

Abstract
Curcumin is the active component of dried rhizome of Curcuma longa, a perennial herb belonging to ginger family, cultivated extensively in south and southeastern tropical Asia. It is widely consumed in the Indian subcontinent, south Asia and Japan in traditional food recipes. Extensive research over last few decades has shown that curcumin is a potent anti-inflammatory agent with powerful therapeutic potential against a variety of cancers. It suppresses proliferation and metastasis of human tumors through regulation of various transcription factors, growth factors, inflammatory cytokines, protein kinases and other enzymes. It induces apoptotic cell death and also inhibits proliferation of cancer cells by cell cycle arrest. Pharmacokinetic data has shown that curcumin undergoes rapid metabolism leading to glucuronidation and sulfation in the liver and excretion in the feces, which accounts for its poor systemic bioavailability. The compound has, therefore, been formulated and administered using different drug delivery systems such as liposomes, micelles, polysaccharides, phospholipid complexes and nanoparticles that can overcome the limitation of bioavailability to some extent. Attempts to avoid rapid metabolism of curcumin until now have been met with limited success. This has prompted researchers to look for new synthetic curcumin analogs in order to overcome the drawbacks of limited bioavailability and rapid metabolism, and gain efficacy with reduced toxicity. In this review we provide a summarized account of novel synthetic curcumin formulations and analogs, and the recent progress in the field of cancer prevention and treatment.
Keywords: Curcumin, curcumin analogs, anticancer, novel formulations
1. INTRODUCTION
Phytochemicals are a broad variety of biologically active compounds produced by plants usually as secondary metabolites that possess either antioxidant or hormone-like actions. They are generally found in fruits, vegetables, beans or grains. Diets rich in fruits, vegetables or whole grains have been shown to reduce the risk of certain cancers, heart diseases, diabetes and other health disorders, and hence there is a continual attempt to identify specific compounds in these foods that may account for their beneficial effects in humans. Since phytochemicals help prevent formation of potential cancer-causing moieties by blocking their action, they are also viewed as chemopreventive agents against cancers [1].
Among many such agents, Curcumin has attracted increased attention. This is a phytochemical which is derived from turmeric (Curcumin longa) and has been used for thousands of years as a healing agent in Indian and Chinese systems of medicine for variety of illnesses. Several research reports over the last few decades have established that curcumin is a potent anti-inflammatory agent [2–6] with powerful therapeutic potential against a variety of cancers [7–11]. It has been shown to suppress transformation, proliferation, and metastasis of tumors [12]. These effects are mediated through regulation of various transcription factors, growth factors, inflammatory cytokines, protein kinases and other enzymes [13]. Curcumin also inhibits proliferation of cancer cells by arresting them in various phases of the cell cycle and by inducing apoptosis [14]. The compound has the ability to inhibit carcinogen bioactivities via suppression of specific cytochrome P450 enzymes, and to induce the activity or expression of phase II carcinogen detoxifying enzymes, which may account for its cancer chemopreventive effects [15].
Although curcumin has been successfully evaluated for wide ranging biological activities, the two main concerns related to its poor bioavailability and rapid metabolism have prompted researchers to look for novel drug delivery systems, and new synthetic curcumin analogs in order to overcome these drawbacks and gain in efficacy with reduced toxicity. In the present review, we provide a short account of some of the new curcumin analogs reported since 2005 which have shown enhanced anticancer activities than curcumin against certain cancers, and hence offer hope for clinical benefits. A summarized account of the reports published earlier is provided in our earlier review published in 2010 [16].
2. CHEMISTRY OF CURCUMIN
Curcumin is an orange–yellow crystalline powder practically insoluble in water and ether but soluble in ethanol, dimethylsulfoxide and acetone. It was first isolated in 1815 by Vogel and Pelletier, while it was isolated in crystalline form in 1870, and identified as 1, 6-heptadiene-3, 5-dione1,7-bis(4-hydroxy-3-methoxyphenyl)-(1E, 6E) or Diferuloylmethane. The feruloylmethane skeleton of curcumin was confirmed in 1910 by Lampe [17, 18]. Chemically, curcumin is a bis-α, β-unsaturated β-diketone and exists in equilibrium with its enol tautomer (Fig. 1). The bis-keto form predominates in acidic and neutral aqueous solutions as well as in the cell membrane due to the heptadienone linkage between two methoxyphenol rings containing a highly activated carbon atom.
Fig. 1.
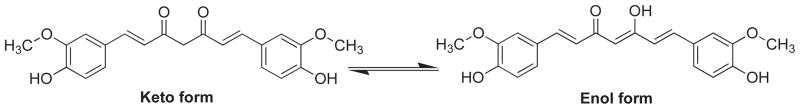
Keto-Enalo tautomerism in curcumin (1).
Tonnesen and Karlsen [19] have studied the hydrolytic degradative reaction kinetics of curcumin over the rage of pH 1–11 by using HPLC and found that at pH < 1, curcumin aqueous solution exhibits red color which is indicative of the protonated form (2). At a pH range 1–7, curcumin molecules are in the neutral state (3) with yellow color and low water solubility, while at pH>7.5 this color turns to red (Fig. 2). In the keto form of curcumin, the heptadienone linkage between the two methoxyphenol rings contains a highly activated carbon atom and the C–H bonds on this carbon are very weak due to delocalization of the unpaired electron on the adjacent oxygen atoms. Curcumin possesses three protons that are ionizable in water, Viz. the enolic proton with a pKa of approximately 8.5 and two phenolic protons with pKa of 10 – 10.5 (in mixed alcoholic/water solvent). The pKa values for the dissociation of the three acidic protons in curcumin (forms 4, 5 and 6) have been determined to be 7.8, 8.5 and 9.0, respectively (Fig. 3). Between the acidic range of pH 3–7, curcumin acts as a potent H-atom donor, while above pH 8 (basic condition), the enolate form of the heptadienone chain predominates and the compound acts mainly as an electron donor similar to many phenolic antioxidants [19]. Tonnesen and Karlsen [19] have further found that the main degradation products of curcumin are ferulic acid and feruloylmethane and have investigated their kinetics of degradation in a MeOH/aqueous buffer medium (1: 9), with phosphate buffer (pH 6 – 9) or carbonate buffer (pH 9 – 10), wherein the rate of degradation was found to be second order for curcumin. It was found that curcumin decomposed 90% within 30 min in 0.1 M phosphate buffer at pH 7.2 at 37°C, while the tentatively identified intermediate decomposition product, viz.trans-6-(4-hydroxy-3-methoxyphenyl)-2,4-dioxo-5-hexenal, decomposed further to yield vanillin as a final product (Fig. 4) along with ferulic acid and feruloylmethane respectively [20].
Fig. 2.
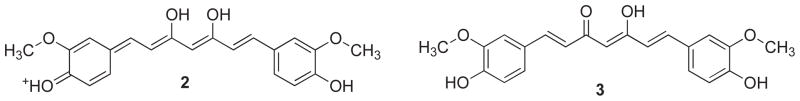
The form of curcumin present at pH < 1 (2) and pH 1–7 (3).
Fig. 3.
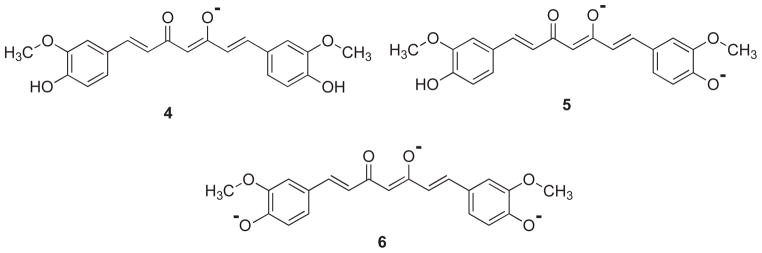
Different forms of curcumin after dissociation with pKa value of 7.8 (4), 8.5 (5) and 9.0 (6).
Fig. 4.

Decomposition products of curcumin.
Bernabe-Pineda et al. [21] have shown that curcumin exhibits a pseudo-zero-order kinetics of decomposition in non-buffered aqueous medium of pH 10–13.5, with a rate constant of 1.39×10−9 M/min. Several in vivo studies have identified number of intestinal metabolites (Fig. 5) in human and rats including curcumin glucuronide (7), curcumin sulfate (8), tetrahydrocurcumin (9) and hexahydrocurcumin (10) [22]. Hence, both metabolic conjugation and reduction were observed along with traces of the aforementioned decomposition products of dihydroferulic acid and ferulic acid as biliary metabolites of oral curcumin administration in rats [23].
Fig. 5.
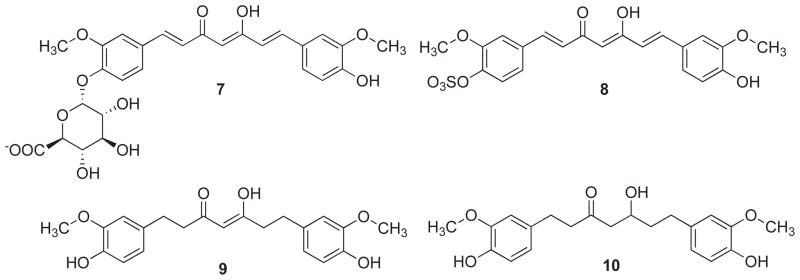
In vivo metabolites of curcumin (7–10).
3. STRATEGIES FOR IMPROVING CURCUMIN AS THERAPEUTIC AGENT
Curcumin has been reported to be very safe because it does not cause any adverse effects, even up to doses as high as 8 g per day in humans and no resistance against curcumin has been reported. However, the bioavailability of Curcumin is a major concern limiting its therapeutic utility, since as much as 75% of Curcumin gets excreted in the feces, indicating its poor absorption in the gut. This suggests that in order to make Curcumin as a viable therapeutic agent one needs to address two shortcomings of Curcumin, one being its low bioavailability and the other concerning its rapid metabolism. This has been tackled by adopting two strategies: (a) Employing novel drug delivery systems and (b) Synthesizing its analogs through modification of its structural motif. Attempts avoiding rapid metabolism until now have been met with limited success except in case of difluorocurcumin (CDF), a novel synthetic analog of curcumin described recently by us [24]. In the discussion below we have summarized a brief account of both.
3.1. Novel Drug Delivery Systems
In an attempt to stabilize curcumin, Wang and co-workers have developed curcumin micelles by aggregation of surfactant molecules, like sodium dodecyl sulfate, cetyltrimethylammonium bromide (CTAB), Tween 80, Triton X-100 and pluronic polymers [25]. Stable self-emulsifying curcumin formulations having particles size of approximately 30 nm and approximately 99% curcumin loading have successfully been developed [26] which showed 10–14-fold greater absorption rate in male Wistarstrain rats.
Curcumin nanosuspension (CUR-NS) was stabilized by d-α-tocopheryl polyethylene glycol 1000 succinate (TPGS), which has been examined for its pharmacokinetics after intravenous administration to rabbits and mice [27]. It was interesting to observe that these formulations increased the plasma concentration of curcumin by 3.8 times, thus increasing its bioavailability. In another study, Pandey et al. [28] developed novel polyethylene glycosylated (PE-Gylated) curcumin analogs for potent nuclear factor erythroid-2 related factor 2 (Nrf2) activators, which positively regulated the antioxidant defense system and could decrease inflammation. These analogs improved curcumin solubility from 0.6 × 10−6 to 0.98 × 10−6 g/ml. This formulation exhibited growth inhibitory effects on a panel of human pancreatic cancer cell lines. Another report has indicated that polyacetal-based curcumin polymers (PCurc 8) are highly cytotoxic to SKOV-3 and OVCAR-3 ovarian cancers, and MCF-7 breast cancer cell lines. These conjugates were selectively taken up by cancer cells, hydrolyzed and released an active form of curcumin in lysosomes. The mechanistic study revealed that inhibition of cancer cell growth was predominant in G (0)/G (1) phase and occurred through induced apoptosis by caspase-3 activation [29]. Another strategy of increasing bioavailability was to prepare micro-emulsions of curcumin which are isotropic nanostructures and stable solutions comprising of surfactant, oil and water. In another study, Ganta and co-workers [30] have reported that curcumin nanoemulsion increases the bioavailability of paclitaxel up to 5.2-fold, and there was a 3.2-fold increase in its accumulation at the tumor site in an oral administration to SKOV-3 tumor-bearing xenografts mice models. This was possible due to down-regulation of intestinal cytochrome P450 3A2 (CYP3A2) protein levels.
