

Abstract
The NR5A1 gene encodes for steroidogenic factor 1, a nuclear receptor that regulates proper adrenal and gonadal development and function. Mutations identified by NR5A1 sequencing have been associated with disorders of sex development (DSD), ranging from sex reversal to severe hypospadias in 46,XY patients and premature ovarian failure (POF) in 46,XX patients. Previous reports have identified four families with a history of both 46,XY DSD and 46,XX POF carrying segregating NR5A1 sequence mutations. Recently, three 46,XY DSD sporadic cases with NR5A1 microdeletions have been reported.
Here, we identify the first NR5A1 microdeletion transmitted in a pedigree with both 46,XY DSD and 46,XX POF. A 46,XY individual with DSD due to gonadal dysgenesis was born to a young mother who developed POF. Array CGH analysis revealed a maternally inherited 0.23 Mb microdeletion of chromosome 9q33.3, including the NR5A1 gene. Based on this finding, we screened patients with unexplained 46,XY DSD (n=11), proximal hypospadias (n=21) and 46,XX POF (n=36) for possible NR5A1 copy-number variations (CNVs) via multiplex ligation-dependent probe amplification (MLPA), but did not identify any additional CNVs involving NR5A1. These data suggest that NR5A1 CNVs are an infrequent cause of these disorders but that array CGH and MLPA are useful genomic screening tools to uncover the genetic basis of such unexplained cases. This case is the first report of a familial NR5A1 CNV transmitting in a pedigree, causing both the male and female phenotypes associated with NR5A1 mutations, and the first report of a NR5A1 CNV associated with POF.
Keywords: 46,XY gonadal dysgenesis; 46,XY disorders of sex development; NR5A1; SF-1 transcription factor; premature ovarian failure; hypospadias
INTRODUCTION
The nuclear receptor subfamily 5, group A, member 1 gene (NR5A1; OMIM 184757) is located on human chromosome 9q33.3 and encodes for steroidogenic factor 1 (SF1), an intracellular transcription factor that regulates the expression of key genes required for proper sexual development. In human and murine development, NR5A1 is expressed in the developing brain, adrenal gland, and both the XX and XY undifferentiated gonadal somatic cells [Hanley et al., 1999; Lin and Achermann, 2008; Ramayya et al., 1997]. During sexual differentiation, NR5A1 plays a crucial role in male sex development by governing the regulatory and hormonal networks of male internal and external genital differentiation. In testicular Sertoli cells, NR5A1 is necessary for the expression of male sexual differentiation genes, such as SRY and SOX9, and cooperates with the Wilms’ tumor gene (WT1) to regulate the expression of Anti-Müllerian hormone (AMH) [Nachtigal et al., 1998; Sekido and Lovell-Badge, 2008]. In testicular Leydig cells, NR5A1 stimulates the expression of the luteinizing hormone receptor (LHR), insulin-like polypeptide 3 (INSL3), the AMH receptor (AMHR2), and enzymes required for testosterone biosynthesis, thus mediating male testicular descent and external genital virilization [Fynn-Thompson et al., 2003; Schimmer and White, 2010; Zimmermann et al., 1998].
In females, NR5A1 expression is persistent from early ovarian development though sex differentiation phases, dictating normal ovarian morphogenesis [Hanley et al., 1999]. However, female internal genital duct and external genital development are not dependent upon NR5A1. After puberty, ovarian somatic cell NR5A1 expression becomes important, governing proper ovarian steroidogenesis and follicular cycling [Lourenco et al., 2009].
Targeted deletion of Nr5a1 in mice results in early postnatal death from failed adrenal development. In xy Nr5a1-null mice, complete testicular dysgenesis and male-to-female sex reversal is present with female external genitalia and persistence of Müllerian structures [Luo et al., 1994]. Conditional Nr5a1 inactivation within ovarian granulosa cells results in sterile XX mice with hypoplastic ovaries lacking corpora lutea and containing hemorrhagic cysts [Jeyasuria et al., 2004].
Disorders of sex development (DSD) are human congenital conditions in which development of the chromosomal, gonadal, or anatomic sex is atypical [Lee et al., 2006]. In cases with 46,XY karyotype, the DSD state can be due to either a disorder of androgen synthesis or action or a disorder of gonadal (testicular) development (46,XY DSD due to gonadal dysgenesis [hereafter 46,XY GD]). 46,XY GD can be either complete GD (previously called ‘sex reversal’), yielding female external genitalia, streak gonads, and Müllerian structures, or partial GD, yielding ambiguous external genitalia, dysgenetic testes, and partial development of Müllerian and Wolffian structures. Due to phenotypic similarity with Nr5a1-null male mice, two 46,XY GD patients with primary adrenal failure were screened for mutations in NR5A1 and both patients were found to carry loss-of-function mutations in this gene [Achermann et al., 1999; Achermann et al., 2002]. Although initially associated with adrenal failure, subsequent studies of NR5A1 mutations have discovered that loss-of-function mutations in NR5A1 are more frequent in 46,XY DSD sex reversal patients without adrenal failure than in 46,XY DSD sex reversal patients with adrenal failure [Tajima et al., 2009].
