrenergic receptors in human platelets by 3H-dihydroergocryptine binding. J Clin Invest. 1978;61:395–402. doi: 10.1172/JCI108950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kaywin P, McDonough M, Insel PA, et al. Platelet function in essential thrombocythemia: decreased epinephrine responsivenesss associated with a deficiency of platelet alpha-adrenergic receptors. N Engl J Med. 1978;299:505–9. doi: 10.1056/NEJM197809072991002. [DOI] [PubMed] [Google Scholar]
- 103.Siess W, Weber PC, Lapetina EG. Activation of phospholipase C is dissociated from arachidonate metabolism during platelet shape change induced by thrombin or platelet-activating factor. Epinephrine does not induce phospholipase C activation or platelet shape change. J Biol Chem. 1984;259:8286–92. [PubMed] [Google Scholar]
- 104.Williams A, Woolkalis MJ, Poncz M, et al. Identification of the pertussis toxin-sensitive G proteins in platelets, megakaryocytes and HEL cells. Blood. 1990;76:721–30. [PubMed] [Google Scholar]
- 105.Woulfe D, Jiang H, Mortensen R, et al. Activation of Rap1B by Gi family members in platelets. J Biol Chem. 2002;277:23382–90. doi: 10.1074/jbc.M202212200. [DOI] [PubMed] [Google Scholar]
- 106.Heemskerk JW, Willems GM, Rook MB, et al. Ragged spiking of free calcium in ADP-stimulated human platelets: regulation of puff-like calcium signals in vitro and ex vivo. J Physiol. 2001;535:625–35. doi: 10.1111/j.1469-7793.2001.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Heemskerk JW, Hoyland J, Mason WT, et al. Spiking in cytosolic calcium concentration in single fibrinogen-bound fura-2-loaded human platelets. Biochem J. 1992;283:379–83. doi: 10.1042/bj2830379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Purvis JE, Chatterjee MS, Brass LF, et al. A molecular signaling model of platelet phosphoinositide and calcium regulation during homeostasis and P2Y1 activation. Blood. 2008;112:4069–79. doi: 10.1182/blood-2008-05-157883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Varga-Szabo D, Braun A, Nieswandt B. STIM and Orai in platelet function. Cell Calcium. 2011;50:270–8. doi: 10.1016/j.ceca.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 110.Crittenden JR, Bergmeier W, Zhang Y, et al. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat Med. 2004;10:982–6. doi: 10.1038/nm1098. [DOI] [PubMed] [Google Scholar]
- 111.Rittenhouse-Simmons S, Deykin D. Release and metabolism of arachidonate in human platelets. In: Gordon JL, editor. Platelets in biology and pathology-2. Elsevier; North-Holland: 1981. pp. 349–72. [Google Scholar]
- 112.Rittenhouse-Simmons S, Russell FA, Deykin D. Mobilization of arachidonic acid in human platelets. Kinetics and Ca2+ dependency. Biochim Biophys Acta. 1977;488:370–80. doi: 10.1016/0005-2760(77)90196-5. [DOI] [PubMed] [Google Scholar]
- 113.Börsch-Haubold AG, Ghomashchi F, Pasquet S, et al. Phosphorylation of cytosolic phospholipase A2 in platelets is mediated by multiple stress-activated protein kinase pathways. Eur J Biochem. 1999;265:195–203. doi: 10.1046/j.1432-1327.1999.00722.x. [DOI] [PubMed] [Google Scholar]
- 114.Börsch-Haubold AG, Kramer RM, Watson SP. Cytosolic phospholipase A2 is phosphorylated in collagen- and thrombin-stimulated human platelets independent of protein kinase C and mitogen-activated protein kinase. J Biol Chem. 1995;270:25885–92. doi: 10.1074/jbc.270.43.25885. [DOI] [PubMed] [Google Scholar]
- 115.Jin JG, Quinton TM, Zhang J, et al. Adenosine diphosphate (ADP)-induced thromboxane A2 generation in human platelets requires coordinated signaling through integrin alphaIIBbeta3 and ADP receptors. Blood. 2002;99:193–8. doi: 10.1182/blood.v99.1.193. [DOI] [PubMed] [Google Scholar]
- 116.Klages B, Brandt U, Simon MI, et al. Activation of G12/G13 results in shape change and Rho/Rho-kinase-mediated myosin light chain phosphorylation in mouse platelets. J Cell Biol. 1999;144:745–54. doi: 10.1083/jcb.144.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fukuhara S, Chikumi H, Gutkind JS. RGS-containing RhoGEFs: the missing link between transforming G proteins and Rho? Oncogene. 2001;20:1661–8. doi: 10.1038/sj.onc.1204182. [DOI] [PubMed] [Google Scholar]