Li et al. [31] have evaluated the anticancer efficacy of liposomes of curcumin on various pancreatic carcinoma cell lines, including ASPC-1, BxPC-3, Capan-1, Capan-2, HS766-T and Mia-paCa-2, respectively. The study revealed that this liposomal formulation could inhibit cancer cells at IC50 values in the range of 2.0–37.8 μM. Nano-formulations based on dextransulfate–chitosan combinations are widely accepted for oral, intravenous and controlled delivery purposes. In a study by Anitha et al. [32], the authors quantified the cellular uptake of curcumin encapsulated in dextransulfate–chitosan NPs using a spectrophotometric method in a panel of cancer cells including L929, MCF-7, PC-3 and MG 63 cells. The results revealed that the anticancer efficacy of this formulation was greater in MCF-7 cells. Another method employed for curcumin formulation was to generate self-assembly complexes with β-cyclodextrin and their derivatives. Such complexes helped in the suppression of curcumin degradation and consequently increased the stability and bioavailability [33, 34]. Employing this strategy, Yadav and co-workers [35] have developed cyclodextrin–curcumin self-assembly, which exhibited higher efficacy compared to curcumin in inhibiting tumor necrosis factor (TNF)-induced expression of NF-κB regulated genes (VEGF, MMP-9 and cyclin D1) and up-regulation of death receptors (DR4 and DR5) in KBM-5 cancer cells. Similar studies by Yallapu et al. [36] showed that self-assembled complex of curcumin with poly-β-cyclodextrin showed anti-proliferative properties against prostate cancer cells. It is presumable that the presence of curcumin in the complex results in down-regulation of anti-apoptotic Bcl-2 and Bcl-xL, and induction of pro-apoptotic Bax family proteins thereby inducing apoptosis. Our group has recently reported a new difluoro curcumin analog commonly referred to as CDF (139) with superior anticancer activity. The CDF conjugate with β-cyclodextrin (CDFCD) [37] in 1: 2 proportions exhibited significantly lower IC50 values when tested against a panel of BXPC-3, MDA-MB-231 and PC-3 cancer cell lines compared to CDF alone. In vivo studies revealed that the conjugate preferentially accumulated in the pancreas and the levels of CDF-β-cyclodextrin conjugate in mice were 10 times higher than in serum, following intravenous administration of an aqueous CDF-β-cyclodextrin preparation. These studies indicated that the self-assembly of β-cyclodextrin and CDF may provide a crucial breakthrough in enhancing the bioavailability and tissue distribution of such analogs.
Although survey of available literature on curcumin-based nano-particulate formulations suggests that this strategy holds some promise in the use of curcumin in cancer therapeutics, the issue of rapid metabolism of curcumin remains a matter of concern.
3.2. Novel Curcumin analogs through Structural modifications
Curcumin is a simple symmetrical β-diketone and incorporates several functional groups. The two aromatic rings containing phenolic groups are connected by two α, β-unsaturated carbonyl groups. These carbonyl groups form a diketone moiety which exists in keto-enolic tautomeric forms, where energetically more stable enol-form exists in the solid phase and in acidic solutions. It can be easily deprotonated under mild alkaline condition to yield enolate moiety. Such facile tautomeric conversions are suspected to contribute to the rapid metabolism of curcumin. In its unmodified form α, β-unsaturated carbonyls in curcumin play the role of a good Michael acceptor and can undergo nucleophilic additions under biological conditions which may enhance its bioavailability. Exploiting this strategy led to limited success in terms of modulating Curcumin’s metabolism, resulting in ill-defined and unstable products. In view of this several research groups have attempted to modify structural motif of Curcumin in order to slow down its metabolism and improvements in its potency and efficacy of anticancer activity. In (Fig. 6), we have summarized the sites used by these workers for structural modifications of curcumin. These include aryl side chain modification (Fig. 7A), modification of diketo functionality (Fig. 7B), modification of double bond (Fig. 7C), modification of active methylene functionality (Fig. 7D), metal complexes of curcumin (Fig. 7E) and structural analogs of curcumin (Fig. 8). Most of these modifications were made on the basis of what was achievable chemically and for tailoring the biological space and hence has remained partially successful. Following is the brief account of each modification and resulting changes in terms of specificity and efficacy for different cancers.
Fig. 6.
Structural possibilities for modification of curcumin - Aryl side chain modification (A), Modification of diketo functionality (B), Modification of double bond (C), Modification of active methylene functionality (D), Metal complexes of curcumin (E) and Appended curcumin mimics/structural analogs of curcumin (F).
Fig. 7.








(A) Aryl side chain modifications (11–23, 27–60); (B) Modification of diketo functionality (24, 61–78); (C) Modification of double bond (9–10, 79–80); (D) Modification of active methylene functionality (81–143) and (E) Metal complexes of curcumin (144–153).
Fig. 8.


Structural analogs of curcumin (154–188, 25–26).
3.2.1. Modifications of Aryl Side Chain
Aggarwal and coworkers [38] have synthesized a bioconjugate of Luteinizing Hormone Releasing Hormone (LHRH)) and Curcumin as shown in 11. The work revealed that MIAPaCa-2, Panc-1 and BxPC-3 pancreatic cancer cell lines express LHRH receptors and consequently the conjugate inhibited cell proliferation and induced apoptotic cell death. Apoptosis was induced by cleavage of polyadenosine-5′-diphosphate-ribose-polymerase and caspase-3. Unlike curcumin, the conjugate is water soluble which allows its intravenous administration. Under in vivo conditions, the conjugate brought about significant reduction in tumor weight and volume (compared to free curcumin), in the nude mouse pancreatic cancer model. Al-Hujaily et al. [39] have investigated the anticancer activities of two novel curcumin analogs EAC (12) and PAC (13) against normal and breast cancer cells wherein the latter was found to be five times more efficient than curcumin in inducing apoptosis at 40 μM dose. This effect was 10-fold higher against ER-negative as compared to ER-positive breast cancer cells. In addition, PAC delayed the cell cycle at G2/M phase with a stronger effect on ER-negative cells. The compound exhibited higher stability in blood and greater bio-distribution and bioavailability than curcumin in mice. Al-Wabli et al. [40] have synthesized and evaluated new curcumin and ethyl-curcumin bioconjugates with various functionalities which are more active than the parent compounds. Many of these compounds are non-toxic to non-cancerous cells. Compound 14 exerted cytotoxicity against SK-MEL (human malignant melanoma) and BT-549 (breast ductal carcinoma) with IC50 values of 12.5 and 10 μM, respectively. The compound 15 showed highest cytotoxic activity against SK-MEL and SK-OV-3 (human ovary carcinoma) cancer cells with IC50 values 4.75 and 2.8 μM, respectively.
Devasena et al. [41] have studied the protective effect of a salicylcurcumin (16) on hepatic lipid peroxidation (LPO) and antioxidant status during 1, 2-dimethylhydrazine (DMH)-induced colon carcinogenesis in male wistar rats. Intra-gastric administration of curcumin and salicylcurcumin (80 mg/kg body weight) to DMH-injected rats significantly reduced the number and size of tumors in the colon, lowered lipid peroxidation and enhanced the activities of GPx, GST, SOD and CAT in the liver. The mechanism of this action was shown to be the modulation of hepatic biotransformation enzymes and antioxidant status which was similar and comparable with that of curcumin. This suggests that the hydroxyl group in the aromatic ring is responsible for the protective effect of these compounds rather than the methoxy group as thought previously [41, 42].
The cytotoxicity of some newly synthesized glycoconjugates, including curcumin β-glucoside (17), has been investigated by Arafa and co-workers [43] in a panel of human colon cancer cells, viz. Caco-2, HT29 and T84 cells. Curcumin-β-glucoside exerted cytotoxicity against Caco-2, HT29 and T84 cells with IC50 value of 10.3, 24.6 and 50.3 μM, respectively. This sensitivity correlates well with β-glucosidase activity which was assessed in these cell lines. The curcumin β-glucoside was only cleaved by beta-glucosidases and β-galactosidases, but not by pancreatic lipase or hepatic esterase, suggesting that these enzymes are crucial for the bioactivation and cytotoxicity of these glycoconjugates. Dubey et al. [44] have synthesized the monoesters of curcumin, a symmetric diphenol with valine (18) and glycine (19) substituents, by solid phase synthesis and its diesters by solution phase method. The assessment of their anti-proliferative activities suggested that the diesters are relatively more active than curcumin itself against KB and HeLa cell lines probably due to their increased solubility, slow metabolism and better cellular uptake.
Ferrari et al. [45] have synthesized some new curcumin derivatives (20–21) in order to improve chemical properties of curcumin. The aromatic ring glycosylation of curcumin provides more water-soluble compounds with greater kinetic stability which is a fundamental feature for drug bioavailability. The acyl-derivative of curcumin (20) and glycosyl-curcuminoid (21) exhibited cytotoxicity against cisplatin (cDDP)-sensitive human ovarian carcinoma cell line and its resistant counterpart, C13* cancer cells, as compared to curcumin. Although binding of glucose to curcumin reduces the cytotoxicity of the derivatives towards cisplatin (cDDP)-sensitive/resistant human ovarian cancer cell lines, the compounds display a good selectivity since they are much less toxic to non-tumorigenic Vero cells. The combination of cDDP with the most active glycosyl-curcuminoid drug against both cDDP-sensitive and -resistant as well as against Vero cell lines showed an improvement of cDDP efficacy with higher selectivity towards cancer cells than non-cancer cells. These studies indicate further need for developing new valid components for drug treatment protocols to cDDP-resistant cells as well. Fuchs and coworkers [46] have synthesized a library of curcumin analogs and evaluated them against prostate and breast cancer cell lines. Compounds 22–26 were the most potent analogs which exerted cytotoxicity against androgen-dependent LNCaP as well as androgen-independent prostate cancer PC-3 cell line. The compounds were also inhibitory against estrogen-dependent MCF-7, and estrogen-independent MDA-MB-231 breast cancer cell lines. The compound 26 was found to be most cytotoxic compound against LNCaP, PC-3, MCF-7 and MDA-MB-231 cancer cell lines. Goto and group [47] have synthesized a novel curcumin conjugate of taxoid and have evaluated its cytotoxic activity against a panel of cancer cell line including A549 (human lung carcinoma), 1A9 (human ovarian carcinoma), HCT-8 (colon adeno-carcinoma), A431 (epidermoid skin carcinoma), KB (human epidermoid carcinoma of the nasopharynx) and multi-drug resistant KB variant expressing P-glycoprotein (KB-VIN) cancer cell lines. The compound was found to be cytotoxic to all cell lines except HCT-8 colon cancer cell line. Han et al. [48] have synthesized 2-hydroxycurcuminoid (HCC-7, 27) based on the structural motif of 2-hydroxy-cinnamaldehyde which strongly inhibited the growth of SW620 colon tumor cells while the parent compounds, HCA and curcumin displayed higher inhibitory concentraions. HCC-7 was found to induce apoptosis through reactive oxygen species–mitochondria pathway and cell cycle arrest at G2/M phase. The compound also exhibited cytotoxicity against other cancer cell lines although at higher concentrations.
Handler et al. [49] have reported synthesis of curcumin analogs 28 and 29 wherein the former was found to be most potent compound inhibiting COX-1 and COX-2 enzymes. Jankun et al. [50] have used a homology model of the three-dimensional structure of human P-12-LOX by performing computational docking studies of synthetic curcumin derivatives to identify inhibitors superior than curcumin. Over 75% of the compounds of interest were successfully docked into the active site of P-12-LOX, many of them sharing similar binding modes. Curcuminoids that did not dock into the active site failed to inhibit P-12-LOX. From a set of curcuminoids that were successfully docked, two compounds [E22C (16) and E26C (30)] were found to inhibit human lipoxygenase better than curcumin. False-positive curcuminoids showed high LogP values, indicating poor water solubility, a possible reason for lack of inhibitory activity and/or non-realistic binding.
Kapoor et al. [51] have linked a deoxy-11-mer oligonucleotide (5′-GTTAGGGTTAG-3′), complementary to a repeat sequence of human telomerase RNA template through phosphate and C-2 linker to the bioactive tetraglycine conjugate of curcumin (31). This molecule was transfected into KB and HeLa cell lines and was found to affect cell growth in the former at a concentration of 25–50 μM. The compound probably acts as a pro-drug being targeted by anti-sense mechanism to telomerase.
Khan and co-workers [52] have synthesized aromatic and heterocyclic aromatic curcuminoids and have evaluated their anti-inflammatory activities (AIA) in vivo model of acute carrageenan-induced paw edema and chronic adjuvant arthritis. At dose of 100 mg/kg, the compounds RK-97 (32), RK-103 (33), RK-104 (34) and RK-106 (35) have exerted potent activity in the anti-arthritic assay with little gastric or systemic toxicity. Amongst the compounds evaluated, RK-106 was the only compound to inhibit the production of TNF-α and IL-1β in a monocytic cell-line THP-1 at 100 μM concentration. Li et al. [53] have prepared a water-soluble polyethylene glycol (PEG)-conjugate of curcumin (36) in which curcumin was covalently linked to PEG (35kD). PEGylated curcumin showed enhanced solubility, targeted delivery and greater reduction of cell growth than free curcumin in pancreatic cancer cells. Cells treated with PEGylated curcumin had increased arrest at the mitotic phase with the formation of abnormal multi-nucleated cells, indicating that this compound affects cell cycle progression, which may contribute to cell growth inhibition. PEGylated curcumin increased protein stability of these proteins in pancreatic cancer cells and directly inhibited the activity of Jab1/CSN-associated kinases. Moreover, the inhibitory effect of PEGylated curcumin on cell proliferation was blunted in pancreatic cancer cells with Jab1 knock-down. The results suggested that PEGylated curcumin inhibits cell proliferation through suppression of Jab1/CSN activity.