The reported frequency of NR5A1 loss-of-function mutations is approximately 20% in 46,XY GD patients with impaired androgenization but normal adrenal function [Kohler et al., 2008; Lin et al., 2007; Philibert et al., 2010] and 15% in patients with severe penoscrotal hypospadias and cryptorchidism [Kohler et al., 2009]. The disease-causing variants reported in NR5A1 range from missense, nonsense, and splicing mutations to small deletions and insertions. In contrast to mutations detectable by sequencing, copy-number variations (CNVs) are deletions or duplications of part of a genomic fragment that change the effective number of DNA copies of a gene. Three NR5A1 CNVs, one partial deletion and two entire gene deletions, have previously been reported in sporadic patients with 46,XY DSD [Barbaro et al., 2011; Schlaubitz et al., 2007; van Silfhout et al., 2009]. However, NR5A1 CNVs have not been comprehensively searched for in a cohort of 46,XY DSD cases.
Variations in NR5A1 extend beyond 46,XY DSD and hypospadias phenotypes, to anorchia, male infertility, and premature ovarian failure. Premature ovarian failure (POF; OMIM 612964) is a condition characterized by amenorrhea for at least 4 months before the age of 40 years. In sporadic cases of POF, the mutation frequency of NR5A1 is approximately 3–8% [Janse et al., 2012; Lourenco et al., 2009]. Four families with histories of both 46,XY DSD and 46,XX POF have been identified in which the affected individuals carry NR5A1 loss-of-function mutations [Lourenco et al., 2009]. However, patients with these phenotypes have not been screened for NR5A1 CNVs.
In this study, we report a pedigree detailing a mother and son with POF and 46,XY GD, respectively, both carrying the same 9q33.3 deletion encompassing NR5A1. This case is the first report of a familial NR5A1 CNV transmitting in a pedigree, causing both the male and female phenotypes associated with NR5A1 mutations, and the first report of a NR5A1 CNV associated with POF. Based on the findings of NR5A1 CNV, we investigated sporadic cases of unexplained 46,XY DSD, proximal hypospadias, and POF for possible NR5A1 CNV.
MATERIALS AND METHODS
Study population
All human subjects were recruited for genetic testing and informed consent obtained per protocol approved by the Institutional Review Board at the University of Texas Southwestern Medical Center. Patients with known chromosomal abnormalities or genetic cause for their disease were excluded from the study. Patients underwent karyotyping and complete phenotyping at the University of Texas Southwestern Medical Center affiliated hospitals and medical records were reviewed. Of the 70 human study subjects, individuals were of varied ethnicity (16 Caucasians, 12 Hispanics, 6 African Americans and 36 Asians) and had either 46,XY DSD (n=12; Table I), 46,XY proximal hypospadias (n=21), 46,XX POF (n=36) or was normal (n=1; Patient I-2). All were unrelated except the three subjects comprising the pedigree (Figure 1: Patients I-2, II-2, III-1). Genomic DNA was extracted from patients’ peripheral blood lymphocytes (Puregene, Gentra Systems, Minneapolis, MN) per manufacturer’s protocols.
Table I.