- 118.Wilde JI, Retzer M, Siess W, et al. ADP-induced platelet shape change: an investigation of the signalling pathways involved and their dependence on the method of platelet preparation. Platelets. 2000;11:286–95. doi: 10.1080/09537100050129305. [DOI] [PubMed] [Google Scholar]
- 119.Pandey D, Goyal P, Bamburg JR, et al. Regulation of LIM-kinase 1 and cofilin in thrombin-stimulated platelets. Blood. 2005;107:575–83. doi: 10.1182/blood-2004-11-4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Woulfe D, Jiang H, Morgans A, et al. Defects in secretion, aggregation, and thrombus formation in platelets from mice lacking Akt2. J Clin Invest. 2004;113:441–50. doi: 10.1172/JCI20267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen J, De S, Damron DS, et al. Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood. 2004;104:1703–10. doi: 10.1182/blood-2003-10-3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.O’Brien KA, Stojanovic-Terpo A, Hay N, et al. An important role for Akt3 in platelet activation and thrombosis. Blood. 2011;118:4215–23. doi: 10.1182/blood-2010-12-323204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jackson SP, Schoenwaelder SM, Goncalves I, et al. PI 3-kinase p110beta: a new target for antithrombotic therapy. Nat Med. 2005;11:507–14. doi: 10.1038/nm1232. [DOI] [PubMed] [Google Scholar]
- 124.Brass LF, Zhu L, Stalker TJ. Minding the gaps to promote thrombus growth and stability. J Clin Invest. 2005;115:3385–92. doi: 10.1172/JCI26869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shattil SJ. The beta3 integrin cytoplasmic tail: protein scaffold and control freak. J Thromb Haemost. 2009;7(Suppl 1):210–3. doi: 10.1111/j.1538-7836.2009.03397.x. [DOI] [PubMed] [Google Scholar]
- 126.Prevost N, Kato H, Bodin L, et al. Platelet integrin adhesive functions and signaling. Meth Enzymol. 2007;426:103–15. doi: 10.1016/S0076-6879(07)26006-9. [DOI] [PubMed] [Google Scholar]
- 127.Stegner D, Nieswandt B. Platelet receptor signaling in thrombus formation. J Mol Med. 2011;89:109–21. doi: 10.1007/s00109-010-0691-5. [DOI] [PubMed] [Google Scholar]
- 128.Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104:1606–15. doi: 10.1182/blood-2004-04-1257. [DOI] [PubMed] [Google Scholar]
- 129.Calderwood DA, Ginsberg MH. Talin forges the links between integrins and actin. Nat Cell Biol. 2003;5:694–7. doi: 10.1038/ncb0803-694. [DOI] [PubMed] [Google Scholar]
- 130.Calderwood DA, Zent R, Grant R, et al. The talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J Biol Chem. 1999;274:28071–4. doi: 10.1074/jbc.274.40.28071. [DOI] [PubMed] [Google Scholar]
- 131.Tadokoro S, Shattil SJ, Eto K, et al. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302:103–6. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- 132.Ratnikov BI, Partridge AW, Ginsberg MH. Integrin activation by talin. J Thromb Haemost. 2005;3:1783–90. doi: 10.1111/j.1538-7836.2005.01362.x. [DOI] [PubMed] [Google Scholar]
- 133.Law DA, DeGuzman FR, Heiser P, et al. Integrin cytoplasmic tyrosine motif is required for outside-in alphaIIbbeta3 signalling and platelet function. Nature. 1999;401:808–11. doi: 10.1038/44599. [DOI] [PubMed] [Google Scholar]
- 134.Law DA, Nannizzi-Alaimo L, Ministri K, et al. Genetic and pharmacological analyses of Syk function in alphaIIbbeta3 signaling in platelets. Blood. 1999;93:2645–52. [PubMed] [Google Scholar]
- 135.Payrastre B, Missy K, Trumel C, et al. The integrin alphaIIb/beta3 in human platelet signal transduction. Biochem Pharmacol. 2000;60:1069–74. doi: 10.1016/s0006-2952(00)00417-2. [DOI] [PubMed] [Google Scholar]
- 136.Philips DR, Prasad KS, Manganello J, et al. Integrin tyrosine phosphorylation in platelet signaling. Curr Opin Cell Biol. 2001;13:546–54. doi: 10.1016/s0955-0674(00)00250-7. [DOI] [PubMed] [Google Scholar]
