

Abstract
Cyanobacteria are valuable organisms for studying the physiology of photosynthesis and carbon fixation, as well as metabolic engineering for the production of fuels and chemicals. This work describes a novel counter selection method for the cyanobacterium Synechococcus sp. PCC 7002 based on organic acid toxicity. The organic acids acrylate, 3-hydroxypropionate, and propionate were shown to be inhibitory towards Synechococcus sp. PCC 7002 and other cyanobacteria at low concentrations. Inhibition was overcome by a loss of function mutation in the gene acsA, which is annotated as an acetyl-CoA ligase. Loss of AcsA function was used as a basis for an acrylate counter selection method. DNA fragments of interest were inserted into the acsA locus and strains harboring the insertion were isolated on selective medium containing acrylate. This methodology was also used to introduce DNA fragments into a pseudogene, glpK. Application of this method will allow for more advanced genetics and engineering studies in Synechococcus sp. PCC 7002 including the construction of markerless gene deletions and insertions. The acrylate counter-selection could be applied to other cyanobacterial species where AcsA activity confers acrylate sensitivity (e.g. Synechocystis sp. PCC 6803).
Introduction
Current research concerning the physiology of cyanobacteria has led to an increased understanding of photosynthesis, CO2 fixation, and metabolism [1–3]. In addition to basic science, researchers are also exploring the application of cyanobacteria for the production of biofuels and chemicals [4,5]. Continued advances in these areas will require more efficient methods for genetic manipulation and gene expression [6,7]. In particular, counter selection methods that are applicable to multiple strains of cyanobacteria would be useful in dealing with the issues of multiple chromosomes and limited antibiotic resistance cassettes. Currently, a sacB counter selection system was demonstrated in a glucose tolerant strain of Synechocystis sp. PCC 6803 (PCC 6803) and a mazF based system in wild type PCC 6803 [8,9]. Unfortunately, attempts to build a sacB counter selection system have been unsuccessful and mazF has not been demonstrated in Synechococcus sp. PCC 7002 (PCC 7002), another model organism that grows rapidly and is widely tolerant to light intensity, temperature and salt conditions. Due to these advantages, PCC 7002 has recently been explored for the production of α-olefins and sugars via metabolic engineering [10,11].
Since a counter selection system was not available for PCC 7002, we developed a new system based on sensitivity to organic acids. In general, organic acids can cause toxicity by depleting proton motive force, accumulating anions, and inhibiting enzymes [12–14]. The organic acids of interest in this work are acrylate, 3-hydroxypropionate, and propionate. Acrylate was shown to inhibit beta oxidation in Pseudomonas and Ralstonia species, 3-hydroxypropionate was shown to inhibit the chorismate pathway in Escherichia coli, and propionate was shown to inhibit gluconeogenesis via metabolism to 2-methylcitrate in Salmonella [15–18]. Additionally, exposure to propionate was shown to cause a decrease in the free Coenzyme-A (CoA) pool in a species of Synechococcus [19].
This work demonstrates that these three organic acids, acrylate, 3-hydroxypropionate, and propionate, are inhibitory to multiple species of cyanobacteria at low concentrations. This inhibition can be overcome in PCC 7002 by a single loss of function mutation in a gene encoding a CoA-ligase (acsA). These observations were used as a basis for the development of a counter selection system using acrylate sensitivity. Acrylate was chosen amongst the organic acids due to its inhibitory activity in multiple species and dramatically increased tolerance in strains without AcsA activity. This counter selection method was used to directly introduce and select for integrations into the acsA locus, as well as generate a scarless integration in a neutral locus. The ability to rapidly introduce heterologous gene constructs and make markerless deletions will advance both basic science and applied research efforts in cyanobacteria.
Materials and Methods
Chemicals and 3-hydroxypropionate quantification
Acrylate was purchased from MP Biomedicals (ICN211387). 3-hydroxypropionate was purchased from TCI America (H0297) as a ca. 3.6 mol/L solution in water. Additional chemicals and reagents were purchased from Fisher Scientific and Sigma Aldrich. The 3-hydroxypropionate solution purchased from TCI had an unknown purity, so the solution was quantified with an enzyme assay using propionyl-CoA synthase. Escherichia coli (E. coli) BL21 harboring pKS1 (expressing propionyl-CoA synthase) was grown in lysogeny broth (LB) to an OD600 of 0.4 and induced with 0.4 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). The induced culture was incubated at 30 °C for 4 h before centrifugation. The resulting cell pellet was resuspended in twice the volume of 100 mM Tris pH 7.8 and lysed by sonication. The cell debris was removed by centrifugation at 16,000 x g for 10 min at 4 °C. The supernatant was incubated at 63 °C for 10 min and the precipitated protein was removed by centrifugation at 20,000 x g. The resulting protein fraction was concentrated 2X using an Amicon Ultra-4 centrifugation column to increase activity. The resulting enzyme was used in an assay to quantify 3-HP by NAPH reduction as described in Schneider et al [20].
Strains, culturing conditions, and media
All strains used in this study are listed in Table 1. PCC 7002, Synechococcus sp. PCC 7942 (PCC 7942), and PCC 6803 were obtained from the Pasteur Culture Collection. PCC 7002 was grown in Medium A+ pH 8.0 [21]. PCC 7942 and PCC 6803 were grown in BG-11 [22]. Unless otherwise noted, liquid cultures were grown in 10 mL volumes at a light intensity of 140 µE/m2/s at 35 °C and bubbled with air. Agar plates were incubated under the same light and temperature conditions. Cultures used to observe the initial mutant population (Figure 1) and harvest RNA were grown in 50 mL volumes without agitation or supplementation with air. Cultures were inoculated using patches grown from a single colony on solid media under the same conditions. Unless otherwise noted, cell growth was monitored by measuring the optical density at 730nm (OD730) using a Spectrophotometer 20 (Milton Roy).
Table 1. Cyanobacterial strains used in this study.
| Strain | Genotype | Comments |
|---|---|---|
| BPSyn_001 | Synechococcus sp. PCC 7002 | Obtained from the Pasteur Culture Collection |
| BPSyn_006 | acsA::aadA | Replacement of acsA with streptomycin resistance marker |
| BPSyn_014 | acsA::BC | Replacement of acsA with a 40 bp bar code |
| BPSyn_015 | acsA::pcpcB_YFP | Replacement of acsA with YFP under a constitutive promoter |
| BPSyn_022 | acsA::BC glpK::acsA aadA | Replacement of glpK of BPSyn014 with acsA and a streptomycin resistance marker |
| BPSyn_026 | acsA::BC glpK::acsAW49L aadA | Replacement of glpK of BPSyn014 with acsAW49L and a streptomycin resistance marker |
| BPSyn_027 | acsA::BC glpK::pcpcB_YFP | Replacement of glpK of BPSyn014 with YFP under a constitutive promoter |
| BP6803_001 | Synechocystis sp. PCC 6803 | Obtained from the Pasteur Culture Collection |
| BP6803_002 | sll0542::KmR | Replacement of sll0542 with kanamycin resistance marker |
| BP7942_001 | Synechococcus sp. PCC 7942 | Obtained from the Pasteur Culture Collection |
Figure 1. Growth of wild type and an adapted strain of PCC 7002 with acrylate.
