

Abstract
Hydroxyapatite (HAp) is often used as a bone-implant material because it is biocompatible and osteoconductive. However, HAp possesses poor rheological properties and it is inactive against disease-causing microbes. To improve these properties, we developed a green method to synthesize multifunctional composites containing: (1) cellulose (CEL) to impart mechanical strength; (2) chitosan (CS) to induce antibacterial activity thereby maintaining a microbe-free wound site; and (3) HAp. In this method, CS and CEL were co-dissolved in an ionic liquid (IL) and then regenerated from water. HAp was subsequently formed in situ by alternately soaking [CEL+CS] composites in aqueous solutions of CaCl2 and Na2HPO4. At least 88% of IL used was recovered for reuse by distilling the aqueous washings of [CEL+CS]. The composites were characterized using FTIR, XRD and SEM. These composites retained the desirable properties of their constituents. For example, the tensile strength of the composites was enhanced 1.9X by increasing CEL loading from 20% to 80%. Incorporating CS in the composites resulted in composites which inhibited the growth of both Gram positive (MRSA, S. aureus and VRE) and Gram negative (E. coli and P. aeruginosa) bacteria. These findings highlight the potential use of [CEL+CS+HAp] composites as scaffolds in bone tissue engineering.
Keywords: Cellulose, Chitosan, Hydroxyapatite, Antimicrobial Activity, Bone Tissue Engineering
INTRODUCTION
Hydroxyapatite (HAp), the main component of teeth and bones has received considerable attention as suitable material for bone tissue engineering because it is both biocompatible and osteoconductive.1,2 Despite these excellent properties, its rheological strength is far less than those required for bone tissue engineering materials.3,4 Moreover, HAp powder tends to migrate from implant sites and it possesses no antimicrobial activity. These limitations can be overcome by blending HAp with organic components thereby mimicking the extracellular matrix of the natural bone.5 The organic matrix acts as a binder to keep HAp at the implant site. The ideal composite materials for bone tissue engineering should be biodegradable, biocompatible, porous, possess high mechanical strength, and antimicrobial. 6–14
Implant-associated infections often limit the use of biomaterials in humans.15 Bacteria adhere to biomaterial surfaces and evade the host’s immune defense by forming a protective biofilm.16 Once the implant has been infected, the only remedy would be to remove the implant and perform another costly and painful surgery. Thus, novel biomaterials possessing antimicrobial activity provide the best option to ensure a bacteria-free implant site. In this regard chitosan (CS)-based materials have received considerable attention in bone tissue engineering.17–19 CS is a linear polysaccharide obtained by deacetylation of naturally abundant chitin, a polysaccharide found in exoskeletons of crustaceans such as crabs and shrimp and cell walls of fungi.20 CS is biocompatible, biodegradable and antibacterial.21 In view of these properties, it is expected that a composite containing both CS and HAp may have properties of both materials, namely, antimicrobial activity (from CS) and osteoconductive (from HAp). However, in spite of its potential use as scaffolds in bone tissue engineering, [CS+HAp] composite is known to have rather poor rheological properties. This is because CS undergoes extensive swelling in water. This undesirable property impedes the use of CS-HAp composites in load bearing applications.
To increase the structural strength of CS products, attempts have been made to covalently bind or graft CS onto man-made polymers or clays to strengthen its structure.21–41 Such modification is not desirable because it may inadvertently alter CS properties, making it not biocompatible and toxic and lessening or removing its unique properties.42 In view of these problems, blending CS with other polymers has emerged as a convenient and effective option to improve the mechanical properties of the resultant composite. Cellulose (CEL), the most abundant biopolymer on earth, has been explored in fabricating strong CS-CEL blend films.43–45 Cellulose is a linear polymer consisting of β-(1→4)-linked D-glucopyranose units. Owing to its high mechanical strength, CEL has also been blended with HAp to yield composites possessing desirable properties derived from both CEL and HAp.6,10,12 Similarly to CS, CEL has an extensive network of intra- and inter-molecular hydrogen bonds which makes it insoluble in water or in common organic solvents.46,47 This lack of solubility makes it difficult to process and functionalize CS and CEL. Until recently, N-methyl morpholine N-oxide (NMMO)/water system23 was widely used to dissolve CEL whilst acetic acid was used to dissolve chitosan. However, NMMO/H2O system may lead to the degradation of cellulose and worse still, the solvent is costly. In addition, none of these two solvent systems (NMMO/H2O and acetic acid) can dissolve both CEL and CS. Thus, there is need for a solvent system which can co-dissolve CEL and CS. It has been reported that trifluoroacetic acid (TFA) can be used to co-dissolve and cast films of chitosan-cellulose.43 The acid was subsequently neutralized using a base. Such a procedure is not only costly and time consuming but may also lead to acid induced changes in the structure of CS. These structural changes may render the composites toxic and therefore unsuitable for biomedical applications. For example, it has been reported that TFA forms salts with chitosan, and if the TFA is not completely removed, the residual TFA in the resultant composite will render the composite toxic.48 Also CS films made by dissolving CS in acetic acid and neutralizing with NaOH have been reported to inhibit the growth of keratinocytes.49
Ionic liquids (ILs) have recently emerged as potential green solvents for biopolymers.50,51–54 For example, 1-butyl-3-methylimidazolium chloride ([BMIm+CL−]), an IL, has been reported to dissolve up to 10% (w/w) of CEL.50 Interestingly, it was found that [BMIm+CL−] can also dissolve other polysaccharides such as CS.51 The fact that the same solvent can effectively dissolve various polysaccharides is of extreme importance as it offers the possibility to develop novel and green method, in one step, to synthesize composite materials containing two or more of these polysaccharides. In fact, recently, we have successfully developed a novel and totally recyclable method based on the use of [BMIm+Cl−] as a solvent to synthesize polysaccharide composite materials from CEL and CS.55 As expected, the [CEL+CS] composite materials obtained have combined advantages of their components, namely superior mechanical and thermal stability (from CEL) and excellent adsorbent for pollutants and toxins (from CS).55
The information presented is indeed provocative and clearly indicate that it is possible to use this simple process, without any chemical modification to synthesize novel three-component composite materials from CS, CEL and HAp for bone tissue engineering. It is expected that the composite material not only is biocompatible but also will possess all features which are needed for bone tissue material, namely mechanical strength (from CEL), excellent antimicrobial activity and ability to deliver growth factors and drugs (from CS) and bone material (from HAp). Such considerations prompted us to initiate this study which aims to hasten the breakthrough by combining our method with biomineralization process to synthesize novel three-component scaffold composite materials from CS, CEL and HAp for bone tissue engineering. Results on the synthesis, spectroscopic characterization and antibacterial activity of these composite materials are reported in the following sections.