- 137.Phillips DR, Nannizzi-Alamio L, Prasad KS. Beta3 tyrosine phosphorylation in alphaIIbbeta3 (platelet membrane GP IIb-IIIa) outside-in integrin signaling. Thromb Haemost. 2001;86:246–58. [PubMed] [Google Scholar]
- 138.Obergfell A, Eto K, Mocsai A, et al. Coordinate interactions of Csk, Src and Syk kinases with alphaIIbbeta3 initiate integrin signaling to the cytoskeleton. J Cell Biol. 2002;157:265–75. doi: 10.1083/jcb.200112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Boylan B, Gao C, Rathore V, et al. Identification of FcgammaRIIa as the ITAM-bearing receptor mediating alphaIIbbeta3 outside-in integrin signaling in human platelets. Blood. 2008;112:2780–6. doi: 10.1182/blood-2008-02-142125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Prevost N, Woulfe D, Tanaka T, et al. Interactions between Eph kinases and ephrins provide a mechanism to support platelet aggregation once cell-to-cell contact has occurred. Proc Natl Acad Sci U S A. 2002;99:9219–24. doi: 10.1073/pnas.142053899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Prevost N, Woulfe DS, Tognolini M, et al. Signaling by ephrinB1 and Eph kinases in platelets promotes Rap1 activation, platelet adhesion, and aggregation via effector pathways that do not require phosphorylation of ephrinB1. Blood. 2004;103:1348–55. doi: 10.1182/blood-2003-06-1781. [DOI] [PubMed] [Google Scholar]
- 142.Prevost N, Woulfe DS, Jiang H, et al. Eph kinases and ephrins support thrombus growth and stability by regulating integrin outside-in signaling in platelets. Proc Natl Acad Sci U S A. 2005;102:9820–5. doi: 10.1073/pnas.0404065102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pasterkamp RJ, Giger RJ. Semaphorin function in neural plasticity and disease. Curr Opin Neurobiol. 2009;19:263–74. doi: 10.1016/j.conb.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Roth L, Koncina E, Satkauskas S, et al. The many faces of semaphorins: from development to pathology. Cell Mol Life Sci. 2009;66:649–66. doi: 10.1007/s00018-008-8518-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gherardi E, Love CA, Esnouf RM, et al. The sema domain. Curr Opin Struct Biol. 2004;14:669–78. doi: 10.1016/j.sbi.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 146.Negishi M, Oinuma I, Katoh H. Plexins: axon guidance and signal transduction. Cell Mol Life Sci. 2005;62:1363–71. doi: 10.1007/s00018-005-5018-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhu L, Bergmeier W, Wu J, et al. Regulated surface expression and shedding support a dual role for semaphorin 4D in platelet responses to vascular injury. Proc Natl Acad Sci U S A. 2007;104:1621–6. doi: 10.1073/pnas.0606344104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wannemacher KM, Zhu L, Jiang H, et al. Diminished contact-dependent reinforcement of Syk activation underlies impaired thrombus growth in mice lacking Semaphorin 4D. Blood. 2010;116:5707–15. doi: 10.1182/blood-2010-04-279943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Furlong B, Henderson AH, Lewis MJ, et al. Endothelium-derived relaxing factor inhibits in vitro platelet aggregation. Br J Pharmacol. 1987;90:687–92. doi: 10.1111/j.1476-5381.1987.tb11221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Whittle BJ, Moncada S. Pharmacological interactions between prostacyclin and thromboxanes. Br Med Bull. 1983;39:232–8. doi: 10.1093/oxfordjournals.bmb.a071825. [DOI] [PubMed] [Google Scholar]
- 151.Weksler BB. Prostacyclin. Prog Hemost Thromb. 1982;6:113–38. [PubMed] [Google Scholar]