Manju et al. [54] have synthesized polyvinyl pyrrolidone–curcumin conjugate (37) to enhance water solubility of curcumin. The drug conjugate self-assembled in aqueous solution to form nano-sized micellar aggregates, which were cationic and stable against hydrolytic degradation. The net zeta potential of these in the pH range 3 −7.4 was +20 to +25 mV, reflecting the potential stability of the conjugate micelle at physiological pH. The cytotoxic potential of the conjugate was evaluated against L929 fibroblast cells indicating that the conjugate has higher cytotoxicity than free curcumin probably due to enhanced aqueous solubility and polymer-mediated drug internalization. Pandey et al. [28] have PEGylated curcumin (38–41) as water soluble drug candidate with enhanced aqueous solubility and bioavailability. Based on the luciferase-based reporter gene assay most of the PEGylated curcumin analogs were found to strongly activate Nrf2 several folds higher than the free curcumin wherein the copolymer 38 was identified as the most potent Nrf2 activator, which induced Nrf2-driven NQO1 expression in a concentration dependent manner.
Mishra et al. [55] have examined pro-oxidant and antioxidant properties of dipiperoyl (42) and diglycinoyl (43) derivatives which showed higher apoptotic activity at lower concentrations than diacetylcurcumin. The apoptotic activity of these derivatives correlated with the generation of ROS by the tumor cells, whereas GSH levels remained unaffected. The studies also indicated down-regulation of Bcl-2 protein and activation of caspase-3 in the apoptotic death of tumor cells. Qui et al. [56] have synthesized a series of curcumin derivatives with modifications in the aryl ring and have evaluated their inhibitory activities on thioredoxin reductase (TrxR) by DTNB assay in vitro. Most of the analogs were found to inhibit TrxR in the low micromolar range. Structure-activity relationship analysis revealed that analogs with furan moiety (44–47) have excellent inhibitory effect on TrxR in an irreversible manner, indicating that the furan moiety may serve as a possible pharmacophore during the interaction of curcumin analogs with TrxR. The compound 46 showed growth inhibitory activity against different TrxR over-expressed A549/R and MCF-7/R cancer cell lines.
Safavy et al. [57] have prepared conjugates of curcumin with two polyethylene glycol (PEG) derivatives in order to improve its lower water solubility and improve its cytotoxicity against human cancer cell lines. The soluble conjugates 48 and 49 exhibited enhanced cytotoxicity against different pancreatic cancer line including LS-174T, MIAPaCa-2 and BxPC-3 cell lines. The half-Life (t1/2) of curcumin drug release from the High- and Low-MW PEG conjugates was found to be 60 and 200 minute, respectively. It was suggested that water-soluble conjugates may provide useful starting point for the development of injectable curcumin conjugates. Another aryl-side chain modified analogue dimethoxycurcumin (23) was found to be more potent in vitro against human HCT116 colon cancer cells in terms of its ability to inhibit proliferation and induce apoptosis. Nearly 100% of curcumin compared to less than 30% of dimethoxycurcumin was degraded in cells treated for 48 h. Incubation with liver microsomes confirmed the limited metabolism of dimethoxycurcumin. Although both compounds were rapidly degraded in vivo, the demethoxycurcumin was more stable. It is likely that differential extent of apoptosis induced by these compounds was associated with their different metabolic profiles [58].
Wichitnithad et al. [59] have synthesized a novel series of succinyl-derivatives of three curcuminoids as potential prodrugs (50–53). Their anti-proliferative activity was evaluated using Caco-2 cells. The succinate pro-drugs were found to possess much lower IC50 values (1.8–9.6 μM) compared to the parent compounds. Curcumin diethyl disuccinate exhibited the highest potency and was chosen for stability studies. Hydrolysis of this compound in phosphate buffer at pH 7.4 and in human plasma followed pseudo first-order kinetics indicating that the compound was much more stable than curcumin and was able to release curcumin in human plasma.
Youssef et al. [60] prepared a series of novel analogues of curcumin, and analyzed their cytotoxic activity against L1210 (murine leukemia) and human lymphoblast Molt4C/8 CEM cell lines where compounds 54–57 were found to be cytostatic. Vyas et al. [61] conjugated curcumin with a recombinant chimeric antibody cPiPP (58), exhibiting high affinity and specificity for human chorionic gona-dotropin-β (hCG-β)/hCG, which was tested for growth inhibitory properties against MOLT-4 and U-937 cells. Interestingly, the antibody did not impair the growth of MOLT-4 and U-937 cells while its conjugate was lethal to both cell lines. The immuno-conjugate killed tumor cells bearing the CD33 marker of an AML patient expressing hCG-β, but not for the patient who was negative for hCG-β.
Yodkeeree et al. [62] have reported that demethoxycurcumin (DMC, 59) inhibits adhesion, migration and invasion of MDA-MB-231 human breast cancer cells. MDA-MB-231 cells treated with DMC had decreased levels of extracellular matrix (ECM) degradation-associated proteins including matrix metalloproteinase-9 (MMP-9), membrane type-1 matrix metalloproteinase (MT1-MMP), urokinase plasminogen activator (uPA) and uPA receptor (uPAR), while the level of uPA inhibitor (PAI-1) was up-regulated. Moreover, DMC also reduced the expression of intercellular adhesion molecule-1 (ICAM-1) and chemokine receptor 4 (CXCR-4), which is involved in the modulation of the tumor metastasis process. It was found that DMC treatment inhibited DNA binding activity of NF-κB, which is known to mediate the expression of MMPs, uPA, uPAR, ICAM-1, and CXCR-4 proteins. Ryu et al. [63] have shown that DMC and bis-demethoxycurcumin (BDMC, 60) suppressed β-catenin response transcription (CRT) that was activated by Wnt3a conditioned-medium (Wnt3a-CM) without altering the level of intracellular β-catenin, and inhibited the growth of various colon cancer cells, with comparable potency to curcumin. Additionally, DMC and BDMC down-regulates p300, which is a positive regulator of the Wnt/β-catenin pathway.
Thus, it may be concluded that some of the modifications made to the aryl side chain result in improving the potency of Curcumin but not their specificity towards type of cancer cells. Similarly data on their in vivo activities in most cases is lacking.
3.2.2. Modifications of Diketo Functionality
Das and coworkers [64] have investigated the role of carbonyl and hydroxyl groups of Curcumin in PKC binding by synthesizing pyrazole (61–62) and isoxazole (63–65) derivatives and studying their interaction with protein kinase-Cδ (PKC-δ), especially with the activator-binding second cysteine-rich C1B sub-domain. All synthesized derivatives showed higher binding with the PKC-θ-C1B compared with PKC-δ-C1B and PKC-ε-C1B. Compound 63 exhibited most potent PKC binding activity. Molecular docking revealed that isoxazole (63) and pyrazole (61) derivatives bind to the activator binding site of PKCs and both carbonyl and hydroxyl groups of curcumin play role in such binding process, depending on the nature of Curcumin derivative and the PKC isotype. Compared with Curcumin, its isoxazole (63) and pyrazole (61) derivatives exhibited increased cell growth inhibitory and pro-apoptotic effects in liver cancer HA22T/VGH cells as well as in other tumor cell types. The anti-tumor effects of isoxazole (63) and pyrazole (61) were not influenced by concomitant administration of N-acetylcysteine, as a source of –SH groups, or buthionine sulfoximine as an inhibitor of glutathione synthesis. Additionally treatment with Curcumin, but not with isoxazole (63) and pyrazole (61), significantly decreased the content of reduced glutathione in the HA22T/VGH cells. Isoxazole (63) and pyrazole (61) lacked the ability of the parent compound to sensitize the HA22T/VGH cells to cisplatin (CIS), an effect which is known to occur through an interaction of curcumin and cisplatin at the level of the thiolic groups. Thus, the ability of interacting with cell thiols might not be required for these curcumin derivatives and they may be devoid of chemo-sensitization capabilities [65].
Poma et al. [66] have also examined the effects of curcumin and compound 63 in the MCF-7 breast cancer cell line, and in its multi-drug resistant variant MCF-7R, which lacks estrogen receptor alpha (ER-α) and exhibits over-expression of P-glycoprotein (P-gp), IAP (inhibitory of apoptosis proteins) and COX-2 proteins. It was observed that the anti-tumor activity of curcumin and compound 63 is equivalent in the MDR cell line. It seems that the overall anti-tumor activities of curcumin or 63 are not hampered by P-gp expression or lack of ER-α in breast cancer cells. Remarkably, the agents appeared to modify their molecular effects according to the diverse gene expression patterns existing in the MDR and parental MCF-7 cells. Sahoo and co-workers [67] have examined the interaction of 63 with human serum albumin (HSA) using various biophysical methods. The observed fluorescence quenching of HSA by 63 is due to complex formation with quenching constant of the order of 105 M−1.
The semicarbazone derivative of curcumin (CRSC, 66), has been described by our group which was evaluated for antioxidant, anti-proliferative, and radical scavenging activities [68]. CRSC was found to inhibit radiation induced lipid peroxidation in the rat liver microsomes at 10 μM concentration. Its interaction with 2, 2′-diphenyl-1-picrylhydrazide (DPPH) was studied using a stopped-flow spectrophotometer. The rate constant for curcumin was found to be 1640 ± 38 M−1 s−1 and the reaction was completed within 10–20s, while the reaction with CRSC was much slower under similar conditions and took more than 200 s. The rate constant for the reaction was determined to be 274 ± 7 M−1 s−1. The difference in their rate constants was attributed to the steric hindrance provided by the bulky semicarbazide side chain and its higher reduction potential. CRSC exhibited potent cytotoxic activity (74% growth inhibition) against breast cancer MCF-7 cell line at 1μg/mL concentration as compared to curcumin (36% growth inhibition). Lal et al. [69] have synthesized 3,4-dihydropyrimidinones of curcumin by multi-component one-pot condensation of curcumin, substituted aromatic aldehydes and urea/thiourea under solvent free conditions using SnCl2.2H2O catalyst and have evaluated cytotoxic profile of the compound. Among all derivatives, compound 67–69 showed better cytotoxic activity than curcumin against human Hep-G2, HCT-116 and QG-56 cancer cell lines, respectively.
Shim et al. [70] have described hydrazinocurcumin (HC, 61) and benzoylhydrazino- curcumin (HBC, 70) derivatives which were found to be inhibitory against proliferation of bovine aortic endothelial cells (BAECs) at nanomolar concentration (IC50=520 nM) without cytotoxicity. In vivo and in vitro angiogenesis experiments showed HC as a new potent candidate for developing anti-angiogenic agents. These researchers have further identified Ca2+/calmodulin (Ca2+/CaM) as a direct target protein of HBC using phage display biopanning [71]. Ca2+/CaM-expressing phages specifically bound to the immobilized benzoylhydrazino-curcumin (HBC) and the binding was found to be Ca2+ dependent. In order to isolate the direct binding protein of HBC, the biotinyl-HBC (71) was synthesized as a molecular probe for target identification and was found to be two fold less potent than HBC against colon cancer cells. Flexible docking demonstrated that HBC was compatible with the binding cavity for a known inhibitor, viz.W7 (N-(6-amino-hexyl)-5-chloro-1-naphthalensulfonamide), in the C-terminal hydrophobic pocket of Ca2+/CaM. It was suggested that HBC-induced prolonged phosphorylation of ERK1/2 and activation of p21WAF1 (p21) expression, resulting in the induction of G0/G-cell cycle arrest in HCT15 colon cancer cells.
Simoni et al. [72] have studied enaminones, oximes, and isoxazole derivatives of curcumin. These were evaluated against hepatocellular HA22T/VGH cancer cells, MCF-7 breast cancer cells and its multi-drug resistant variant MCF-7R, respectively. The compounds 63, 72 and 73 showed growth inhibitory activity against all cancer cell lines. Enhanced anti-tumor activity on all cell lines was found with isoxazole (63) and benzyloxime (73) derivatives. Swarts et al. [73] have investigated semicarbazone and pyrazole derivatives of curcumin as potential mitigation agents to treat acute radiation syndrome (ARS). Pyridyl (74–75), furyl (76), and phenyl (77) derivatives of curcumin semicarbazone were found to provide the highest dose modifying factors (DMF) with respect to survival in sub-TBI (bone marrow sparing) exposures in mouse models. In order to investigate the basis for the mitigating effects of these agents on ARS, their oxidation potentials and radical scavenging properties were compared with other semicarbazone and pyrazole derivatives of curcumin. Such comparison did not show sufficient differences in their reducing or hydrogen atom donating properties to be the basis of dose-modifying activities of these compounds. Therefore, their DMFs probably reflect the structure-activity relationship, wherein the interaction with key receptors/enzyme expression results in modifications of cellular or tissue responses to radiation, rather than on the ability of these derivatives to modify radiation-induced flux of free radicals.