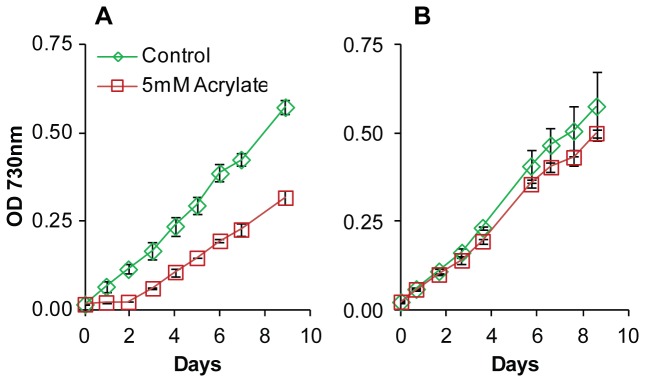
(A) Growth of wild type PCC 7002 in unmodified medium (green diamonds) or 5 mM acrylate (red squares). (B) Growth of acrylate adapted cultures of PCC 7002 in unmodified medium (green diamonds) or 5 mM acrylate (red squares). Data points are the mean of biological triplicates and error bars represent the standard deviation.
Determining minimum inhibitory concentrations
Inocula were prepared by growing cultures of wild type and mutant cyanobacteria to an OD730 of 0.3. These cultures were used to inoculate, to an initial OD730 of 0.03, a 96 well plate containing buffered media with increased concentrations of the compound of interest. Stocks of the compounds of interest were prepared in Medium A+ and adjusted to a pH of 8.0. Wild type PCC 7002 was challenged with organic acid concentration increments of 5 µM for acrylate and 1 mM for 3-hydroxypropionate and propionate. Mutant strains with increased tolerance were further challenged with concentration increments of 10 mM acrylate, 5 mM 3-hydroxypropionate, and 100 mM propionate. Plates were incubated at 35 °C at a light intensity of 140 µE/m2/s. OD730 was measured periodically using a plate reader (Tecan M1000). The minimum inhibitory concentration (MIC) was determined as the concentration at which no increase in OD730 was observed after 48 h.
Quantification of mutation frequency
Wild type PCC 7002 was grown to an OD730 of 1.0 and serially diluted in triplicate on both unmodified and acrylate-containing plates. Acrylate concentrations of 50 µM, 500 µM, 5 mM, and 50 mM were used. Plates were incubated for 5 days and counted for colonies. Mutation frequency is defined as the number of acrylate tolerant colony forming units, divided by the total number of colony forming units.
RNA-sequencing and analysis
Cultures of PCC 7002 were grown in 5 mM acrylate or unmodified medium A+ in quadruplet to an OD730 of 0.5-0.6. Duplicate cultures from each condition were harvested for RNA sequencing. RNA was isolated from PCC 7002 using the Trizol 95 method and a RNeasy kit (Qiagen) [23]. RNA purity was verified using an Agilent RNA 600 Nano Kit and Agilent 2100 Bioanalyzer. Ribosomal RNA was removed using the Epicentre rRNA removal kit for Gram negative bacteria (RZNB1056). RNA was sequenced by the University of Wisconsin Gene Expression Center using an Illumina HiSeq 2000. The resulting RNA sequencing reads were aligned to the PCC 7002 genome (Genbank ID CP000951.1-CP000957.1) and analyzed using CLC Genomics Workbench software. Reads per kilobase per million total reads (RPKM) was used to quantify expression values [24]. Genes identified as being differentially regulated were defined as having greater than 2 fold changes in RPKM and a P-value less than 0.05. Single nucleotide polymorphisms (SNPs) were identified using the CLC Genomics Workbench software. Genes of interest were identified as having SNPs in acrylate cultures but not control cultures. These data have been deposited in NCBI’s Gene Expression Omnibus (REF) and are accessible through the GEO Series accession number GSE48981 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48981).
Mutant construction
All plasmids used in this study are listed in Table 2. A PCC 7002 ΔacsA mutant (BPSyn_006) was constructed by double homologous recombination as previously described [25]. Briefly, a linear DNA fragment was constructed containing a streptomycin resistance marker derived from pSRA81 (aadA) flanked by approximately 600 bp regions that are homologous to regions directly 5’ and 3’ of the acsA (SYNPCC7002_A1838) gene. The acsA upstream and acsA downstream pieces were individually amplified using the primers in Table 3, digested, and then ligated together with the digestion fragment of pSRA81. The resulting fragment was PCR amplified and transformed into wild type PCC 7002. A similar protocol was used to replace acsA with a 40 bp barcode sequence, resulting in strain BPSyn_014. A linear fragment was created with 600 bp homologous regions and a 40 bp barcode sequence in between. This fragment was transformed into PCC 7002 and mutants were selected on plates containing 50 µM acrylate. Positive clones were streaked out on plates containing 10 mM acrylate to achieve complete chromosomal segregation. The resulting mutant had a deletion of acsA without a residual antibiotic resistance cassette. Complementation of acsA was performed through the introduction of acsA under the native acsA promoter into glpK (SYNPCC7002_A2842). Due to a frameshift mutation, glpK is a pseudogene in PCC 7002. For this reason, glpK was chosen as a neutral insertion site. Plasmids pGLPK_acsA_SpR and pGLPK_acsAW49L_SpR were constructed with homologous flanking regions to replace glpK with either (1) a wild type copy of acsA or (2) acsAW49L under the native acsA promoter along with a streptomycin resistance marker, using the in vitro recombination method using primers in Table 3 [26]. Plasmids pACSA_pcpcB_YFP and pGLPK_pcpcB_YFP were constructed in a similar manner using the cpcB promoter from PCC 6803 and yellow florescent protein (YFP) gene from plasmid pAQ1_Exp_YFP built by Xu et al. [27]. All cyanobacterial strains were screened by colony PCR for proper integration and full segregation of all chromosomes.
Table 2. Plasmids used in this study.
| Plasmid | Genotype | Comments |
|---|---|---|
| pET28b_acsA | PT7 acsA KmR | Cloned acsA into the MCS of pet28b for His-tag purification |
| pET28b_acsAW49L | PT7 acsAW49L KmR | Cloned acsAW49L into the MCS of pet28b for His-tag purification |
| pGLPK_acsA_SpR | PacsA acsA SpR | Replaced the flanking regions of pAQ1-Exp-PcpcBYFP (Xu et al) with 500 bp flanking regions of glpK |
| pGLPK_acsAW49L_SpR | PacsA acsAW49L SpR | Used Quick Change PCR to make the W49L mutation in pGLPK_acsA_SpR |
| pACSA_pcpcB_YFP | PcpcBYFP | Replaced the flanking regions of pAQ1-Exp-PcpcBYFP (Xu et al) with 500 bp flanking regions of acsA |
| pGLPK_pcpcB_YFP | PcpcBYFP | Replaced the insert of pGLPK_acsA_SpR with YFP |
Table 3. Oligonucleotides used in this study.