Clinical, anatomic, and genetic characteristics of 12 identified patients with 46,XY disorders of sex development
| Case | Sex of Rearing | External Genital Phenotype | Müllerian Structures | Wollfian Structures | Gonadal Position | Gonadal Histology | aCGH Results | Normal Genetic Testing |
|---|---|---|---|---|---|---|---|---|
| 1 | Female | Clitoromegaly, mild labial fusion, short UGS | Hemiuterus; vaginal duplication | No vas bilaterally | IA | Normal testes | Normal |
SRY NR5A1 WT1 DHH |
| 2 | Female | Normal clitoris, mild labial fusion, short UGS | None | None | IA | Dysgenetic gonads | Normal |
SRY AR WT1 SRD5A2 |
| 3 | Female | Clitoromegaly, labial fusion, short UGS | None | Normal | Inguinal | Testicular atrophy bilaterally | Normal | N.D. |
| 4 | Female | Clitoromegaly, labial fusion, UGS | Uterus, vagina | Normal | IA | Testicular atrophy bilaterally | N.D. | N.D. |
| 5 | Female | Mild clitoromegaly, mild labial fusion, short UGS | No uterus; vagina present | Normal | Inguinal | Fibrotic testes with germ cell hypoplasia | Normal | AR |
| 6 | Male | Perineal hypospadias, microphallus, penoscrotal transposition, bifid scrotum with right hypoplasia | Hemiuteru s; large vagina | Mixed | Left descended ; Right absent | Left testicular dysgenesis with absent right testis | N.D. | N.D. |
| 7 | Female | Severe clitoromegaly, no labial fusion | No uterus; short vagina | Normal | Inguinal | Right normal testis | Normal | N.D. |
| 8 | Female | Clitoromegaly, UGS | No uterus; short vagina | Unknown | Unknown | Unknown | N.D. | N.D. |
| 9 | Female | Clitoromegaly, labial fusion, UGS | No uterus; short vagina | Normal | IA | Dysgenetic testes | N.D. | AR |
| 10 | Male | Microphallus | No uterus; no vagina | Normal | Inguinal | Microtestes with paucity of germ cells | N.D. | N.D. |
| 11 | Female | Clitoromegaly, UGS | Hemiuteru s; vagina | Normal | IA | Right dysgenetic testis; left dysgenetic gonad | N.D. |
SRY NR5A1 WT1 DHH |
| Index Case(III-1) | Male | Clitoromegaly, UGS | None | Normal | Labioscrotal | Dysgenetic testes | Microdel (9q33.3) | NR5A1 |
aCGH, array comparative genomic hybridization; UGS, urogenital sinus; IA, intraabdominal; N.D., not done; SRY, sex-determining region Y; NR5A1, nuclear receptor subfamily 5 group A member 1; AR, androgen receptor; WT1, Wilms tumor 1; DHH, desert hedgehog; SRD5A2, 5α-reductase type 2 gene
Figure 1.
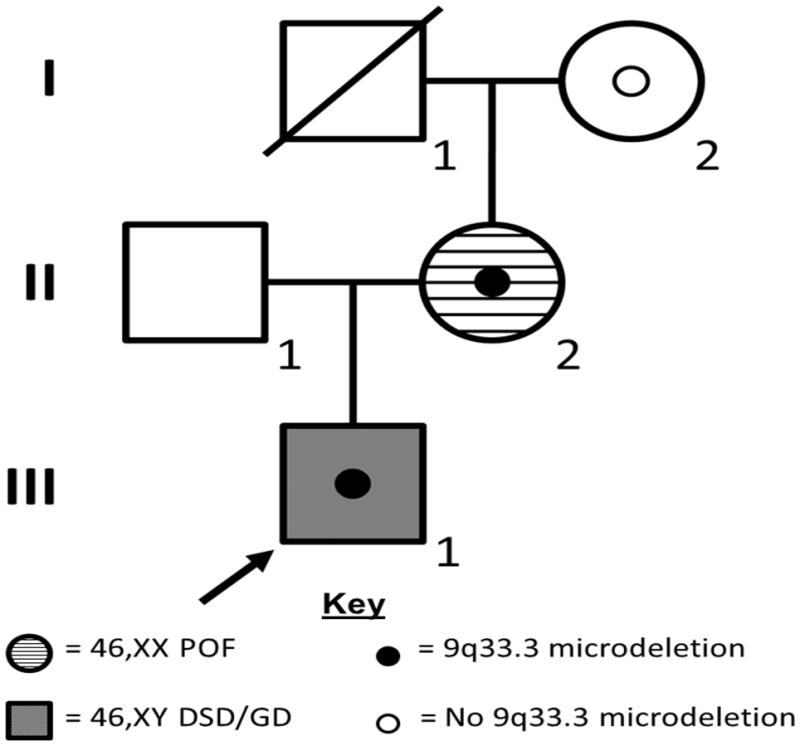
A XY GD and XX POF pedigree with a familial 9q33.3 deletion. In the pedigree, squares represent male family members and circles represent female family members. Grey shaded squares represent 46,XY patient affected with GD. Horizontal-striped circles represent 46,XX patient affected with POF. A solid circle within an individual indicates that family member was found to harbor a 9q33.3 microdeletion including NR5A1. An empty circle within an individual indicates that family member does not carry the 9q33.3 microdeletion. Individuals without an interior circle were not tested for deletion. The index patient is indicated with an arrow.
Clinical array comparative genomic hybridization/FISH
Array CGH was performed using a custom, whole genome 180,000 oligonucleotide array, “V8.1 OLIGO”, designed by the Medical Genetics Laboratories at BCM and manufactured by Agilent Technologies (Santa Clara, CA). Digestion, labeling, and hybridization were completed per protocols. BCM web-based software was used for genomic copy number analysis. The computational methods have been described previously [Schlaubitz et al., 2007]. Findings were confirmed via FISH probe RP11-164H5 per standard protocol.
