

Abstract
Smith-Lemli-Opitz syndrome (SLOS) is a neurodevelopmental disorder caused by inborn errors of cholesterol metabolism resulting from mutations in 7-dehydrocholesterol reductase (DHCR7). There are only a few studies describing the brain imaging findings in SLOS. This study examines the prevalence of magnetic resonance imaging (MRI) abnormalities in the largest cohort of patients with SLOS to date. Fifty-five individuals with SLOS (27M, 28F) between age 0.17 years and 25.4 years (mean = 6.2, SD = 5.8) received a total of 173 brain MRI scans (mean = 3.1 per subject) on a 1.5T GE scanner between September, 1998 and December, 2003, or on a 3T Philips scanner between October 2010 and September 2012; all exams were performed at the Clinical Center of the National Institutes of Health. We performed a retrospective review of these imaging studies for both major and minor brain anomalies. Aberrant MRI findings were observed in 53 of 55 (96%) SLOS patients, with abnormalities of the septum pellucidum the most frequent (42/55, 76%) finding. Abnormalities of the corpus callosum were found in 38 of 55 (69%) patients. Other findings included cerebral atrophy, cerebellar atrophy, colpocephaly, white matter lesions, arachnoid cysts, Dandy-Walker variant, and Type I Chiari malformation. Significant correlations were observed when comparing MRI findings with sterol levels and somatic malformations. Individuals with SLOS commonly have anomalies involving the midline and para-midline structures of the brain. Further studies are required to examine the relationship between structural brain abnormalities and neurodevelopmental disability in SLOS.
Keywords: Smith-Lemli-Opitz syndrome, SLOS, MRI, magnetic resonance imaging, brain
INTRODUCTION
Smith-Lemli-Opitz syndrome (SLOS) is an autosomal recessive, multiple malformation, neurodevelopmental disorder caused by mutations in the gene encoding 7-dehydrocholesterolreductase (DHCR7), resulting in impaired cholesterol synthesis [Smith et al., 1964; Tint et al., 1994; Irons et al., 1993; Wassif et al., 1998; Porter, 2000]. Individuals with SLOS have decreased cholesterol, and excess of the precursor 7-dehydrocholesterol (7DHC) and its isomer 8-dehydrocholesterol (8DHC). The incidence is estimated to be 1 in 20,000 to 60,000 live births [Kelley and Hennekam, 2000; Lowry and Yong, 1980; Ryan et al., 1998; Bzduch et al., 2000]. Neuropathologic studies demonstrate that individuals with SLOS can have numerous brain abnormalities including microcephaly, holoprosencephaly, atelen/aprosencephaly, abnormal gyri, aqueductal stenosis with hydrocephalus, incomplete separation of the mammillary bodies, corpus callosum agenesis, dorsal fusion of the dorsomedial nuclei and pulvinar of the thalamus, hypoplasia of the cerebral peduncles, dysplasia of inferior olivary nuclei, deformity of the cerebellum and atrophy of the folia, Dandy-Walker malformation, hippocampal hypoplasia, and hypothalamic hamartoma [Garcia et al., 1973; Cherstvoy et al., 1975; Fierro et al., 1977; Cherstvoy et al., 1984; Curry et al.,1987; Lanoue et al., 1997; Angle et al., 1998; Kelley and Hennekam, 2000; Nowaczyk et al., 2001; Opitz et al., 2002; Hennekam, 2005; Putman et al., 2005; Weaver et al., 2010; Opitz and Furtado, 2012, Quélin et al., 2012; Grynspan et al., 2013]. Spinal cord abnormalities such as hydromyelia, holomyelia, and hypoplasia of the spinothalamic and spinocerebellar tracts have been described [Opitz and Furtado, 2012; Quélin et al., 2012]. Histologic examination has shown loss of Purkinje cells and neuronal degeneration in the cerebellum, exotopic Purkinje cells in the inner granular layer and white matter of the cerebellum, abnormal cell migration in the cerebral cortex showing neuronal presence in the first layer and white matter and reduced neurons in the third layer, lack of myelination, patchy two-layered structure of the hippocampus fascia dentate, disturbed orientation in the H2 field of Ammon’s horn, dysplastic dentate nucleus, dysplastic olivary nuclei of the medulla oblongata, and astrocyte gliosis [Fierro et al., 1977; Cherstvoy et al., 1984; Opitz et al., 2002; Opitz and Furtado, 2012]. Kelley et al. [1996] reported diagnosing SLOS in four patients with holoprosencephaly, and a report describing a 24-week gestation fetus with cyclopia further supports SLOS as a cause of holoprosencephaly [Weaver et al., 2010]. In general, neuropathological studies have been conducted on severely affected patients. Structural MRI studies are able to characterize mild and classical cases. There is one published study involving MRI in 18 patients with SLOS showing abnormal CNS findings in five patients including callosal abnormalities (n = 4), Dandy-Walker variant (n = 1), holoprosencephaly (n = 1), and an arachnoid cyst (n = 1) [Caruso et al., 2004].
While the SLOS behavioral and physical phenotype has been published, there are few studies describing the range of structural brain malformations [Cunniff et al., 1997; Porter, 2000; Tierney et al., 2001; Sikora et al., 2006; Tierney et al., 2006]. The aim of this study was to examine the prevalence of MRI-detectable brain abnormalities in SLOS, and to determine the relationship of these abnormalities to other markers of severity in these patients.
