

Abstract
With the ability to fully sequence tumor genomes/exomes, the quest for cancer driver genes can now be undertaken in an unbiased manner. However, obtaining a complete catalog of cancer genes is difficult due to the heterogeneous molecular nature of the disease and the limitations of available computational methods. Here we show that the combination of complementary methods allows identifying a comprehensive and reliable list of cancer driver genes. We provide a list of 291 high-confidence cancer driver genes acting on 3,205 tumors from 12 different cancer types. Among those genes, some have not been previously identified as cancer drivers and 16 have clear preference to sustain mutations in one specific tumor type. The novel driver candidates complement our current picture of the emergence of these diseases. In summary, the catalog of driver genes and the methodology presented here open new avenues to better understand the mechanisms of tumorigenesis.
The identification of the genes that drive carcinogenesis has been regarded in the past 35 years as the first step to understand the mechanisms of tumor emergence and evolution. Since the identification of the first somatic mutation in a human cancer gene – G12V in HRAS in a human bladder carcinoma cell line1,2 – almost 500 cancer genes have been identified and are now included in the Cancer Gene Census (CGC)3. More recently, fueled by Next Generation Sequencing technologies, large international consortia, like the TCGA and the ICGC have undertaken whole exome sequencing of thousands of tumor samples. These initiatives share the explicit goal of detecting all genes and molecular mechanisms underlying tumorigenesis in every major cancer type4,5.
Tumor genomes contain from tens to thousands of somatic mutations. However, only a few of them “drive” tumorigenesis by affecting genes –drivers– which upon alteration confer selective growth advantage to tumor cells6,7,8,9. While only few driver genes are frequently mutated in cancer, many others are altered in a small fraction of tumors. Due to these lowly recurrent drivers and to the underlying molecular heterogeneity of cancer, large number of tumor samples must be sequenced –and the results analyzed employing bioinformatics methods– to thoroughly detect driver genes in the quest to fully understand the mechanisms of tumorigenesis. Bioinformatics analyses of exome sequence data from large cohorts of tumor samples produced by these projects are not trivial. Current approaches are based on identifying genes that exhibit signals of positive selection across a cohort of tumor samples, all showing particular shortcomings and specific biases9.
Most common methods identify genes that are mutated more frequently than expected from the background mutation rate (recurrence)10,11. Their biggest challenge is to correctly estimate this background rate to keep the number of false positives to a minimum9,11. Nevertheless, driver genes mutated at very low frequency are still difficult to detect with this approach. Other methods attempt to identify genes that exhibit other signals of positive selection across tumor samples, such as a high rate of non-silent mutations compared to silent mutations16,17, or a bias towards the accumulation of functional mutations (FM bias)12. One advantage of this latest approach is its independent of the background mutation rate, although its performance could be affected by drawbacks of the metrics used to score the putative impact of somatic mutations on protein function13,14,15. Some metrics, for instance, underestimate functional changes in poorly conserved positions46. Still, other methods exploit the tendency to sustain mutations in certain regions of the protein sequence (CLUST bias)18, based on the knowledge that whereas inactivating mutations are distributed along the sequence of the protein, gain-of-function mutations tend to occur specifically in particular residues or domains18. Finally, other approaches exploit the overrepresentation of mutations in specific functional residues, such as phosphorylation sites (ACTIVE bias)19. Intuitively, different types of driver genes will exhibit the signals of positive selection exploited by these approaches in varying degrees. For example, mutations are known to cluster in specific residues in oncogenes more strongly than in tumor suppressors. Therefore, one should expect that different subsets of candidate drivers will rank at the top of lists of driver candidates identified by each method. Moreover, the implementation of each method will probably influence its results. For example, frequency-based methods with looser background mutation rates will detect longer lists of driver candidates probably with a high rate of false positives. On the other hand, methods implementing stricter models will identify shorter, more specific lists but might miss some true cancer driver genes.
Here, we describe the analysis of somatic mutations obtained via exome sequencing of 3,205 tumor from 12 tumor types by the Cancer Genome Atlas (TCGA) research network 47 (Supplementary Table 1). This analysis results in the comprehensive detection of the mutational cancer driver genes acting in these tumors. To this aim, we employed five complementary methods that search for genes showing the signals of positive selection described in the previous paragraph. We combined the lists of driver candidates identified by these five methods both across the whole pan-cancer dataset and in each individual tumor type using a two-step rule-based approach. First, gene lists from four methods (MuSiC, OncodriveFM, OncodriveCLUST and ActiveDriver) each one considering a different of positive selection signal are intersected looking for genes exhibiting several signals of positive selection (see Results for details), thus composing a list of high-confidence drivers. Second, MutSig significantly mutated genes, which are probed for three signals of positive selection are incorporated to the list of high-confidence drivers.
We demonstrate that the combination of approaches based on complementary signals of positive selection outperforms the use of individual methods. As a result, the use of this novel approach provides a comprehensive and reliable list of mutational drivers acting across 12 tumor types.