MATERIALS AND METHODS
Materials
Cellulose (microcrystalline powder or Avicel, DP≈300), chitosan (MW≈310–375 kDa, 75% degree of deacetylation), ammonium persulfate and potassium antimonyl tartrate were obtained from Sigma-Aldrich, and used as received. Ammonium molybdate tetrahydrate was supplied by J.T. Baker. [BMIm+Cl−] was synthesized from freshly distilled 1-chlorobutane and 1-methylimidazole (both from Alfa Aesar) using a method developed previously by our group.56
Instrumentation
X-ray photoelectron spectra were taken on a HP 5950A ESCA spectrometer with Al monochromatic source and a flood gun was used for charge suppression. X-ray diffraction (XRD) measurements were conducted on a Rigaku MiniFlex II diffractometer using the Ni filtered Cu Kα radiation (λ=1.54059Å). The voltage and current of the X-ray tube were 30 kV and 15 mA respectively. The samples were measured within the 2θ angle range from 2.0 to 40.00. The scan rate was 50 per minute. Data processing procedures were performed with the Jade 8 program package.57 Near-infrared (NIR) spectra of the dried films and [BMIm+CL−], in transmission mode, were collected on a home-built NIR spectrometer.58,59 Normally, each spectrum was an average of 30 spectra taken at 1-nm intervals from 1450 to 2450 nm. FTIR spectra were measured on a PerkinElmer 100 spectrometer at 2 cm−1 resolution with either KBr or by a ZnSe single reflection ATR accessory (Pike Miracle ATR). Each spectrum was an average of 64 spectra. UV-visible absorption spectra were taken on a Cary 5000 UV-Vis-NIR spectrophotometer. The surface morphologies of the composite films were examined using a scanning electron microscope (SEM) (JSM-35, JEOL). The films were made conductive by sputter-coating with palladium prior to SEM analysis. Tensile strength measurements were carried out on a Universal Tensile Tester (Instron 5500R) using 1 kN load cell and crosshead speed 5 mm/min.
Methods
Preparation of CEL, CS and [CEL+CS] composite materials
[CEL+CS] composite materials were synthesized using the same procedure that was previously developed in our laboratory.55 Essentially, as shown in Scheme 1, an ionic liquid, [BMIm+Cl−], was used as a solvent to dissolve CEL, CS, and to facilitate regeneration of composite materials containing CEL and CS with different compositions. [BMIm+Cl−], used in the dissolution process, was removed from the films by washing the films in deionized water for 3 days. Specifically, a composite film of about 10cm×10cm was washed with 2.0 L of distilled water. The washing water was replaced with fresh water after every 24 hrs for 72 hrs. Subsequent distillation of the washing water rendered recovery of the IL for reuse.
Scheme 1.

Procedure used to prepare the cellulose-chitosan-hydroxyapatite composite materials.
Mineralization of Polysaccharide Films
Calcium phosphate was deposited in situ on the CEL, CS and [CEL+CS] composite films by the alternate soaking procedure described elsewhere.60 Typically, as shown in Scheme 1, the film (7.0 cm × 3.5 cm L × W) was dipped in 50.0 mL of 200.0 mM CaCl2 for 60 seconds during which time the cations were diffusing into the film matrix. The film was then rinsed twice in doubly distilled water (18 MΩ) to remove unbound Ca2+. A solution of 50.0 mL of 120.0 mM Na2HPO4 was substituted for the calcium solution. The film with bound Ca2+ was then dipped in the phosphate solution for 60 seconds. It was rinsed twice with doubly distilled water as before. This constituted one cycle. The whole series of operations was repeated 20 times. The wet film was then air dried at room temperature in a home-designed drier.
Procedure Used to Determine Molar Ratio of Calcium and Phosphate in [CEL+HAp] [CS+HAp] and [CEL+CS+HAp] Composite Films
The amount of calcium (Ca) and phosphorus (P) in the [CEL+HAp] [CS+HAp] and [CEL+CS+HAp] composite films were determined by flame atomic absorption spectrometry61 and colorimetry via ascorbic acid method62 respectively. The film samples were digested by suspending 50.0 mg of sample in 50.0 mL of double distilled water. One milliliter of 11.0 N sulfuric acid and 0.400 g ammonium persulfate were added successively. The mixture was boiled gently on a hot plate until the final volume reached 10 mL. This took ca. 1 ½ hours. [CEL+CS] composite film dissolved completely during this digestion process. The solution was allowed to cool before being adjusted to 30.0 mL with double distilled water. One drop of phenolphthalein was added after which the acid was neutralized to a faint pink color using 1.0 N NaOH. The solution was transferred quantitatively to a 100 mL volumetric flask and the volume adjusted to the mark using double distilled water. This sample solution was then used for the determination of P and Ca.
For P determination, the following protocol was followed. One milliliter of the sample solution was further diluted to 100 mL with double distilled water. Ten milliliters of this dilute sample were measured into each of the six 25mL volumetric flasks. Different volumes (0.0–10.0 mL) of 2.50 ppm P were added into each flask before the volume was adjusted to the mark with double distilled water. These solutions were transferred to six Erlenmeyer flasks before 4.0 mL of the combined reagent was added to each solution. The combined reagent was prepared by mixing 50.0 mL 5 N H2SO4, 5.0 mL 8.2 M potassium antimonyl tartrate, 15.0 mL 32.4 mM ammonium molybdate and 30.0 mL 0.1 M ascorbic acid solutions. A blue colored complex was formed within a minute of addition of the combined reagent. After 10minutes, absorbance of each sample was measured at 880 nm using a Perkin Elmer Lambda 35 UV/Vis spectrometer. By using the standard addition calibration curve, the percentage phosphorus content in the original film sample was calculated.
Calcium was determined by the widely used flame atomic absorption spectrometry. Ten milliliters of the sample solution was diluted to 100 mL with double distilled water. To each of six 25 mL volumetric flasks, 10 mL of this dilute sample solution were added. Varying amounts (0.0–5.0 mL) of 10 ppm Ca2+ were added to these flasks. Three milliliters of 0.18 M La2O3 were added to each flask. The volumes were adjusted to the marks using 0.2 M HNO3. The absorbance of each solution, using 422.7 nm excitation wavelength was measured on a flame atomic absorption spectrometer (Perkin Elmer AAnalyst 100). Air and acetylene were used as oxidant and fuel respectively. A standard addition calibration curve was constructed and used to calculate the percentage of calcium content in the film sample. The Ca/P mole ratio in each composite film was then calculated from the determined Ca and P percentages.
in vitro Antibacterial Assays
Bacterial killing assays were performed in the presence and absence of HAp-based composite materials. The model bacterial strains used in this protocol included Escherichia coli (ATCC 8739), Staphylococcus aureus (ATCC 25923), methicillin resistant S. aureus (ATCC 33591) and vancomycin resistant Enterococcus faecalis (ATCC 51299). The strains were maintained on blood agar at 4°C. By following a modified protocol,63 bacterial cells were grown overnight in nutrient broth for 18–20 hr at 37°C with gentle agitation. The cells were diluted in fresh medium and incubated for 24 hr at 37°C in the presence of the membrane composites. Serial dilutions of the bacteria were plated onto nutrient agar and incubated for 24 hr. Bacterial colony forming units (CFUs) were quantified and compared to bacteria grown in the absence of composites materials.