MATERIALS AND METHODS
Study Population
This study was approved by the Institutional Review Board of the Eunice Kennedy Shriver National Institute of Child Health and Human Development in Bethesda, Maryland. Patients or their parents/legal guardians signed informed consent in order to participate in the study. This study included 55 individuals with SLOS between 0.17 and 25.4 years of age on whom MRI scans of the brain were performed. The diagnosis of SLOS was made by physical exam then confirmed biochemically by an expert evaluator at the NIH Clinical Center (FDP). Sagittal T1 weighted, axial T1 weighted, and axial T2 weighted images were acquired as part of all exams on all patients. The exams also included either axial proton density weighted images or axial FLAIR images. Some exams also included 3D T1 weighted images (SPGR or MPRAGE), and some included post-contrast axial T1 weighted images. The examinations were performed using a 1.5T GE scanner between September 1998 and December 2003, or a 3T Philips scanner between October 2010 and September 2012; all exams were performed at the Clinical Center of the National Institutes of Health, and all exams were performed under sedation monitored by an anesthesiologist. A single neuroradiologist at the NIH Clinical Center evaluated and documented the imaging findings for all subjects. Review of medical records and parent interviews were used to acquire clinical data.
Severity Measures
The SLOS severity score is based on organ system findings in 10 embryological development fields [Bialer et al., 1987; Kelley and Hennekam, 2000]. Absolute scores range from 0–20; scores are frequently normalized to the maximal score based on the domains ascertained, but for this study we used the absolute scores. In order to examine the relationship between the presence of brain malformations detected in this study and somatic malformations we have calculated a somatic severity score (SSS) that removes the brain score from the anatomical severity score. We have developed a brain severity score (BSS) based on categories of brain abnormalities observed in this and other studies. Total severity score (TSS) was calculated as the sum of SSS and BSS. Serum 7-dehydrocholesterol (7DHC) (mg/dl) and total cholesterol (mg/dl) levels were determined at the time of diagnosis or represent the earliest value that we have been able to obtain. This avoids, to the extent possible, variation due to active dietary cholesterol supplementation. Linear regression analysis and Pearson correlation (r) statistics were performed to determine the degree of correlation between the BSS and the other measures of severity (SSS, 7DHC, and total cholesterol).
RESULTS
Fifty-five individuals with SLOS (27 males, 28 females) between 0.17 and 25.4 years old (mean = 6.2 +/− 5.8) had a total of 173 brain MRI scans performed (mean = 3.1). The patients were Caucasian (94.5%), Hispanic (3.6%), and Asian (1.8%). The group mean SSS was 3.22 (SD 1.95, range 1 – 9), mean serum cholesterol was 76.7 mg/dl (SD 44.8, range 8 – 174), and mean 7DHC was 10.1 mg/dl (SD 9.7, range 0.07 – 49.7). Demographics and group sample data are shown in Table I. A Supplementary Data Table with detailed data of demographics, severity scores, sterol levels, MRI findings, and genetic mutation status is provided in the supplemental data section (in supporting information online).
Table I.
Demographics and sample data of the SLOS patients (N=55).
| Mean (Standard Deviation) | Range | ||
|---|---|---|---|
| Age at scan (years) | 6.2 | (+/−5.8) | 0.17 to 25.40 |
| Gender | 27 M | 28 F | — |
| Somatic severity score (SSS) | 3.22 | (+/−1.9) | 1 to 9 |
| Brain severity score (BSS) | 9.13 | (+/−4.5) | 2 to 24 |
| Total severity score (TSS) | 12.35 | (+/−5.7) | 3 to 30 |
| Serum cholesterol (mg/dl) | 76.7 | (+/−44.8) | 8 to 174 |
| Serum 7DHC (mg/dl) | 10.1 | (+/−9.7) | 0.07 to 49.70 |
Most of the abnormalities found on the brain images of the SLOS patients involved midline structures, such as the corpus callosum, intraventricular septum, and inferior cerebellar vermis. There were also abnormalities of the ventricles, whose formation is related to the corpus callosum, and there were abnormalities of the cerebellar tonsils, whose formation is related to the inferior cerebellar vermis. We also found an unusually high frequency of arachnoid cysts, half of which were in the midline. The minor brain anomalies and their prevalence are detailed in Table II.
Table II.
Brain anatomical abnormalities in SLOS patients (N=55) detected by MRI.
| Abnormality | N | % |
|---|---|---|
| Cavum | 42 | 76 |
| Large communicating cavum | 21 | 38 |
|
| ||
| Incomplete corpus callosum | 12 | 22 |
| Hypoplastic or absent rostrum | 8 | 15 |
| Hypoplastic or absent splenium | 6 | 11 |
| Hypoplastic or absent splenium and posterior body | 4 | 7 |
|
| ||
| Thick and/or thin segments of the corpus callosum * | 31 | 56 |
| Thick genu | 9 | 16 |
| Thick body | 13 | 24 |
| Thick all segments | 2 | 4 |
| Thin genu | 6 | 11 |
| Thin body | 9 | 16 |
| Thin splenium | 15 | 27 |
| Thin all segments * | 5 | 9 |
|
| ||
| Abnormal shape of the corpus callosum * | 26 | 47 |
| Foreshortening | 13 | 24 |
| Arching * | 11 | 20 |
| Flattening | 2 | 4 |
|
| ||
| Colpocephaly | 35 | 64 |
| Bilateral | 21 | 38 |
| Left only | 7 | 13 |
| Right only | 7 | 13 |
| Minimal ** | 16 | 15 |
| Mild ** | 30 | 27 |
| Moderate ** | 8 | 7 |
| Marked ** | 2 | 2 |
|
| ||
| Posterior fossa abnormalities | 17 | 31 |
| Mild Dandy-Walker variant | 11 | 20 |
| Cerebellar tonsil ectopia | 2 | 4 |
| Type I Chiari malformation | 2 | 4 |
| Abnormal shape of the cerebellum (not hypoplastic) | 1 | 2 |
| Small gliotic cerebellum | 1 | 2 |
|
| ||
| Arachnoid cysts (28 total cysts in 17 patients) | 17 | 31 |
| Midline location, all types § | 14 | 50 |
| Quadrigeminal plate cistern § | 12 | 43 |
| Middle cranial fossa § | 7 | 25 |
| Posterior cranial fossa § | 5 | 18 |
| Anterior cranial fossa § | 2 | 7 |
| Torcular fossa § | 1 | 4 |
| Parietal § | 1 | 4 |
|
| ||
| Atrophy | 8 | 15 |
| Diffuse cerebral atrophy | 2 | 4 |
| Central pattern cerebral atrophy | 5 | 9 |
| Generalized cerebellar atrophy | 2 | 4 |
|
| ||
| White matter lesions | 2 | 4 |
The numbers reported do not include cases where the finding is secondary to an enlarged third ventricle.