Results
We applied methods based on the aforementioned approaches to detect signals of positive selection (MuSiC10, OncodriveFM12, OncodriveCLUST18 and ActiveDriver19) to the unified analysis of all tumors (see methods) (Fig. 1a and b). To evaluate the quality of the lists of driver candidates produced by each method, and the combinations thereof, we computed their content of known cancer genes. To that end, we employed the Cancer Gene Census, CGC3 as the most reliable catalog of known cancer genes to date. Nevertheless, due to its biased nature and the fact that arguably, many cancer genes are yet to be uncovered, we consider the rate of CGC genes in each list simply as a surrogate estimator of the actual positive predictive value of each method or combination (see Discussion). Applying this principle, we found that the four methods prioritized lists of genes highly enriched for known cancer drivers. Moreover, increasing the cutoff of statistical significance increased the proportion of known cancer genes retrieved (Fig. 2a). This proportion was higher among genes exhibiting more than one signal of positive selection. In other words, the likelihood that a gene is involved in tumorigenesis increased proportionally with the number of methods that identified it (Fig. 2b and c), probably because the false positives of one method are likely to be discarded by the others. For example, only 84 out of 232 recurrently mutated genes (MuSiC) –or 87 out of 259 FM biased genes– are also identified by other methods (Fig. 2b). However, the proportion of genes in the CGC rises from 22% and 25%, respectively to 54%. On the other hand, genes missed by one method may be identified by others designed to detect other signals of positive selection, as exemplified in Figure 1c. For instance, while RB1 possesses both clear recurrence and FM bias, it has undetectable CLUST or ACTIVE biases. Mutations in HRAS are both significantly clustered and biased towards high functional impact, but are neither significantly recurrent nor ACTIVE biased. BRAF, on the other hand shows all signals of positive selection, except FM bias.
Figure 1.

(A) Illustration of the four signals of positive selection used to identify driver genes and the methods that implement them. (B) Venn diagram showing the contribution of each method in number of genes that it detects to the list of HCDs. The names of the genes detected by 3 or more methods are shown. (C) Mutational patterns of 4 HCDs. Circles represent protein affecting mutation across pan-cancer samples, and are colored according to their functional impact calculated by the Mutation Assessor method. Triangles indicate active residues of the protein in which mutation occurs. Protein domains retrieved from Pfam are depicted. Label boxes indicate which of the methods identifies the gene as significant.
Figure 2. Validation of methods' output lists of driver candidates and the approach taken to combine them.

(A) Proportion of genes included in the Cancer Gene Census (CGC) depending on the number of top-ranking genes from the list retrieved by each method from the pan-cancer analysis. Note that OncodriveCLUST retrieves only 72 genes, therefore it does not appear in the last two histograms, and ActiverDriver retrieves 95 genes, and thus it doesn't appear in the last histogram. (B) Venn diagram showing the overlap between the genes selected by each method in the pan-cancer analysis. The numbers in parenthesis represent the CGC genes rate in each group. Note that the CGC rates of groups of genes exhibiting more than one signal of positive selection range from 13% (OncodriveCLUST-MuSiC) to 92% (genes with the four signals). On the other hand, these rates are rather low in genes that posses only one signal, ranging between 4% (MuSiC) and 11% (OncodriveFM). Based on these results we decided to establish the quasi-majority vote described in Methods to select the genes in the core of the HCD list. In other words, genes with at least two signals of positive selection either in the pan-cancer analysis and/or any per-project analysis were nominated as high-confidence drivers. (C) Bar graph detailing the proportion of CGC depending on the number of signals of positive selection identified in the genes.
Pooling all pan-cancer samples together (pan-cancer analysis) increases the statistical power to detect drivers acting across tumor types, thus facilitating the identification of driver genes that are not detected when each tumor is analyzed individually. However, the pan-cancer analysis may also diminish the relevance of mutations in some drivers acting only in certain tumor types (Supplementary Fig. 1). To overcome this issue, we also analyzed each tumor type separately (per-project analysis) and added the genes identified in each project to those detected across all pan-cancer tumors (see Methods).
Next, we decided to combine the resulting 48 (four pan-cancer and 44 per-project) lists of driver candidates. We discarded the direct combination of pvalues or rankings of the genes across the lists, because they reflect different signals of positive selection in different tumor-types. For example, a gene exhibiting the four signals of positive selection to a mild degree across several tumor types is not necessarily a better candidate than other with one stronger signal in an individual tumor type. More elaborate combination approaches based, for example on Bayesian classifiers or other machine learning methods are unfeasible due to the lack of a gold standard dataset of drivers and passengers to optimize the combination. Instead, we used a rule-based approach exploiting our current knowledge of the features of cancer genes (Supplementary Fig. 2). To construct a list of high-confidence drivers (HCDs) we first selected 130 genes that exhibit more than one signal of positive selection in the pan-cancer (or any per-project) analysis. This may leave out drivers with only one signal of positive selection. To rescue some of those while keeping the false-positive rate as low as possible within HCDs, we included 40 CGC genes with one signal of positive selection. Furthermore, we upgraded to the HCD list 81 genes detected by a single approach which functionally interact –considering all Pathway Commons20 database connections, except those less specific direct protein-protein interactions– with at least one HCD. In addition, we populated a list of Candidate Drivers (CDs) with 144 one-signal genes that participate in protein-protein interactions with HCDs. (See Methods and Supplementary Fig. 2 for details.) Finally, we included in the HCD list another 40 significantly mutated genes identified by MutSig's most recent version –also combining three signals of positive selection— (Supplementary Fig. 3). (Note that because these genes are already selected based on a combination of signals of positive selection, we unite rather than intersect the list of MutSig significantly mutated list with our own HCD list.) In summary, we provide a very reliable list of 291 HCDs and a second one, of 144 CDs, more comprehensive but with an expectedly higher false-positives rate (Supplementary Table 2).