RESULTS AND DISCUSSION
Synthesis and Characterization
Shown in Figure 1A are XRD patterns of starting materials (microcrystalline CEL and CS powder), regenerated CEL, CS and CS50CEL50 films. As illustrated, microcrystalline CEL exhibits diffraction peaks at 2θ= 14.9°, 22.6° and 34.6° for (101), (002) and (040) planes respectively. Diffraction peaks of regenerated polysaccharides were found to be different from those of corresponding starting polysaccharides, e.g. for CEL film, the diffraction peaks were not just shifted from 2θ= 14.9° and 22.7° (for microcrystalline CEL) to ~10.9° and 20.0°, respectively, but also have much lower intensity than those of microcrystalline CEL. These results suggest that the degree of crystallinity of regenerated CEL is relatively lower than that of corresponding starting microcrystalline CEL. While diffraction bands of regenerated CS film also had lower intensity and shifted compared to those of starting material (i.e., CS powder), the shift in this case is relatively less than that in CEL, e.g., the diffraction peaks for the (101) and (002) were shifted just from 11.2° and 20.1° to ~10.9° and 18.8°, respectively. This may be due to the fact that compared to CS; CEL has relatively highly ordered structure. Specifically, an extensive network of intra- and inter-hydrogen bonds by -OH groups in CEL enables it to adopt a highly ordered structure, whereas, hydrogen bond network is much less extensive in CS because a majority of O-H groups are replaced by -NH2 groups. Consequently, loss of crystallinity was much higher in CEL than in CS when the polysaccharides were dissolved and regenerated from the ionic liquid [BMIm+ Cl−]. Figure 1A also shows XRD spectrum of CS50CEL50 composite material. As expected, the spectrum of this composite is a combination of that of 100% CEL and 100% CS. See reference 55 for more detailed information on spectroscopic characterization of regenerated CEL, CS and [CEL+CS] composite materials.
Figure 1.

X-ray diffraction spectra of (A) microcrystalline cellulose, chitosan powder, regenerated CEL and chitosan film and CEL50CS50 composite material; and (B) CEL100HAp, CS100HAp, CEL50CS50HAp and CEL60CS40HAp. See text for detailed information.
When calcium and phosphate were deposited onto these composite materials the XRD of the materials underwent substantial changes (Fig 1B). As illustrated, in addition to bands corresponding to CEL and CS, the composite materials also exhibit additional sharp bands at ~25° and 31°. Apparently, the calcium and phosphate ions arranged themselves into HAp structure because according to literature,64 these bands can be attributed to diffraction bands for the (002) and (211) planes of the hydroxyl apatite. As will be described in the following sections, in addition to XRD spectra, results from FTIR, elemental analysis and SEM also provide further confirmation that hydroxyl apatite was successfully deposited onto these composite materials.
Recently, there have been some reports on toxicity of ILs. However, the IL used in this work, [BMIm+Cl−], is relatively nontoxic compared to other ILs (its EC-50 and LD50 values are 897.47 ppm and 550mg/kg, respectively).65,66 Nevertheless, it is desirable to completely remove the IL from regenerated polysaccharide materials to ensure the materials are biocompatible. Since [BMIm+ Cl−] is totally miscible with water (the logP, its octanol-water partition coefficient, is −2.4 [42]),67 it was removed from the composite materials by washing the materials with water. Washing water (2L for a composite film of about 10cm×10cm) was repeatedly replaced with fresh water every 24 hrs until it was confirmed that IL was not detected in the washed water (by monitoring UV absorption of the IL at 290 nm). It was found that after washing for 72 hours, no IL was detected in the washing water by UV measurements. Since the limit of detection of the spectrophotometer used in this work was estimated to be about 3×10−5 AU, and the molar absorptivity of [BMIm+Cl−] at 290 nm is 2.6 M−1cm−1, it is estimated that if any [BMIm+Cl−] remains, its concentration would be smaller than 2 μg/mL of the washed water and 2 μg/g of the composite film. Since this concentration is two orders of magnitude lower than the LD50 value of the [BMIm+Cl−], if any IL remains in the composite films, it would not pose any harmful effect. UV-vis, FTIR and NIR techniques were used to: (1) confirm that when the composite films were washed with water, [BMIm+Cl−] was removed from the films to a level not detectable by these techniques; and (2) determine chemical composition of composite materials. Shown in Figure 2 is spectrum of [BMIm+Cl−]. As illustrated, overtone and combination bands of aliphatic C-H groups of the [BMIm+Cl−] can be clearly observed at 1388 nm and 1720 nm.68 Since these bands are specific for [BMIm+Cl−], they can be used as indicators to determine if the IL is present. Also shown in Figure 2 are NIR spectra of regenerated 100%CEL, 100%CS as well as 40:60 CS:CEL and 50:50 CS:CEL. NIR spectra of these regenerated materials exhibit none of the indicator bands specific for [BMIm+Cl−]. Thus, it is clear that washing with water effectively and completely removed the IL from the composite materials. Further confirmation of removal of the ionic liquid from the films can also be seen in FTIR spectra of the same materials shown in 2, namely, [BMIm+Cl−] and regenerated 100%CEL, 100%CS as well as 40:60 CS:CEL and 50:50 CS:CEL (Figure SI-1 in Supporting Information). Again, none of the FTIR bands due to [BMIm+Cl−] were present in the spectra of the regenerated materials.
Figure 2.

NIR spectra of regenerated CEL100, CEL60CS40, CEL50CS50 and CS100 films and [BMIm+CL−]. See text for detailed information
The IL used was recovered by distilling the washed aqueous solution (the IL remained because it is not volatile). The recovered [BMIm+Cl−] was dried under vacuum at 70°C overnight before reuse. It was found that at least 88% of [BMIm+Cl−] was recovered for reuse. As such, the method developed here is not only green but recyclable because [BMIm+Cl−] is the only solvent used in the preparation and it is fully recovered for reuse.
Chemically, the regeneration of both CEL and CS was confirmed by FTIR spectroscopy. As illustrated in Figure 3, the FT-IR spectrum of regenerated CEL film (blue spectrum) exhibits three pronounced bands at around 3400 cm−1, 2850 – 2900 cm−1 and 890 – 1150 cm−1. These bands can be tentatively assigned to stretching vibrations of O-H, C-H and -O- groups, respectively.64,69,70 The fact that the starting material (microcrystalline CEL) (spectrum not shown) also exhibits these three bands and is very similar to those of the regenerated CEL clearly indicates that CEL was completely regenerated, by this synthetic method. Similarly, the FTIR spectrum of regenerated CS film (black curve in Fig 3) is similar to the FTIR spectrum of the CS powder (spectrum not shown) from which it was made. These spectra display characteristic CS bands around 3400 cm−1 (O-H stretching vibrations), 3250 – 3350 cm−1 (symmetric and asymmetric N-H stretching), 2850 – 2900 cm−1 (C-H stretching), 1657 cm−1 (C=O, amide 1), ~1580 cm−1 (N-H deformation), 1380 cm−1 (CH3 symmetrical deformation), 1319 cm−1 (C-N stretching, amide III) and 890 – 1150 cm−1 (ether bonding)8,9,71 (Left insert figure shows detailed absorption in region from 1500–1800 cm−1 where amide and amide groups of chitosan absorb). These results indicate that both CEL and CS were successfully regenerated by the synthetic method developed here without any chemical transformation. Also shown in the figure are spectra of composite materials containing both CEL and CS (CS50CEL50, CS40CEL60). As expected, spectra of these composite materials contain bands corresponding to both CEL and CS.
Figure 3.

FTIR spectra of CEL100, CS50CEL50, CEL60CS40, CS100HAp, CEL50CS50HAp, CEL60CS40HAp. See text for detailed information.
Substantial changes in the FTIR spectra were observed when calcium and phosphate were deposited onto the CEL, CS and [CEL+CS] materials. Perhaps the most pronounced one are two new bands at 563 cm−1 and 604 cm−1 (right insert figure shows detailed absorption in the region from 520 cm−1 to 650 cm−1). According to literature,8,9,71,72 the ν4 or bending vibrational mode of O=P=O group is responsible for the bands at 604 cm−1 and 563 cm−1. In addition to these bands, other smaller bands including the band at around 960 cm−1 band can be tentatively attributed to ν1 (symmetric P=O stretching mode), band in the region around 1037–1095 cm−1 attributable to the antisymmetric P=O stretching mode, and several bands at around 1400 cm−1 which are probably due to vibration of CO3− group were also observed.8,9,71,72 Again, the presence of these bands further confirms that hydroxyl apatite was successfully deposited onto the composite materials.