The percentage is calculated relative to 110 total ventricles.
The percentage is calculated relative to 28 total arachnoid cysts.
The most common abnormalities found in the SLOS patients were abnormalities of the septum pellucidum, including cavum of the septum pellucidum (CSP), cavum Vergae, or cavum of the velum interpositum; 42 patients (76%) had one or more type of cavum. The most common variant was a large CSP that communicated with the third ventricle, present in 21 (38%) of the patients. A striking feature of the patients who had a large communicating CSP was that by itself, each lamina of the divided septum pellucidum was thicker than the total thickness of a normal undivided septum pellucidum. One patient had an anomalous white matter tract that crossed the large communicating CSP. Examples of abnormalities of the septum pellucidum are shown in Fig. 1.
Fig. 1.

The most obvious abnormalities involved the corpus callosum, found in 38 patients (69%). Twelve patients (22%) lacked a corpus callosum. There were 8 patients in whom the rostrum was hypoplastic or absent and 6 in whom the splenium was hypoplastic or absent; of these 6, there were 4 in whom the posterior body of the corpus callosum was also hypoplastic or absent. It was also a common occurrence to have segments that were either too thick or too thin. There were 31 patients (56%) who had at least one segment of the corpus callosum that was either unusually thick (17 patients) or unusually thin (19 patients), and there were 5 patients who had both. The most common findings were a thick body of the corpus callosum (13 patients) and a thin splenium (15 patients), but most other variations were present, including thick genu (6 patients), thin body (9 patients), thin genu (6 patients), generalized thinning (5 patients), and generalized thickening (2 patients). The overall shape of the corpus was abnormal in 26 patients (47%). The most common shape abnormality was shortening of the anterior-posterior dimension of the corpus callosum, which occurred in 13 patients. Almost as common was elongation in the superior-inferior dimension, producing excessive arching of the corpus callosum; this was found in 11 patients who did not have concurrent enlargement of the third ventricle (arching and thinning of the corpus callosum is a normal consequence of third ventricle enlargement). The arch was abnormally flat in two patients, and elongated in one other patient (secondary to enlargement of the third ventricle). Examples of abnormalities of the corpus callosum are shown in Fig. 2.
Fig. 2.

Colpocephaly, which we defined as any enlargement of the occipital horns that was disproportionate to the size of the remainder of the lateral ventricles, was present to some degree in 35 patients (64%). Both sides were involved in 21 patients, seven cases involved only the left side, and seven cases involved only the right side. Enlargement was rated as minimal in 16 occipital horns, mild in 30 occipital horns, moderate in eight occipital horns, and marked in two occipital horns. The one case that had marked enlargement of both occipital horns also had absent splenium and posterior body of the corpus callosum, and parallel orientation of the lateral ventricles.
Many patients had abnormalities of the fourth ventricle and cerebellum. There were 14 patients who had a large fourth ventricle and a large foramen of Magendie; of these, there were 11 (20%) who also had high position of the cerebellar tonsils and inferior vermis together with hypoplasia of the cerebellar tonsils and/or inferior vermis, resulting in a Dandy-Walker variant. Two patients (4%) had ectopia of the cerebellar tonsils (lower than normal position of the cerebellar tonsils without crowding of structures in the foramen magnum), and two patients (4%) had a Type I Chiari malformation. One patient’s cerebellum had a very unusual shape (short and flat, but completely formed and of normal volume). This patient also had a tethered spinal cord; de-tethering surgery was performed prior to our study, but there was no history of abnormal position of the tonsils in this patient. One patient had a small cerebellum with diffuse abnormal signal that suggested gliosis. Examples of abnormalities of the fourth ventricle and cerebellum are shown in Fig. 3.
Fig. 3.

An unusually high number of arachnoid cysts was present in this cohort of SLOS patients. Half of these arachnoid cysts were located in the midline. A total of 28 cysts were found in 17 patients (31%). There were 12 cysts in the quadrigeminal plate cistern, seven in the middle cranial fossa, five in the posterior cranial fossa, two in the anterior cranial fossa, one adjacent to the parietal lobe, and one in the torcular fossa. Examples of arachnoid cysts are shown in Fig. 4.
Fig. 4.

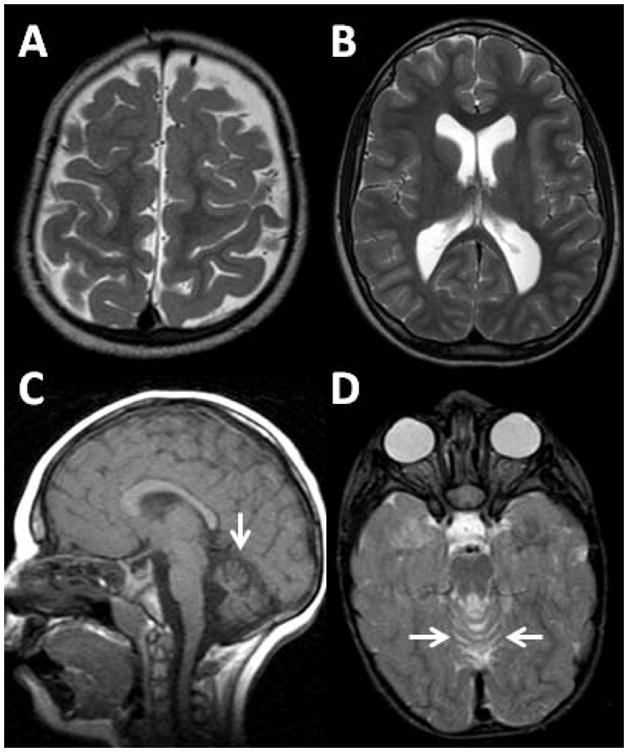
Few patients had cerebral atrophy. Two patients had prominent sulci, indicating diffuse cerebral atrophy, while five patients had a central atrophy pattern characterized by a thinner than normal layer of white matter with normal thickness of the cortical ribbon and normal width of the sulci. Two patients had atrophy of the cerebellum that preferentially (but not exclusively) involved the superior vermis. One patient had both cerebral and cerebellar atrophy, so there were a total of 8 patients (15%) who had atrophy of any type. Fig. 5 shows examples of the atrophy patterns that were seen in the SLOS patients.