| Primer Name | Sequence (5'–3') |
|---|---|
| Construction of BPSyn_006 | |
| acsA-a1 | ATGATCATCGGGGAATGCTCTTGATTC |
| acsA-a2 | AAAACTGCAGTCGTGGGATTTATTTCACCCCATTGTC |
| acsA-b1 | AAAAGGATCC GTCTTAATGTATGAAGGCGCACCC |
| acsA-b2 | CCTCTGGACATCTCCCTCAAGG |
| Construction of BPSyn_014 | |
| acsA UpF | GATTTTCAAGCCCAGGTG |
| acsA UpR-BC | CCCGCATGCCCGGTCTACCTGTACACGAGTTCGTTTCAATGAAGGCGAAAC |
| acsA DwnF-BC | CCCGCATGCCTTCGTAATAAGGATGCGCTCAAGTTGATCGAATGTATG |
| acsA DwnR | CTATATCTGGCAAACAACTTTGGC |
| acsA Amplify F (A1) | AGGTGACTGCCGCACTCA |
| acsA Amplify R (A2) | CTGGCAAACAACTTTGGCTGCC |
| internal acsA F (B1) | ATTTTTGCGCCAGGTTTTGAG |
| acsA screen R (B2, C2) | CCGTAACCTCCTAGGATTGGG |
| ΔacsA BC F (C1) | AACTGTGTACAGGTAGACCG |
| Construction of pGLPK_acsA_SpR | |
| glpKUp Fwd (E1) | TGAAGCGATTGGCTATGATCTACCAAAG |
| glpKUp Rev | CTCAGGGAACCATAAGAATTCTTTTTTAAATGGGTTAAATTAGGTC |
| pacsA Fwd (G1) | GAATTCTTATGGTTCCCTGAGGCGATC |
| T7 Rev (G2) | CAAAAAACCCGTCAAGACCCGTTTAG |
| glpKDwn Fwd | GGGTCTTGACGGGTTTTTTGTTACTGCTCCATGACCAACATTATTCCC |
| glpKDwn Rev (E2) | GAAACGAGATTATCTAAAACAGAAGCATGG |
| pGLPK Fwd | GTTTTAGATAATCTCGTTTCGCATGCGACGTCGGGCCC |
| pGLPK Rev | GATCATAGCCAATCGCTTCAATGCATAGCTTGAGTATTCTATAGTGTCACC |
| Construction of pGLPK_acsAW49L_SpR | |
| acsAW49L QC Forward | AAAATGACCCCGAAGGCTTTTTGGGGGAACTCG |
| acsAW49L QC Reverse | CGAGTTCCCCCAAAAAGCCTTCGGGGTCATTTT |
| Construction of pET28b_acsA | |
| NdeIacsAForward | AAAAAACATATGTCCGAACAAAACATTGAATCCATCCTC |
| BamHIacsAReverse | AAAAAAGGATCCTTAGCCCCGGAGTTGATCGAGGA |
| Construction of pGLPK_pcpcB_YFP | |
| cpcBFwdGib | GAATTCGTTATAAAATAAACTTAACAAATCTATACCC |
| T7RevGib | CAAAAAACCCGTCAAGACCCGTTTAG |
| glpKUp Rev | CTCAGGGAACCATAAGAATTCTTTTTTAAATGGGTTAAATTAGGTC |
| glpKDwn Fwd | GGGTCTTGACGGGTTTTTTGTTACTGCTCCATGACCAACATTATTCCC |
| YFP Screen Fwd (D1) | GACGACGGCAACTACAAGAC |
| YFP Screen Rev (D2) | GGTGTTCTGCTGGTAGTG GT |
| glpK internal Fwd (F1) | CACCGCTGGGGCTTGTATCC |
| glpK outside Rev (F2) | GAGAGA AAGGCTTCATGATCAAGGG |
| Construction of pACSA_pcpcB_YFP | |
| cpcBFwdGib | GAATTCGTTATAAAATAAACTTAACAAATCTATACCC |
| pAQ1 into pACSA Rev | CATACATTCGATCAACTTGAGCGCTCTAGACAAAAAACCCGTCAAG |
| acsA Down Fwd | GCGCTCAAGTTGATCGAATGTATG |
| acsA Up Rev | GTTCGTTTCAATGAAGGCGAAAC |
AcsA activity assay
Wildtype acsA and the acsAW49L mutant were cloned into the pET-28a plasmid (Novagen) with an N-terminal His-tag. Strains of E. coli DH10B harboring these plasmids were grown in 50 mL LB at 37 °C to an OD600 of 0.6-0.8 and induced with 1 mM IPTG. Induced cultures were incubated at 37 °C for 3 h. The cultures were centrifuged at 3,000 x g for 15 min and the resulting cell pellets were frozen at -20 °C. BugBuster reagent (Novagen) was used to lyse cells and collect the soluble protein fraction. AcsA protein was purified from the soluble fraction using Ni-NTA His-tag affinity chromatography. Reactions containing 500 nM enzyme and 2 mM organic acid were performed at 37 °C for 6 min. Specific activity was determined by measuring the loss of free Coenzyme A over time using Ellman’s reagent. Specific activity is defined as µmol free CoA min-1 µmol-1 AcsA. 3-hydroxypropionate is abbreviated to 3-HP.
Counter selection
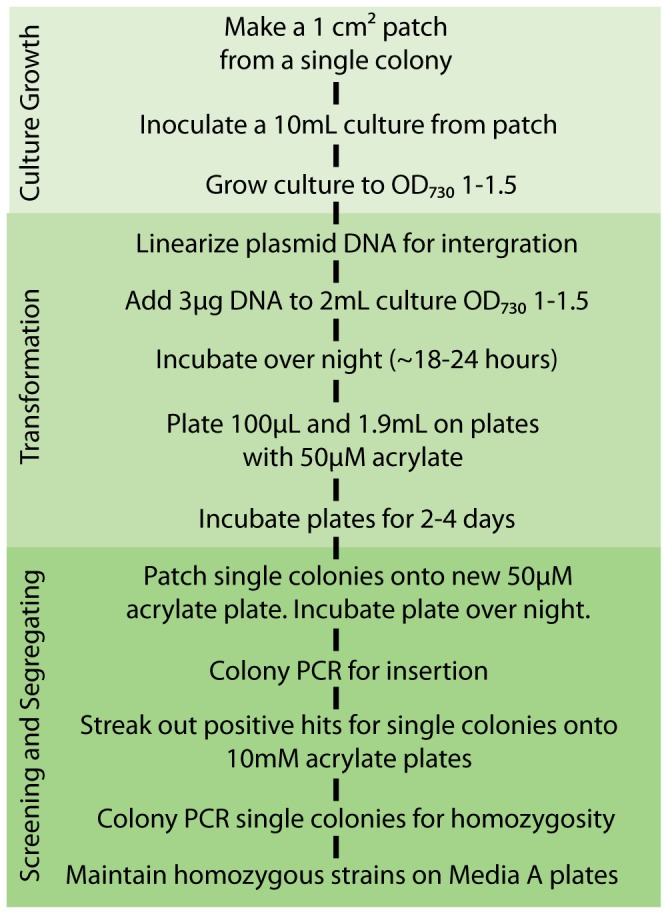
PCC 7002 was transformed by adding 3 µg of linearized plasmid to 2 mL of culture at OD730 1-1.5. The cultures were incubated without agitation overnight under the previously mentioned light and temperature conditions. Transformants were selected by plating both 100 µL and the remaining 1.9 mL transformation suspension, concentrated, on solid media supplemented with 50 μM acrylate. Plates were incubated in the light for 2-3 days at 35 °C. A successful transformation usually had significantly more colonies than the no-DNA, negative control. Single colonies were patched onto solid media supplemented with 50 μM acrylate. This intermediate step was used to avoid colony PCR contamination by excess DNA used in the transformation. After 1 day of growth at standard conditions, the presence of the insertion was confirmed by colony PCR. Single colonies of positive transformants were isolated on solid media containing 10 mM acrylate. Incubation for 3-5 days allowed for complete segregation of the transformants. Segregation was confirmed by colony PCR, using primers flanking the insertion site, specific to the insertion sequence, and specific to the sequence being replaced. Confirmed mutants were maintained on unmodified medium.