Analysis of the film materials by SEM reveals some interesting features about the texture and morphology of these materials. As expected, CEL100 and CS100 materials (Figure 4, top left and top right, respectively) are homogeneous. Chemically, the only difference between CS and CEL is the –NH2 groups in the former. However, their structures, as recorded by the SEM, are substantially different. Specifically, while CS seems to exhibit smooth structure, CEL arranges itself into fibrous structure with fibers having diameter of about ~0.5 – 1.0 micron. This may be due to the fact that, as described in previous section, CEL has relatively higher ordered structure than CS because of the extensive network of inter- and intra-hydrogen bond network in the former. Interestingly, a CS50CEL50 composite material (Fig 4 top center) is not only homogeneous but it is more similar to structure of CS than that of CEL, namely, it has a rather smooth structure without any fibrous forms.
Figure 4.

Scanning electron microscope images of CEL100, CS100, CS50CEL50 and their corresponding hydroxyapatite composites.
SEM images of corresponding polysaccharide-hydroxyapatite film are shown in the lower row of Figure 4. As illustrated, hydroxyapatite formed layers of densely and homogeneously distributed spherical particles on these polysaccharide films when calcium and phosphate were deposited onto these films. This is expected as it was also reported by other groups that hydroxyapatite formed spherical particles on different biopolymers.8,9,71,72 Of particular interest is the fact that hydroxyapatite particles are not of the same size on these polysaccharide films. Rather, it seems that the particles on the CEL100 film are largest with the smallest being on the CEL50CS50 film with those on the CS100 being of the intermediate size.
It is well recognized that the formation of HAp involves initial nucleation and subsequent growth.60,71 The soaking in the CaCl2 solution is believed to provide the supersaturation of Ca2+ ions around CEL and/or CS through ionic interaction between calcium ions and the negatively charged OH groups available on the polysaccharides and/or physical entrapment due to the 3-D network structure of the polysaccharides with tiny hollow spaces.60,71 Then the incorporated calcium ions can bind phosphate ions to form the initial nuclei. Once the apatite nuclei are formed, they grow by uptake of calcium and phosphate ions from the surroundings. As described above, while CS seems to exhibit smooth structure, CEL arranges itself into fibrous structure. Because of its structure, there would be more tiny hollow spaces in CEL films. As a consequence, nucleation is relatively easier on CEL with its fibrous surface and tiny hollow spaces than on smooth surface of CS. This, in turn, will enable hydroxyapatite to grow more and to form relatively larger size crystals on CEL than on CS. Nucleation and crystal growth are probably the most difficult on the CEL50CS50 composite film because of presence of two different polysaccharides with different structures. This will lead to formation of hydroxyapatite crystals with smallest size.
The exact structure of Ca and P in the composite materials can also be reliably predicted on the basis of the ratio of calcium and phosphorous in the materials. Initially, concentration of calcium and phosphorous in the composite materials were determined by flame atomic absorption and spectrophotometric method, respectively. Molar ratios of Ca/P in different composite materials were then calculated from concentrations of Ca and P in the materials. Each measurement was performed in triplicate, and averaged values together with standard deviation are listed in Table I. As listed in the table, Ca/P values for all four composite materials (CS100HAp, CEL100HAp, CEL50CS50HAp and CEL60CS40HAp) measured, are, within experimental error, in agreement with hydroxyapatite stoichiometric value of 1.67.
X-ray photoelectron spectroscopy (XPS) was also used to determine the elemental composition and the chemical structure of the composite materials. Shown in figure 6 are XPS spectra of CEL100HAp and CS100HAp composites. Both composites contain Ca2+ and P5+ as evidenced by the presence of Ca bands at 350 eV (Ca2p) and 439 eV (Ca2s) and P bands at 133 eV (P2p) and 191eV (P2s) in their spectra (see Table 2 for band assignments). Bands correspond to P2p (133 eV) and O1s (532 eV) were further deconvoluted in order to determine bond structure of the phosphate. As shown in insert B of Figure 6 and listed in Table 2, the O1s bands for both CEL100HAp and CS100HAp can be resolved into two bands. The band at 532.4 eV for CEL100HAp (and 532.2 eV for CS100HAp) was tentatively assigned to the C-O bond.73 The band at 531.1eV for CEL100HAp (and 531.0 eV for CS100HAp) could be assigned to PO43−. These results confirm that PO43− is the structure of oxygen and phosphorous in the composites. Additional confirmation can also be gained when the P2p band at 133 eV was resolved into two components one assigned to PO43− (132.9 and 132.8 eV for CEL100HAp and CS100HAp) and the other assigned to HPO43− (133.6 and 133.8 eV for CEL100HAp and CS100HAp).
Figure 6.

X-ray photoelectron spectra of CEL100HAp and CS100HAp. Inserts: Deconvolution of (A): O1s band at 532eV and (B) P2p band at 133eV.
Table 2.
Assignments of XPS bands of CEL100HAp and CS100HAp
| Element | Binding Energy, eV | Assignments | |
|---|---|---|---|
| CEL100HAp | CS100HAp | ||
| P2p3/2 | 132.9 | 132.8 | PO43− |
| 133.6 | 133.8 | HPO43− | |
| O1s | 531.1 | 531.0 | PO43− |
| 532.4 | 532.2 | HPO43−/-C-O- | |
| Ca2p3/2 | 347.2 | 347.0 | Ca2+ |
| Ca2p1/2 | 350.7 | 350.5 | |
| C1s | 284.9 | 284.8 | -C-C- |
| 286.3 | 286.2 | C-O (CEL or CS backbone) | |
| 287.9 | 287.8 | -O-C-O- | |
Ratio of calcium and phosphorous in the composites can also be determined from XPS spectra. As listed in Table 1, Ca/P values were found to be 1.28±0.09 and 1.4±0.1 for CS100HAp and CEL100HAp, respectively. These values are relatively smaller than values determined by AA technique for the same composites. The discrepancy stems from structure of the HAp composites and the nature of the AA and XPS measurements. Specifically, it was reported that when HAp materials prepared by alternatively depositing layers of Ca and P, the surface layers are compositionally different from the bulk material.74,75 This could be due to the initial formation of octacalcium phosphate (Ca/P = 1.33) which is later transformed to the more thermodynamically stable form, HAp. Since the precipitation would occur on the surface, the layers beneath the surface would transform to HAp before the layers at the top. XPS measurements are only on the surface top few angstroms of the composites whereas AA was measured on digested samples, namely, it measured Ca and P contents, not on outermost layers but rather on the entire body of the composites. As a consequence, Ca/P values obtained by XPS method are relatively smaller than those by the AA method.
Table 1.