Fig. 5.
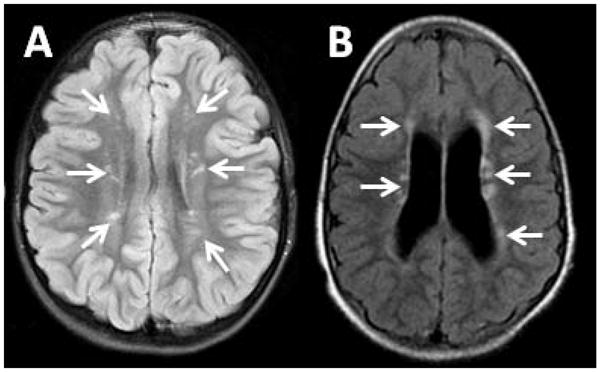
There were two patients who had discrete lesions in the white matter. These patients each had multiple lesions, but the pattern of distribution of the lesions was not the same for the two patients. In one patient, the lesions were all periventricular and were mostly located in the frontal and parietal lobes. In the other patient, the lesions were all in the centrum semiovale and were generally oriented with their long axis perpendicular to the lateral ventricles. We could not identify any history of a concurrent illness that may have explained the presence of these lesions in these particular patients. The lesions are shown in Fig. 6.
Fig. 6.
All brains on all patients were judged to have an appropriate degree of myelination for the patient’s age; therefore, all patients under age 4 who had follow-up imaging (25 of 29) demonstrated normal progression of myelination. We also did not detect heterotopia, microgyria, pachygyria, or holoprosencephaly variants. Abnormal orientation of gyri was seen only in the context of absence of the posterior corpus callosum, which necessarily results in incomplete cingulate gyri and development of radial gyri posterior to the defect. Two patients did not have any of the abnormalities described above, and therefore had structurally normal brains according to our evaluation.
Linear regression analysis for correlations between the types of abnormalities described above and the other severity measures (SSS, 7DHC at diagnosis, and total cholesterol at diagnosis) indicated that certain types of abnormalities correlated more strongly than others. The findings that correlated best with the other severity measures were the degree of colpocephaly and the position of the cerebellar tonsils (higher position correlating with greater severity). Atrophy and morphological abnormalities of the corpus callosum had more moderate degrees of correlation. Lobar anomalies and arachnoid cyst count had weak correlation. White matter lesions and septum pellucidum anomalies did not seem to correlate.
The anatomical findings were combined into a brain anatomy severity score (BSS); the method for combining the findings into the BSS is detailed in Table III. Statistically significant correlations were found for the BSS relative to the somatic severity score (SSS) (p < 0.001, r = 0.47), earliest available measurement of the total cholesterol (p < 0.001, r = −0.47), and earliest available measurement of 7DHC (p = 0.049, r = 0.27). Strength of correlation to 7DHC was increased by using the base 10 log of the 7DHC (p = 0.006, r = 0.37), demonstrating a log-linear relationship between these two factors, which is as expected, since the relationship between cholesterol and 7DHC was log-linear (p < 0.001, r = −0.71). Similarly, correlations were found between the SSS and the earliest available measurement of the total cholesterol (p = 0.002, r = −0.42), and earliest available measurement of the 7DHC (p = 0.02, r = 0.32). The total severity score (TSS, equal to the sum of BSS and SSS) also correlated with the earliest available measurement of the total cholesterol (p < 0.001, r = −0.52), earliest available measurement of the 7DHC (p = 0.016, r = 0.33), and the log of the 7DHC (p = 0.012, r = 0.34). Plots of these relationships are shown in Fig. 7.
Table III.
Brain severity score (BSS) for congenital brain anatomical abnormalities in SLOS.
| Abnormality | Score | Criteria |
|---|---|---|
| Colpocephaly (0–8) | Sum of the points for each side | |
| 0 | None | |
| 1 | Minimal | |
| 2 | Mild | |
| 3 | Moderate | |
| 4 | Severe | |
|
| ||
| Cerebellar tonsils (2–8) | Position relative to the foramen magnum | |
| 2 | Below the foramen magnum | |
| 4 | Within the foramen magnum | |
| 6 | Posterior fossa, not high | |
| 8 | High | |
|
| ||
| Corpus callosum (0–16) | Sum of 3 sub-score domains below | |
| 0 – 10 | Number of absent segments, multiply by 2 * | |
| 0 – 5 | Number of thick or thin segments * | |
| 0 – 1 | Arching, flattening, or foreshortening | |
|
| ||
| Atrophy (0–3) | Rate the severity of the atrophy | |
| 0 | None | |
| 1 | Mild | |
| 2 | Moderate | |
| 3 | Severe | |
|
| ||
| Lobar anomalies (0–1) | 0 | Hypoplastic lobe of the cerebrum or cerebellum |
| 1 | Malformed lobe of cerebrum or cerebellum | |
|
| ||
| Arachnoid cysts (0–4) | 0 – 4 | Number of arachnoid cysts ** |
|
| ||
| Septum pellucidum (0–3) | Cavum septum pellucidum, cavum Vergae, or cavum velum interpositum | |
| 0 | No cavum | |
| 1 | Non-communicating cavum, 1 segment | |
| 2 | Non-communicating cavum, 2 or 3 segments | |
| 3 | Communicating cavum | |
|
| ||
| Holoprosencephaly (0–4) | Rate the severity of the holoprosencephaly variant | |
| 0 | None | |
| 1 | Absent septum pellucidum | |
| 2 | Lobar holoprosencephaly | |
| 3 | Semilobar holoprosencephaly | |
| 4 | Alobar holoprosencephaly | |
|
| ||
| Other malformations (0–3) | Rate the severity of congenital brain abnormalities not listed above§ | |
| 0 | None | |
| 1 | Minor | |
| 2 | Intermediate | |
| 3 | Major | |
The body is split into anterior and posterior segments, for a total of 5 segments.