Recently, Arif et al. [74] have shown inhibition of the histone acetyltransferase (HAT) activity of p300 by a water-soluble hydrazinocurcumin (78, CTK7A), which substantially reduced the oral tumor growth in a xenograft mouse model. The compound was found to inhibit HAT p300/CBP and PCAF which suggest that this water-soluble derivative inhibits histone acetylation in the cellular system, at least, partially through the inhibition of p300 auto-acetylation. The compound was found to be non-toxic to the mice after intra-peritoneal (i.p.) administration. There was no observed weight loss at doses up to 100 mg/kg body weight twice a day during a month long treatment. KB cells-derived tumors were about 50% smaller in mice treated with this compound than in control mice.
3.2.3. Modifications of Double Bond
Chun et al. [75] have evaluated the anti-tumor promoting potential of Yakuchinone-A (79) and -B (80), major pungent ingredients of Alpinia oxyphylla Miquel, which can be considered as curcumin analogs where the double bond between the aryl rings are reduced. The topical application of Yakuchinone A or B significantly suppressed TPA-induced epidermal ornithine decarboxylase activity while higher concentration reduced TPA-stimulated production of tumor necrosis factor-α (TNF-α) in cultured human pro-myelocytic leukemia (HL-60) cells. Both compounds blunted the TPA-induced superoxide generation in differentiated HL-60 cells in a concentration-dependent manner as well as inhibited lipid peroxidation in rat brain homogenates. Furthermore, both compounds nullified activation of the activator protein-1 (AP-1) in immortalized mouse fibro-blast cells in culture. Overall Yakuchinone-B showed better activity than Yakuchinone-A, but less than curcumin.
Wu et al. [76] have found that treatment with tetrahydrocurcumin (THC, 9), which is the double-bond reduced derivative of curcumin, results in autophagic cell death in human HL-60 pro-myelocytic leukemia cells by increasing the formation of auto-phagic cell death marker, acidic vascular organelle (AVO). THC significantly down-regulated phosphatidylinositol 3-kinase/protein kinase B and mitogen-activated protein kinase signaling, decrease in the phosphorylation of mammalian target of rapamycin, viz. glycogen synthase kinase 3β and p70 ribosomal protein S6 kinase respectively. Yodkeeree et al. [77] have shown that treatment with THC reduced HT1080 cell invasion and migration in a dose-dependent manner. The inhibition of cancer cell invasion was associated with down-regulation of ECM degrading enzymes as well as inhibition of cell adhesion to ECM proteins. Curcumin and THC were also found to attenuate pathological features of angiogenesis including micro-vascular dilatation, and hyper-permeability. Treatment with curcumin and THC resulted in a significant decrease in the capillary vascularity (CV). However, the beneficial effects of THC treatment on CV were observed from day 21 onwards [78]. Chen et al. [79] have studied the cytotoxicity of hexahydrocurcumin (10) and its effect on cell cycle in human colorectal SW480 cancer cells. Treatment of these cells with compound 10 resulted in a massive accumulation of the cells in the G1/G0 phase of the cell cycle.
3.2.4. Modifications at Active Methylenic Group
Ohtsu et al. [80] have prepared curcumin analogs by modifying the active methylenic group and have evaluated their potential as androgen receptor (AR) antagonists against two human prostate cancer cell lines, PC-3 and DU-145 transfected with AR and androgen receptor co-activator, ARA70 constructs. Compounds 23 and 81–83 showed potent anti-androgenic activities superior to hydroxyflutamide, which is the standard anti-androgenic compound currently available for the treatment of prostate cancer. The compound 23 has shown reduction of dihydrotestosterone (DHT)-induced AR activity in PC-3 cells, while in DU-145 cells transfected with wild-type AR and ARA70 compounds 81 and 82 were almost equipotent and were slightly more active than 23. Structure-activity relationship (SAR) studies indicated that the bis-(3, 4-dimethoxyphenyl) conjugated β-diketone moiety and the intra-molecular symmetry of molecules seem to be important factors related to the anti-androgenic activity of these curcumin analogs. The data further suggested that co-planarity of the β-diketone moiety and presence of strong hydrogen bond donor groups was crucial for the anti-androgenic activity, which was consistent with previous SAR results for Hydroxyflutamide analogs. When the pharmacophoric elements of DHT and compound 23 are superimposed, the resulting construct implies that the curcumin analogs may function as a 17α-substituted DHT.
Lin et al. [81] have designed 4-fluoro-4-ethoxycarbonylethyl curcumin (83) and 4-ethoxycarbonylethylenyl curcumin (84) to overcome the problems inherent in the tautomerism of 4-Ethoxycarbonylethyl curcumin (ECECur) (82). These compounds and their synthetic intermediates (including compound 85) were evaluated for their inhibitory activity against LNCaP and PC-3 prostate cancer cell lines at a concentration of 3 μM. While the keto-enol analogs showed varying anti-androgen potencies, the diketo compounds showed no activity. Tetrahydropyranylation of the phenoxy groups had a positive impact on the anti-androgenic activity of 4-ethoxycarbonylethylenyl curcumin. Di-tetrapyranylated-4-ethoxycarbonylethylenyl curcumin (85), which exists only in the keto-enol form, has potent anti-androgenic activity, which may be a good lead compound for further structural modifications. Based on the SAR information, five new compounds were designed and subsequently synthesized. Among them, compound 86 was found to be the most potent anti-androgenic agent and is considered to be a promising drug candidate for the treatment of prostate cancer. Lin et al. [82] have synthesized further curcumin analogs containing monophenyl and substituted phenyl/heterocyclic moieties at the methylenic position of curcumin with various linkers. These new compounds were tested for cytotoxicity against two human prostate cancer cell lines, viz. androgen-dependent LNCaP and androgen-independent PC-3 cells. Anti-androgenic activity was also evaluated on LNCaP cells and PC-3 cells transfected with wild-type AR. The compounds 87–92 showed most potent activity against PC-3 and LNCaP cells. The anti-androgenic activity of compounds 90 and 92 were studied and found to exert transactivation of LNCaP and PC-3 cells transfected with wild-type AR. Amolins and coworkers [83] have synthesized another group of curcumin derivatives substituted at methylenic group with pyrazole and isoxazole pharmacophores and have studied their anticancer activity against MCF-7 and SKBR3 cancer cell lines. Compounds 93–95 were the most potent cytotoxic compounds against MCF-7 and SKBR3 cancer cell lines, while their pyrazole (96–98) and isoxazole (99–100) counterpart were found to be less active.
Arezki et al. [84] have synthesized ferrocenyl conjugates of curcumin which were evaluated for their cytotoxicity on B16 melanoma cells and normal NIH 3T3 cells as well as for inhibition of tubulin polymerization and effect on the morphology of endothelial cells. The compounds 101–105 were found to induce cytotoxicity against B16 melanoma cells, while compound 102 was found to inhibit tubulin polymerization. Lin et al. [85] have developed signal transducer and activator of transcription-3 (STAT3) inhibitors, known as FLLL31 (106) and FLLL32 (107), which are derived by functionalization of the methylenic position in curcumin. These compounds are designed to bind selectively to Janus kinase-2 and the STAT3 Src homology-2 domain, which serve crucial role in STAT3 dimerization and signal transduction. It was found that these compounds are effective inhibitors of STAT3 phosphorylation, DNA-binding activity, and transactivation leading to the impediment of multiple oncogenic processes and induction of apoptosis in pancreatic and breast cancer cell lines. It was observed that administration of FLLL32 can inhibit tumor growth and vascularity in chicken embryo xenografts as well as substantial reduction in tumor volumes in mouse xenografts. The findings indicated the potential of these new compounds in targeting pancreatic and breast cancers that exhibit constitutive STAT3 signaling. These workers have further shown that the phosphorylated or activated form of STAT3 was expressed in colon cancer stem-like cells, which was attenuated by STAT3-selective inhibitor FLLL32 [86]. The compound also inhibited the expression of STAT3 downstream target genes including survivin, Bcl-xL, as well as Notch-1, -3, and -4 in colon cancer stem-like cells, which may be involved in stem cell function. It exerted anti-proliferative activity against SW480, HCT116 and DLD-11 colon cancer cell lines with lower IC50 values compared to curcumin, respectively. FLLL32 specifically reduced STAT3 phosphorylation at Tyr705 (pSTAT3) and induced apoptosis at micro-molar concentration in human melanoma cell lines. The compound displayed specificity for STAT3 over other homologous STAT proteins [87].
Chakraborti et al. [88] have studied a series of curcumin analogs for their tubulin binding affinities and tubulin self-assembly inhibition. It was found that curcumin acts as a bifunctional ligand and analogs with substitution at the diketone functionality or acetylation of the terminal phenolic groups are less effective. The benzylidiene derivative 108 was more effective than curcumin in inhibiting tubulin self-assembly. It was shown that curcumin binds tubulin away from the colchicine-binding site. Docking studies suggested that the curcumin-binding site was closer to the vinblastine-binding site. Structure-activity studies suggested that the tridentate nature of compound 108 was responsible for its higher affinity for tubulin compared to curcumin.
Chen et al. [89] have studied the effects of C086 (compound 109) on growth inhibition and NF-κB regulation in colon cancer cells and xenograft tumors. The compound exhibited potent anti-proliferative activity against HT29, SW480, KM12, SW1116, WiDr and Colon26 cancer cell lines. In a xenograft model of SW480 in nude mice, the oral administration of C086 showed significant growth suppression of SW480 tumors and decreased NF-κB expression in tumor tissues. Using TNF-α to induce NF-κB activation in SW480 cells, it was revealed that C086 inhibited IκB-α phosphorylation and its subsequent degradation thereby suppressing the nuclear translocation and DNA binding activity of NF-κB. It was also observed that NF-κB-regulated gene products C-myc, cyclin D1 and Bcl-2, involved in cellular proliferation and anti-apoptosis, were down-regulated in C086 treated group.
Fadda et al. [90] have synthesized curcumin analogues 110–116 and have evaluated them for in vitro and in vivo cytotoxicities against ehrlich ascites carcinoma (EAC). In vitro results revealed that compounds 115 and 116 were the most potent analogs. Ferrari et al. [91] have synthesized curcumin analogs (ester and acid series) which showed IC50 values lower than curcumin when evaluated their against human ovarian carcinoma cells (A2780, C13*, and A2780/CP), and human colon carcinoma cells (HCT116 and LoVo). The compounds exhibited selectivity against colon carcinoma cells which could be ascribed to their high lipophilicity that favors a greater and faster cellular uptake and thereby overcoming their apparently higher instability under physiological condition. Qiu et al. [92] have synthesized a series of new 4-arylidene curcumin analogs which inhibited growth of a panel of lung cancer cells at sub-micromolar concentrations. Compounds 117–126 were found to exert superior growth inhibitory activities against A549 Cells through NF-κB inhibition which can be explained, at least, in part by inhibition of IκB phosphorylation and degradation via IKK blockage.
Sertel et al. [93] have analyzed the cytotoxic activity of curcumin and its four derivatives (127–130), among which the ethoxy-curcumin-tri-thiadiazol-amino-methyl-carbonate (128) compound was found to be the most potent one. The compounds were not cross-resistant to standard anticancer drugs and were not involved in ATP-binding cassette transporter-mediated multi-drug resistance. Zhou et al. [94] have prepared β-ionone-curcumin conjugates which led to the identification of a novel anti-androgen compound 131 having two bulky side chains, which was a pure antagonist of the wild-type and T877A, W741C, and H874Y mutated AR cells, showing no cross-reactivity with progesterone receptor. Compounds 131 and 132 have been found to exert cytotoxicity towards LNCaP, PCa-2b, 22Rv1, C4-2B and PC-3 prostate cancer cells. Molecular modeling indicated that compound 131 adopts “Y”-shape conformation and forms multiple hydrogen bonds with androgen receptor (AR) backbone.
In our group, we have synthesized some novel curcumin analogs using Knoevenagel condensation (133–135) to convert enolic diketones of curcumin into non-enolizable moieties. Their Schiff bases (136–138) were prepared using a bioactive thiosemicarbazone pharmacophore. The compounds were evaluated for their potential in inhibiting TNF-induced NF-κB activation and proliferation in human leukemic KBM-5 cells [95]. Later on, we extended our studies to include fluoro-substituted Knoevenagel condensates of Curcumin (139–143) and evaluated their growth inhibitory activities against colon and pancreatic cancer cell lines [96]. Out of these efforts emerged a compound known as difluorinated-curcumin commonly referred to as CDF (139) with superior anticancer activity in colon, prostate and pancreatic cancer cell lines. The compound was found to have inhibitory effect on purified rabbit 26S proteasome. Molecular docking studies into COX-2 protein cavity revealed that CDF did not introduce any major steric changes in the parent Curcumin molecule and facilitated more H-bonding interactions, resulting in higher binding energy of −7.91 Kcal/mol compared to curcumin (−5.71 Kcal/mol). We showed that Curcumin and CDF both down-regulated the expression of NF-κB in MIAPaCa-2 cells but the latter did it at minimal concentration of 2 μM or less. Both CDF and Curcumin decreased the PGE2 levels in MIAPaCa-2 cells. However, in BxPC-3 cells, only CDF- treated cells had a decrease in PGE2 level. These results clearly suggested that COX-2 is a better target of CDF compared to Curcumin [96].