- 152.Yuhki K, Kojima F, Kashiwagi H, et al. Roles of prostanoids in the pathogenesis of cardiovascular diseases: novel insights from knockout mouse studies. Pharmacol Ther. 2011;129:195–205. doi: 10.1016/j.pharmthera.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 153.Marcus AJ, Broekman MJ, Drosopoulos JH, et al. The endothelial cell ecto-ADPase responsible for inhibition of platelet function is CD39. J Clin Invest. 1997;99:1351–60. doi: 10.1172/JCI119294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Abramow-Newerly M, Roy AA, Nunn C, et al. RGS proteins have a signalling complex: interactions between RGS proteins and GPCRs, effectors, and auxiliary proteins. Cell Signal. 2006;18:579–91. doi: 10.1016/j.cellsig.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 155.Yowe D, Weich N, Prabhudas M, et al. RGS18 is a myeloerythroid lineage-specific regulator of G-protein-signalling molecule highly expressed in megakaryocytes. Biochem J. 2001;359:109–18. doi: 10.1042/0264-6021:3590109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nagata Y, Oda M, Nakata H, et al. A novel regulator of G-protein signaling bearing GAP activity for Galphai and Galphaq in megakaryocytes. Blood. 2001;97:3051–60. doi: 10.1182/blood.v97.10.3051. [DOI] [PubMed] [Google Scholar]
- 157.Kim SD, Sung HJ, Park SK, et al. The expression patterns of RGS transcripts in platelets. Platelets. 2006;17:493–7. doi: 10.1080/09537100600758123. [DOI] [PubMed] [Google Scholar]
- 158.Gagnon AW, Murray DL, Leadley RJ. Cloning and characterization of a novel regulator of G protein signalling in human platelets. Cell Signal. 2002;14:595–606. doi: 10.1016/s0898-6568(02)00012-8. [DOI] [PubMed] [Google Scholar]
- 159.Garcia A, Prabhakar S, Hughan S, et al. Differential proteome analysis of TRAP-activated platelets: involvement of DOK-2 and phosphorylation of RGS proteins. Blood. 2004;103:2088–95. doi: 10.1182/blood-2003-07-2392. [DOI] [PubMed] [Google Scholar]
- 160.Berthebaud M, Riviere C, Jarrier P, et al. RGS16 is a negative regulator of SDF-1-CXCR4 signaling in megakaryocytes. Blood. 2005;106:2962–8. doi: 10.1182/blood-2005-02-0526. [DOI] [PubMed] [Google Scholar]
- 161.Ma P, Cierniewska A, Signarvic R, et al. A newly-identified complex of spinophilin and the tyrosine phosphatase, SHP-1, modulates platelet activation by regulating G protein-dependent signaling. Blood. 2012;119:1935–45. doi: 10.1182/blood-2011-10-387910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gegenbauer K, Elia G, Blanco-Fernandez A, et al. Regulator of G-protein signaling protein 18 integrates activating and inhibitory signaling in platelets. Blood. 2012;119(16):3799–807. doi: 10.1182/blood-2011-11-390369. [DOI] [PubMed] [Google Scholar]
- 163.Haslam RJ, Dickinson NT, Jang EK. Cyclic nucleotides and phosphodiesterases in platelets. Thromb Haemost. 1999;82:412–23. [PubMed] [Google Scholar]
- 164.Keularts IM, Van Gorp RM, Feijge MA, et al. Alpha2A-adrenergic receptor stimulation potentiates calcium release in platelets by modulating cAMP levels. J Biol Chem. 2000;275:1763–72. doi: 10.1074/jbc.275.3.1763. [DOI] [PubMed] [Google Scholar]
- 165.Murata T, Ushikubi F, Matsuoka T, et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388:678–82. doi: 10.1038/41780. [DOI] [PubMed] [Google Scholar]
- 166.Quinton TM, Dean WL. Cyclic AMP-dependent phosphorylation of the inositol-1,4,5-trisphosphate receptor inhibits Ca21 release from platelet membranes. Biochem Biophys Res Commun. 1992;184:893–9. doi: 10.1016/0006-291x(92)90675-b. [DOI] [PubMed] [Google Scholar]
- 167.Grunberg B, Kruse HJ, Negrescu EV, et al. Platelet rap1B phosphorylation is a sensitive marker for the action of cyclic AMP- and cyclic GMP-increasing platelet inhibitors and vasodilators. J Cardiovasc Pharmacol. 1995;25:545–51. doi: 10.1097/00005344-199504000-00006. [DOI] [PubMed] [Google Scholar]
- 168.Grunberg B, Negrescu E, Siess W. Synergistic phosphorylation of platelet rap1B by SIN-1 and iloprost. Eur J Pharmacol. 1995;288:329–33. doi: 10.1016/0922-4106(95)90045-4. [DOI] [PubMed] [Google Scholar]
- 169.Smolenski A. Novel roles of cAMP/cGMP dependent signaling in platelets. J Thromb Haemost. 2011;10(2):167–76. doi: 10.1111/j.1538-7836.2011.04576.x. [DOI] [PubMed] [Google Scholar]