Florescence Detection
Cultures of PCC 7002 containing YFP were grown in triplicate to an OD730 0.5-1. Cultures were normalized to 4 OD730 * mL and centrifuged at 5,000 x g for 10 min. The resulting pellets were aspirated, resuspended in 300 µL BugBuster Protein Extraction Reagent (Novagen), rocked at room temperature for 30 min, and centrifuged at 16,000 x g for 25 min at 4 °C. The florescence of the resulting supernatant was measured (excitation 514 nm, emission 527 nm) using a Tecan M1000 plate reader.
Quantification of acetate
After 75 h of growth, 1 mL samples taken from triplicate cultures were centrifuged at 16,000 x g for 10 min. Each supernatant was passed through a 0.22 µm filter prior to analysis via gas chromatography and flame ionization detection using a Shimadzu GC-2010 oven with a Restek Stabilwax-DA fused silica column (11052) and AOC-20i auto-injector. The limit of detection for acetate was 10 µM.
Results and Discussion
Cyanobacteria are sensitive to several organic acids
Two components are necessary for a successful counter selection method. First, a compound must be identified that is inhibitory at low concentrations. Second, this inhibition must be removed by the loss of a genetic element. Here, the toxicity, as judged by MIC, of organic acids towards PCC 7002 PCC 6803, and PCC 7942 was assessed by exposing each strain to increasing concentrations of organic acids in buffered media. The organic acids acrylate, 3-hydroxypropionate, and propionate were each inhibitory to all three cyanobacteria at relatively low concentrations (Table 4). Acrylate generated the lowest MIC for each strain (<50 µM) followed by propionate (0.25-4 mM) and 3-hydroxypropionate (2-70 mM).
Table 4. Minimum inhibitory concentration (MIC) of organic acids.
| Strain | acrylate (mM) | 3-HP (mM) | Propionate (mM) |
|---|---|---|---|
| Synechococcus sp. PCC 7942 | 0.003 | 2 | 0.25 |
| Synechocystis sp. PCC 6803 | 0.05 | 70 | 0.25 |
| BP6803_002 (sll0542::KmR) | 70 | 70 | No Data |
| Synechococcus sp. PCC 7002 | 0.025 | 10 | 4 |
| BPSyn_006 (acsA::aadA) | 70 | 260 | >400 |
| BPSyn_014 (acsA::BC) | 70 | 260 | No Data |
| BPSyn_022 (acsA::BC glpK::acsA aadA) | 0.01 | 15 | No Data |
| BPSyn_026 (acsA::BC glpK::acsAW49L aadA) | 7 | No Data | No Data |
Mutations increase organic acid tolerance
Wild type PCC 7002 was inoculated into medium containing 5 mM acrylate, a concentration several orders of magnitude above the MIC. The OD730 was observed relative to control cultures for a period of 200 h. For the first 50 h of incubation, no increase in OD730 was observed in acrylate containing cultures. After this long lag the OD730 increased at rate similar to the control culture (Figure 1A). Sub-culturing of the acrylate exposed cultures into fresh medium containing 5 mM acrylate resulted in no growth inhibition (Figure 1B). Additionally, acrylate exposed cultures that were serially sub-cultured in medium without acrylate for four iterations maintained the ability to grow in an acrylate concentration above the wild type MIC. These data suggested that a mutation arose within the population that resulted in increased tolerance to acrylate. When wild type PCC 7002 was plated onto solid media containing 5 mM acrylate, mutant colonies were observed at a frequency of around 10-6. Mutants were observed on plates containing 500 µM and 50 µM acrylate at 5 and 10 times the frequency of 5 mM, respectively. No mutants were observed on plates containing 50 mM acrylate. This rate of mutation frequency was suggestive of a loss of function mutation [28]. Additionally, the correlation between mutation frequency and concentration of acrylate suggested that gene dosage or the presence of partial loss of function mutations existed. Interestingly, mutants obtained on acrylate containing medium were also more tolerant of 3-hydroxpropionate and propionate.
RNA-Sequencing was used to identify mutations
RNA was harvested from the cultures described in Figure 1A. Samples were processed and sequenced to identify changes in gene expression caused by exposure of wild type PCC 7002 to acrylate above the MIC. The transcriptome of cultures grown with acrylate had significantly reduced transcript levels for genes associated with oxidative stress (Table 5) compared to cells grown in unsupplemented media. In particular, isiA and sufA were down-regulated >10- and >2-fold, respectively. This was not surprising, as acrylate was shown to act as an antioxidant in eukaryotic algae [29]. Due to the starting OD730 (0.03) and the OD730 at which the cultures were harvested (0.5), it was assumed that the majority of each population carried a mutation for acrylate tolerance. An analysis of single nucleotide polymorphisms (SNPs) relative to the published genome sequence (GenBank CP000951.1) was performed (Table 6). Four genes were identified with SNPs in all acrylate samples but not present in any of the control samples. The gene acsA (GenBank NC_010475.1) was of interest because some acetyl-CoA ligases have been shown to have activity towards acrylate [30]. In acrylate exposed samples, the reads spanning bp 152 of acsA, 60% (32/53) had a point mutation of a G to a T. The point mutation results in an amino acid change of Trp49 to Leu. This amino acid is part of a conserved region in the acsA of PCC 7002, PCC 6803 (GenBank AP012278.1) and Escherichia coli K12 (GenBank NP_418493.1), suggesting it is integral to a functional protein. These results led to the hypothesis that loss of function of acsA would result in the observed increase in organic acid tolerance.
Table 5. Genes differentially expressed in the presence of 5 mM acrylate.