We further studied the pharmacokinetics of Curcumin and CDF compound by administering 250 mg/kg orally to female ICR-SCID mice. Compared to Curcumin, CDF achieved a similar Cmax (0.21 Ig/mL) with a relatively slow oral absorption with Tmax of 8 h. However, CDF had 2.7-fold higher systemic drug level than Curcumin (AUClast, 1.22 vs. 0.44 Ig/mL*h). Serendipitously CDF was found to accumulate preferentially in pancreas (10.6-fold higher then Curcumin), suggesting that it had improved bioavailability and preferential affinity towards pancreatic tissue. These findings make CDF an ideal candidate for cancer prevention and treatment of pancreatic cancers in conjunction with other cytotoxic agents.
We have studied the combination of CDF (1 μM/L) with Gemcitabine (10nM/L) in BxPC-3 cell lines [97]. The results indicated that the combination therapy exhibited far more efficacy than CDF alone. The combination significantly induced apoptosis in pancreatic BxPC-3, MIAPaCa-E and MIAPaCa-M cancer cell lines. Combination of CDF with Gemcitabine brought about a significant reduction of cell viability and almost eliminated pancreatosphere formation after four weeks of treatment suggesting that CDF could be a useful therapeutic agent for elimination of Cancer Stem-like Cells (CSCs). CDF also attenuated CD44 and EpCAM expressions in pancreatospheres. The in vivo subcutaneous xenograft tumor model induced by MIAPaCa-2 cells in CB17-SCID mice was helpful in showing that the combination of CDF with Gemcitabine significantly inhibited tumor growth in MIAPaCa-2 tumors much more than either of the individual agents. There was no significant weight loss in mice during the treatment period (30 days), which suggested that these treatments had no major adverse effects on animals. CDF treatment with or without Gemcitabine showed increased expression of tumor suppressive micro-RNAs (miRNAs), viz. miR-200b and miR-200c, respectively [98].
In another investigation carried out with an orthotropic xenograft model of human pancreatic cancer cells, CDF inhibited tumor growth through reduced expression of histone methyltransferase EZH2, Notch-1, CD44, EpCAM, and Nanog signaling pathways, which was consistent with increased expression of let-7, miR-26a, and miR-101 respectively [98]. The effects of CDF in combination with 5-fluorouracil and oxaliplatin (5-FU + Ox) against colon cancer were evaluated against chemo-resistant HCT-116 and HT-29 cells [99] which showed a significant inhibition of cellular growth when the cells were incubated for 48 h. These results clearly suggest that combination of CDF with conventional chemotherapeutics agents might have a potential in preventing the emergence of chemo-resistant colon cancer cells. Recent study in our laboratory has shown, for the first time, that Ras expression and its activity are significantly higher in MIAPaCa-2 cells as compared to COLO-357 and BxPC-3 cell lines [100]. It was found that CDF treatment either in vitro studies or MIAPaCa-2 induced tumors in vivo results in the re-expression of let-7 and miR-143 along with down-regulation of miR-21 expression, which is consistent with decrease of Ras expression and inhibition of tumor growth. It was concluded that the loss of let-7 and miR-143 expression and increased expression of miR-21 leads to increased expression of Ras and its GTPase activity, which could be attenuated by CDF treatment in pancreatic cancer cell lines.
We have also assessed the effects of CDF on the regulation of AR/TMPRSS2-ERG/Wnt signaling [101]. It was observed that activation of AR resulted in the induction of ERG expression through TMPRSS2-ERG fusion. It was found that ERG over-expression and nuclear translocation activated the activity of Wnt signaling. Interestingly, we observed that CDF inhibited the signal transduction in AR/TMPRSS2-ERG/Wnt signaling network, leading to inactivation of the Wnt signaling consistent with inhibition of prostate cancer cell invasion.
3.2.5. Metal Complexation
It has been well established that metal complexation can improve and enhance the biological activity of the parent drug molecule [102, 103]. Since the enolic tautomers as well as the diketo functionality of the modified Curcumin are capable of complexing with biologically relevant metals, it can be considered as one of the methods for improving its bioactivity. Recently, curcumin has been found to alter iron metabolism by chelating iron and suppressing the protein hepcidin, causing iron deficiency in susceptible patients [104]. Curcumin interaction with copper and iron has also been suggested as one possible mechanism of action in animal models of Alzheimer’s disease. With this in mind, we summarize here recent reports on metal complexation of curcumin and its analogs which may have some bearing on its anticancer activity.
Alcalde et al. [105] have investigated a new curcuminoid ligand, viz. 9Accm (Compound 144) and its corresponding copper-complex, [Cu(phen)Cl(9Accm)] for their interactions with calf thymus DNA which indicate a weak electrostatic interaction with the double helix. Annaraj and co-workers [106] have examined the binding mode of ternary copper (II) complexes of Curcumin Schiff bases (145–146) involving phenanthroline/bipyridyl as ancillary ligands with herring sperm DNA by spectral and electrochemical techniques. These studies revealed that interaction with DNA slightly improves reversibility of the quasi-reversible Cu +2/+1 redox couple with considerable reduction in current intensity. The results indicate that these ternary copper complexes (146) can intercalate more effectively into the DNA base pairs than usual bipyridyl (145) counterparts.
Barik et al. [107] have studied the mononuclear copper complex of Curcumin (147) for its superoxide dismutase (SOD) activity. It was found that the copper atom was coordinated through the oxygen donor atoms of Curcumin along with an acetate anion and one water molecule. The compound could scavenge superoxide radicals and was found to regenerate completely, indicating catalytic activity in neutralizing superoxide radicals. The complex also inhibited radiation-induced lipid peroxidation.
Caruso et al. [108] have evaluated the anti-proliferative activity of novel Ruthenium-Curcumin complex (148) on five cancer cell lines which showed compound’s preference for the colorectal HCT-116 cancer cells followed by breast MCF-7 and ovarian A2780 cell lines. The human glioblastoma U-87 and lung carcinoma A549 cells were found to be less sensitive. John and coworkers [109] have synthesized copper complexes of four new Curcumin analogs (149–152) wherein the metal complexes were found to be remarkably active.
Valentini et al. [110] have evaluated a novel water soluble ternary Pd (II) complex, 153, containing Curcumin and bipyridyl ligands against human prostate LNCaP, PC-3, and DU-145 cancer cells. ROS production induced by the treatment with this complex activated apoptosis through mitochondrial membrane de-polarization in all prostate cancer cells, accompanied by up-regulation of Bax and down-regulation of Bcl-2 proteins. While Curcumin produced cell growth delay due to the G2 phase arrest in these prostate cancer cells, an apoptosis was observed in cells treated with the metal complex mediated through JNK activation and mitochondrial membrane depolarization.
3.2.6. Appended Curcumin mimics
Appended Curcumin mimics refers to those compounds that have partial structural resemblance to Curcumin motif but are appended with different pharmacophores for targeting proteins that are crucial for survival of tumor cells in question. In the following discussion, we summarize some of these analogs which offer some advantages in terms of tumor specificity, pharmacokinetics or non-toxicity towards normal cells.
Ruan and co-workers [111] have recently reported on the synthesis and anti-proliferative activity of a series of resveratrol analogs bearing curcumin motif (154) against a panel of cancer cell lines including murine melanoma B16-F10, human hepatoma HepG2 and human lung carcinoma A549 cells. Amongst the several compounds that were evaluated for their anticancer activity, the compound possessing methoxy substitution at the aromatic C-ring (154) exhibited highest activity against murine B16-F10 cells. SAR studies indicated that the anti-proliferative activity was influenced by the substitutions on C-ring. These results demonstrated that hydrophobic and electron-donating methyl/methoxy groups can increase the anti-proliferative activity considerably. Mechanistic investigation showed that these compounds inhibited tubulin polymerization efficiently. Cell cycle analysis of the most potent methoxy analog showed the induction of G2/M-phase cell cycle arrest in B16-F10 cancer cells. The molecular modeling studies of the potent compound in the tubulin-colchicine binding site of β-tubulin revealed that hydrophobic pockets of the tubulin-colchicine binding site are occupied by the methoxy group on C-ring and the monocarboxyl group of the Curcumin moiety. It indicates that these substituents have relevance in the anti-tubulin activity.
Recently Cao and co-workers [112] have reported pyridinyl analogs of dibenzylideneacetone (155–156) by the condensation of substituted nicotinaldehyde and acetone. These curcumin analogs were evaluated for their anti-inflammatory activity by NF-κB inhibition assay and also for their cytotoxicity against colorectal carcinoma HCT-116 cells. Almost all synthesized, analogs exhibited better cytotoxicity than curcumin, but one analog 155 showed highest efficacy in inhibiting NF-κB, and more potency in suppressing growth of colon cancer cells.
Somers-Edgar and co-workers [113] have synthesized a series of novel cyclohexanone curcumin analogs containing heterocyclic substitutions at the terminal benzene rings. These analogs exhibited anti-cancer activity towards ER-negative MDA-MB-231 human breast cancer cells. Amongst the analogs screened, 157 and 158 were remarkably potent against MDA-MB-231 cells. Mechanistically these compounds induced G2/M-phase cell cycle arrest and caused apoptosis. They also modulated the expression of key cell signaling proteins including Her-2, Akt and NF-κB in a time-dependent manner. In case of MDA-MB-231 cells, EGFR protein levels were decreased, while β-catenin protein level and the activity of P38 MAPK, Akt and JNK1/2 were increased. Thus, replacement of the phenyl group with heterocyclic rings turned out to be beneficial in evolving anti-cancer agents with some advantages against ER-negative breast cancer cells.
In another study, Kudo et al. [114] have evaluated large number of structural curcumin analogs, including acyclic 1, 5-diaryl-3-oxo-1, 4-pentadiene analogs, 1, 7-diaryl-1, 6-heptadiene-3, 5-dione, and acyclic 1, 5-diaryl-3-oxo-1, 4-pentadiene curcumin analogs (159–163) which have been examined for their anti-proliferative activity. The growth-inhibition potential of these compounds was studied on a panel of cancer cell lines. All compounds had at least 10 times higher growth-inhibitory potential than curcumin. Cell cycle analysis revealed growth inhibition of HCT-116 cells correlated with increase of the sub-G1 fraction.
Kalai and co-workers [115] have reported a series of 3, 5-bis-(arylidene)-4-piperidone (DAP) curcumin analogs. The anticancer efficacy of these analogs was evaluated against A2780, MCF-7 cancer cell lines and H9c2 cardiomyoblast cells. Compounds without nitroxide moiety demonstrated a substantial cytotoxic effect against A2780 and MCF-7 cells. The electron-withdrawing fluoro- and trifluoro-methane substituents (164, 165) exhibited greater cytotoxicity than trimethoxy derivative. Molecular modeling studies demonstrated that these analogs have high binding affinity for STAT3 at the deoxyribonucleic acid (DNA) binding domain of the molecule.
Curcumin analogs possessing indan-2-one and dibenzylidene-piperidin-4-one motif (166 and 167) have been recently described by Karthikeyan et al. [116] which have been evaluated against a panel of cancer cell lines including HCT-116, Panc-1, H460, Calu-1 and ACHN cells respectively. The monocarbonyl analog with the ethoxy substituent (166) on the phenyl ring exhibited highest efficacy overall. Dayton’s group [117] studied the cellular uptake, retention and bio-absorption potential of monocarbonyl fluorocurcumin, viz. HO-3867 (168), against a series of cancer cells including cisplatin-resistant A2780R cells. The cellular uptake of HO-3867 was 10-fold greater than Curcumin at 60 min of incubation at concentrations of 10–100 μM. The cellular absorption was highest in MCF-7 cells. In vivo studies indicated that the compound was absorbed significantly in the liver compared to serum and kidney.
Lin and co-workers [118] have studied the efficacy of synthetic monocarbonyl curcumin analogues, FLLL11 (169) and FLLL12 (170), in eight different breast and prostate cancer cell lines. The compounds were substantially more effective than curcumin in inhibiting AKT phosphorylation and down-regulating the expression of HER2/neu. In addition, they also inhibited phosphorylation of signal transducer and transactivator STAT3, an oncogene frequently found activated in many cancer types. These results suggest that FLLL11 and FLLL12 may have translational potential as chemopreventive or therapeutic agents for breast and prostate cancers. Yamakoshi and co-workers [119] reported novel curcumin analogs of the type, 1,5-bis(4-hydroxy-3-methoxyphenyl)-penta-(1E,4E)-1,4-dien-3-one, and evaluated their cytotoxicities against human colon cancer HCT-116 cell line. Amongst the three series of compounds reported by his group, compounds belonging to the symmetric 3, 5-substituted series, (GO-YO30 (171) and GO-YO31 (172) were found to be the most potent analogs. Similar monoketo-curcumin analogs having cyclopentanone (173), acetone (174–176) and cyclohexanone (177) central motifs have also been synthesized [120]. Their cytotoxicity was evaluated against panel of cancer cell lines which revealed that analogs from series B and C were more potent than series A. The dimethoxy 174 analog exhibited potent inhibitory activity against PC-3 and LNCaP cells. Structure-activity relationship studies indicated that the presence of strong electron withdrawing substituents at 2′ position may be important for the enhancement of cytotoxicity.