Ca/P values of composite materials determined by Atomic Absorption (AA) and X-ray Photoelectron Spectroscopy (XPS)
| Composite Material | Ca/P by AA | Ca/P by XPS |
|---|---|---|
|
| ||
| CS100HAp | 1.7±0.2 | 1.28±0.09 |
| CEL100HAp | 1.60±0.03 | 1.4±0.1 |
| CEL50CS50HAp | 1.7±0.2 | |
| CEL60CS40HAp | 1.5±0.2 | |
The mechanical strength of CS is so poor that, practically, it cannot be used by itself for any applications. Adding cellulose to CS-based material is expected to increase mechanical strength to the materials. To confirm this possibility, measurements were made to determine the tensile strength of [CEL+CS+HAp] composite films with different CEL concentrations. Results obtained, shown in figure 5, clearly indicate that adding CEL into [CS+HAp] substantially increase its tensile strength. For example, tensile strength of the [CEL+CS+HAp] composite with 80% CEL is 1.9X higher than that of the [CEL+CS+HAp] composite with 20% cellulose and that the tensile strength of the composite material can be adjusted by adding judicious amount of CEL. Thus it is evidently clear that the [CEL+CS+HAp] composite materials developed here have overcome the main limitation currently imposed on utilizations of the materials, namely they have superior mechanical strength and still are able to retain their biocompatible and unique properties.
Figure 5.

Plot of tensile strength as a function of cellulose concentration in [CEL+CS+HAp] composite materials.
in vitro Antibacterial Assays
Antimicrobial infections often limit the success of implants. Therefore, it is plausible to design a composite that possesses intrinsic antibacterial activity. Chitosan is known to possess innate antimicrobial properties.21,76,77 The material has been used in the food and agricultural industries and in wound dressings because of its characteristic antibacterial activity toward both Gram positive and Gram negative organisms.76 Figure 7 shows the bactericidal effects of the novel CS100, [CEL+HAp], [CS+HAp], and [CEL+CS+HAp] composites synthesized using method reported in this study. The activities were largely dependent on the presence of chitosan within the composite and it is noted that the presence of HAp did not hinder the ability to reduce bacterial growth. The composite made solely of CS and HAp (CS100 HAp) exhibited much more efficiency and substantial bacterial killing ability than the other composites and CS alone for all strains of bacteria tested. While VRE and MRSA were also affected by the composite CEL50CS50HAp, only VRE growth was inhibited by CEL60CS40HAp. The composite materials with the greater amounts of cellulose also showed at least one log reduction in growth of S. aureus. It should be noted that except CS100HAp, all other composites were nearly ineffective against P. aeruginosa. This organism is well-known for its resistance to antimicrobials and antibacterial substances. The fact that CS100 HAp did show antimicrobial action against P. aeruginosa is particularly promising.
Figure 7.
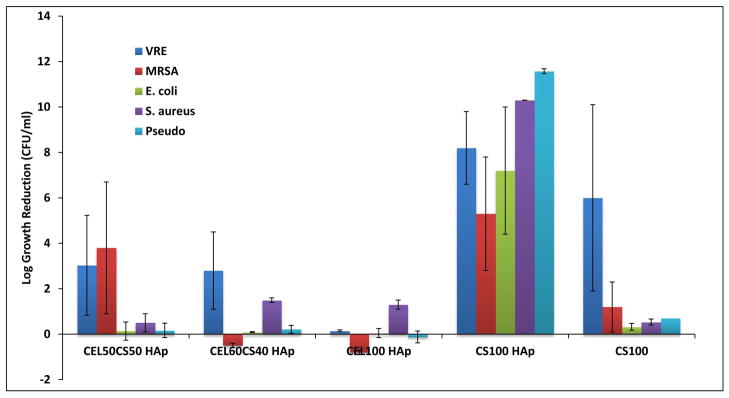
Bactericidal activity of membrane composites. VRE (blue), MRSA (red), E. coli (green), S. aureus (purple) and P. aeruginosa (light blue) were cultured in the presence and absence of varying concentrations of CS, CEL HAp compared to CS alone (CS100). The log reduction represents the log of the CFU/ml of bacteria in the absence of composite minus the log of the CFU/ml after exposure to composite. Each experiment was performed at least three independent times and the error bars represent standard error of the mean.
Modified chitosan material, such as quarternized or those supplemented with silver nanoparticles have more of an effect against microorganisms than chitosan alone.63,78 Generally, the presence of HAp alone does not lead to antimicrobial effects. HAp is utilized because of the bioactivity of the material, especially in the field of orthopedics. Existing methods based on chemical modifications of chitosan to synthesize HAp composite materials can be expensive, potentially toxic and complicated. The method developed here is simple, nontoxic and inexpensive as it is based on dissolution of CEL and CS with an ionic liquid, a green solvent, followed by depositing HAp onto the CEL and/or CS materials. This method enables the use chitosan and HAp in their natural states in wound dressings. As such, it will be beneficial in regards to biodegradability, innate antimicrobial activity and scaffolding for tissue regeneration. The antimicrobial properties of the composites synthesized using the method reported here showed the inhibition of growth of both Gram positive (MRSA, S. aureus and VRE) and Gram negative (E. coli and P. aeruginosa) bacteria by the CS, CEL and HAp composites over 24 hr. Previous antibacterial studies have shown different effects based on the type of bacteria tested. In one study, a chitosan-based composite, specifically chitosan-silk fibroin composite, inhibited the growth of Gram negative bacteria but not Gram positive79 whereas in a different study, using chitosan-dextran composite, only Gram positive bacteria were inhibited.80 We have shown an inhibition effect on multiple organisms, both Gram positive and Gram negative with the composite CS100HAp. Only a single study with HAp was reported which shows that it exhibited antimicrobial results against E. coli and Staphylococcus epidermidis within a 4-hr time period. The bacteria began to lose their integrity when exposed to a membrane composed of HAp and silver particles.81 Compared to these studies which show that [CEL+CS] and HAp composites exhibit antimicrobial activity to only a few organisms, the [CEL+CS+HAp] composites prepared using method reported here are superior as they showed inhibition of growth of a wide range of Gram positive and Gram negative. Bacteriostatic and bactericidal properties are important for wound healing applications in preventing infection and even possible sepsis. These effects of the chitosan and HAp composites reported here on the wound pathogens illustrate their great potential as components in wound dressings to provide both antibacterial protection and scaffolding for tissue and bone growth.
CONCLUSIONS
In this study, [BMIm+CL−] was used as a powerful, nonderivatizing solvent to fabricate [CEL+CS] composite materials which were subsequently mineralized by a modified alternate soaking method. Unlike the methods reported in literature, the method developed in this study is simple, environmentally and inexpensive. It enables, for the first time, to use CEL, CS and HAp in their natural states. The recovery of the solvent, [BMIm+CL−], by distillation adds economic potential to the whole process. Interestingly, the tri-component composite material, [CEL+CS+HAp] produced had desirable properties derived from its individual components. As expected, adding CEL to [CS+HAp] increased the tensile strength of resultant composites, [CEL+CS+HAp]. In addition, the composite materials exhibited antibacterial activity (presumably due to CS) against a wide range of both Gram positive (MRSA, S. aureus and VRE) and Gram negative (E. coli and P. aeruginosa) bacteria over 24 hr than existing HAp composites. Specifically, the composite made solely from CS and HAp (CS100 HAp) exhibited the highest efficiency and substantial bacterial killing ability than the other composites for all strains of bacteria tested. VRE and MRSA were also affected by the composite CEL50CS50HAp and VRE with CEL60CS50HAp. Interestingly, except CS100HAp, all other composites were nearly ineffective with P. aeruginosa. This organism is well-known for its resistance to antimicrobials and antibacterial substances. The fact that CS100HAp did show antimicrobial action against P. aeruginosa is of particular significance. Taken together, the results presented are very encouraging and indicate that the [CEL+CS+HAp] composite material may be able to successfully serve as scaffold for tissue engineering.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award number R15GM099033.