When HCDs are mapped to a functional interaction network (see Methods), they appear enriched for biological processes within 5 broad modules –Chromatin remodeling, mRNA processing, Cell signaling/proliferation, Cell adhesion, DNA repair/Cell cycle– which loosely correspond to both established and emergent cancer hallmarks (Fig. 3 and Supplementary Table 3). Thirteen selected non-CGC, or novel cancer genes are depicted in Figure 4 within their functional interaction context. These novel driver candidates appear alongside other well-established cancer genes. One may thus hypothesize that as more tumor genomes are sequenced, new lowly recurrent mutational drivers in these modules will emerge. This idea is further illustrated in Figure 4a, where, for example well-known cancer genes within the Cell cycle pathway are schematically represented together with not well established HCDs. Examples of novel cell cycle driver candidates include ATR, a kinase which phosphorylates p53 and other proteins, such as CHK1 and RAD1721 and has been associated to tumors with hypermutator phenotypes when defective. ATR is included in the HCD list because it is both recurrently mutated and FM biased in UCEC (Fig. 4b). CDKN1A and CDKN1B, inhibitors of cyclin-dependent kinase activity22,23 which mediate the role of TP53 in the arrest of cellular proliferation after DNA damage, also appear to drive tumorigenesis in several pan-cancer samples alongside other well-known cell cycle genes. CDKN1A is recurrently mutated and FM biased in BLCA and in the pan-cancer analysis, whereas CDKN1B is recurrently mutated and FM biased in BRCA. Both genes are also detected by MutSig (Fig. 4b). On the other hand, in the broad module of signal transduction and proliferation, PIK3CG and PIK3CB, within the PIK3-AKT signaling pathway appear to complement the tumorigenic role of PIK3CA. Collectively, these kinases are key in the transduction of information from receptors on the outer membrane of eukaryotic cells to effectors in the nucleus24,25,26. They receive their names after their catalytic subunit. De-regulation of PIK3CG and PIK3CB had been previously linked to tumor progression27,28,29,30. PIK3CB exhibits a significant FM bias and PIK3CG, a significant mutational recurrence, both in the pan-cancer analysis. Thus, they are both included in the HCD list based on their functional interactions with other HCDs, such as PIK3CA (Fig. 4b). Finally, FOXA1 and FOXA2 are general transcriptional regulators, involved in opening the chromatin to make DNA accessible to the entry of other regulators31,32. They are both missregulated in several malignancies33,34,35,36,37. While FOXA1 is both recurrently mutated and FM biased in BRCA, FOXA2 is recurrently mutated and FM biased in UCEC and recurrently mutated and CLUST biased in the pan-cancer analysis. In summary, these non-CGC likely driver candidates –25 are detailed in Supplementary Table 4– help to complete the landscape of tumor-causing mechanisms in known cancer pathways.
Figure 3.

(A) Network representation of HCDs. Trimmed version of the functional interaction network integrated by 124 HCDs that either map to the five broad biological modules enriched among HCDs or connect them. Genes annotated in the CGC are represented as round squares, HCDs not in CGC are represented as circles and non-HCDs used as linkers between HCDs as diamonds. Circles with thicker border are ‘novel’ candidate drivers discussed in supplementary Table 4 and shown in Figure 3. Genes with a clear preference for bearing PAMs in one tumor type (Fisher's odds ratio > 25) are colored following the project code shown in the figure legend. Colored shadows encircle genes within five enriched biological modules. (B) Frequency of PAMs observed HCDs in panel A across samples of each cancer type, following the tumor type color code. The annotations below indicate methods that identify each gene signals of positive selection. Genes with clear preference for bearing PAMs in one tumor type are indicated with a colored square below the histogram, using the tumor type color code. ‘Novel’ driver candidates are shown in bold font.
Figure 4.

(A) Diagram showing 13 selected candidate cancer genes within their functional interaction context. (B) Heat-map depicting the frequency and number of samples with PAMs of the 13 selected ‘novel’ cancer genes in each tumor type and in the complete pan-cancer dataset. Colored circles indicate methods identifying each gene either in the per-project analyses or in the pan-cancer analysis. Note that six of the genes in the Figure show two signals of positive selection and are therefore not included within the HCDs due to their connections with other drivers.