| Feature ID | Annotation | Fold Change | P-value |
|---|---|---|---|
| hliA | high light/nutrient deprived stress response | 5.2 | 9.75E-03 |
| SYNPCC7002_A1476 | high light inducible | 3.46 | 1.67E-05 |
| nirA | nitrite reductase | 3.42 | 0 |
| SYNPCC7002_A2493 | conserved hypothetical protein | 3.37 | 1.13E-04 |
| narK | nitrate transporter | 3.33 | 0 |
| narB | nitrate reductase | 3.29 | 2.00E-15 |
| SYNPCC7002_G0056 | hypothetical protein | 3.09 | 0.03 |
| SYNPCC7002_A1237 | hypothetical protein | 2.97 | 1.67E-05 |
| sigC | group II sigma-70 type sigma factor | 2.89 | 2.75E-12 |
| SYNPCC7002_A0782 | conserved hypothetical membrane protein | 2.89 | 0 |
| SYNPCC7002_G0109 | hypothetical protein | 2.79 | 0 |
| SYNPCC7002_A1733 | ABC 3 transport family protein (Mn2+) | 2.78 | 2.44E-15 |
| rubA | rubredoxin (photosynthesis lipid) | 2.77 | 0 |
| SYNPCC7002_A2433 | galactosyl-1-phosphate transferase | 2.68 | 0.06 |
| SYNPCC7002_A2086 | conserved hypothetical protein | 2.66 | 0.02 |
| rpaB | two-component response regulator | 2.64 | 8.39E-11 |
| SYNPCC7002_A2692 | hypothetical protein | 2.6 | 6.89E-03 |
| ndhL | NADH dehydrogenase subunit L | 2.58 | 8.58E-11 |
| SYNPCC7002_A2772 | conserved hypothetical | 2.55 | 0.01 |
| SYNPCC7002_A0125 | formate/nitrite transporter | 2.53 | 7.38E-12 |
| SYNPCC7002_A1238 | predicted ATPase/GTPase | 2.53 | 2.22E-16 |
| SYNPCC7002_A0472 | conserved hypothetical protein | 2.53 | 8.69E-04 |
| SYNPCC7002_A2240 | CobQ/CobB nucleotide binding protein | 2.51 | 1.02E-13 |
| SYNPCC7002_G0125 | hypothetical protein | 2.39 | 1.97E-04 |
| nblA | putative phycobilisome degradation protein | 2.37 | 1.78E-05 |
| SYNPCC7002_A0468 | bacterial domain of unknown function | 2.33 | 8.85E-06 |
| SYNPCC7002_A0793 | AhpC/TSA family protein | 2.29 | 0 |
| hliA | high light/nutrient deprived stress response | 2.28 | 8.32E-03 |
| SYNPCC7002_A1618 | glycosyl transferase family | 2.26 | 7.11E-14 |
| rplE | 50S ribosomal protein L5 | 2.24 | 0 |
| SYNPCC7002_A2607 | probable Rieske iron-sulfur protein | 2.24 | 6.43E-04 |
| SYNPCC7002_A1619 | conserved hypothetical protein | 2.21 | 6.11E-14 |
| SYNPCC7002_A2241 | conserved hypothetical protein | 2.21 | 0.03 |
| SYNPCC7002_D0026 | conserved hypothetical protein | 2.19 | 3.90E-04 |
| SYNPCC7002_A0470 | sodium-dependent bicarbonate transporter | 2.19 | 3.24E-06 |
| cupA | CO2 hydration protein | 2.19 | 3.66E-08 |
| SYNPCC7002_A2535 | conserved hypothetical protein | 2.18 | 2.30E-05 |
| SYNPCC7002_A1908 | conserved hypothetical protein | 2.14 | 0.02 |
| SYNPCC7002_A0429 | conserved hypothetical protein | 2.14 | 3.23E-03 |
| SYNPCC7002_A0397 | ABC transporter | 2.14 | 4.62E-06 |
| SYNPCC7002_F0033 | hypothetical protein | 2.13 | 1.24E-13 |
| SYNPCC7002_A0469 | conserved hypothetical protein | 2.1 | 3.40E-06 |
| SYNPCC7002_A0467 | putative cysteine protease superfamily | 2.08 | 2.47E-08 |
| rplP | ribosomal protein L16 | 2.07 | 1.58E-11 |
| hslO | Hsp33-like chaperonin | 2.05 | 0 |
| SYNPCC7002_A2244 | conserved hypothetical protein | 2.05 | 7.33E-08 |
| rplN | ribosomal protein L14 | 2.04 | 0 |
| SYNPCC7002_C0016 | hypothetical protein | 2.04 | 5.35E-06 |
| SYNPCC7002_A0582 | precorrin isomerase | 2.03 | 0 |
| SYNPCC7002_A0175 | conserved hypothetical protein | 2.03 | 3.26E-08 |
| SYNPCC7002_A0421 | ABC-type transport protein | 2.03 | 7.64E-03 |
| rplR | ribosomal protein L18 | 2.03 | 0 |
| rdgB | non-canonical purine NTP pyrophosphatase | 2.03 | 6.86E-14 |
| SYNPCC7002_A0395 | ABC transporter | 2.02 | 9.73E-10 |
| SYNPCC7002_A1477 | conserved hypothetical protein | 2.02 | 0 |
| SYNPCC7002_A0396 | ABC transporter | 2.02 | 3.88E-04 |
| SYNPCC7002_A0102 | hypothetical protein | 2.01 | 5.70E-05 |
| SYNPCC7002_A0381 | conserved hypothetical protein | 2.01 | 4.38E-11 |
| SYNPCC7002_A1368 | conserved hypothetical protein | 2.01 | 0 |
| isiA | photosystem II chlorophyll-binding protein | -14.42 | 6.16E-04 |
| SYNPCC7002_A1291 | flavodoxin | -8.27 | 6.13E-04 |
| SYNPCC7002_G0019 | siderophore biosynthesis protein | -8.12 | 7.80E-06 |
| SYNPCC7002_G0018 | esterase/lipase | -7.21 | 7.75E-07 |
| SYNPCC7002_G0083 | ABC transporter | -6.17 | 6.41E-04 |
| SYNPCC7002_A2659 | conserved hypothetical | -5.05 | 4.01E-03 |
| SYNPCC7002_G0021 | siderophore biosynthesis protein | -4.48 | 0.02 |
| SYNPCC7002_A2660 | conserved hypothetical | -4.39 | 0.02 |
| fecC | FecCD transport (permease) family | -4.37 | 0.02 |
| SYNPCC7002_G0079 | ABC transporter, permease, FecCD family | -4.1 | 2.08E-03 |
| SYNPCC7002_G0090 | TonB family C-terminal domain protein | -4.08 | 0.03 |
| SYNPCC7002_A0933 | hypothetical protein | -4.07 | 2.07E-37 |
| SYNPCC7002_G0089 | MORN repeat protein | -4.02 | 0.01 |
| SYNPCC7002_G0086 | ATP-binding protein of ABC transporter | -4.01 | 7.98E-03 |
| SYNPCC7002_G0082 | glyoxalase family protein family | -4 | 3.64E-03 |
| SYNPCC7002_G0022 | siderophore biosynthesis protein | -3.78 | 0.03 |
| SYNPCC7002_A2657 | hypothetical protein | -3.09 | 0.02 |
| fecD | FecCD transport family | -3.06 | 1.24E-03 |
| SYNPCC7002_G0017 | hypothetical protein | -2.97 | 0.03 |
| sufA | iron transport protein | -2.96 | 0.02 |
| SYNPCC7002_A0612 | tRNA-Arg | -2.9 | 5.05E-03 |
| SYNPCC7002_G0023 | siderophore biosynthesis protein | -2.85 | 0.06 |
| SYNPCC7002_A0871 | ABC transporter | -2.78 | 0.04 |
| SYNPCC7002_G0084 | conserved hypothetical protein | -2.78 | 0.02 |
| SYNPCC7002_E0003 | hypothetical protein | -2.7 | 1.35E-72 |
| ardA | antirestriction protein | -2.45 | 1.65E-19 |
| SYNPCC7002_G0080 | iron compound ABC transporter | -2.41 | 3.26E-04 |
| SYNPCC7002_A1858 | conserved hypothetical | -2.4 | 2.41E-04 |
| SYNPCC7002_E0004 | hypothetical protein | -2.35 | 1.16E-39 |
| SYNPCC7002_A0934 | hypothetical protein | -2.32 | 2.30E-03 |
| SYNPCC7002_A2658 | oxidoreductase | -2.07 | 0.02 |
| nrdA | ribonucleotide reductase subunit alpha | -2.01 | 5.95E-04 |
| SYNPCC7002_A2117 | conserved hypothetical protein | -2.01 | 2.34E-03 |
Table 6. Genes identified in SNP analysis.