- 170.Margarucci L, Roest M, Preisinger C, et al. Collagen stimulation of platelets induces a rapid spatial response of cAMP and cGMP signaling scaffolds. Mol Biosyst. 2011;7:2311–9. doi: 10.1039/c1mb05145h. [DOI] [PubMed] [Google Scholar]
- 171.Wangorsch G, Butt E, Mark R, et al. Time-resolved in silico modeling of finetuned cAMP signaling in platelets: feedback loops, titrated phosphorylations and pharmacological modulation. BMC Syst Biol. 2011;5:178. doi: 10.1186/1752-0509-5-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gambaryan S, Kobsar A, Rukoyatkina N, et al. Thrombin and collagen induce a feedback inhibitory signaling pathway in platelets involving dissociation of the catalytic subunit of protein kinase A from an NFkappaB-IkappaB complex. J Biol Chem. 2010;285:18352–63. doi: 10.1074/jbc.M109.077602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hayashi H, Sudo T. Effects of the cAMP-elevating agents cilostamide, cilostazol and forskolin on the phosphorylation of Akt and GSK-3beta in platelets. Thromb Haemost. 2009;102:327–35. doi: 10.1160/TH08-12-0781. [DOI] [PubMed] [Google Scholar]
- 174.Zahedi RP, Lewandrowski U, Wiesner J, et al. Phosphoproteome of resting human platelets. J Proteome Res. 2008;7:526–34. doi: 10.1021/pr0704130. [DOI] [PubMed] [Google Scholar]
- 175.Newman PJ, Newman DK. Signal transduction pathways mediated by PECAM-1: new roles for an old molecule in platelet and vascular cell biology. Arterioscler Thromb Vasc Biol. 2003;23:953–64. doi: 10.1161/01.ATV.0000071347.69358.D9. [DOI] [PubMed] [Google Scholar]
- 176.Jackson DE, Kupcho KR, Newman PJ. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of platelet/endothelial cell adhesion molecule-1 (PECAM-1) that are required for the cellular association and activation of the protein-tyrosine phosphatase, SHP-2. J Biol Chem. 1997;272:24868–75. doi: 10.1074/jbc.272.40.24868. [DOI] [PubMed] [Google Scholar]
- 177.Moraes LA, Barrett NE, Jones CI, et al. PECAM-1 regulates collagen-stimulated platelet function by modulating the association of PI3 Kinase with Gab1 and LAT. J Thromb Haemost. 2010;8:2530–41. doi: 10.1111/j.1538-7836.2010.04025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Patil S, Newman DK, Newman PJ. Platelet endothelial cell adhesion molecule-1 serves as an inhibitory receptor that modulates platelet responses to collagen. Blood. 2001;97:1727–32. doi: 10.1182/blood.v97.6.1727. [DOI] [PubMed] [Google Scholar]
- 179.Falati S, Patil S, Gross PL, et al. Platelet PECAM-1 inhibits thrombus formation in vivo. Blood. 2006;107:535–41. doi: 10.1182/blood-2005-04-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Huber M, Izzi L, Grondin P, et al. The carboxyl-terminal region of biliary glycoprotein controls its tyrosine phosphorylation and association with proteintyrosine phosphatases SHP-1 and SHP-2 in epithelial cells. J Biol Chem. 1999;274:335–44. doi: 10.1074/jbc.274.1.335. [DOI] [PubMed] [Google Scholar]
- 181.Beauchemin N, Kunath T, Robitaille J, et al. Association of biliary glycoprotein with protein tyrosine phosphatase SHP-1 in malignant colon epithelial cells. Oncogene. 1997;14:783–90. doi: 10.1038/sj.onc.1200888. [DOI] [PubMed] [Google Scholar]
- 182.Wong C, Liu Y, Yip J, et al. CEACAM1 negatively regulates platelet-collagen interactions and thrombus growth in vitro and in vivo. Blood. 2009;113:1818–28. doi: 10.1182/blood-2008-06-165043. [DOI] [PubMed] [Google Scholar]
- 183.Stalker TJ, Wu J, Morgans A, et al. Endothelial cell specific adhesion molecule (ESAM) localizes to platelet-platelet contacts and regulates thrombus formation in vivo. J Thromb Haemost. 2009;7:1886–96. doi: 10.1111/j.1538-7836.2009.03606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Naik MU, Stalker TJ, Brass LF, et al. JAM-A protects from thrombosis by suppressing integrin alphaIIbbeta3-dependent outside-in signaling in platelets. Blood. 2012;119:3352–60. doi: 10.1182/blood-2011-12-397398. [DOI] [PMC free article] [PubMed] [Google Scholar]