Custom array comparative genomic hybridization
A custom, high-resolution 9q33-q34 array was designed using a 60,000 oligonucleotide array format (Agilent Technologies, Inc., Santa Clara, CA, USA). Average probe spacing was ~1,250 bp per probe. Chromosomal sex-matched DNA samples were used as hybridization reference normal controls. After sample processing and hybridization, data were analyzed using Agilent Genomic Workbench Software (Agilent Technologies, Inc, CA, USA) and plotted using the R Statistical Computing Package (R Core Development Team).
Deletion breakpoint determination
Long range PCR was employed to amplify the deletion-specific junction fragment utilizing the manufacturer’s protocol (Takara Bio Inc, Otsu, Japan). PCR primers were designed to flank the familial deletion breakpoints based on array CGH data. PCR products were Sanger sequenced (Lone Star, Houston, TX) and compared to the reference human genome sequence using the UCSC Genome Browser (version hg19).
Inheritance determination
PCR primers were designed to flank HapMap SNPs (listed in Supplemental Table I) contained within the deleted region and to produce amplicons containing the SNPs of the mother (II-1) and grandmother (I-2). Products were Sanger sequenced (Lone Star, Houston, TX) to determine the genotype at each locus.
Multiplex ligation-dependent probe amplification analysis
DNA was screened for NR5A1 copy-number variations using the SALSA P185-B2 Intersex MLPA kit (MRC-Holland, Amsterdam, The Netherlands), per manufacturer’s protocol. This assay includes probes for 5 of the 6 coding exons of NR5A1, in addition to probes for other causal DSD genes. Amplified products were separated by size on an ABI3100 genetic analyzer (Applied Biosystems, Foster City, California, USA) and the data were analyzed using GeneMarker V2.2.0 (SoftGenetics, State College, Pennsylvania, USA).
RESULTS
Clinical presentation of child with 46,XY GD and mother with 46,XX POF
A 15-day-old full term “female” Caucasian infant (Fig 1: Patient III-1) was evaluated emergently for fever and found to have ambiguous genitalia, with clitoromegaly, a urogenital sinus, and bilateral labioscrotal masses. Testing identified a urinary tract infection, 46,XY karyotype with neonatal female testosterone levels (testosterone <3 ng/dl), normal adrenal steroidogenic function, no internal Müllerian structures and bilateral labioscrotal testes. At 6 months age, bilateral testicular biopsies demonstrated closely packed seminiferous tubules mostly containing germ cells. Some of the germ cells were bi- or tri-nucleated and located more centrally within the tubules. Neither Leydig cells nor Müllerian structures were observed, consistent with partial gonadal dysgenesis (46,XY GD). The family elected to reassign the child as a male and after testosterone supplementation to stimulate phallic growth, bilateral orchidopexy and proximal hypospadias repair were performed at 6 months of age. At age 11, he has normal intelligence, asthma, attention-deficit-hyperactivity disorder and bipolar disorder. He was retreated with testosterone therapy for lack of spontaneous male puberty and small phallic size.
The proband’s mother (Fig 1: Patient II-2) had menarche at age 12 years and irregular menses prior to her first conception. At age 19, she conceived and delivered Patient III-1. Following the birth, she had menses every 3 months over the next 3–4 years and thereafter became less frequent. She developed secondary amenorrhea at age 28 unresponsive to multiple hormonal therapies. She had normal genitalia, vagina, cervix and uterus with post-menopausal hormonal values. Computed tomography revealed normal adrenal glands, moderate hepatomegaly and diverticulosis of the sigmoid colon. Her comorbidities included bipolar disorder, dyslexia, obesity, obstructive sleep apnea, gastroesophageal reflux disease, hiatal hernia, sigmoid diverticulosis and type II diabetes. Her past surgical history included appendectomy, cholecystectomy and knee repair. Family history is negative for disorders of sex development or premature ovarian failure and subject II-2’s mother (Fig 1; Patient I-2) did not have POF. Family history is positive for colon cancer and coronary artery disease in paternal grandparents and a strong bilineal family history of bipolar disorder.
Microdeletion of NR5A1 in child with 46,XY GD and mother with 46,XX POF
Clinical chromosomal microarray analysis of the 46,XY GD proband (Patient III-1) revealed a 0.232 Mb microdeletion of chromosome band 9q33.3, involving NR5A1 and four additional RefSeq genes. No disease-associated NR5A1 mutations were found in the other allele by sequencing the coding region (exons 2–7) in the proband. FISH testing revealed that the deletion present in the 46,XY GD proband (Patient III-1) was also harbored by his mother (Patient II-2) but was absent from the maternal grandmother (Patient I-2).