The maximum number of arachnoid cysts observed in our cohort was 4.
Potentially includes abnormalities such as pachygyria, microgyria, heterotopia, schizencephaly, aqueductal stenosis, Dandy-Walker malformation, meningocele, encephalocele, etc. Abnormalities occurring with a prevalence of less than about 1% in the SLOS population would have a 50% chance of going undetected in a study of 55 patients. Prenatal or perinatal lethal abnormalities would also not be detected, as this study included only patients who were at least 2 months old.
Fig. 7.

DISCUSSION
In 55 individuals with SLOS, we found that there were only two who did not have a structural brain abnormality when evaluated by brain MRI. The most common abnormalities involved the septum pellucidum. Other common abnormalities involved the corpus callosum, occipital horns, cerebellum, and arachnoid mater. Atrophy was found in some patients, whereas white matter lesions were found in a few patients. In contrast to the previous study of Caruso et al. [2004] we did not find any cases in which the septum pellucidum was absent. We also found an overall higher rate of anomalies (53 of 55 in our study, versus 5 of 18 in their study). However, the anomalies that we report involved the same anatomical structures that were reported previously. Like the previous study, we did not find any abnormal patterns of myelination.
Non-union of the leaflets (laminae) of the septum pellucidum is a normal stage of development in the fetus [Shaw and Alvord, 1969]. The laminae are fused in about half of term infants, and most of the rest fuse within the first few months after birth [Shaw and Alvord, 1969; Farruggia and Babcock, 1981]. The rate of persistence of CSP into adulthood is estimated at 12% [Shaw and Alvord, 1969]. The rate of persistence of cavum Vergae into adulthood is estimated at 20% [Schwidde, 1952]. Our finding of a 78% rate of non-fusion of the leaflets of the septum pellucidum is clearly different from the rate in normal subjects, but the clinical significance is unclear, as a cavum is not generally thought to produce symptoms. We were unable to find a previously published description of the communicating cavum with thickened laminae, so the consequences of this variation are also unknown.
Arachnoid cysts are not generally thought to produce symptoms. The frequency of arachnoid cysts (31%) in our population is clearly different from that in normal adults, which is about 1% [Vernooij et al., 2007]. Additionally, the most common location in the SLOS patients, the quadrigeminal plate cistern, is unusual in the general population; the most common location in the normal population is in the middle cranial fossa, and the next most common location is in the posterior cranial fossa [Vernooij et al., 2007].
In contrast to a cavum or arachnoid cyst, abnormalities of the corpus callosum and colpocephaly clearly have the potential to produce symptoms, as both affect the large interhemispheric white matter tracts. The Dandy-Walker variant and Type I Chiari malformation also have the potential to produce symptoms, as the former involves hypoplasia of portions of the cerebellum, and the latter can cause pressure-related injury to parts of the cerebellum and spinal cord. The prevalence of Type I Chiari malformation in the general population is about 1%, while our prevalence was 4% [Vernooij et al., 2007]. We were unable to find any published reports on the prevalence of Dandy-Walker variant; review of the images of 101 normal volunteers recruited for other NIH brain imaging studies turned up 0 instances of Dandy-Walker variant, suggesting that the prevalence in the normal population is (conservatively) less than 1%, while the prevalence in our SLOS patients was 20%.
Atrophy and white matter lesions most often do not represent congenital structural abnormalities, except in cases of genetic predisposition toward inability to form myelin. Acquired atrophy and white matter abnormalities arise as a consequence of injury (diffuse injury in the case of atrophy, and localized injury in the case of discrete white matter lesions). Atrophy in the context of a metabolic disease such as SLOS implies a mechanism of injury related to accumulation of abnormal or toxic metabolites, or deficiency of the normal end product of the affected metabolic pathway. The potential connection between SLOS and discrete white matter lesions is unclear.
Our finding of an appropriate degree of myelination for age was to some extent unexpected given the high cholesterol content of myelin. We are not aware of any human autopsy studies that have systematically characterized myelin content and structure in SLOS patients, and the current mouse models are limited with respect to characterizing myelin by either early perinatal death prior to significant myelination in the mouse or by a very mild biochemical phenotype. Thus, it is not clear as to what degree dehydrocholesterol is able to functionally substitute for cholesterol in myelin, and, in addition, the patients in this study have residual cholesterol synthetic activity. Thus, the mild to classical patients ascertained in this study may have sufficient functional sterol levels with respect to myelin formation. It is also possible that structural MRI may not be sensitive enough to identify mild disturbances in myelin formation. These questions can be explored further using advanced MRI methods, such as diffusion tensor imaging which provides information on macromolecular structure, and using magnetic resonance spectroscopy which provides information on other biochemical factors.
The significant correlations found between the brain severity score (BSS) and other measures of SLOS severity (SSS, 7DHC, and total cholesterol) demonstrates a relationship between disease severity and brain structure. Although some abnormalities are more common than others, a theme of multiple minor abnormalities affecting a limited set of structures is noteworthy. These abnormalities do not seem to occur in a particular order, but they are more severe and numerous in more severely affected individuals.
The degree to which the brain malformations give prognostic insight into cognitive and behavioral impairment is an area that will need to be explored in more detail. In a rare disease with a heterogeneous phenotype, power to detect such associations is limited. However, we have recently correlated lower corpus callosum length and cross sectional area with both lower developmental quotient in gross motor and language domains in SLOS [Lee et al., 2013].