Amongst HCDs, only TP53 and PIK3CA have protein affecting mutations, or PAMs (non-synonymous, stop, splice site and frameshift indels), in more than 10% of pan-cancer samples (Fig. 3). Another 51 genes –some of which are not well-established drivers– bear PAMs in more than 10% of samples of at least one tumor type (Supplementary Fig. 4). Interestingly, 16 HCDs have a clear bias (Fisher's odds-ratio > 25) towards sustaining PAMs in one tumor type with respect to others (Fig. 3 and Supplementary Fig. 5). (We checked that Fisher's results were not biased towards tumor types with higher mutation rates; see Methods and Supplementary Fig. 7).
Further support of the mutational drivers identified by our combined methodology stems from the analysis of copy number changes (CNAs) across pan-cancer samples. Many HCDs are also affected by CNAs, and 38 of them are significantly altered according to GISTIC38 and/or highly biased towards misregulation due to CNAs according to OncodriveCIS39 (Supplementary Fig. 6). Therefore, these are also likely involved in tumorigenesis upon deletion (tumor suppressors) or amplification (oncogenes).
It has previously been suggested that tumorigenesis requires 5–7 driver mutations in common epithelial cancers, while hematological and pediatric malignancies may require fewer8,40,41. Even under the assumption that the HCD list is not complete, it allows us to explore this question. Pan-cancer tumors have a median of 4 PAMs in HCDs (Fig. 5), although this number varies widely depending on the cancer type; OV and AML tumors exhibit the lowest rate (median of 2), whereas BLCA (9.5), LUSC (9) and LUAD (9) have the highest. Most tumors (94%) have at least one HCD bearing a PAM (Fig. 5). Again, AML tumors present the highest rate of samples without PAMs in HCDs (16%), highlighting the possible relevance of other alterations in this cancer type.
Figure 5.
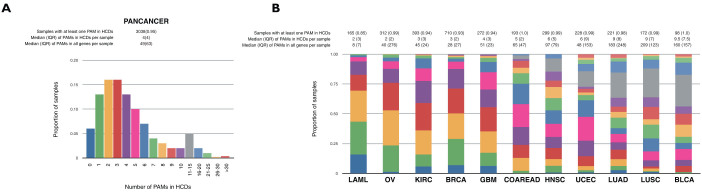
(A) Histogram of the proportion of samples in the pancancer dataset with PAMs in HCDs. (B) Proportion of samples in each cancer type with PAMs in HCDs.
Discussion
In this manuscript we provide a comprehensive catalog of driver candidates acting across the 3,205 tumor samples within the pan-cancer cohort. One hundred and sixty-five of these candidates are novel findings not included in the CGC. Hypotheses regarding their involvement in cancer emergence and evolution can be experimentally tested and might subsequently lead to new insights into this process in the 12 cancer types included within the pan-cancer dataset.
We designed a novel approach to elaborate the catalog of high-confidence drivers, HCDs, across the pan-cancer dataset combining the results of multiple methods to identify cancer driver genes. In this regard, although the newest version of MutSig incorporates other criteria on top of the frequency assessment, the list of significantly mutated genes obtained with this method is clearly different to the one obtained with the other methods (see Supplementary Fig. 3), further stressing the value of their complementarity. The five methods whose outputs we combined constitute the state-of-the-art of the detection of mutational drivers based on the four signals of positive selection described in this work, however new methods exploiting the same or other signals of positive selection, or improved versions of existing ones will likely appear in the near future. Nevertheless, the rationale of the approach presented here could be used to combine a different set of methods. The rule based approach employed to combine the 48 lists of driver candidates obtained from the four initial methods (and the list of MutSig significantly mutated genes) must be regarded as a first and probably imperfect approach to this problem. More sophisticated combinations based on the p-values or rankings of genes from different methods in different tumor types would be cumbersome and not necessarily more optimal. It is easy to see that genes showing few signals of positive selection in one specific tumor type would have a disadvantage compared to genes exhibiting mild varied signals of positive selection across several tumor types. Addressing this issue would require more laborious approaches involving optimization methods, such as Bayesian classifiers, which will suffer of one common caveat: the lack of a proper training set of true drivers and passengers to perform the optimization. Therefore, in summary, we decided to carry out the heuristic approach described in this work, although more sophisticated combinations could be assessed once more complete and unbiased datasets of drivers and passenger genes become available.
Another challenge to combine gene lists from different methods was related to assessing the quality of both individual and combined lists of driver candidates. Because there is currently no gold-standard dataset of driver and passenger genes to correctly compute the specificity and sensitivity of each prediction method, we computed a proxy positive predictive value as the rate of known cancer genes (CGC genes) in each list. Although the CGC is undoubtedly biased and incomplete, it is to date the most thorough catalog of bona fide cancer drivers. Nevertheless, due to its incompleteness, the rate of CGC genes is always a low estimator of the positive predictive value of the lists of drivers uncovered by each method or the combinations thereof. As a consequence, the goal of any predictive method is to maintain a relatively high rate of CGC genes in its list of predicted drivers, but still identify some non-CGC genes. In practical terms, we used this idea to determine the cutoff to apply to the list of driver candidates identified by each method and also to assess whether the combined lists were more accurate than individual ones. It is also important to stress that our statement that the combination of methods outperforms individual methods in the quality of the lists of driver candidates they produce is based on the increase of this proxy positive predictive value in the former.