Footnotes
Supporting Information. Additional Supporting Information may be found in the online version of this article.
References
- 1.He P, Sahoo S, Ng KS, Chen K, Toh SL, Goh JCH. Enhanced osteoinductivity and and osteoconductivity through hydroxyapatite coating of silk-based tissue-engineered ligament scaffold. J Biomed Mater Res Part A. 2012 doi: 10.1002/jbm.a.34333. [DOI] [PubMed] [Google Scholar]
- 2.Pallela R, Venkatesan J, Janapala VR, Kim SK. Biophysicochemical evaluation of chitosan-hydroxyapatite-marine sponge collagen composite for bone tissue engineering. J Biomed Mater Res Part A. 2012;100A:486–495. doi: 10.1002/jbm.a.33292. [DOI] [PubMed] [Google Scholar]
- 3.Watanabe Y, Eryu H, Matsuura K. Evaluation of three-dimensional orientation of Al3Ti platelet in Al-based functionally graded materials fabricated by a centrifugal casting technique. Acta Mater. 2001;5:775–783. [Google Scholar]
- 4.Itoh S, Kikuchi K, Takakuda K, Koyama Y, Matsumoto HN, Ichinose S, Tanaka J, Kawauchi T, Shinomiya K. The biocompatibility and osteoconductive activity of a novel hydroxyapatite/collagen composite biomaterial, and its function as a carrier of rhBMP-2. J Biomed Mater Res Part A. 2001;54A:445–453. doi: 10.1002/1097-4636(20010305)54:3<445::aid-jbm190>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 5.Muzzarelli C, Muzzarelli RAA. Natural and artificial chitosan–inorganic composites. J Inorg Biochem. 2002;92:89–94. doi: 10.1016/s0162-0134(02)00486-5. [DOI] [PubMed] [Google Scholar]
- 6.Tsioptsias C, Panayiotou C. Preparation of cellulose-nanohydroxyapatite composite scaffolds from ionic liquid solutions. Carbohydrate Pol. 2008;74:99–105. [Google Scholar]
- 7.Liuyun J, Yubao L, Xuejiang W, Li Z, Jiqiu W, Mei G. Preparation and properties of nanohydroxyapatite/chitosan/carboxymethyl cellulose composite scaffold. Carbohydrate Pol. 2008;74:680–684. [Google Scholar]
- 8.Teng SH, Lee EJ, Yoon BH, Shin DS, Kim HE, Oh JS. Chitosan/nanohydroxyapatite composite membranes via dynamic filtration for guided bone regeneration. J Biomed Mat Res Part A. 2009;88A:569–580. doi: 10.1002/jbm.a.31897. [DOI] [PubMed] [Google Scholar]
- 9.Zhang H, Zhu Q. Syntheis of nanospherical and ultralong fibrous hydroxyapatite and reinforcement of biodegradable chitosan/hydroxyapatite composite. Mod Phys Lett B. 2009;23:3967–3976. [Google Scholar]
- 10.Hong L, Wang YL, Jia SR, Huang Y, Gao C, Wanm YZ. Hydroxyapatite/bacterial cellulose composites synthesized via biomimetic route. Mat Lett. 2006;60:1710–1713. [Google Scholar]
- 11.Chen J, Nan K, Yin S, Wang Y, Wu T, Zhang Q. Characterization and biocompatibility of nanohybrid scaffold prepared via in situ crystallization of hydroxyapatite in chitosan matrix. Colloids Surf B. 2010;81:640–647. doi: 10.1016/j.colsurfb.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 12.Wan YZ, Hong L, Jia SR, Huang Y, Zhu Y, Wang YL, Jiang HJ. Synthesis and characterization of hydroxyapatite-bacterial cellulose nanocomposites. Composites Sci Tech. 2006;66:1825–1832. [Google Scholar]
- 13.Venkatesan J, Kim SK. Chitosan composites for bone tissue engineering- An overview. Mar Drugs. 2010;8:2252–2266. doi: 10.3390/md8082252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pinheiro AG, Pereira FFM, Santos MRP, Freire FNA, Goes JC, Sombra ASB. Chitosan-hydroxyapatite-BIT composite films. Polym Compos. 2007;28:582–587. [Google Scholar]
- 15.Aviv M, Berdicevsky I, Zilberman M. Gentamicin-loaded bioresorbable films for prevention of bacterial infections associated with orthopedic implants. J Biomed Mater Res Part A. 2007;83A:10–19. doi: 10.1002/jbm.a.31184. [DOI] [PubMed] [Google Scholar]
- 16.Wu P, Grainger DW. Drug/device combinations for local drug therapies and infection prophylaxis. Biomaterials. 2006;27:2450–2467. doi: 10.1016/j.biomaterials.2005.11.031. [DOI] [PubMed] [Google Scholar]
- 17.Khor E, Lim LY. Implantable applications of chitin and chitosan. Biomaterials. 2003;24:2339–2349. doi: 10.1016/s0142-9612(03)00026-7. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Ni M, Zhang M, Ratner B. Calcium Phosphate-Chitosan Composite Scaffolds for Bone Tissue Engineering. Tissue Eng. 2003;9:337–345. doi: 10.1089/107632703764664800. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Zhang M. Synthesis and characterization of macroporous chitosan/calcium phosphate composite scaffolds for tissue engineering. J Biomed Mater Res. 2001;55:304–312. doi: 10.1002/1097-4636(20010605)55:3<304::aid-jbm1018>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 20.Phisalaphong M, Jatupaiboon N, Kingkaew J. Biosynthesis of Cellulose-Chitosan Composite. In: Kim S, editor. Chitin, Chitosan, Oligosaccharides and their Derivatives: Biological Activities and Applications. New York: CRC Press; 2011. pp. 53–65. [Google Scholar]
- 21.Rabea EI, Badawy MET, Stevens CV, Smagghe G, Steurbaut W. Chitosan as Antimicrobial Agent: Applications and Mode of Action. Biomacromolecules. 2003;4:1457–1465. doi: 10.1021/bm034130m. [DOI] [PubMed] [Google Scholar]
- 22.Cai J, Liu Y, Zhang L. Dilute solution properties of cellulose in LiOH/urea aqueous system. J Polym Sci B Pol Phys. 2006;44:3093–3101. [Google Scholar]
- 23.Fink HP, Purz P, Ganster HJ. Structure formation of regenerated cellulose materials from NMMO-solutions. Prog Polym Sci. 2001;26:1473–1524. [Google Scholar]
- 24.Dai T, Tegos GP, Burkatovskaya M, Castano AP, Hamblin MR. Chitosan acetate bandage as a topical antimicrobial dressing for infected burns. Antimicrobial Agents Chemo. 2009;53:393–400. doi: 10.1128/AAC.00760-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bordenave N, Grelier S, Coma V. Hydrophobization and Antimicrobial Activity of Chitosan and Paper-Based Packaging Material. Biomacromolecules. 2010;11:88–96. doi: 10.1021/bm9009528. [DOI] [PubMed] [Google Scholar]
- 26.Altiok D, Altiok E, Tihminlioglu F. Physical, antibacterial and antioxidant properties of chitosan films incorporated with thyme oil for potential wound healing applications. J Mater Sci: Mater Med. 2010;21:2227–2236. doi: 10.1007/s10856-010-4065-x. [DOI] [PubMed] [Google Scholar]