| Gene | Annotation | Base Change | Frequency (%)a | Coverageb | Amino Acid Change |
|---|---|---|---|---|---|
| acsA | acetate-CoA ligase | G/T | 60 | 53 | Trp49Leu |
| SYNPCC7002_A0421 | ABC-type transport protein | C/T | 43 | 65 | Arg186Cys |
| SYNPCC7002_A2260 | conserved hypothetical protein | C/A | 65 | 37 | Gln7Lys |
| SYNPCC7002_A2570 | 4Fe-4S binding domain protein | T/G | 38 | 26 | Val1Gly |
a Percentage of reads with the specified mutation
b The total number of reads covering the specified base position
AcsA has activity towards acrylate and other organic acids
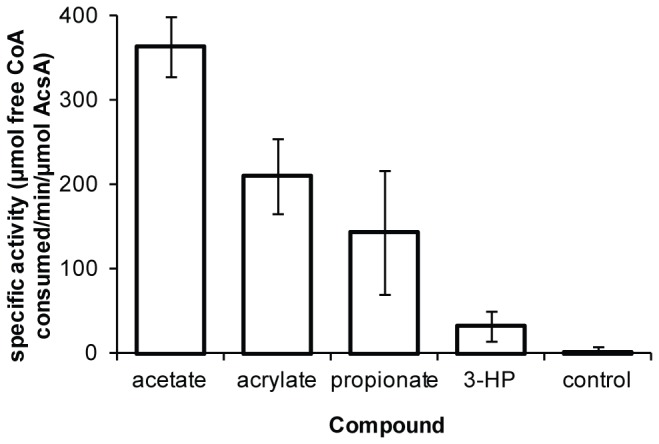
To examine the function of AcsA, His-tagged versions of the wild type gene and an acsAW49L mutant were heterologously expressed in E. coli. The wild type AcsA protein was found in the soluble protein fraction and was purified using Ni-NTA His-tag affinity chromatography. Expression of the acsAW49L mutant gene generated an insoluble protein. Attempts to purify the AcsAW49L protein from the soluble fraction with Ni-NTA affinity chromatography failed. The activity of purified AcsA protein towards various organic acids was assessed in vitro. CoA ligase activity was observed for acetate, acrylate, 3-hydroxypropionate, and propionate (Figure 2). No activity was observed for succinate under these assay conditions (data not shown). The activity of AcsA towards these organic acids supports the results that a single mutation increases tolerance to all three compounds. While the presence of acyl-CoA thioesters may be toxic, we hypothesize that the observed toxicity of acrylate and other organic acids was caused because the CoA thioesters were metabolic dead-ends and the continuous activity of AcsA depleted the free CoA pool. This hypothesis is supported by prior work in which a species of Synechococcus grown in the presence of propionate was no longer able to import acetate and was observed to have decreased levels of free CoA [19].
Figure 2. Specific activity of AcsA towards the organic acids acetate, propionate, acrylate, and 3HP.
Loss of function of acsA increases organic acid tolerance
A PCC 7002 acsA deletion mutant, BPSyn_006, was constructed by double homologous recombination to replace acsA with the streptomycin resistance marker aadA. BPSyn_006 had dramatically higher MIC values for acrylate, 3-hydroxypropionate, and propionate (Table 4). Specifically, this mutant had 2,800-fold, 26-fold, and >100 fold increase in MIC to acrylate, 3-hydroxypropioante, and propionate, respectively. A similar observation was made in PCC 6803 by replacing acsA (sll0542) with a kanamycin resistance cassette, resulting in strain BP6803_002. BP6803_002 has a 3,500-fold increased tolerance to acrylate, but no increased tolerance to 3-hydroxypropionate. This suggests that the mechanism of toxicity of 3-hydroxypropionate is different in PCC 7002 and PCC 6803. This difference between species and the order or magnitude increase in acrylate tolerance for both species makes acrylate a more attractive compound for a universal counter selection.
The organic acid tolerance phenotype was complemented in PCC 7002
Attempts to complement the PCC 7002 acsA deletion strain (BPSyn_006) by introducing and expressing acsA on the native plasmid pAQ1 or in a chromosomal loci with a kanamycin resistance cassette resulted in heterogeneous strains that could not be made homozygous, and thus could not be used in MIC determination experiments. In order to reuse the aadA cassette, a markerless deletion of the native acsA locus via counter selection and then reinsert acsA and the aadA resistance marker into a newly validated neutral insertion site. The acsA markerless deletion strain (BPSyn_014) was created by transforming wild type PCC 7002 with a linear DNA fragment to replace acsA with a 40 bp barcode (Figure 3A). Transformants were selected on solid medium containing 50 µM acrylate, a low concentration that allows for the growth of heterozygous transformants. Mutants positive for the barcode insertion were streaked onto medium containing 10 mM acrylate. The increase in acrylate concentrations allowed for selection of homozygous clones. Single colonies were demonstrated to be homozygous via colony PCR (Figure 3B). This strain, BPSyn_014, had MIC values identical to BPSyn_006. When compared to wild type PCC 7002, strain BPSyn_014 did not have a significant defect in growth up to 75 h (Figure 4A), but was observed to secrete acetate (370 ± 20 µM for BPSyn_014 vs. none detected for wild type). The markerless deletion allowed for the reuse of the aadA selection marker to introduce acsA into another locus on the chromosome.
Figure 3. Homozygous mutants were obtained in both the acsA and glpK loci.

(A) Diagram of the primers used to screen for homozygous strains in the acsA locus. Primer set A amplifies from the up and down5' and 3' stream flanking regions of acsA and should be amplify 3.2 kb, 1.1 kb, and 2.6 kb in fragments for WT 7002, BPSyn_014, and BPSyn_015, respectively. Primer set B amplifies 1.2 kb from inside acsA to the region downstream of acsA. Primer set C amplifies 0.9 kb from the barcode (BC) region to downstream of acsA. Primer set D amplifies a 0.25 kb region internal to YFP. (B) 1% agarose gel showing colony PCR products amplified using the primer sets A, B, C, and D on WT PCC 7002, BPSyn_014, and BPSyn_015. Lane 1 contains a 2-log ladder from New England Biolabs. Lanes 2-5, 6-9, and 10-13 are primers sets A, B, C, and D amplifying WT PCC 7002, BPSyn_014, and BPSyn_015, respectively. (C) Diagram of the primers used to screen for homozygous strains in the glpK locus. Primer set E amplifies from the up and down stream5' and 3' flanking regions of glpK and should be amplify 2.7 kb, 4.8 kb, and 2.6 kb fragments from BPSyn_014, BPSyn_022, and BPSyn_027, respectively. Primer set F amplifies 0.72 kb from inside glpK to the region downstream of glpK. Primer set G amplifies a 2.3 kb region of the acsA cassette that was introduced into the glpK locus. (D) 1% agarose gel showing colony PCR products amplified using the primer sets E, F, G, and D on BPSyn_014, BPSyn_022, and BPSyn_027. Lane 1 contains a 2-log ladder from New England Biolabs. Lanes 2-5, 6-9, and 10-13 are primers sets E, F, G, and D amplifying BPSyn_014, BPSyn_022, and BPSyn_027, respectively.
Figure 4. Comparison of acsA and glpK mutant phenotypes.