To decide how many signals of positive selection a gene should exhibit to be considered a driver candidate, we followed the same compromise between a high rate of CGC genes and the appearance of some non-CGC genes. While a list of genes bearing three signals of positive selection (30; Fig. 1c) possesses a higher rate of CGC genes (80%) than the equivalent list of two-signals genes (51%), the latter has a higher chance to include yet unknown drivers. (It is important to point out here that genes with clearer signals of positive selection are probably more likely to have been detected to date and thus be included in the CGC.) In summary, this is the reason why we decided to use this criterion to do the final combination. Different combinatorial rules, such as selecting more stringent cutoffs to produce individual lists and subsequently uniting them instead of intersecting them may be attempted and produce slightly different sets of driver candidates. Note that to incorporate MutSig significantly mutated genes we carried out a union instead of an intersection with the results of the other methods, because MutSig already integrates several signals of positive selection.
We found that 57 of the novel –non CGC– driver candidates actually map to well-known cancer pathways, thus complementing our current knowledge of the emergence of the disease. They therefore support the viewpoint that the main subjects of alterations resulting in tumorigenesis are not individual genes, but rather modules of functionally related proteins. But their appearance at very low frequencies also imply that our knowledge of the heterogeneity of cancer –specially the diversity of molecular alterations underlying diseases that are very similar in histology and phenotype– is still incomplete. Future projects that undertake the systematic identification of mutational drivers of new tumor types using a combinatorial approach like the one we have described will probably expand this picture even further. On the other hand, several novel driver candidates don't fall within our compiled knowledge on functional interactions. This likely means that –as in the case of the recent findings of chromatin regulatory proteins and splicing factors– still new pathways associated with tumorigenesis remain to be discovered.
In addition, our finding that the median of mutated HCDs across tumor types is close to numbers previously hypothesized might imply that the detection of mutational drivers acting on this set of tumors is close to saturation when a thorough combinatorial approach is followed. The variability in the number of mutated HCDs in samples of the same cancer type could be attributed to a mixture of different stages in the samples that form the cohort or, alternatively to a variety of mechanisms underlying tumorigenesis.
We have demonstrated for the first time that the combination of methods based on the detection of complementary signals of positive selection outperforms the use of a single approach. This improvement relies on two facts: first, driver genes exhibit different signals of positive selection and thus the use of multiple criteria allows detecting a more comprehensive list of drivers. Second, combining the results obtained by several methods also allows estimating their reliability permitting the retrieval of a list of driver candidates highly enriched by bona fide cancer genes.
In summary, here we provide a comprehensive catalog of putative mutational drivers acting in 3,205 tumors from 12 different cancer types of high societal importance, which opens new avenues to better understand the mechanisms of tumorigenesis. All results of the analyses described here are available at www.intogen.org/tcga and can be browsed using www.gitools.org/tcga42. The presence within this catalog of several novel candidate drivers occurring in very few tumors could help extend our knowledge of tumor emergence to more patients of these diseases.
Methods
Initial mutation data
Samples with at least one mutation from the pan-cancer dataset available at Synapse (syn1729383) were retrieved after excluding 71 considered as hypermutators. Hypermutators of a tumor type contained more than (Q3 + 4.5 * IQR) somatic mutations, where Q3 and IQR are the third quartile and the interquartile range of the distribution of mutations across all samples of the tumor type, respectively. After filtering, the dataset was composed of 3,205 samples with 287,822 protein affecting mutations.
Mutational cancer drivers
Somatic mutations generated by all projects within the pan-cancer were analyzed using four methods based on complementary criteria to detect likely driver genes.
The input of the four methods were the Mutation Annotation Files (maf) produced by each tumor type Analysis Working Group carefully filtered as explained in syn1729383. (The colon adenocarcinoma and rectum adenocarcinoma datasets were combined into a single colorectal adenocarcinoma dataset for all analyses.) The first step of each execution consisted in excluding mutations in hypermutated samples from the input files, as explained in the previous section. MuSiC, a method based on recurrence is thoroughly described in Dees et al., 201210, and it was employed on the pan-cancer datasets as described in Kandoth et al, personal communication. The statistical model and implementation of OncodriveFM12, which identifies genes with a bias towards accumulation of mutations with high functional impact appear in Gonzalez-Perez and Lopez-Bigas 201212. Those of OncodriveCLUST, which identifies genes with significantly clustered mutations are described in detail in Tamborero et al, 201318. These two methods were executed as described in Gonzalez-Perez et al 48, (see below a brief description of this process). The rationale beneath ActiveDriver, which pinpoints genes whose mutations occur predominantly in protein active sites, and its implementation are described in full in Reimand and Bader 201319. It was applied as described in Reimand et al. 46.
Each method was applied to all pan-cancer samples, pooled together to increase the statistical power to detect mutational driver genes across several tumor types (pan-can analysis). In addition, we analyzed the samples of each cancer type separately to overcome any potential dilution effect resulting from merging samples from different projects (per-project analyses).