Most of the anatomical abnormalities we saw involved midline and para-midline structures. Formation of midline brain structures occurs between 3 and 12 weeks gestation and involves molecular signals that regulate neural tube patterning. Cholesterol serves as a co-factor for an important ventral patterning protein, sonic hedgehog [Ericson et al., 1995; Rubenstein and Beachy, 1998]. Sonic hedgehog may also play a role in patterning of dorsal midline structures [Himmelstein et al., 2010]. Holoprosencephaly, a failure of the forebrain to separate into two hemispheres, occurs around the 5th week of gestation [Golden, 1999]. Although holoprosencephaly was not found in our cohort, it has been reported in several previous papers [Cunniff et al., 1997; Kelley et al., 1996; Lanoue et al., 1997; Nowaczyk et al., 2001; Weaver et al., 2010]. Abnormal sterol levels during embryologic brain development have been shown to disrupt early midline patterning processes, and may represent a mechanism of disease for the malformations reported in our study. Cholesterol is essential for processing and signaling of sonic hedgehog, a morphogen involved in midline development [Beachy et al., 1997; Cooper et al., 2003; Porter et al., 1996]. Cholesterol is also involved in myelin structure, membrane lipid raft distribution, activity-dependent synaptic plasticity, and neurosteroid formation [Korade and Kenworthy, 2008; Linetti et al., 2010; Mellon and Griffin, 2002; Rakheja and Boriack, 2008; Simons and Ehehalt, 2002; Singh et al., 2007; Fantini and Barrantes, 2009]. Our findings of diffuse atrophy in some patients may have been the result of disturbances in these cholesterol-dependent processes.
In summary, our study suggests that midline brain abnormalities are commonly found in individuals with SLOS, and SLOS should be included in the spectrum of midline malformation disorders. Further studies are needed to determine the role of cholesterol metabolism during formation of midline structures. In addition, studies examining the relationship between brain malformations and clinical outcome measures may lead to a better understanding of neurologic disease in individuals with SLOS and other sterol disorders.
Supplementary Material
Acknowledgments
This work was supported by the intramural research program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, a Bench to Bedside award from the NIH Clinical Center, the NIH Office of Rare Diseases, and a research grant from the RSH/Smith-Lemli-Opitz Foundation. We would like to thank the MRI technologists who acquired the images for this project, including Mary Busse, RTR(MR), Lisa Catron, RTR(MR), Bonita Damaska, RTR(MR), Sandra Hess, RT, Sandra McKee, RTR(MR), Mastaneh Owhadi, RTR(MR), Hugo Sandoval, RT, James Sedlacko, RTR(MR), Maryanne Russell, RT, Ronald White, RT, and Betty Wise, RTR(MR). We thank our research coordinator, Halima Goodwin, CPNP, and we also thank the patients (and their families) who participated in this study.
Footnotes
The authors report no conflicts of interest.
References
- Angle B, Tint GS, Yacoub OA, Clark A. Atypical case of Smith-Lemli-Opitz syndrome: Implications for diagnosis. Am J Med Genet. 1998;80:322–326. [PubMed] [Google Scholar]
- Beachy PA, Cooper MK, Young KE, von Kessler DP, Park WJ, Hall TM, Leahy DJ, Porter JA. Multiple roles of cholesterol in hedgehog protein biogenesis and signaling. Cold Spring Harb Symp Quant Biol. 1997;62:191–204. [PubMed] [Google Scholar]
- Bialer MG, Penchaszadeh VB, Kahn E, Libes R, Krigsman G, Lesser ML. Female external genitalia and Müllerian duct derivatives in a 46,XY infant with the Smith-Lemli-Opitz syndrome. Am J Med Genet. 1987;28:723–731. doi: 10.1002/ajmg.1320280320. [DOI] [PubMed] [Google Scholar]
- Bzduch V, Behulova D, Skodova J. Incidence of Smith-Lemli-Opitz syndrome in Slovakia. Am J Med Genet. 2000;90:260. doi: 10.1002/(sici)1096-8628(20000131)90:3<260::aid-ajmg17>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- Caruso PA, Poussaint TY, Tzika AA, Zurakowski D, Astrakas LG, Elias ER, Irons MB. MRI and 1H MRS findings in Smith-Lemli-Opitz syndrome. Neuroradiology. 2004;46:3–14. doi: 10.1007/s00234-003-1110-1. [DOI] [PubMed] [Google Scholar]
- Cherstvoy ED, Lazjuk GI, Lurie IW, Nedzved MK, Usoev SS. The pathological anatomy of the Smith-Lemli-Opitz syndrome. Clin Genet. 1975;7:382–7. doi: 10.1111/j.1399-0004.1975.tb00345.x. [DOI] [PubMed] [Google Scholar]
- Cherstvoy ED, Lazjuk GI, Ostrovskaya TI, Shved IA, Kravtzova GI, Lurie IW, Gerasimovich AI. The Smith-Lemli-Opitz syndrome. Virchows Arch. 1984;404:413–425. doi: 10.1007/BF00695225. [DOI] [PubMed] [Google Scholar]
- Cooper MK, Wassif CA, Krakowiak PA, Taipale J, Gong R, Kelley RI, Porter FD, Beachy PA. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat Genet. 2003;33:508–513. doi: 10.1038/ng1134. [DOI] [PubMed] [Google Scholar]
- Cunniff C, Kratz LE, Moser A, Natowicz MR, Kelley RI. Clinical and biochemical spectrum of patients with RSH/Smith-Lemli-Opitz syndrome and abnormal cholesterol metabolism. Am J Med Genet. 1997;68:263–269. [PubMed] [Google Scholar]