To determine the extent of the familial 9q33.3 deletion, we performed custom, high-resolution 9q33-q34 array CGH followed by long range PCR and Sanger sequencing (Fig 2; Supplemental eFigure 1 see supporting information online). Breakpoint analysis revealed the deletion of chr9:127,251,113–127,487,356 (GRChr37/hg19), impacting five RefSeq genes: NR5A1 (exons 1–6), NR6A1 (exons 3–10), MIR181A2HG (whole gene), MIR181A2 (whole gene), and MIR181B2 (whole gene).
Figure 2.

Genomic extent of the familial 9q33.3 deletion. A) Custom-designed high-resolution aCGH analysis of the 9q33.3 region. Array CGH probes with log2 subject:control ratios less than −0.5 are colored red for ease of visualization. The locations of the RefSeq genes in the region are indicated along the top of the graph as black rectangles. B) Breakpoint sequence of the patient from this study. The reference sequence of the proximal intact segment of the chromosome harboring the deletion allele is shown in red while the distal intact segment is shown in blue. The patient’s sequence is shown in between. A single thymidine base of microhomology is shown in green. All coordinates and reference sequences are provided in the GRChr37/hg19 2009 assembly.
To confirm that the NR5A1 deletion was not inherited from the deceased maternal grandfather (subject I-1) we performed genotyping of HapMap (hapmap.org) SNPs contained within the deleted region (Supplemental eTable I in supporting information online) to assure that the NR5A1 CNV did not segregate with this phenotypically normal individual who was unavailable for testing. Sanger sequencing of one of the SNPs, rs10986358, revealed a genotype in the mother (Patient II-2) that was incompatible with the genotype of the grandmother (Patient I-2) (Fig 3). Since the mother has only one allele at the rs10986358 locus (on the intact chromosome) and it is incompatible with the grandmother’s genotype, the intact chromosome is most likely inherited from the grandfather. Thus, the chromosome harboring the deletion in the mother is most likely inherited from the grandmother. A de novo point mutation between generations resulting in an rs10986358 T allele on a chromosome inherited from the grandmother cannot be excluded. However, given low locus-specific mutation rates and the fact that the haplotype of the mother’s intact chromosome is the most common among chromosomes of individuals with Northern and Western European ancestry (91/234 HapMap Phase III CEU phased chromosomes, data not shown), the probability of a mutation explaining the data is extremely low.
Figure 3.

Determination of parent of origin of the chromosome harboring the NR5A1 deletion. Genotyping of SNPs from the HapMap project reveals that the mother’s intact chromosome is inherited from her father because the genotype of rs10986358 is incompatible with her mother’s genotype. Top, schematic representation of the genotypes of the mother and grandmother. The mother is hemizygous for the minor allele (T), while the grandmother is homozygous for the reference allele (G). Thus, the mother’s intact chromosome is most likely inherited from the grandfather. Therefore, the chromosome harboring the deletion is most likely inherited from the grandmother. Bottom, Sanger sequencing traces of the rs10986358 locus in both individuals. Twelve other HapMap SNPs were also genotyped but were uninformative. No sample was available from the grandfather.
To test for the possibility of low-level mosaicism in the grandmother, we also subjected DNA isolated from her peripheral blood leukocytes to PCR testing. There was no evidence of the deletion in the grandmother (Supplementary eFigure 1 in supporting information online). Since the deletion is most likely located on the chromosome nine inherited from the grandmother and we were unable to detect evidence of the deletion in her peripheral blood genomic DNA, we concluded that the deletion arose de novo in generation II.
Screening of NR5A1 CNVs in patients with 46,XY DSD, 46,XY proximal hypospadias, and 46,XX POF
To assess the frequency of NR5A1 CNVs in DSD states, we screened 11 patients with 46,XY DSD phenotypes (described in Table I), 21 patients with proximal hypospadias (with or without cryptorchidism), and 35 patients with POF for NR5A1 CNV via NR5A1 MLPA. The MLPA assay did not identify any additional NR5A1 CNVs in the DSD, proximal hypospadias, or POF patient groups but did confirm the familial NR5A1 microdeletion (exons 1–6) in subjects II-2 and III-1. The MLPA kit did not include a probe for the unaffected NR5A1 exon 7.