To execute OncodriveFM and OncodriveCLUST on these datasets, we employed the IntOGen-mutations pipeline (http://www.intogen.org/mutations/analysis), described in detail in Gonzalez-Perez et al 48. Briefly, we defined configuration files for each tumor type and one for the pan-cancer analysis. The minimum number of mutated samples to analyze a gene was set at 12 for both OncodriveFM and OncodriveCLUST in the pan-cancer analysis. The limit for OncodriveFM in the per-project analysis was set at 1% of the samples in the case of datasets with median below 100 mutations per sample, and at 5 otherwise. For OncodriveCLUST, these numbers were 3 and 5, respectively. After completion of the IntOGen-mutations pipeline, both OncodriveFM and OncodriveCLUST produced twelve results files –one from the pan-cancer analysis and the other eleven from per-project analyses– comprising FDR values of the respective statistical tests for each analyzed gene. We received similar results files from the MuSiC and ActiveDriver teams, totaling 48 files.
Genes that are not expressed in any pan-cancer tumor type were excluded from the resulting list of candidate drivers; this filter was based on pan-cancer data and criteria included in syn1734155. Finally, TTN and OBSCN were also excluded from the list as they have been proposed as likely false positives from the methods identifying drivers.
Lists of putative mutational drivers
After running the four methods on the pan-cancer dataset (pan-cancer analysis) as explained above, we retrieved the four corresponding lists of putative driver genes reported. The cutoff of each method (MuSiC, OncodriveFM, ActiveDriver, FDR < 0.01; OncodriveCLUST FDR < 0.05) was selected ad hoc after visual inspection of the lists' enrichment for CGC genes (Fig. 2a). The methods were also run on the datasets of each individual tumor type (per-project analysis), and the same cutoffs were set to select the lists of putative drivers. This process thus produced 48 lists of putative driver genes which were then combined following an elaborate rule-based approach, as follows.
Genes with several signals of positive selection formed the Various-Signals Genes (VSG) group, while genes exhibiting only one signal of positive selection were classified as One-Signal Genes, or OSGs. (Genes detected within per-project analyses were required to posses the signals in the same tumor type to be considered VSGs.) We assumed that detection by two methods (a quasi-majority vote) was sufficient to nominate cancer drivers. This thought was supported by the observation that genes detected by more than one method had much higher rates of CGC genes than those exhibiting only one signal (Fig. 1c). Nevertheless, true cancer genes may possess only one signal of positive selection and may have been left out of the VSG list. In order to rescue them –but at the same time keep the false positives rate under control– we performed two actions.
First, we pooled OSGs included in the Cancer Gene Census, CGC3, within a separate group referred to as Known Cancer Genes, or KCG. Second, we made the assumption that genes exhibiting one signal of positive selection which in addition are known to functionally interact with VSGs and KCGs were more likely to be involved in tumorigenesis than otherwise ‘disconnected’ genes. This would include in our high-confidence candidates list likely bona fide drivers still not uncovered by genetic or genomic cancer studies. Therefore, we retrieved from the Pathway Commons database20 the subset of candidates which either directly interact, take part in the same molecular process, or are enzyme or substrate/product of a biochemical reaction with genes included in the VSG and KCG groups. VSGs, KCGs and their functional interaction partners finally integrated the list of high-confidence drivers (HCDs).
Second, genes exhibiting one signal of positive selection and connected with VSGs and/or KCGs by protein-protein interactions populated a separate list of candidates drivers (CDs), given that the potential promiscuity of such physical interactions may undermine the elucidation of relevant events. Finally, note that the remaining genes, i.e. those picked up by a single method and with no interaction with VSGs or KCGs, are presumed to contain a higher proportion of false positives and thus were excluded from any further analysis. In order to complete the HCDs list, we considered the results of MutSig, a well-established method to detect mutational drivers11. Initially, it was developed to detect frequently mutated genes11, but at present includes additional criteria to detect other signals of positive selection, such as the accumulation of mutations in specific regions and in conserved residues. The MutSig results on the pan-cancer data set were retrieved from syn1715784. Forty genes stated as significant according to this method but not by our aforementioned analysis were considered as valid additional findings, and as thus were included in the list of putative drivers of the present analysis.
HCDs biased towards mutations in one tumor type
Fisher's exact test was used to check whether mutations on a certain gene were evenly distributed across tumor types. Mutations in a gene were defined as biased towards a certain cancer type if the Fisher's odds ratio of their occurrence in samples of that tumor type, with respect to the expected frequency was greater than an arbitrary cutoff of 25. We checked that Fisher's results were not biased towards tumor types with higher mutation rates by comparing the number of specific HCDs with the mutation rate in each tumor type (Supplementary Fig. 7). Tumors with lower mutation rates, such as LAML and KIRC actually possess longer lists of specific HCDs. On the other hand, both lung cancer types, with very high mutation rates only contribute one specific HCD. This is probably because, independently of the mutation rate of the tumor type, driver mutations tend to concentrate in genes that drive tumorigenesis in that specific tumor type.