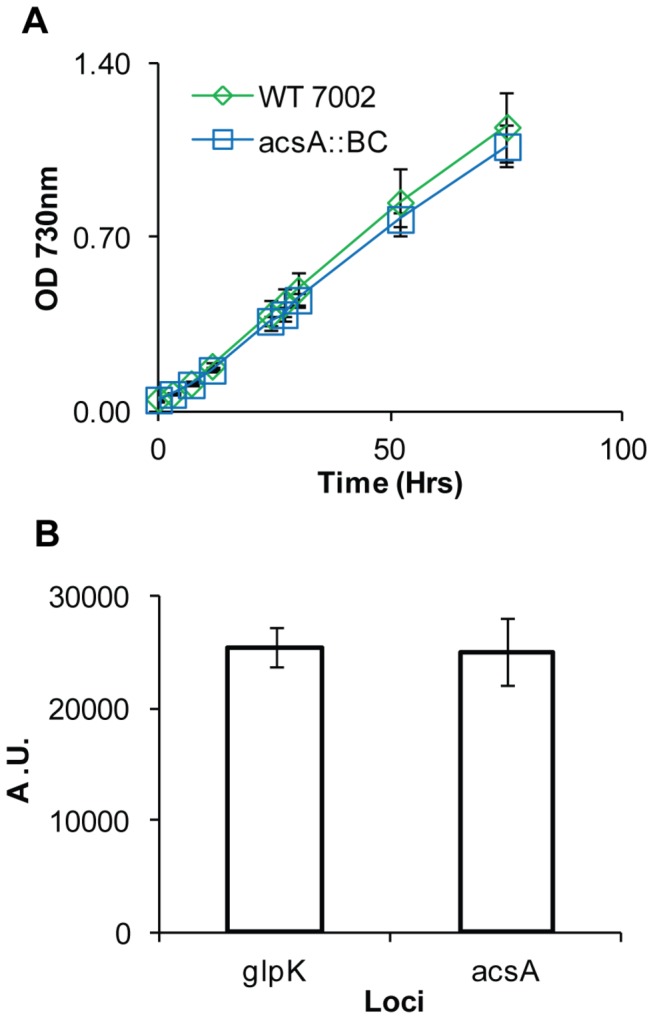
(A) Growth of wild type PCC 7002 (green diamonds) and BPSyn_014 (blue squares) in Medium A+ supplemented with air. (B) Fluorescence measurements of strains BPSyn_015 and BPSyn_027, which contain YFP under the cpcB promoter in the acsA or glpK locus, respectively. Fluorescence is measured using arbitrary units (A.U.) at an excitation of 514 nm and an emission of 527 nm. Data points are the mean of biological triplicates and error bars represent the standard deviation.
The locus glpK was chosen as a neutral insertion site for introduction of genes for complementation of the increased organic acid phenotype. In PCC 7002, glpK has a frameshift mutation and is thus considered a pseudogene. Since glpK cannot produce a functional protein, it was hypothesized that replacement of glpK would not affect physiology. Strain BPSyn_014 was complemented by introducing either (1) a wild type copy of acsA or (2) a W49L acsA mutant under the native acsA promoter along with the aadA selection cassette, into the glpK locus of the chromosome, resulting in strains BPSyn_022 and BPSyn_026, respectively. The MIC for acrylate and 3-hydroxypropionate for BPSyn_022 were 10µM and 15mM, respectively, similar to wild type PCC 7002 (Table 4). The MIC for acrylate for BPSyn_026 is 7mM, which suggests that the W49L mutant results in only a partial loss of function (Table 4). Additionally, the frequency of acrylate tolerance mutations of BYSyn_022 and BPSyn_026 observed on 50 µM acrylate plates were the same as wildtype PCC 7002.
Acrylate counter selection can be used for heterologous gene expression
To optimize the counter selection method, the 40 bp barcode sequence used to create strain BPSyn_014 and a linearized plasmid containing His-tagged yellow fluorescent protein (YFP) under the high expression cpcB constitutive promoter (pACS_ pcpcB_YFP) were transformed into wild type PCC 7002 and selected for on 50, 100, and 500 µM acrylate. After 3 days, colonies containing integrations were only observed on plates containing 50 µM acrylate. The percentage of colonies that were positive for integrations was 49% (n = 42) for the barcode and 30% for YFP (n = 39). A t-test of these data shows no significant difference in integration frequency (p = 0.11) indicating that insertion size had little impact on integration efficiency. Colonies not containing integrations were assumed to be spontaneous mutants of acsA. The number of background colonies is consistent with the loss of function mutation frequency previously observed. A homozygous strain for the integration of YFP (BPSyn_015) was obtained by streaking onto plates containing 10 mM acrylate and confirmed by colony PCR (Figure 3B). As expected, strain BPSyn_015 was highly fluorescent (Figure 4B). YFP was successfully purified from BPSyn_015 using Ni-NTA affinity chromatography and visualized with PAGE.
Next, the acrylate counter selection method was used to introduce heterologous genes into other loci. Strain BPSyn_022 (acsA::BC glpK::acsA aadA) was transformed with the linearized plasmid pGLPK_ pcpcB_YFP to replace acsA and aadA with YFP under the expression of the cpcB promoter in the glpK locus (Figure 3C). 29% (n=14) of colonies were identified as positive transformants via colony PCR, which is similar to transformations into the acsA locus. Positive transformants (BPSyn_027) were verified as homozygous using the same protocol outlined for integration directly into the acsA locus (Figure 3D). As expected, YFP was successfully purified from BPSyn_027 and visualized using Ni-NTA affinity chromatography and PAGE. When compared to BPSyn_015, no significant difference was observed in the level of YFP fluorescence (Figure 4B). These results demonstrate that acrylate counter selection can be used to make modifications beyond the native acsA locus and that the glpK locus has utility as a neutral insertion site. The counter selection methodology is summarized in Figure 5.
Figure 5. Outline of counter selection protocol based on acrylate sensitivity.
Conclusion
Counter selection is an important tool for genetic manipulation of microorganisms. Counter selection is of particular importance in cyanobacterial systems due to the presence of multiple copies of the chromosome and the difficultly of achieving homozygous strains using antibiotic resistance genes. The results presented in this work describe the basis and methodology for using organic acid sensitivity for counter selection in the cyanobacterium PCC 7002. It was shown that the organic acids acrylate, 3-hydroxypropionate, and propionate cause growth inhibition and that this inhibition can be overcome by loss of the gene acsA. Acrylate was further pursued for use in counter selection because of the low concentration required for sensitivity, the dramatic increase in tolerance due to loss of acsA, and the potential to be used in other cyanobacteria (e.g. PCC 6803). A method was optimized for introducing genes of interest directly into the acsA locus. Loss of acsA did not result in a growth defect under the conditions used in this experiment, but did result in an increase in the secretion of acetate by PCC 7002. A neutral site, glpK, was identified as a chromosomal locus that is amenable to acrylate counter selection and can be used for expression of heterologous genes. These results suggest that acrylate counter selection could also be used to make markerless deletions or insertions elsewhere on the chromosome. Additionally, a loss of function of acsA in PCC 6803 was shown to dramatically increase the tolerance to acrylate, suggesting that this method has utility in multiple species. Application of the method presented in this work will be used in future physiology studies and metabolic engineering efforts.
Acknowledgments
The pKS1 plasmid was a gift from the Birgit Alber lab at the Ohio State University. The pAQ1_Exp_YFP plasmid was a gift from the Donald Bryant lab at Pennsylvania State University.