DISCUSSION
We identified a novel 0.232 Mb microdeletion on 9q33.3 in a 46,XY GD patient with partial gonadal dysgenesis. Breakpoint mapping revealed that the deletion involves chr9:127,251,113–127,487,356 (GRChr37/hg19), impacting five RefSeq genes (NR5A1, NR6A1, MIR181A2HG, MIR181A2, and MIR181B2). The patient’s mother, diagnosed with POF, was found to carry the same 9q33.3 microdeletion. Subsequent studies utilizing SNP genotyping and PCR analysis determined that the deletion arose de novo between generations I and II (Fig 1). A single thymidine base pair of microhomology was detected at the breakpoint (Fig 2B). Microhomology is the hallmark of replicative mechanisms of CNV formation such as microhomology mediated break-induced replication or fork stalling and template switching; however non-homologous end joining cannot be excluded [Hughes et al., 2006].
One of the genes implicated in the familial 9q33.3 microdeletion is NR5A1, also known as steroidogenic factor 1 (SF1), a transcription factor that regulates the expression of key genes required for proper sexual development, such as SRY, SOX9, and AMH. NR5A1 is a confirmed causal gene for DSD as NR5A1 sequence mutations account for approximately 20% of 46,XY GD cases [Kohler et al., 2008; Lin et al., 2007; Philibert et al., 2010]. Additionally, three NR5A1 CNV have previously been reported in cases of 46,XY DSD [Barbaro et al., 2011; Schlaubitz et al., 2007; van Silfhout et al., 2009]. NR5A1 sequence mutations were previously identified in affected members from four families with histories of both 46,XY DSD and 46,XX POF [Lourenco et al., 2009]. We report the first pedigree with both 46,XY DSD and 46,XX POF due to NR5A1 CNV.
Aside from NR5A1, four other genes are located in the 9q33.3 deletion reported in this pedigree. Of these four genes, only NR6A1 is expressed in tissues related to sexual development. However, NR6A1 has not been associated with sex reversal in humans and is not deleted in a 46,XY GD patient reported by Barbaro et al with only partial NR5A1 deletion, suggesting that NR5A1 haploinsufficiency alone is sufficient to cause the patient’s abnormal sexual development [Barbaro et al., 2011].
Mutations in known sex-determination genes, such as SRY, SOX9, WNT4, DAX1, and NR5A1 account for almost 50% of 46,XY GD, suggesting the genetic basis for the remaining 50% of patients is unknown [White et al., 2011]. However, screening for mutations in the known sex-determining genes typically only identifies point mutations, while genomic rearrangements, such as CNV, are often missed by sequencing and are too small to be detected by karyotyping. Two recent publications have identified causal CNV in 13–30% patients with unexplained, syndromic or non-syndromic 46,XY GD [Ledig et al., 2010; White et al., 2011], rates in range with our observed 17% in a similar patient cohort (Harrison et al., In review). Additionally, 22% of DSD patients with phenotypes including ambiguous genitalia, hypospadias, and cryptorchidism, had a clinically relevant CNV, 74% of which were undetectable by karyotype [Tannour-Louet et al., 2010].
Making the correct molecular diagnosis can have significant implications for gender assignment and thus the likely response to hormone treatment as well as genetic counseling for the family. As only 15–20% of 46,XY DSD and proximal hypospadias cases and 3% of POF cases have NR5A1 sequence mutations, copy-number variations in NR5A1, such as the deletion reported in this pedigree, are another genomic cause to consider. These data suggest that NR5A1 CNVs are an infrequent cause of these disorders; however, copy number tests, such as array CGH and MLPA, are useful genomic screening tools to uncover the genetic basis of such unexplained 46,XY DSD cases.
Supplementary Material
PCR analysis of the familial 9q33.3 deletion. Top) Family tree illustrating the source of peripheral blood genomic DNA used in PCR reactions with primers specific for the familial 9q33.3 deletion. Bottom) Electrophoresis of PCR products through a 1% agarose gel with ethidium bromide reveals the presence of the deletion specific amplicon in the patient and mother. The same junction fragment could not be amplified from the maternal grandmother.
Acknowledgments
This investigation was supported by the NIH National Research Service Award T32GM083831 (SMH) from the National Institute of General Medical Sciences, the Baylor College of Medicine Medical Scientist Training Program 5T32GM007330 (IMC), NICHD award R01HD048690 (DHC), and the Seay Endowment (LAB).
Footnotes
The authors have no conflicts of interest to declare.