Ohori et al. [121] have reported symmetrical 1, 5-diaryl-pentadienone curcumin analogs whose aromatic rings possess an alkoxy substitution at each of the 3 and 5 positions. The most active compound was (GO-035, 178) which strongly suppressed the growth of DLD-1colon cancer cell line. Ravindran et al. [122] have reported anti-proliferative potential of hispolon group of compounds (179–185) which are the monoaryl curcumin analogs. Amongst these, hispolon (180) was the most cytotoxic against human leukemia KBM-5 cell line. It also exhibited highest cytotoxicity against HCT-116 and MCF-7 cells whereas dehydroxy-hispolon (182) showed maximum cytotoxicity against prostate PC-3 cells. Youssef and co-workers [123] have reported synthesis of monocarbonyl cyclic and non-cyclic curcumin analogs (186–188) amongst which 188 elicited highest activity toward non-small lung cancer cell line.
4. CONCLUSION
Curcumin is one of the most potent and multi-targeting phytochemicals against a variety of cancers. In the past few decades, hundreds of research papers have been devoted to elucidating its role in inhibiting proliferation, angiogenesis and metastasis of different cancers. These studies clearly indicated that the use of curcumin as a therapeutic agent was limited due to its low bioavailability and rapid metabolism. Various formulations including nano-particulates although enhanced the bioavailability of curcumin to varying extent were not successful in slowing down the rapid metabolism of the compound. Our review of existing literature clearly indicates that researches are now devoting their efforts to this aspect by structurally modifying the curcumin motif systematically. These efforts have led to some serendipitous discoveries of some new compounds which not only display slowing down of the metabolism but selective accumulation in pancreatic as well as prostate tissues. Some of the structural derivatives described in the present review indicate exciting possibilities of developing curcumin analogs selective towards some specific cancers giving hope of finding use of Curcumin or its analogs in the clinics for the prevention and/or treatment of these cancers.
Acknowledgments
PD and AV would like to thank Abhijit Takawale and Khadija Syeda for their timely help and support. We are thankful to Dr. E. M. Khan and Mr. P. A. Inamdar for their keen interest and encouragement.
GRANT SUPPORT
Part of the work cited in this article was funded by National Cancer Institute, NIH grant 1R01CA154321-02 (F.H. Sarkar).
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
Send Orders of Reprints at reprints@benthamscience.net
References
- 1.Craig WJ. Health-promoting properties of common herbs. Am J Clin Nutr. 1999;70:491S–9S. doi: 10.1093/ajcn/70.3.491s. [DOI] [PubMed] [Google Scholar]
- 2.Ammon HP, Safayhi H, Mack T, Sabieraj J. Mechanism of antiinflammatory actions of curcumine and boswellic acids. J Ethnopharmacol. 1993;38:113–9. doi: 10.1016/0378-8741(93)90005-p. [DOI] [PubMed] [Google Scholar]
- 3.Reddy AC, Lokesh BR. Studies on anti-inflammatory activity of spice principles and dietary n-3 polyunsaturated fatty acids on carrageenan-induced inflammation in rats. Ann Nutr Metab. 1994;38:349–58. doi: 10.1159/000177833. [DOI] [PubMed] [Google Scholar]
- 4.Xu YX, Pindolia KR, Janakiraman N, et al. Curcumin, a compound with anti-inflammatory and anti-oxidant properties, down-regulates chemokine expression in bone marrow stromal cells. Exp Hematol. 1997;25:413–22. [PubMed] [Google Scholar]
- 5.Banerjee M, Tripathi LM, Srivastava VM, Puri A, Shukla R. Modulation of inflammatory mediators by ibuprofen and curcumin treatment during chronic inflammation in rat. Immunopharmacol Immunotoxicol. 2003;25:213–24. doi: 10.1081/iph-120020471. [DOI] [PubMed] [Google Scholar]
- 6.Lantz RC, Chen GJ, Solyom AM, Jolad SD, Timmermann BN. The effect of turmeric extracts on inflammatory mediator production. Phytomedicine. 2005;12:445–52. doi: 10.1016/j.phymed.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 7.Han SS, Chung ST, Robertson DA, Ranjan D, Bondada S. Curcumin causes the growth arrest and apoptosis of B cell lymphoma by downregulation of egr-1, c-myc, bcl-XL, NF-kappa B, and p53. Clin Immunol. 1999;93:152–61. doi: 10.1006/clim.1999.4769. [DOI] [PubMed] [Google Scholar]
- 8.Huang MT, Lou YR, Xie JG, et al. Effect of dietary curcumin and dibenzoylmethane on formation of 7,12-dimethylbenz[a] anthracene-induced mammary tumors and lymphomas/leukemias in Sencar mice. Carcinogenesis. 1998;19:1697–700. doi: 10.1093/carcin/19.9.1697. [DOI] [PubMed] [Google Scholar]
- 9.Jaiswal AS, Marlow BP, Gupta N, Narayan S. Beta-catenin-mediated transactivation and cell-cell adhesion pathways are important in curcumin (diferuylmethane)-induced growth arrest and apoptosis in colon cancer cells. Oncogene. 2002;21:8414–27. doi: 10.1038/sj.onc.1205947. [DOI] [PubMed] [Google Scholar]
- 10.Bhaumik S, Anjum R, Rangaraj N, Pardhasaradhi BV, Khar A. Curcumin mediated apoptosis in AK-5 tumor cells involves the production of reactive oxygen intermediates. FEBS Lett. 1999;456:311–4. doi: 10.1016/s0014-5793(99)00969-2. [DOI] [PubMed] [Google Scholar]
- 11.Ranjan D, Chen C, Johnston TD, Jeon H, Nagabhushan M. Curcumin inhibits mitogen stimulated lymphocyte proliferation, NFkappaB activation, and IL-2 signaling. J Surg Res. 2004;121:171–7. doi: 10.1016/j.jss.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 12.Kunnumakkara AB, Anand P, Aggarwal BB. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008;269:199–225. doi: 10.1016/j.canlet.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 13.Shishodia S, Singh T, Chaturvedi MM. Modulation of transcription factors by curcumin. Adv Exp Med Biol. 2007;595:127–48. doi: 10.1007/978-0-387-46401-5_4. [DOI] [PubMed] [Google Scholar]
- 14.Aggarwal S, Takada Y, Singh S, Myers JN, Aggarwal BB. Inhibition of growth and survival of human head and neck squamous cell carcinoma cells by curcumin via modulation of nuclear factor-kappaB signaling. Int J Cancer. 2004;111:679–92. doi: 10.1002/ijc.20333. [DOI] [PubMed] [Google Scholar]
- 15.Choi H, Chun YS, Shin YJ, et al. Curcumin attenuates cytochrome P450 induction in response to 2,3,7,8-tetrachlorodibenzo-p-dioxin by ROS-dependently degrading AhR and ARNT. Cancer Sci. 2008;99:2518–24. doi: 10.1111/j.1349-7006.2008.00984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Padhye S, Chavan D, Pandey S, et al. Perspectives on chemopreventive and therapeutic potential of curcumin analogs in medicinal chemistry. Mini Rev Med Chem. 2010;10:372–87. doi: 10.2174/138955710791330891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milobedeska J, Kostanecki V, Lampe V. Structure of curcumin. Ber Dtsch Chem Ges. 1910;43:2163–70. [Google Scholar]
- 18.Lampe V, Milobedeska J. Studien uber curcumin. Ber Dtsch Chem Ges. 1913;46:2235–40. [Google Scholar]
- 19.Tonnesen HH, Karlsen J. Studies on curcumin and curcuminoids. VI. Kinetics of curcumin degradation in aqueous solution. Z Lebensm Unters Forsch. 1985;180:402–4. doi: 10.1007/BF01027775. [DOI] [PubMed] [Google Scholar]
- 20.Wang YJ, Pan MH, Cheng AL, et al. Stability of curcumin in buffer solutions and characterization of its degradation products. J Pharm Biomed Anal. 1997;15:1867–76. doi: 10.1016/s0731-7085(96)02024-9. [DOI] [PubMed] [Google Scholar]
- 21.Bernabe-Pineda M, Ramirez-Silva MT, Romero-Romo M, Gonzalez-Vergara E, Rojas-Hernandez A. Determination of acidity constants of curcumin in aqueous solution and apparent rate constant of its decomposition. Spectrochim Acta A Mol Biomol Spectrosc. 2004;60:1091–7. doi: 10.1016/S1386-1425(03)00342-1. [DOI] [PubMed] [Google Scholar]
- 22.Ireson CR, Jones DJ, Orr S, et al. Metabolism of the cancer chemopreventive agent curcumin in human and rat intestine. Cancer Epidemiol Biomarkers Prev. 2002;11:105–11. [PubMed] [Google Scholar]
- 23.Holder GM, Plummer JL, Ryan AJ. The metabolism and excretion of curcumin (1,7-bis-(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione) in the rat. Xenobiotica. 1978;8:761–8. doi: 10.3109/00498257809069589. [DOI] [PubMed] [Google Scholar]
- 24.Padhye S, Banerjee S, Chavan D, et al. Fluorocurcumins as cyclooxygenase-2 inhibitor: molecular docking, pharmacokinetics and tissue distribution in mice. Pharm Res. 2009;26:2438–45. doi: 10.1007/s11095-009-9955-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z, Leung MH, Kee TW, English DS. The role of charge in the surfactant-assisted stabilization of the natural product curcumin. Langmuir. 2010;26:5520–6. doi: 10.1021/la903772e. [DOI] [PubMed] [Google Scholar]
- 26.Setthacheewakul S, Mahattanadul S, Phadoongsombut N, Pichayakorn W, Wiwattanapatapee R. Development and evaluation of self-microemulsifying liquid and pellet formulations of curcumin, and absorption studies in rats. Eur J Pharm Biopharm. 2010;76:475–85. doi: 10.1016/j.ejpb.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 27.Gao Y, Li Z, Sun M, et al. Preparation, characterization, pharmacokinetics, and tissue distribution of curcumin nanosuspension with TPGS as stabilizer. Drug Dev Ind Pharm. 2010;36:1225–34. doi: 10.3109/03639041003695139. [DOI] [PubMed] [Google Scholar]
- 28.Pandey MK, Kumar S, Thimmulappa RK, et al. Design, synthesis and evaluation of novel PEGylated curcumin analogs as potent Nrf2 activators in human bronchial epithelial cells. Eur J Pharm Sci. 2011;43:16–24. doi: 10.1016/j.ejps.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 29.Tang H, Murphy CJ, Zhang B, et al. Curcumin polymers as anti-cancer conjugates. Biomaterials. 2010;31:7139–49. doi: 10.1016/j.biomaterials.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Ganta S, Devalapally H, Amiji M. Curcumin enhances oral bioavailability and anti-tumor therapeutic efficacy of paclitaxel upon administration in nanoemulsion formulation. J Pharm Sci. 2010;99:4630–41. doi: 10.1002/jps.22157. [DOI] [PubMed] [Google Scholar]
- 31.Li L, Braiteh FS, Kurzrock R. Liposome-encapsulated curcumin: in vitro and in vivo effects on proliferation, apoptosis, signaling, and angiogenesis. Cancer. 2005;104:1322–31. doi: 10.1002/cncr.21300. [DOI] [PubMed] [Google Scholar]
- 32.Anitha A, Deepagan VG, Divya Rani VV, et al. Preparation, characterization, in vitro drug release and biological studies of curcumin loaded dextran sulphate-chitosan nanoparticles. Carbohydrate Polymers. 2011;84:1158–64. [Google Scholar]
- 33.Tonnesen HH. Studies on curcumin and curcuminoids. XVI. Effect of curcumin analogs on hyaluronic acid degradation in vitro. International Journal of Pharmaceutics. 1989;51:259–61. [Google Scholar]
- 34.Tomren MA, Masson M, Loftsson T, Tonnesen HH. Studies on curcumin and curcuminoids: XXXI. Symmetric and asymmetric curcuminoids: Stability, activity and complexation with cyclodextrin. International Journal of Pharmaceutics. 2007;338:27–34. doi: 10.1016/j.ijpharm.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 35.Yadav VR, Prasad S, Kannappan R, et al. Cyclodextrin-complexed curcumin exhibits anti-inflammatory and antiproliferative activities superior to those of curcumin through higher cellular uptake. Biochem Pharmacol. 2010;80:1021–32. doi: 10.1016/j.bcp.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Yallapu MM, Jaggi M, Chauhan SC. Poly(beta-cyclodextrin)/curcumin self-assembly: a novel approach to improve curcumin delivery and its therapeutic efficacy in prostate cancer cells. Macromol Biosci. 2010;10:1141–51. doi: 10.1002/mabi.201000084. [DOI] [PubMed] [Google Scholar]