- 27.Burkatovskaya M, Tegos G, Swietlik E, Demidova TN, Castano AP, Hamblin MR. Use of chitosan bandage to prevent fatal infections developing from highly contaminated wounds. Biomaterials. 2006;27:4157–4164. doi: 10.1016/j.biomaterials.2006.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gustafson SB, Fulkerson P, Bildfell R, Aguilera L, Hazzard TM. Chitosan dressing provides hemostasis in swine femoral arterial injury model. Prehospital Emergency Care. 2007;11:172–178. doi: 10.1080/10903120701205893. [DOI] [PubMed] [Google Scholar]
- 29.Pusateri AE, McCarthy SJ, Gregory KW, Harris RA, Cardenas L, McManus AT, Goodwin CW. Effect of a chitosan-based haemostatic dressing on blood loss and survival in a model of severe venous hemorrhage and hepatica injury in swine. J Trauma Injury Infect Crit Care. 2003;54:177–182. doi: 10.1097/00005373-200301000-00023. [DOI] [PubMed] [Google Scholar]
- 30.Keong LC, Halim AS. In vitro models in biocompatibility assessment for biomedical-grade chitosan derivatives in woung management. International J Mol Sci. 2009;10:1300–1313. doi: 10.3390/ijms10031300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kiyozumi T, Kanatani Y, Ishihara M, Saitoh D, Shimizu J, Yura H, Suzuki S, Okada Y, Kikuchi M. Medium (DMEM/F-12)-containing chitosan hydrogel as adhesive and dressing in autologous skin grafts and acceleration in the healing process. J Biomed Mat Res B: Appl Biomat. 2006;79B:129–136. doi: 10.1002/jbm.b.30522. [DOI] [PubMed] [Google Scholar]
- 32.Rossi S, Sandri G, Ferrari F, Bonferoni MC, Caramella C. Buccal delivery of acyclovir from films based on chitosan and polyacrylic acid. Pharm Dev Tech. 2003;8:199–208. doi: 10.1081/pdt-120018490. [DOI] [PubMed] [Google Scholar]
- 33.Jain D, Banerjee R. Comparison of ciprofloxacin hydrochloride-loaded protein, lipid, and chitosan nanoparticles for drug delivery. J Biomed Mat Res B: Appl Biomat. 2008;86B:105–112. doi: 10.1002/jbm.b.30994. [DOI] [PubMed] [Google Scholar]
- 34.Varshosaz J, Tabbakhian M, Salmani Z. Designing of a thermosensitive chitosan/poloxamer in situ gel for ocular delivery of ciprofloxacin. Open Drug Delivery J. 2008;2:61–70. [Google Scholar]
- 35.Elmotasem H. Chitosan-alginate blend films for the transdermal delivery of meloxicam. Asian J Pharm Sci. 2008;3:12–29. [Google Scholar]
- 36.Naficy S, Razal JM, Spinks GM, Wallace GG. Modulated release of dexamethasone from chitosan-carbon nanotube films. Sensors and Actuators A: Physical. 2009;A155:120–124. [Google Scholar]
- 37.Tirgar A, Golbabaei F, Hamedi J, Nourijelyani K, Shahtaheri SJ, Moosavi SR. Removal of airborne hexavalent chromium mist using chitosan gel beads as a new control approach. Int J Environ Sci Tech. 2006;3:305–313. [Google Scholar]
- 38.Nishiki M, Tojima T, Nishi N, Sakairi N. β-Cyclodextrin-linked chitosan beads: Preparation and application to removal of bisphenol A from water. Carbohydrate Lett. 2000;4:61–67. [PubMed] [Google Scholar]
- 39.Hassan MAA, Hui LS, Noor ZZ. Removal of boron from industrial wastewater by chitosan via chemical precipitation. J Chem Nat Res Eng. 2009;4:1–11. [Google Scholar]
- 40.Dhakal RP, Oshima T, Baba Y. Synthesis of unconventional materials using chitosan and crown ether for selective removal of precious metal ions. World Acad Sci Eng Tech. 2009;56:204–208. [Google Scholar]
- 41.Ngah WWS, Isa IM. Comparison study of copper ion and adsorption on chitosan. J Appl Pol Sci. 1998;67:1067–1070. [Google Scholar]
- 42.Mi YF, Tan HL, Sung H. In vivo biocompatibility and degradability of a novel injectable-chitosan-based implant. Biomaterials. 2002;23:181–191. doi: 10.1016/s0142-9612(01)00094-1. [DOI] [PubMed] [Google Scholar]
- 43.Wu YB, Yu SH, Mi FL, Wu CW, Shyu SS, Peng CK, Chao AC. Preparation and characterization on mechanical and antibacterial properties of chitosan/cellulose blends. Carbohydr Polym. 2004;57:435–440. [Google Scholar]
- 44.Lima IS, Lazarin AM, Airoldi C. Favorable chitosan/cellulosefilm combinations for copper removal from aqueous solutions. Int J Biol Macromol. 2005;36:79–83. doi: 10.1016/j.ijbiomac.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 45.Hasegawa M, Isogai A. Characterization of cellulose–chitosan blend films. J Appl Polym Sci. 2003;45:1873–1879. [Google Scholar]
- 46.Finkenstadt VL, Millane RP. Crystal Structure of Valonia Cellulose 1β. Macromolecules. 1998;31:7776–7783. [Google Scholar]
- 47.Kurita K. Chitin and Chitosan: Functional Biopolymers from Marine Crustaceans. Mar Biotechnol. 2006;8:203–226. doi: 10.1007/s10126-005-0097-5. [DOI] [PubMed] [Google Scholar]
- 48.Chen JP, Chen SH, Lai GJ. Preparation and Characterization of Biomimetic Silk Fibroin/Chitosan Composite Nanofibers by Electrospinning for Osteoblasts Culture. Nanoscale Res Lett. 2012;8:170–180. doi: 10.1186/1556-276X-7-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tchemtchoua VT, Atanasova G, Aqil A, Filee P, Garbacki N, Vanhooteghem O, Deroanne C, Noel A, Jerome C, Nusgens B, Poumay Y, Colige A. Development of a Chitosan Nanofibrillar Scaffold for Skin Repair and Regeneration. Biomacromolecules. 2011;12:3194–3204. doi: 10.1021/bm200680q. [DOI] [PubMed] [Google Scholar]
- 50.Swatlowski M, Spear S, Holbrey JD, Rogers RD. Dissolution of cellulose with ionic liquids. J Am Chem Soc. 2002;124:4974–4975. doi: 10.1021/ja025790m. [DOI] [PubMed] [Google Scholar]
- 51.El Seould OA, Koschella A, Fidale LC, Dom S, Heinze T. Applications of ionic liquids in carbohydrate chemistry. Biomacromolecules. 2007;8:2629–2647. doi: 10.1021/bm070062i. [DOI] [PubMed] [Google Scholar]
- 52.Pinkert A, Marsh KN, Pang S, Staiger MP. Ionic liquids and their interaction with cellulose. Chem Rev. 2009;109:6712–6728. doi: 10.1021/cr9001947. [DOI] [PubMed] [Google Scholar]
- 53.Mora-Pale M, Meli L, Doherty TV, Linhardt RJ. Room temperature ionic liquids as emerging solvents for the pretreatment of lignocellulosic biomass. Biotechnol Bioeng. 2011;108:1229–1245. doi: 10.1002/bit.23108. [DOI] [PubMed] [Google Scholar]
- 54.Qian L, Zhang H. Green synthesis of chitosan-based nanofibers and their applications. Green Chem. 2010;12:1207–1214. [Google Scholar]