References
- Achermann JC, Ito M, Hindmarsh PC, Jameson JL. A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat Genet. 1999;22(2):125–126. doi: 10.1038/9629. [DOI] [PubMed] [Google Scholar]
- Achermann JC, Ozisik G, Ito M, Orun UA, Harmanci K, Gurakan B, Jameson JL. Gonadal determination and adrenal development are regulated by the orphan nuclear receptor steroidogenic factor-1, in a dose-dependent manner. J Clin Endocrinol Metab. 2002;87(4):1829–1833. doi: 10.1210/jcem.87.4.8376. [DOI] [PubMed] [Google Scholar]
- Barbaro M, Cools M, Looijenga LH, Drop SL, Wedell A. Partial deletion of the NR5A1 (SF1) gene detected by synthetic probe MLPA in a patient with XY gonadal disorder of sex development. Sex Dev. 2011;5(4):181–187. doi: 10.1159/000328821. [DOI] [PubMed] [Google Scholar]
- Fynn-Thompson E, Cheng H, Teixeira J. Inhibition of steroidogenesis in Leydig cells by Mullerian-inhibiting substance. Mol Cell Endocrinol. 2003;211(1–2):99–104. doi: 10.1016/j.mce.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Hanley NA, Ball SG, Clement-Jones M, Hagan DM, Strachan T, Lindsay S, Robson S, Ostrer H, Parker KL, Wilson DI. Expression of steroidogenic factor 1 and Wilms’ tumour 1 during early human gonadal development and sex determination. Mech Dev. 1999;87(1–2):175–180. doi: 10.1016/s0925-4773(99)00123-9. [DOI] [PubMed] [Google Scholar]
- Hughes IA, Houk C, Ahmed SF, Lee PA. Consensus statement on management of intersex disorders. J Pediatr Urol. 2006;2(3):148–162. doi: 10.1016/j.jpurol.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Janse F, de With LM, Duran KJ, Kloosterman WP, Goverde AJ, Lambalk CB, Laven JS, Fauser BC, Giltay JC. Limited contribution of NR5A1 (SF-1) mutations in women with primary ovarian insufficiency (POI) Fertil Steril. 2012;97(1):141–146. e142. doi: 10.1016/j.fertnstert.2011.10.032. [DOI] [PubMed] [Google Scholar]
- Jeyasuria P, Ikeda Y, Jamin SP, Zhao L, De Rooij DG, Themmen AP, Behringer RR, Parker KL. Cell-specific knockout of steroidogenic factor 1 reveals its essential roles in gonadal function. Mol Endocrinol. 2004;18(7):1610–1619. doi: 10.1210/me.2003-0404. [DOI] [PubMed] [Google Scholar]
- Kohler B, Lin L, Ferraz-de-Souza B, Wieacker P, Heidemann P, Schroder V, Biebermann H, Schnabel D, Gruters A, Achermann JC. Five novel mutations in steroidogenic factor 1 (SF1, NR5A1) in 46,XY patients with severe underandrogenization but without adrenal insufficiency. Hum Mutat. 2008;29(1):59–64. doi: 10.1002/humu.20588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler B, Lin L, Mazen I, Cetindag C, Biebermann H, Akkurt I, Rossi R, Hiort O, Gruters A, Achermann JC. The spectrum of phenotypes associated with mutations in steroidogenic factor 1 (SF-1, NR5A1, Ad4BP) includes severe penoscrotal hypospadias in 46,XY males without adrenal insufficiency. Eur J Endocrinol. 2009;161(2):237–242. doi: 10.1530/EJE-09-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledig S, Hiort O, Scherer G, Hoffmann M, Wolff G, Morlot S, Kuechler A, Wieacker P. Array-CGH analysis in patients with syndromic and non-syndromic XY gonadal dysgenesis: evaluation of array CGH as diagnostic tool and search for new candidate loci. Hum Reprod. 2010;25(10):2637–2646. doi: 10.1093/humrep/deq167. [DOI] [PubMed] [Google Scholar]
- Lee PA, Houk CP, Ahmed SF, Hughes IA. Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics. 2006;118(2):e488–500. doi: 10.1542/peds.2006-0738. [DOI] [PubMed] [Google Scholar]
- Lin L, Achermann JC. Steroidogenic factor-1 (SF-1, Ad4BP, NR5A1) and disorders of testis development. Sex Dev. 2008;2(4–5):200–209. doi: 10.1159/000152036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Philibert P, Ferraz-de-Souza B, Kelberman D, Homfray T, Albanese A, Molini V, Sebire NJ, Einaudi S, Conway GS, Hughes IA, Jameson JL, Sultan C, Dattani MT, Achermann JC. Heterozygous missense mutations in steroidogenic factor 1 (SF1/Ad4BP, NR5A1) are associated with 46,XY disorders of sex development with normal adrenal function. J Clin Endocrinol Metab. 2007;92(3):991–999. doi: 10.1210/jc.2006-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenco D, Brauner R, Lin L, De Perdigo A, Weryha G, Muresan M, Boudjenah R, Guerra-Junior G, Maciel-Guerra AT, Achermann JC, McElreavey K, Bashamboo A. Mutations in NR5A1 associated with ovarian insufficiency. N Engl J Med. 2009;360(12):1200–1210. doi: 10.1056/NEJMoa0806228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77(4):481–490. doi: 10.1016/0092-8674(94)90211-9. [DOI] [PubMed] [Google Scholar]