- 37.Dandawate PR, Vyas A, Ahmad A, et al. Inclusion Complex of Novel Curcumin Analogue CDF and beta-Cyclodextrin (1: 2) and Its Enhanced In vivo Anticancer Activity Against Pancreatic Cancer. Pharm Res. 2012 doi: 10.1007/s11095-012-0700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aggarwal S, Ndinguri MW, Solipuram R, et al. [DLys(6)]-luteinizing hormone releasing hormone-curcumin conjugate inhibits pancreatic cancer cell growth in vitro and in vivo. Int J Cancer. 2011;129:1611–23. doi: 10.1002/ijc.26132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Al-Hujaily EM, Mohamed AG, Al-Sharif I, et al. PAC, a novel curcumin analogue, has anti-breast cancer properties with higher efficiency on ER-negative cells. Breast Cancer Res Treat. 2011;128:97–107. doi: 10.1007/s10549-010-1089-3. [DOI] [PubMed] [Google Scholar]
- 40.Al-Wabli R, AboulWafa O, Youssef KM. Synthesis of curcumin and ethylcurcumin bioconjugates as potential antitumor agents. Medicinal Chemistry Research. 2012;21:874–90. [Google Scholar]
- 41.Devasena T, Rajasekaran KN, Menon VP. Bis-1,7-(2-hydroxyphenyl)-hepta-1,6-diene-3,5-dione (a curcumin analog) ameliorates DMH-induced hepatic oxidative stress during colon carcinogenesis. Pharmacol Res. 2002;46:39–45. doi: 10.1016/s1043-6618(02)00043-9. [DOI] [PubMed] [Google Scholar]
- 42.Devasena T, Menon VP, Rajasekharan KN. Prevention of 1,2-dimethylhydrazine-induced circulatory oxidative stress by bis-1,7-(2-hydroxyphenyl)-hepta-1,6-diene-3,5-dione during colon carcinogenesis. Pharmacol Rep. 2006;58:229–35. [PubMed] [Google Scholar]
- 43.Arafa HM. Possible contribution of beta-glycosidases and caspases in the cytotoxicity of novel glycoconjugates in colon cancer cells. Invest New Drugs. 2010;28:306–17. doi: 10.1007/s10637-009-9248-2. [DOI] [PubMed] [Google Scholar]
- 44.Dubey SK, Sharma AK, Narain U, Misra K, Pati U. Design, synthesis and characterization of some bioactive conjugates of curcumin with glycine, glutamic acid, valine and demethylenated piperic acid and study of their antimicrobial and antiproliferative properties. Eur J Med Chem. 2008;43:1837–46. doi: 10.1016/j.ejmech.2007.11.027. [DOI] [PubMed] [Google Scholar]
- 45.Ferrari E, Lazzari S, Marverti G, et al. Synthesis, cytotoxic and combined cDDP activity of new stable curcumin derivatives. Bioorg Med Chem. 2009;17:3043–52. doi: 10.1016/j.bmc.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 46.Fuchs JR, Pandit B, Bhasin D, et al. Structure-activity relationship studies of curcumin analogues. Bioorg Med Chem Lett. 2009;19:2065–9. doi: 10.1016/j.bmcl.2009.01.104. [DOI] [PubMed] [Google Scholar]
- 47.Nakagawa-Goto K, Yamada K, Nakamura S, et al. Antitumor agents. 258. Syntheses and evaluation of dietary antioxidant--taxoid conjugates as novel cytotoxic agents. Bioorg Med Chem Lett. 2007;17:5204–9. doi: 10.1016/j.bmcl.2007.06.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han YM, Shin DS, Lee YJ, et al. 2-Hydroxycurcuminoid induces apoptosis of human tumor cells through the reactive oxygen species-mitochondria pathway. Bioorg Med Chem Lett. 2011;21:747–51. doi: 10.1016/j.bmcl.2010.11.114. [DOI] [PubMed] [Google Scholar]
- 49.Handler N, Jaeger W, Puschacher H, Leisser K, Erker T. Synthesis of novel curcumin analogues and their evaluation as selective cyclooxygenase-1 (COX-1) inhibitors. Chem Pharm Bull (Tokyo) 2007;55:64–71. doi: 10.1248/cpb.55.64. [DOI] [PubMed] [Google Scholar]
- 50.Jankun J, Aleem AM, Malgorzewicz S, et al. Synthetic curcuminoids modulate the arachidonic acid metabolism of human platelet 12-lipoxygenase and reduce sprout formation of human endothelial cells. Mol Cancer Ther. 2006;5:1371–82. doi: 10.1158/1535-7163.MCT-06-0021. [DOI] [PubMed] [Google Scholar]
- 51.Kapoor N, Sharma AK, Dwivedi V, et al. Telomerase targeted anticancer bioactive prodrug by antisense-based approach. Cancer Lett. 2007;248:245–50. doi: 10.1016/j.canlet.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 52.Khan MA, El-Khatib R, Rainsford KD, Whitehouse MW. Synthesis and anti-inflammatory properties of some aromatic and heterocyclic aromatic curcuminoids. Bioorg Chem. 2012;40:30–8. doi: 10.1016/j.bioorg.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 53.Li J, Wang Y, Yang C, et al. Polyethylene glycosylated curcumin conjugate inhibits pancreatic cancer cell growth through inactivation of Jab1. Mol Pharmacol. 2009;76:81–90. doi: 10.1124/mol.109.054551. [DOI] [PubMed] [Google Scholar]
- 54.Manju S, Sreenivasan K. Synthesis and characterization of a cytotoxic cationic polyvinylpyrrolidone-curcumin conjugate. J Pharm Sci. 2011;100:504–11. doi: 10.1002/jps.22278. [DOI] [PubMed] [Google Scholar]
- 55.Mishra S, Kapoor N, Mubarak AA, et al. Differential apoptotic and redox regulatory activities of curcumin and its derivatives. Free Radic Biol Med. 2005;38:1353–60. doi: 10.1016/j.freeradbiomed.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 56.Qiu X, Liu Z, Shao WY, et al. Synthesis and evaluation of curcumin analogues as potential thioredoxin reductase inhibitors. Bioorg Med Chem. 2008;16:8035–41. doi: 10.1016/j.bmc.2008.07.054. [DOI] [PubMed] [Google Scholar]
- 57.Safavy A, Raisch KP, Mantena S, et al. Design and development of water-soluble curcumin conjugates as potential anticancer agents. J Med Chem. 2007;50:6284–8. doi: 10.1021/jm700988f. [DOI] [PubMed] [Google Scholar]
- 58.Tamvakopoulos C, Dimas K, Sofianos ZD, et al. Metabolism and anticancer activity of the curcumin analogue, dimethoxycurcumin. Clin Cancer Res. 2007;13:1269–77. doi: 10.1158/1078-0432.CCR-06-1839. [DOI] [PubMed] [Google Scholar]
- 59.Wichitnithad W, Nimmannit U, Wacharasindhu S, Rojsitthisak P. Synthesis, characterization and biological evaluation of succinate prodrugs of curcuminoids for colon cancer treatment. Molecules. 2011;16:1888–900. doi: 10.3390/molecules16021888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Youssef D, Nichols CE, Cameron TS, et al. Design, synthesis, and cytostatic activity of novel cyclic curcumin analogues. Bioorg Med Chem Lett. 2007;17:5624–9. doi: 10.1016/j.bmcl.2007.07.079. [DOI] [PubMed] [Google Scholar]
- 61.Vyas HK, Pal R, Vishwakarma R, Lohiya NK, Talwar GP. Selective killing of leukemia and lymphoma cells ectopically expressing hCGbeta by a conjugate of curcumin with an antibody against hCGbeta subunit. Oncology. 2009;76:101–11. doi: 10.1159/000188665. [DOI] [PubMed] [Google Scholar]
- 62.Yodkeeree S, Ampasavate C, Sung B, Aggarwal BB, Limtrakul P. Demethoxycurcumin suppresses migration and invasion of MDA-MB-231 human breast cancer cell line. Eur J Pharmacol. 2010;627:8–15. doi: 10.1016/j.ejphar.2009.09.052. [DOI] [PubMed] [Google Scholar]
- 63.Ryu MJ, Cho M, Song JY, et al. Natural derivatives of curcumin attenuate the Wnt/beta-catenin pathway through down-regulation of the transcriptional coactivator p300. Biochem Biophys Res Commun. 2008;377:1304–8. doi: 10.1016/j.bbrc.2008.10.171. [DOI] [PubMed] [Google Scholar]
- 64.Das J, Pany S, Panchal S, Majhi A, Rahman GM. Binding of isoxazole and pyrazole derivatives of curcumin with the activator binding domain of novel protein kinase C. Bioorg Med Chem. 2011;19:6196–202. doi: 10.1016/j.bmc.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 65.Labbozzetta M, Baruchello R, Marchetti P, et al. Lack of nucleophilic addition in the isoxazole and pyrazole diketone modified analogs of curcumin; implications for their antitumor and chemosensitizing activities. Chem Biol Interact. 2009;181:29–36. doi: 10.1016/j.cbi.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 66.Poma P, Notarbartolo M, Labbozzetta M, et al. The antitumor activities of curcumin and of its isoxazole analogue are not affected by multiple gene expression changes in an MDR model of the MCF-7 breast cancer cell line: analysis of the possible molecular basis. Int J Mol Med. 2007;20:329–35. [PubMed] [Google Scholar]
- 67.Sahoo BK, Ghosh KS, Dasgupta S. Molecular interactions of isoxazolcurcumin with human serum albumin: spectroscopic and molecular modeling studies. Biopolymers. 2009;91:108–19. doi: 10.1002/bip.21092. [DOI] [PubMed] [Google Scholar]
- 68.Dutta S, Padhye S, Priyadarsini KI, Newton C. Antioxidant and antiproliferative activity of curcumin semicarbazone. Bioorg Med Chem Lett. 2005;15:2738–44. doi: 10.1016/j.bmcl.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 69.Lal J, Gupta SK, Thavaselvam D, Agarwal DD. Design, synthesis, synergistic antimicrobial activity and cytotoxicity of 4-aryl substituted 3,4-dihydropyrimidinones of curcumin. Bioorg Med Chem Lett. 2012;22:2872–6. doi: 10.1016/j.bmcl.2012.02.056. [DOI] [PubMed] [Google Scholar]
- 70.Shim JS, Kim DH, Jung HJ, et al. Hydrazinocurcumin, a novel synthetic curcumin derivative, is a potent inhibitor of endothelial cell proliferation. Bioorg Med Chem. 2002;10:2987–92. doi: 10.1016/s0968-0896(02)00129-3. [DOI] [PubMed] [Google Scholar]
- 71.Shim JS, Lee J, Park HJ, Park SJ, Kwon HJ. A new curcumin derivative, HBC, interferes with the cell cycle progression of colon cancer cells via antagonization of the Ca2+/calmodulin function. Chem Biol. 2004;11:1455–63. doi: 10.1016/j.chembiol.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 72.Simoni D, Rizzi M, Rondanin R, et al. Antitumor effects of curcumin and structurally beta-diketone modified analogs on multidrug resistant cancer cells. Bioorg Med Chem Lett. 2008;18:845–9. doi: 10.1016/j.bmcl.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 73.Swarts SG, Zhang M, Yin L, et al. Antioxidant properties of select radiation mitigators based on semicarbazone and pyrazole derivatives of curcumin. Adv Exp Med Biol. 2011;701:291–7. doi: 10.1007/978-1-4419-7756-4_39. [DOI] [PubMed] [Google Scholar]
- 74.Arif M, Vedamurthy BM, Choudhari R, et al. Nitric oxide-mediated histone hyperacetylation in oral cancer: target for a water-soluble HAT inhibitor, CTK7A. Chem Biol. 2010;17:903–13. doi: 10.1016/j.chembiol.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 75.Chun KS, Sohn Y, Kim HS, et al. Anti-tumor promoting potential of naturally occurring diarylheptanoids structurally related to curcumin. Mutat Res. 1999;428:49–57. doi: 10.1016/s1383-5742(99)00031-9. [DOI] [PubMed] [Google Scholar]
- 76.Wu JC, Lai CS, Badmaev V, et al. Tetrahydrocurcumin, a major metabolite of curcumin, induced autophagic cell death through coordinative modulation of PI3K/Akt-mTOR and MAPK signaling pathways in human leukemia HL-60 cells. Mol Nutr Food Res. 2011;55:1646–54. doi: 10.1002/mnfr.201100454. [DOI] [PubMed] [Google Scholar]
- 77.Yodkeeree S, Garbisa S, Limtrakul P. Tetrahydrocurcumin inhibits HT1080 cell migration and invasion via downregulation of MMPs and uPA. Acta Pharmacol Sin. 2008;29:853–60. doi: 10.1111/j.1745-7254.2008.00792.x. [DOI] [PubMed] [Google Scholar]
- 78.Yoysungnoen P, Wirachwong P, Changtam C, Suksamrarn A, Patumraj S. Anti-cancer and anti-angiogenic effects of curcumin and tetrahydrocurcumin on implanted hepatocellular carcinoma in nude mice. World J Gastroenterol. 2008;14:2003–9. doi: 10.3748/wjg.14.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen CY, Yang WL, Kuo SY. Cytotoxic activity and cell cycle analysis of hexahydrocurcumin on SW 480 human colorectal cancer cells. Nat Prod Commun. 2011;6:1671–2. [PubMed] [Google Scholar]