- Curry CJ, Carey JC, Holland JS, Chopra D, Fineman R, Golabi M, Sherman S, Pagon RA, Allanson J, Shulman S, Barr M, McGravey V, Dabiri C, Schimke N, Ives E, Hall BD. Smitih-Lemli-Opitz syndrome – type II: Multiple congenital anomalies with male pseudohermaphroditism and frequent early lethality. Am J Med Genet. 1987;26:45–57. doi: 10.1002/ajmg.1320260110. [DOI] [PubMed] [Google Scholar]
- Ericson J, Muhr J, Placzek M, Lints T, Jessell TM, Edlund T. Sonic hedgehog induces the differentiation of ventral forebrain neurons: a common signal for ventral patterning within the neural tube. Cell. 1995;81:747–756. doi: 10.1016/0092-8674(95)90536-7. [DOI] [PubMed] [Google Scholar]
- Fantini J, Barrantes FJ. Sphingolipid/cholesterol regulation of neurotransmitter receptor conformation and function. Biochim BiophysActa. 2009;1788:2345–2361. doi: 10.1016/j.bbamem.2009.08.016. [DOI] [PubMed] [Google Scholar]
- Farruggia S, Babcock DS. The cavum septi pellucidi: its appearance and incidence with cranial ultrasonography in infancy. Radiology. 1981;139:147–150. doi: 10.1148/radiology.139.1.7208915. [DOI] [PubMed] [Google Scholar]
- Fierro M, Martinez AJ, Harbison JW, Hay SH. Smith-Lemli-Opitz syndrome: neuropathological and ophthalmological observations. Dev Med Child Neurol. 1977;19:57–62. doi: 10.1111/j.1469-8749.1977.tb08021.x. [DOI] [PubMed] [Google Scholar]
- Garcia CA, McGarry PA, Voirol M, Duncan C. Neurological involvement in the Smith-Lemli-Opitz syndrome: clinical and neuropathological findings. Dev Med Child Neurol. 1973;15:48–55. doi: 10.1111/j.1469-8749.1973.tb04865.x. [DOI] [PubMed] [Google Scholar]
- Golden JA. Towards a greater understanding of the pathogenesis of holoprosencephaly. Brain Dev. 1999;21:513–521. doi: 10.1016/s0387-7604(99)00067-4. [DOI] [PubMed] [Google Scholar]
- Grynspan D, Michaud J, Nikkel SM, Creede E, Staines WA. Hippocampal Hypoplasia in Smith-Lemli-Opitz Syndrome. Pediatr Dev Pathol. 2013 May 20; doi: 10.2350/12-09-1252-LET.1. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Hennekam RC. Congenital brain anomalies in distal cholesterol biosynthesis defects. J Inherit Metab Dis. 2005;28:385–392. doi: 10.1007/s10545-005-7055-2. [DOI] [PubMed] [Google Scholar]
- Himmelstein DS, Bi C, Clark BS, Bai B, Kohtz JD. Balanced Shh signaling is required for proper formation and maintenance of dorsal telencephalic midline structures. BMC Dev Biol. 2010;10:118. doi: 10.1186/1471-213X-10-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irons M, Elias ER, Salen G, Tint GS, Batta AK. Defective cholesterol biosynthesis in Smith-Lemli-Opitz syndrome. Lancet. 1993;341:1414. doi: 10.1016/0140-6736(93)90983-n. [DOI] [PubMed] [Google Scholar]
- Kelley RL, Roessler E, Hennekam RC, Feldman GL, Kosaki K, Jones MC, Palumbos JC, Muenke M. Holoprosencephaly in RSH/Smith-Lemli-Opitz syndrome: does abnormal cholesterol metabolism affect the function of Sonic Hedgehog? Am J Med Genet. 1996;66:478–484. doi: 10.1002/(SICI)1096-8628(19961230)66:4<478::AID-AJMG22>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Kelley RI, Hennekam RC. The Smith-Lemli-Opitz syndrome. J Med Genet. 2000;37:321–335. doi: 10.1136/jmg.37.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korade Z, Kenworthy AK. Lipid rafts, cholesterol, and the brain. Neuropharmacology. 2008;55:1265–1273. doi: 10.1016/j.neuropharm.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanoue L, Dehart DB, Hinsdale ME, Maeda N, Tint GS, Sulik KK. Limb, genital, CNS, and facial malformations result from gene/environment-induced cholesterol deficiency: further evidence for a link to sonic hedgehog. Am J Med Genet. 1997;73:24–31. [PubMed] [Google Scholar]
- Lee RW, Yoshida S, Jung ES, Mori S, Baker EH, Porter FD. Corpus Callosum measurements correlate with developmental delay in Smith-Lemli-Opitz syndrome. Pediatr Neurol. 2013 doi: 10.1016/j.pediatrneurol.2013.03.015. [in press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linetti A, Fratangeli A, Taverna E, Valnegri P, Francolini M, Cappello V, Matteoli M, Passafaro M, Rosa P. Cholesterol reduction impairs exocytosis of synaptic vesicles. J Cell Sci. 2010;123:595–605. doi: 10.1242/jcs.060681. [DOI] [PubMed] [Google Scholar]
- Lowry RB, Yong SL. Borderline normal intelligence in the Smith-Lemli-Opitz (RSH) syndrome. Am J Med Genet. 1980;5:137–143. doi: 10.1002/ajmg.1320050205. [DOI] [PubMed] [Google Scholar]
- Mellon SH, Griffin LD. Neurosteroids: biochemistry and clinical significance. Trends Endocrinol Metab. 2002;13:35–43. doi: 10.1016/s1043-2760(01)00503-3. [DOI] [PubMed] [Google Scholar]
- Nowaczyk MJ, Farrell SA, Sirkin WL, Velsher L, Krakowiak PA, Waye JS, Porter FD. Smith-Lemli-Opitz (RHS) syndrome: holoprosencephaly and homozygous IVS8–1G-->C genotype. Am J Med Genet. 2001;103:75–80. doi: 10.1002/1096-8628(20010915)103:1<75::aid-ajmg1502>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Opitz JM, Gilbert-Barness E, Ackerman J, Lowichik A. Cholesterol and development: the RSH (“Smith-Lemli-Optiz”) syndrome and related conditions. Pediatr Pathol Mol Med. 2002;21:158–181. doi: 10.1080/15227950252852078. [DOI] [PubMed] [Google Scholar]