- Nachtigal MW, Hirokawa Y, Enyeart-VanHouten DL, Flanagan JN, Hammer GD, Ingraham HA. Wilms’ tumor 1 and Dax-1 modulate the orphan nuclear receptor SF-1 in sex-specific gene expression. Cell. 1998;93(3):445–454. doi: 10.1016/s0092-8674(00)81172-1. [DOI] [PubMed] [Google Scholar]
- Philibert P, Leprieur E, Zenaty D, Thibaud E, Polak M, Frances AM, Lespinasse J, Raingeard I, Servant N, Audran F, Paris F, Sultan C. Steroidogenic factor-1 (SF-1) gene mutation as a frequent cause of primary amenorrhea in 46,XY female adolescents with low testosterone concentration. Reprod Biol Endocrinol. 2010;8:28. doi: 10.1186/1477-7827-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramayya MS, Zhou J, Kino T, Segars JH, Bondy CA, Chrousos GP. Steroidogenic factor 1 messenger ribonucleic acid expression in steroidogenic and nonsteroidogenic human tissues: Northern blot and in situ hybridization studies. J Clin Endocrinol Metab. 1997;82(6):1799–1806. doi: 10.1210/jcem.82.6.3967. [DOI] [PubMed] [Google Scholar]
- Schimmer BP, White PC. Minireview: steroidogenic factor 1: its roles in differentiation, development, and disease. Mol Endocrinol. 2010;24(7):1322–1337. doi: 10.1210/me.2009-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaubitz S, Yatsenko SA, Smith LD, Keller KL, Vissers LE, Scott DA, Cai WW, Reardon W, Abdul-Rahman OA, Lammer EJ, Lifchez CA, Magenis E, Veltman JA, Stankiewicz P, Zabel BU, Lee B. Ovotestes and XY sex reversal in a female with an interstitial 9q33.3–q34.1 deletion encompassing NR5A1 and LMX1B causing features of Genitopatellar syndrome. Am J Med Genet A. 2007;143A(10):1071–1081. doi: 10.1002/ajmg.a.31685. [DOI] [PubMed] [Google Scholar]
- Sekido R, Lovell-Badge R. Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature. 2008;453(7197):930–934. doi: 10.1038/nature06944. [DOI] [PubMed] [Google Scholar]
- Tajima T, Fujiwara F, Fujieda K. A novel heterozygous mutation of steroidogenic factor-1 (SF-1/Ad4BP) gene (NR5A1) in a 46, XY disorders of sex development (DSD) patient without adrenal failure. Endocr J. 2009;56(4):619–624. doi: 10.1507/endocrj.k08e-380. [DOI] [PubMed] [Google Scholar]
- Tannour-Louet M, Han S, Corbett ST, Louet JF, Yatsenko S, Meyers L, Shaw CA, Kang SH, Cheung SW, Lamb DJ. Identification of de novo copy number variants associated with human disorders of sexual development. PLoS One. 2010;5(10):e15392. doi: 10.1371/journal.pone.0015392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Silfhout A, Boot AM, Dijkhuizen T, Hoek A, Nijman R, Sikkema-Raddatz B, van Ravenswaaij-Arts CM. A unique 970kb microdeletion in 9q33.3, including the NR5A1 gene in a 46,XY female. Eur J Med Genet. 2009;52(2–3):157–160. doi: 10.1016/j.ejmg.2009.02.009. [DOI] [PubMed] [Google Scholar]
- White S, Ohnesorg T, Notini A, Roeszler K, Hewitt J, Daggag H, Smith C, Turbitt E, Gustin S, van den Bergen J, Miles D, Western P, Arboleda V, Schumacher V, Gordon L, Bell K, Bengtsson H, Speed T, Hutson J, Warne G, Harley V, Koopman P, Vilain E, Sinclair A. Copy number variation in patients with disorders of sex development due to 46,XY gonadal dysgenesis. PLoS One. 2011;6(3):e17793. doi: 10.1371/journal.pone.0017793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann S, Schwarzler A, Buth S, Engel W, Adham IM. Transcription of the Leydig insulin-like gene is mediated by steroidogenic factor-1. Mol Endocrinol. 1998;12(5):706–713. doi: 10.1210/mend.12.5.0107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PCR analysis of the familial 9q33.3 deletion. Top) Family tree illustrating the source of peripheral blood genomic DNA used in PCR reactions with primers specific for the familial 9q33.3 deletion. Bottom) Electrophoresis of PCR products through a 1% agarose gel with ethidium bromide reveals the presence of the deletion specific amplicon in the patient and mother. The same junction fragment could not be amplified from the maternal grandmother.