Funding Statement
This work was funded by the National Science Foundation (EFRI-1240268, CBET-1149678, SEES 12-15871), the US Air Force Office of Scientific Research (FA9550-11-1-0038), the US Department of Energy Genomics: GTL and SciDAC Programs (DE-FG02-04ER25627) and the University of Wisconsin-Madison graduate school. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Umena Y, Kawakami K, Shen J-R, Kamiya N (2011) Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9[thinsp]A. Nature 473: 55-60. doi:10.1038/nature09913. PubMed: 21499260. [DOI] [PubMed] [Google Scholar]
- 2. Zarzycki J, Axen SD, Kinney JN, Kerfeld CA (2013) Cyanobacterial-based approaches to improving photosynthesis in plants. J Exp Bot 64: 787-798. doi:10.1093/jxb/ers294. PubMed: 23095996. [DOI] [PubMed] [Google Scholar]
- 3. Zhang S, Bryant DA (2011) The Tricarboxylic Acid Cycle in Cyanobacteria. Science 334: 1551-1553. doi:10.1126/science.1210858. PubMed: 22174252. [DOI] [PubMed] [Google Scholar]
- 4. Machado IMP, Atsumi S (2012) Cyanobacterial biofuel production. J Biotechnol 162: 50-56. doi:10.1016/j.jbiotec.2012.03.005. PubMed: 22446641. [DOI] [PubMed] [Google Scholar]
- 5. Ducat DC, Way JC, Silver PA (2011) Engineering cyanobacteria to generate high-value products. Trends Biotechnol 29: 95-103. doi:10.1016/j.tibtech.2010.12.003. PubMed: 21211860. [DOI] [PubMed] [Google Scholar]
- 6. Wang B, Wang J, Zhang W, Meldrum DR (2012) Application of synthetic biology in cyanobacteria and algae. Frontiers in Microbiology 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang H-H, Camsund D, Lindblad P, Heidorn T (2010) Design and characterization of molecular tools for a Synthetic Biology approach towards developing cyanobacterial biotechnology. Nucleic Acids Res 38: 2577-2593. doi:10.1093/nar/gkq164. PubMed: 20236988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu X, Sheng J, Curtiss R III (2011) Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci USA 108: 6899-6904. doi:10.1073/pnas.1103014108. PubMed: 21482809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cheah YE, Albers SC, Peebles CAM (2013) A novel counter-selection method for markerless genetic modification in Synechocystis sp. PCC 6803. Biotechnol Prog 29: 23-30. doi:10.1002/btpr.1661. PubMed: 23124993. [DOI] [PubMed] [Google Scholar]
- 10. Mendez-Perez D, Begemann MB, Pfleger BF (2011) Modular Synthase-Encoding Gene Involved in α-Olefin Biosynthesis in Synechococcus sp. Strain PCC 7002. Appl Environ Microbiol 77: 4264-4267. doi:10.1128/AEM.00467-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu Y, Tiago Guerra L, Li Z, Ludwig M, Charles Dismukes G et al. (2013) Altered carbohydrate metabolism in glycogen synthase mutants of Synechococcus sp. strain PCC 7002: Cell factories for soluble sugars. Metab Eng 16: 56-67. doi:10.1016/j.ymben.2012.12.002. PubMed: 23262095. [DOI] [PubMed] [Google Scholar]
- 12. Russell JB (1992) Another explanation for the toxicity of fermentation acids at low pH: anion accumulation versus uncoupling. J Appl Microbiol 73: 363-370. doi:10.1111/j.1365-2672.1992.tb04990.x. [Google Scholar]
- 13. Zaldivar J, Ingram LO (1999) Effect of organic acids on the growth and fermentation of ethanologenic Escherichia coli LY01. Biotechnol Bioeng 66: 203-210. doi:10.1002/(SICI)1097-0290(1999)66:4. PubMed: 10578090. [DOI] [PubMed] [Google Scholar]
- 14. Horswill AR, Dudding AR, Escalante-Semerena JC (2001) Studies of Propionate Toxicity in Salmonella enterica Identify 2-Methylcitrate as a Potent Inhibitor of Cell Growth. J Biol Chem 276: 19094-19101. doi:10.1074/jbc.M100244200. PubMed: 11376009. [DOI] [PubMed] [Google Scholar]
- 15. Thijsse GJ (1964) Fatty-acid Accumulation by acrylate inhibition of beta-oxidation in alkane-oxidizing Pseudomonas. Biochim Biophys Acta 84: 195-197. PubMed: 14181298. [DOI] [PubMed] [Google Scholar]
- 16. Green PR, Kemper J, Schechtman L, Guo L, Satkowski M et al. (2001) Formation of Short Chain Length/Medium Chain Length Polyhydroxyalkanoate Copolymers by Fatty Acid β-Oxidation Inhibited Ralstonia eutropha. Biomacromolecules 3: 208-213. [DOI] [PubMed] [Google Scholar]
- 17. Warnecke TE, Lynch MD, Karimpour-Fard A, Lipscomb ML, Handke P et al. (2010) Rapid dissection of a complex phenotype through genomic-scale mapping of fitness altering genes. Metab Eng 12: 241-250. doi:10.1016/j.ymben.2009.12.002. PubMed: 20060059. [DOI] [PubMed] [Google Scholar]
- 18. Rocco CJ, Escalante-Semerena JC (2010) In Salmonella enterica, 2-Methylcitrate Blocks Gluconeogenesis. J Bacteriol 192: 771-778. doi:10.1128/JB.01301-09. PubMed: 19948794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ihlenfeldt MJA, Gibson J (1977) Acetate uptake by the unicellular cyanobacteria Synechococcus and Aphanocapsa . Arch Microbiol 113: 231-241. doi:10.1007/BF00492030. PubMed: 18124. [DOI] [PubMed] [Google Scholar]
- 20. Schneider K, Asao M, Carter MS, Alber BE (2012) Rhodobacter sphaeroides Uses a Reductive Route via Propionyl Coenzyme A To Assimilate 3-Hydroxypropionate. J Bacteriol 194: 225-232. doi:10.1128/JB.05959-11. PubMed: 22056933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stevens SE, Patterson COP, Myers J (1973) The production of hydrogen peroxide by blue-green algae: a survey. J Phycol 9: 427-430. doi:10.1111/j.0022-3646.1973.00427.x. [Google Scholar]
- 22. Rosmarie R (1988) [1] Isolation and purification of cyanobacteria. In: Lester Packer ANG. Methods in Enzymology. Academic Press; pp. 3-27. [DOI] [PubMed] [Google Scholar]
- 23. Pinto F, Thapper A, Sontheim W, Lindblad P (2009) Analysis of current and alternative phenol based RNA extraction methodologies for cyanobacteria. BMC Mol Biol 10: 79. doi:10.1186/1471-2199-10-79. PubMed: 19660145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621-628. doi:10.1038/nmeth.1226. PubMed: 18516045. [DOI] [PubMed] [Google Scholar]
- 25. Frigaard N-U, Sakuragi Y, Bryant DA (2004) Gene Inactivation in the Cyanobacterium Synechococcus sp. PCC 7002 and the Green Sulfur Bacterium Chlorobium tepidum Using In Vitro-Made DNA Constructs and Natural Transformation. In: Carpentier R. Photosynthesis Research Protocols. Humana Press; pp. 325-340. [DOI] [PubMed] [Google Scholar]
- 26. Gibson DG (2011) Enzymatic assembly of overlapping DNA fragments. Methods Enzymol 498: 349-361. doi:10.1016/B978-0-12-385120-8.00015-2. PubMed: 21601685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu Y, Alvey RM, Byrne PO, Graham JE, Shen G et al. (2011) Expression of Genes in Cyanobacteria: Adaptation of Endogenous Plasmids as Platforms for High-Level Gene Expression in Synechococcus sp. PCC 273 In: Carpentier R. Photosynthesis Research Protocols. Humana Press; pp. 293. [DOI] [PubMed] [Google Scholar]
- 28. Joset F, Buchou T, Zhang CC, Jeanjean R (1988) Physiological and genetic analysis of the glucose-fructose permeation system in two Synechocystis species. Arch Microbiol 149: 417-421. doi:10.1007/BF00425581. [Google Scholar]
- 29. Sunda W, Kieber DJ, Kiene RP, Huntsman S (2002) An antioxidant function for DMSP and DMS in marine algae. Nature 418: 317-320. doi:10.1038/nature00851. PubMed: 12124622. [DOI] [PubMed] [Google Scholar]
- 30. Hashimoto Y, Hosaka H, Oinuma K-I, Goda M, Higashibata H et al. (2005) Nitrile Pathway Involving Acyl-CoA Synthetase. J Biol Chem 280: 8660-8667. PubMed: 15632196. [DOI] [PubMed] [Google Scholar]