- Opitz JM, Furtado LV. The RSH/”Smith-Lemli-Opitz” syndrome: historical footnote. Am J Med Genet C Semin Med Genet. 2012;160C:242–249. doi: 10.1002/ajmg.c.31341. [DOI] [PubMed] [Google Scholar]
- Porter FD. RSH/Smith-Lemli-Opitz syndrome: a multiple congenital anomaly/mental retardation syndrome due to an inborn error of cholesterol biosynthesis. Mol Genet Metab. 2000;71:163–174. doi: 10.1006/mgme.2000.3069. [DOI] [PubMed] [Google Scholar]
- Porter JA, Young KE, Beachy PA. Cholesterol modification of hedgehog signaling proteins in animal development. Science. 1996;274:255–259. doi: 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]
- Putman AR, Szakacs JG, Opitz JM, Byrne JLB. Prenatal death in Smith-Lemli-Opitz/RSH syndrome. Am J Med Genet A. 2005;138:61–65. doi: 10.1002/ajmg.a.30246. [DOI] [PubMed] [Google Scholar]
- Quélin C, Loget P, Verloes A, Bazin A, Bessières B, Laquerrière A, Patrier S, Grigorescu R, Encha-Razavi F, Delahaye S, Jouannic JM, Carbonne B, D’Hervé D, Aubry MC, Macé G, Harvey T, Ville Y, Viot G, Joyé N, Odent S, Attié-Bitach T, Wolf C, Chevy F, Benlian P, Gonzales M. Phenotypic spectrum of fetal Smith-Lemli-Opitz syndrome. Eur J Med Genet. 2012;55:81–90. doi: 10.1016/j.ejmg.2011.12.002. [DOI] [PubMed] [Google Scholar]
- Rakheja D, Boriack RL. Precholesterol sterols accumulate in lipid rafts of patients with Smith-Lemli-Opitz syndrome and X-linked dominant chondrodysplasia punctata. Pediatr Dev Pathol. 2008;11:128–132. doi: 10.2350/06-10-0179.1. [DOI] [PubMed] [Google Scholar]
- Ryan AK, Bartlett K, Clayton P, Eaton S, Mills L, Donnai D, Winter RM, Burn J. Smith-Lemli-Opitz syndrome: a variable clinical and biochemical phenotype. J Med Genet. 1998;35:558–565. doi: 10.1136/jmg.35.7.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Beachy PA. Patterning of the embryonic forebrain. Curr Opin Neurobiol. 1998;8:18–26. doi: 10.1016/s0959-4388(98)80004-4. [DOI] [PubMed] [Google Scholar]
- Schwidde JT. Incidence of cavum septi pellucidi and cavum Vergae in 1,032 human brains. AMA Arch Neurol Psychiatry. 1952;67:625–632. doi: 10.1001/archneurpsyc.1952.02320170043006. [DOI] [PubMed] [Google Scholar]
- Shaw CM, Alvord EC., Jr Cava septi pellucidi et vergae: their normal and pathological states. Brain. 1969;92:213–223. doi: 10.1093/brain/92.1.213. [DOI] [PubMed] [Google Scholar]
- Sikora DM, Pettit-Kekel K, Penfield J, Merkens LS, Steiner RD. The near universal presence of autism spectrum disorders in children with Smith-Lemli-Opitz syndrome. Am J Med Genet Part A. 2006;140:1511–1518. doi: 10.1002/ajmg.a.31294. [DOI] [PubMed] [Google Scholar]
- Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. J Clin Invest. 2002;110:597–603. doi: 10.1172/JCI16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Paila YD, Chattopadhyay A. Differential effects of cholesterol and 7-dehydrocholesterol on the ligand binding activity of the hippocampal serotonin(1A) receptor: implications in SLOS. Biochem Biophys Res Commun. 2007;358:495–499. doi: 10.1016/j.bbrc.2007.04.135. [DOI] [PubMed] [Google Scholar]
- Smith DW, Lemli L, Opitz JM. A newly recognized syndrome of multiple congenital anomalies. J Pediatr. 1964;64:210–217. doi: 10.1016/s0022-3476(64)80264-x. [DOI] [PubMed] [Google Scholar]
- Tierney E, Nwokoro NA, Porter FD, Freund LS, Ghuman JK, Kelley RI. Behavior phenotype in the RSH/Smith-Lemli-Opitz syndrome. Am J Med Genet. 2001;98:191–200. doi: 10.1002/1096-8628(20010115)98:2<191::aid-ajmg1030>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Tierney E, Bukelis I, Thompson RE, Ahmed K, Aneja A, Kratz L, Kelley RI. Abnormalities of cholesterol metabolism in autism spectrum disorders. Am J Med Genet Part B. 2006;141B:666–668. doi: 10.1002/ajmg.b.30368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tint GS, Irons M, Elias ER, Batta AK, Frieden R, Chen TS, Salen G. Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N Engl J Med. 1994;330:107–113. doi: 10.1056/NEJM199401133300205. [DOI] [PubMed] [Google Scholar]
- Vernooij MW, Ikram MA, Tanghe HL, Vincent AJ, Hofman A, Krestin GP, Niessen WJ, Breteler MM, van der Lugt A. Incidental findings on brain MRI in the general population. N Engl J Med. 2007;357:1821–1828. doi: 10.1056/NEJMoa070972. [DOI] [PubMed] [Google Scholar]
- Wassif CA, Maslen C, Kachilele-Linjewile S, Lin D, Linck LM, Conner WE Steiner RD, Porter FD. Mutations in the human sterol delta7-reductase gene at 11q12–13 cause Smith-Lemli-Opitz syndrome. Am J Hum Genet. 1998;63:55–62. doi: 10.1086/301936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver DD, Solomon BD, Akin-Samson K, Kelley RI, Muenke M. Cyclopia (synophthalmia) in Smith-Lemli-Opitz syndrome: First reported case and consideration of mechanism. Am J Med Genet Part C. 2010;154C:142–145. doi: 10.1002/ajmg.c.30241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.