Biological modules analysis
We constructed a functional interactions (FI) network with the 291 HCDs employing the Cytoscape FI plugin43. We then clustered the resulting network into modules and analyzed these for their enrichment for Biological Processes of the Gene Ontologies44 (GOBPs). Several very significantly enriched GOBPs (FDR < 0.001) and other connector genes were manually selected to construct the trimmed version of the Functional Interactions network of Figure 2a. The broad biological modules depicted in the figure were constructed by grouping the genes in similar GOBPs. We then selected 13 HCDs, whose role as mutational drivers had not been previously established to highlight their possible involvement in tumor emergence via their contribution to the alteration of well-studied cellular pathways. (Their corresponding nodes are marked with a thicker border in Fig. 2a.) Using KEGG pathways diagrams45, along with the information collected in Supplementary Table 2, and the functional interactions retrieved from the Pathway Commons database20, we built a schematic network of interactions linking these 13 genes to others in several well-studied pathways (shown in Figure 3).
Cancer drivers due to amplifications and deletions
Copy number alteration (CNA) data was retrieved from syn1703335, retaining only multi-copy amplifications and homozygous deletions. In addition, platform-corrected RNA-seq data retrieved from syn1834628 was used to assess whether these CNAs significantly changed the expression of affected genes. To this end, we used OncodriveCIS39. Briefly, this method ranks genes according to their bias towards overexpression (or underexpression) due to changes of their copy numbers. The OncodriveCIS analysis was performed solely for all tumor samples pooled together. Therefore, and to avoid tissue specific expression bias, the expression impact score caused by CNAs was calculated per each individual by taking into account only samples of the same cancer type; thereafter, the bias of the gene towards misregulation was calculated across all the tumors, and we have evaluated the top-ranking genes of this method. Finally, genes identified by Gistic38 as bearing recurrent CNAs were retrieved from Synapse (syn1703357).
Navigation of pan-cancer mutational drivers
The results of the analysis described here –including the functional impact of mutations, their frequency in different tumor types, the detected signals of positive selection in each gene and the classification of genes as HCDs or CDs– were loaded into a website (available at http://www.intogen.org/tcga) using Onexus. The website is designed following the lines described in the paper describing the IntOGen-mutations platform (http://www.nature.com/nmeth/journal/vaop/ncurrent/full/nmeth.2642.html), and allows navigation to and from other IntOGen-like webservers. In addition, all pan-cancer mutations data can be interactively navigated employing our Gitools 2.0 enhanced heatmap browser42. To that purpose, we have prepared multidimensional data matrices, data annotations files and video tutorials available at www.gitools.org/tcga. The results are also available in Synapse (syn1962006).
Author Contributions
N.L.-B., D.T. and A.G.-P. designed the project. Primary analysis to identify drivers were carried out by D.T. (OncodriveCLUST), A.G.-P. (OncodriveFM), C.K. and L.D. (MuSiC-SMG), M.S.L. and G.G. (MutSig) and J.R. and G.B. (ActiveDriver). D.T. and A.G.-P. combined the results from different methods and carried out the rest of analyses described in the manuscript. J.D.-P. prepared the Onexus web site with the results. C.P.-L. provided technical assistance for running the analyses. D.T., A.G.-P. and N.L.-B. drafted the manuscript and prepared the figures. N.L.-B. supervised the project. All authors read and approved the final draft.
Supplementary Material
Supplementary Note 1
Supplementary Table 2
Acknowledgments
We acknowledge funding from the Spanish Ministry of Science and Technology (grant number SAF2009-06954 and SAF2012-36199) and the Spanish National Institute of Bioinformatics (INB). This work was supported by NRNB (U.S. National Institutes of Health, National Center for Research Resources grant number P41 GM103504). We gratefully acknowledge the contributions from the TCGA Research Network and its TCGA Pan-Cancer Analysis Working Group (contributing consortium members are listed in Supplementary Note 1).
References
- Reddy E. P., Reynolds R. K., Santos E. & Barbacid M. A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene. Nature 300, 149–152 (1982). [DOI] [PubMed] [Google Scholar]
- Tabin C. J. et al. Mechanism of activation of a human oncogene. Nature 300, 143–149 (1982). [DOI] [PubMed] [Google Scholar]
- Futreal P. A. et al. A census of human cancer genes. Nature Reviews. Cancer 4, 177–183 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium T. C. G. A. et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- ICGC. International network of cancer genome projects. Nature 464, 993–998 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway L. A. & Lander E. S. Lessons from the Cancer Genome. Cell 153, 17–37 (2013). [DOI] [PubMed] [Google Scholar]
- Vogelstein B. et al. Cancer Genome Landscapes. Science 339, 1546–1558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton M. R., Campbell P. J. & Futreal P. A. The cancer genome. Nature 458, 719–724 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez A. et al. Computational approaches to identify functional genetic variants in cancer genomes. Nature Methods 10, 723–729 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dees N. D. et al. MuSiC: Identifying mutational significance in cancer genomes. Genome Research (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 10–14 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez A. & Lopez-Bigas N. Functional impact bias reveals cancer drivers. Nucleic acids research 1–10 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P. C. & Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Research 31, 3812–3814 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva B., Antipin Y. & Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic acids research 39, e118 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei I. A. et al. A method and server for predicting damaging missense mutations. Nature Methods 7, 248–249 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodis E. et al. A Landscape of Driver Mutations in Melanoma. Cell 150, 251–263 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenman C. et al. Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamborero D., Gonzalez-Perez A. & Lopez-Bigas N. OncodriveCLUST: exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics (Oxford, England) 7, 1–7 (2013). [DOI] [PubMed] [Google Scholar]