- 80.Ohtsu H, Xiao Z, Ishida J, et al. Antitumor agents. 217. Curcumin analogues as novel androgen receptor antagonists with potential as anti-prostate cancer agents. J Med Chem. 2002;45:5037–42. doi: 10.1021/jm020200g. [DOI] [PubMed] [Google Scholar]
- 81.Lin L, Shi Q, Su CY, Shih CC, Lee KH. Antitumor agents 247. New 4-ethoxycarbonylethyl curcumin analogs as potential antiandrogenic agents. Bioorg Med Chem. 2006;14:2527–34. doi: 10.1016/j.bmc.2005.11.034. [DOI] [PubMed] [Google Scholar]
- 82.Lin L, Shi Q, Nyarko AK, et al. Antitumor agents. 250. Design and synthesis of new curcumin analogues as potential anti-prostate cancer agents. J Med Chem. 2006;49:3963–72. doi: 10.1021/jm051043z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Amolins MW, Peterson LB, Blagg BS. Synthesis and evaluation of electron-rich curcumin analogues. Bioorg Med Chem. 2009;17:360–7. doi: 10.1016/j.bmc.2008.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Arezki A, Chabot GG, Quentin L, et al. Synthesis and biological evaluation of novel ferrocenyl curcuminoid derivatives. Med Chem Commun. 2011;2:190–5. doi: 10.1039/c0md00231c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lin L, Hutzen B, Zuo M, et al. Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer Res. 2010;70:2445–54. doi: 10.1158/0008-5472.CAN-09-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin L, Fuchs J, Li C, et al. STAT3 signaling pathway is necessary for cell survival and tumorsphere forming capacity in ALDH/CD133 stem cell-like human colon cancer cells. Biochem Biophys Res Commun. 2011;416:246–51. doi: 10.1016/j.bbrc.2011.10.112. [DOI] [PubMed] [Google Scholar]
- 87.Bill MA, Fuchs JR, Li C, et al. The small molecule curcumin analog FLLL32 induces apoptosis in melanoma cells via STAT3 inhibition and retains the cellular response to cytokines with anti-tumor activity. Mol Cancer. 2010;9:165. doi: 10.1186/1476-4598-9-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chakraborti S, Das L, Kapoor N, et al. Curcumin recognizes a unique binding site of tubulin. J Med Chem. 2011;54:6183–96. doi: 10.1021/jm2004046. [DOI] [PubMed] [Google Scholar]
- 89.Chen C, Liu Y, Chen Y, Xu J. C086, a novel analog of curcumin, induces growth inhibition and down-regulation of NFkappaB in colon cancer cells and xenograft tumors. Cancer Biol Ther. 2011;12:797–807. doi: 10.4161/cbt.12.9.17671. [DOI] [PubMed] [Google Scholar]
- 90.Fadda A, Badria F, El-Attar K. Synthesis and evaluation of curcumin analogues as cytotoxic agents. Medicinal Chemistry Research. 2010;19:413–30. [Google Scholar]
- 91.Ferrari E, Pignedoli F, Imbriano C, et al. Newly synthesized curcumin derivatives: crosstalk between chemico-physical properties and biological activity. J Med Chem. 2011;54:8066–77. doi: 10.1021/jm200872q. [DOI] [PubMed] [Google Scholar]
- 92.Qiu X, Du Y, Lou B, et al. Synthesis and Identification of New 4-Arylidene Curcumin Analogues as Potential Anticancer Agents Targeting Nuclear Factor-kappaB Signaling Pathway. J Med Chem. 2010 doi: 10.1021/jm1004545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sertel S, Eichhorn T, Bauer J, et al. Pharmacogenomic determination of genes associated with sensitivity or resistance of tumor cells to curcumin and curcumin derivatives. J Nutr Biochem. 2011 doi: 10.1016/j.jnutbio.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 94.Zhou J, Geng G, Shi Q, Sauriol F, Wu JH. Design and synthesis of androgen receptor antagonists with bulky side chains for overcoming antiandrogen resistance. J Med Chem. 2009;52:5546–50. doi: 10.1021/jm801218k. [DOI] [PubMed] [Google Scholar]
- 95.Zambre AP, Kulkarni VM, Padhye S, Sandur SK, Aggarwal BB. Novel curcumin analogs targeting TNF-induced NF-kappaB activation and proliferation in human leukemic KBM-5 cells. Bioorg Med Chem. 2006;14:7196–204. doi: 10.1016/j.bmc.2006.06.056. [DOI] [PubMed] [Google Scholar]
- 96.Padhye S, Yang H, Jamadar A, et al. New difluoro Knoevenagel condensates of curcumin, their Schiff bases and copper complexes as proteasome inhibitors and apoptosis inducers in cancer cells. Pharm Res. 2009;26:1874–80. doi: 10.1007/s11095-009-9900-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ali S, Ahmad A, Banerjee S, et al. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res. 2010;70:3606–17. doi: 10.1158/0008-5472.CAN-09-4598. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 98.Bao B, Ali S, Kong D, et al. Anti-tumor activity of a novel compound-CDF is mediated by regulating miR-21, miR-200, and PTEN in pancreatic cancer. PLoS One. 2011;6:e17850. doi: 10.1371/journal.pone.0017850. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 99.Kanwar SS, Yu Y, Nautiyal J, et al. Difluorinated-curcumin (CDF): a novel curcumin analog is a potent inhibitor of colon cancer stem-like cells. Pharm Res. 2011;28:827–38. doi: 10.1007/s11095-010-0336-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ali S, Ahmad A, Aboukameel A, et al. Increased Ras GTPase activity is regulated by miRNAs that can be attenuated by CDF treatment in pancreatic cancer cells. Cancer Lett. 2012;319:173–81. doi: 10.1016/j.canlet.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 101.Li Y, Kong D, Wang Z, et al. Inactivation of AR/TMPRSS2-ERG/Wnt signaling networks attenuates the aggressive behavior of prostate cancer cells. Cancer Prev Res (Phila) 2011;4:1495–506. doi: 10.1158/1940-6207.CAPR-11-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bertini I, Gray HB, Lippard SJ, Valentine JS. Bioinorganic chemistry. New Delhi: Viva Books Pvt Ltd; 1998. [Google Scholar]
- 103.Gielen M, Tiekink E. Metallotherapeutic drugs and metal-based diagnostic agents: the use of metals in medicine. Wiley Publications; 2005. [Google Scholar]
- 104.Jiao Y, Wilkinson J, Di X, et al. Curcumin, a cancer chemopreventive and chemotherapeutic agent, is a biologically active iron chelator. Blood. 2009;113:462–9. doi: 10.1182/blood-2008-05-155952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Aliaga-Alcalde N, Marques-Gallego P, Kraaijkamp M, et al. Copper curcuminoids containing anthracene groups: fluorescent molecules with cytotoxic activity. Inorg Chem. 2010;49:9655–63. doi: 10.1021/ic101331c. [DOI] [PubMed] [Google Scholar]
- 106.Annaraj J, Srinivasan S, Ponvel KM, Athappan P. Mixed ligand copper(II) complexes of phenanthroline/bipyridyl and curcumin diketimines as DNA intercalators and their electrochemical behavior under Nafion and clay modified electrodes. J Inorg Biochem. 2005;99:669–76. doi: 10.1016/j.jinorgbio.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 107.Barik A, Mishra B, Shen L, et al. Evaluation of a new copper(II)-curcumin complex as superoxide dismutase mimic and its free radical reactions. Free Radic Biol Med. 2005;39:811–22. doi: 10.1016/j.freeradbiomed.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 108.Caruso F, Rossi M, Benson A, et al. Ruthenium-arene complexes of curcumin: X-ray and density functional theory structure, synthesis, and spectroscopic characterization, in vitro antitumor activity, and DNA docking studies of (p-cymene)Ru(curcuminato)chloro. J Med Chem. 2012;55:1072–81. doi: 10.1021/jm200912j. [DOI] [PubMed] [Google Scholar]
- 109.John VD, Krishnankutty K. Antitumour activity of synthetic curcuminoid analogues (1,7-diaryl-1,6-heptadiene-3,5-diones) and their copper complexes. Appl Organometal Chem. 2006;20:477–82. [Google Scholar]
- 110.Valentini A, Conforti F, Crispini A, et al. Synthesis, oxidant properties, and antitumoral effects of a heteroleptic palladium(II) complex of curcumin on human prostate cancer cells. J Med Chem. 2009;52:484–91. doi: 10.1021/jm801276a. [DOI] [PubMed] [Google Scholar]
- 111.Ruan BF, Lu X, Li TT, et al. Synthesis, biological evaluation and molecular docking studies of resveratrol derivatives possessing curcumin moiety as potent antitubulin agents. Bioorg Med Chem. 2012;20:1113–21. doi: 10.1016/j.bmc.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 112.Cao B, Wang Y, Ding K, Neamati N, Long YQ. Synthesis of the pyridinyl analogues of dibenzylideneacetone (pyr-dba) via an improved Claisen-Schmidt condensation, displaying diverse biological activities as curcumin analogues. Org Biomol Chem. 2012;10:1239–45. doi: 10.1039/c1ob06773g. [DOI] [PubMed] [Google Scholar]
- 113.Somers-Edgar TJ, Taurin S, Larsen L, et al. Mechanisms for the activity of heterocyclic cyclohexanone curcumin derivatives in estrogen receptor negative human breast cancer cell lines. Invest New Drugs. 2011;29:87–97. doi: 10.1007/s10637-009-9339-0. [DOI] [PubMed] [Google Scholar]
- 114.Kudo C, Yamakoshi H, Sato A, et al. Synthesis of 86 species of 1,5-diaryl-3-oxo-1,4-pentadienes analogs of curcumin can yield a good lead in vivo. BMC Pharmacol. 2011;11:4. doi: 10.1186/1471-2210-11-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kalai T, Kuppusamy ML, Balog M, et al. Synthesis of N-substituted 3,5-bis(arylidene)-4-piperidones with high antitumor and antioxidant activity. J Med Chem. 2011;54:5414–21. doi: 10.1021/jm200353f. [DOI] [PubMed] [Google Scholar]
- 116.Karthikeyan NS, Sathiyanarayanan KI, Aravindan PG, Giridharan P. Synthesis, crystal structure, and anticancer properties of cyclic monocarbonyl analogs of curcumin. Medicinal Chemistry Research. 2011;20:81–7. [Google Scholar]
- 117.Dayton A, Selvendiran K, Kuppusamy ML, et al. Cellular uptake, retention and bioabsorption of HO-3867, a fluorinated curcumin analog with potential antitumor properties. Cancer Biol Ther. 2010;10:1027–32. doi: 10.4161/cbt.10.10.13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lin L, Hutzen B, Ball S, et al. New curcumin analogues exhibit enhanced growth-suppressive activity and inhibit AKT and signal transducer and activator of transcription 3 phosphorylation in breast and prostate cancer cells. Cancer Sci. 2009;100:1719–27. doi: 10.1111/j.1349-7006.2009.01220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yamakoshi H, Ohori H, Kudo C, et al. Structure-activity relationship of C5-curcuminoids and synthesis of their molecular probes thereof. Bioorg Med Chem. 2010;18:1083–92. doi: 10.1016/j.bmc.2009.12.045. [DOI] [PubMed] [Google Scholar]
- 120.Liang G, Shao L, Wang Y, et al. Exploration and synthesis of curcumin analogues with improved structural stability both in vitro and in vivo as cytotoxic agents. Bioorg Med Chem. 2009;17:2623–31. doi: 10.1016/j.bmc.2008.10.044. [DOI] [PubMed] [Google Scholar]
- 121.Ohori H, Yamakoshi H, Tomizawa M, et al. Synthesis and biological analysis of new curcumin analogues bearing an enhanced potential for the medicinal treatment of cancer. Mol Cancer Ther. 2006;5:2563–71. doi: 10.1158/1535-7163.MCT-06-0174. [DOI] [PubMed] [Google Scholar]
- 122.Ravindran J, Subbaraju GV, Ramani MV, Sung B, Aggarwal BB. Bisdemethylcurcumin and structurally related hispolon analogues of curcumin exhibit enhanced prooxidant, anti-proliferative and anti-inflammatory activities in vitro. Biochem Pharmacol. 2010;79:1658–66. doi: 10.1016/j.bcp.2010.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Youssef KM, El-Sherbeny MA. Synthesis and antitumor activity of some curcumin analogs. Arch Pharm (Weinheim) 2005;338:181–9. doi: 10.1002/ardp.200400939. [DOI] [PubMed] [Google Scholar]