- 55.Duri S, El-Zahab B, Tran CD. Polysaccharide Ecocomposite Materials: Synthesis, Characterization and Application for Removal of Pollutants and Bacteria. ECS Trans. 2012;50:573–594. doi: 10.1149/05011.0573ecst. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frez C, Diebold G, Tran CD, Yu S. Determination of thermal physical properties of room temperature ionic liquids by transient grating technique. J Chem Eng Data. 2006;54:1250–1255. [Google Scholar]
- 57.Duri S, Majoni S, Hossenlopp JM, Tran CD. Determination of chemical homogeneity of fire retardant polymeric nanocomposite materials by near-infrared multispectral imaging microscopy. J Mater Sci: Mater Med. 2010;21:1781–1787. [Google Scholar]
- 58.Baptista MS, Gao GH. Near infrared detection of flow injection analysis by acousto-optic tunable filter based spectrophotometry. Anal Chem. 1996;68:971–976. doi: 10.1021/ac951025x. [DOI] [PubMed] [Google Scholar]
- 59.Tran CD, Kong X. Determination of identity and sequences of tri- and tetrapeptides by near-infrared spectrometry. Anal Biochem. 2000;286:67–74. doi: 10.1006/abio.2000.4768. [DOI] [PubMed] [Google Scholar]
- 60.Furuzono T, Taguchi T, Kishida A, Akashi M, Tamada Y. Preparation and characterization of apatite deposited on silk fabric using an alternate soaking process. J Biomed Mater Res. 2000;50:344–352. doi: 10.1002/(sici)1097-4636(20000605)50:3<344::aid-jbm8>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 61.Cali JP, Bowers GN, Young DS. A Refree method for the determination of total calcium in serum. Clin Chem. 1973;19:1208–1213. [PubMed] [Google Scholar]
- 62.Drummond L, Maher W. Determination of phosphorus in aqueous solution via formation of the phosphoantimonylmolybdenum blue complex Re-examination of optimum conditions for the analysis of phosphate. Anal Chim Acta. 1995;302:69–74. [Google Scholar]
- 63.Pinto RJB, Fernandes SCM, Freire CSR, Sadoco P, Causio J, Eto CP, Trindade T. Antibacterial activity of optically transparent nanocomposite films based on chitosan or its derivatives and silver nanoparticles. Carbohydr Res. 2012;348:77–83. doi: 10.1016/j.carres.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 64.Toth JM, Lynch KL. Mechanical and biological characterization of calcium phosphate for use as biomaterials. In: Wise DL, Trantolo DJ, Altobelli DE, Yaszemski MJ, Gresser JD, Schwartz ER, editors. Encyclopedic Handbook of Biomaterials and Bioengineering Part A. New York: Marcel Dekker; 1995. pp. 1465–1499. [Google Scholar]
- 65.Docherty KM, Kulpa CF. Toxicity and antimicrobial activity of imidazolium and pyridinium ionic liquids. Green Chem. 2005;7:185–189. [Google Scholar]
- 66.Landry TD, Brooks K, Poche D, Woolhiser M. Acute Toxicity Profile of 1-Butyl-3-Methylimidazolium Chloride. Bull Environ Contam Toxicol. 2005;74:559–565. doi: 10.1007/s00128-005-0620-4. [DOI] [PubMed] [Google Scholar]
- 67.Ropel L, Belveze LS, Aki SNVK, Stadtherr MA, Brennecke JF. Octanol-water partition coefficients of imidazolium-based ionic liquids. Green Chem. 2005;7:83–90. [Google Scholar]
- 68.Tran CD, Lacerda SP, Oliveira D. Absorption of water by room-temperature ionic liquids: Effect of anions on concentration and states of water. Appl Spectrosc. 2003;57:152–157. doi: 10.1366/000370203321535051. [DOI] [PubMed] [Google Scholar]
- 69.Da RAL, Leite FL, Pereiro LV, Nascente PAP, Zucolotto V, Oliveira ONJ, Carvalho AJF. Adsorption of chitosan on spin-coated cellulose films. Carbohydr Pol. 2010;80:65–70. [Google Scholar]
- 70.Dreve S, Kacso I, Bratu I, Indrea E. Chitosan-based delivery systems for diclofenac delivery: Preparation and characterizatio. J Phys: Conference Series. 2009;182:1–4. [Google Scholar]
- 71.Ge H, Zhao B, Lai Y, Hu X, Zhang D, Hu K. From crabshell to chitosan-hydroxyapatite composite material via a biomorphic mineralization synthesis method. J MAter Sci: Mater Med. 2010;21:1781–1787. doi: 10.1007/s10856-010-4045-1. [DOI] [PubMed] [Google Scholar]
- 72.Yamaguchi I, Tokuchi K, Fukuzaki H, Koyama Y, Takakuda K, Monma H, Tanaka J. Preparation and microstructure analysis of chitosan/hydroxyapatite nanocomposites. J Biomed Mat Res A. 2001;55A:20–27. doi: 10.1002/1097-4636(200104)55:1<20::aid-jbm30>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 73.Dupraz A, Nguyen TP, Richard M, Daculsi G, Passuti N. Influence of a Cellulosic Ether Carrier on the Structure of Biphasic Calcium Phosphate Ceramic Particles in an Injectable Composite Material. Biomaterials. 1999;20:663–673. doi: 10.1016/s0142-9612(98)00222-1. [DOI] [PubMed] [Google Scholar]
- 74.Wang L, Nancollas GH. Calcium Orthophosphates: Crystallization and Dissolution. Chem Rev. 2008;108:4628–4669. doi: 10.1021/cr0782574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cai Y, Liu Y, Yan W, Hu Q, Tao J, Zhang M, Shi Z, Tang R. Role of Hydroxyapatite Nanoparticle Size in Bone Cell Proliferation. J Mater Chem. 2007;17:3780–3787. [Google Scholar]
- 76.Kong M, Chen XG, Xing K, Park HJ. Antimicrobial properties of chitosan and mode of action: a state of the art review. Int J Food Microbiol. 2010;144:51–63. doi: 10.1016/j.ijfoodmicro.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 77.Jayakumar R, Prabaharan M, Sudheesh KPT, Nair SV, Tamura H. Biomaterials based on chitin and chitosan in wound dressing applications. Biotechnol Adv. 2011;29:322–337. doi: 10.1016/j.biotechadv.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 78.Ignatova M, Starbova K, Markova N, Naolova N, Rashkov I. Electrospun nano-fibre mats with antibacterial properties from quarternized chitosan and poly(vinyl alcohol) Carbohydr Res. 2006;341:2098–2107. doi: 10.1016/j.carres.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 79.Cai ZX, Mo XM, Zhang KH, Fan LP, Yin AL, He CL, Wang HS. Fabrication of chitosan/silk fibroin composite nanofibers for wound dressing applications. Int J Mol Sci. 2010;11:3529–3539. doi: 10.3390/ijms11093529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aziz MA, Cabral JD, Brooks HJL, Moratti SC, Hanton LR. Antimicrobial properties of a chitosan dextran-based hydrogel for surgical use. Antimicrob Agents Chemother. 2012;56:280–287. doi: 10.1128/AAC.05463-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Afzal MA, Kalmodia S, Kesarwani P, Basu B, Balani K. Bactericidal effect of silver-reinforced carbon nanotube and hydroxyapatite composites. J Bomater Appl. 2012 doi: 10.1177/0885328211431856. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.