- Reimand J. & Bader G. D. Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Molecular Systems Biology 9, 637 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E. G. et al. Pathway Commons, a web resource for biological pathway data. Nucleic acids research 39, D685–90 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dart D. A., Adams K. E., Akerman I. & Lakin N. D. Recruitment of the cell cycle checkpoint kinase ATR to chromatin during S-phase. The Journal of biological chemistry 279, 16433–40 (2004). [DOI] [PubMed] [Google Scholar]
- Insinga A. et al. DNA damage in stem cells activates p21, inhibits p53, and induces symmetric self-renewing divisions. Proceedings of the National Academy of Sciences of the United States of America 110, 3931–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. & Kim S. S. The function of p27 KIP1 during tumor development. Experimental & molecular medicine 41, 765–71 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A. et al. Nuclear but not cytosolic phosphoinositide 3-kinase beta has an essential function in cell survival. Molecular and cellular biology 31, 2122–33 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marqués M. et al. Phosphoinositide 3-kinases p110alpha and p110beta regulate cell cycle entry, exhibiting distinct activation kinetics in G1 phase. Molecular and cellular biology 28, 2803–14 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelmann R. et al. TGFbeta-induced downregulation of E-cadherin-based cell-cell adhesion depends on PI3-kinase and PTEN. Journal of cell science 118, 4901–12 (2005). [DOI] [PubMed] [Google Scholar]
- Hill K. M. et al. The role of PI 3-kinase p110beta in AKT signally, cell survival, and proliferation in human prostate cancer cells. The Prostate 70, 755–64 (2010). [DOI] [PubMed] [Google Scholar]
- Wee S. et al. PTEN-deficient cancers depend on PIK3CB. Proceedings of the National Academy of Sciences of the United States of America 105, 13057–62 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba S. et al. Down-regulation of PIK3CG, a catalytic subunit of phosphatidylinositol 3-OH kinase, by CpG hypermethylation in human colorectal carcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research 8, 3824–31 (2002). [PubMed] [Google Scholar]
- Sasaki T. et al. Colorectal carcinomas in mice lacking the catalytic subunit of PI(3)Kgamma. Nature 406, 897–902 (2000). [DOI] [PubMed] [Google Scholar]
- Bernardo G. M. & Keri R. A. FOXA1: a transcription factor with parallel functions in development and cancer. Bioscience reports 32, 113–30 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausa F. M., Tan Y. & Costa R. H. Association between hepatocyte nuclear factor 6 (HNF-6) and FoxA2 DNA binding domains stimulates FoxA2 transcriptional activity but inhibits HNF-6 DNA binding. Molecular and cellular biology 23, 437–49 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson E. A. et al. BRCA1 and FOXA1 proteins coregulate the expression of the cell cycle-dependent kinase inhibitor p27(Kip1). Oncogene 25, 1391–9 (2006). [DOI] [PubMed] [Google Scholar]
- Imamura Y. et al. FOXA1 promotes tumor progression in prostate cancer via the insulin-like growth factor binding protein 3 pathway. PloS one 7, e42456 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch L. et al. Opposite roles of FOXA1 and NKX2-1 in lung cancer progression. Genes, chromosomes & cancer 51, 618–29 (2012). [DOI] [PubMed] [Google Scholar]
- Mirosevich J. et al. Expression and role of Foxa proteins in prostate cancer. The Prostate 66, 1013–28 (2006). [DOI] [PubMed] [Google Scholar]
- Liu M. et al. IKKα activation of NOTCH links tumorigenesis via FOXA2 suppression. Molecular cell 45, 171–84 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome biology 12, R41 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamborero D., Lopez-Bigas N. & Gonzalez-Perez A. Oncodrive-CIS: a method to reveal likely driver genes based on the impact of their copy number changes on expression. PloS One 8(2): e55489. 10.1371/journal.pone.0055489 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinzel A. C. & Hahn W. C. Oncogenic transformation and experimental models of human cancer. Frontiers in bioscience: a journal and virtual library 13, 71–84 (2008). [DOI] [PubMed] [Google Scholar]
- Address T. P. On the Nature of Susceptibility to Cancer. (1953).
- Perez-Llamas C. & Lopez-Bigas N. Gitools: Analysis and Visualisation of Genomic Data Using Interactive Heat-Maps. PLoS ONE 6, e19541 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G., Feng X. & Stein L. A human functional protein interaction network and its application to cancer data analysis. Genome Biology 11, R53 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M. et al. Gene Ontology: tool for the unification of biology. Nature genetics 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S., Furumichi M., Tanabe M. & Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Research 38, D355–D360 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J., Wagih O. & Bader G. D. The mutational landscape of phosphorylation signaling in cancer. Sci. Rep. 3, 2651; 10.1038/srep02651 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart M. J., et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45, 1113–1120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez A. et al. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 10.1038/nmeth.2642 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Note 1
Supplementary Table 2