

Abstract
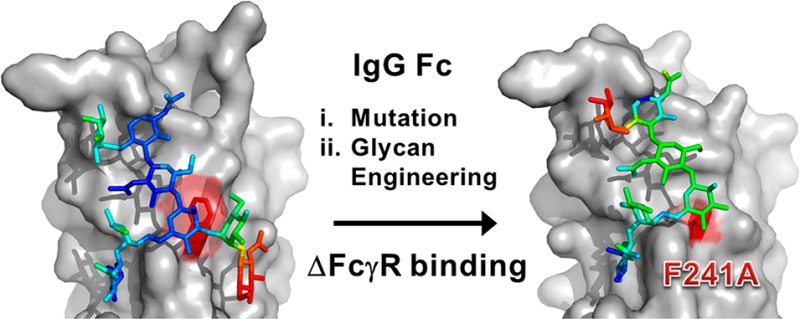
Biologically active conformations of the IgG1 Fc homodimer are maintained by multiple hydrophobic interactions between the protein surface and the N-glycan. The Fc glycan modulates biological effector functions, including antibody-dependent cellular cytotoxicity (ADCC) which is mediated in part through the activatory Fc receptor, FcγRIIIA. Consistent with previous reports, we found that site-directed mutations disrupting the protein–carbohydrate interface (F241A, F243A, V262E, and V264E) increased galactosylation and sialylation of the Fc and, concomitantly, reduced the affinity for FcγRIIIA. We rationalized this effect by crystallographic analysis of the IgG1 Fc F241A mutant, determined here to a resolution of 1.9 Å, which revealed localized destabilization of this glycan–protein interface. Given that sialylation of Fc glycans decreases ADCC, one explanation for the effect of these mutants on FcγRIIIA binding is their increased sialylation. However, a glycan-engineered IgG1 with hypergalactosylated and hypersialylated glycans exhibited unchanged binding affinity to FcγRIIIA. Moreover, when we expressed these mutants as a chemically uniform (Man5GlcNAc2) glycoform, the individual effect of each mutation on FcγRIIIA affinity was preserved. This effect was broadly recapitulated for other Fc receptors (FcγRI, FcγRIIA, FcγRIIB, and FcγRIIIB). These data indicate that destabilization of the glycan–protein interactions, rather than increased galactosylation and sialylation, modifies the Fc conformation(s) relevant for FcγR binding. Engineering of the protein–carbohydrate interface thus provides an independent parameter in the engineering of Fc effector functions and a route to the synthesis of new classes of Fc domain with novel combinations of affinities for activatory and inhibitory Fc receptors.
Introduction
The binding of IgG Fc to cell surface and soluble serum ligands triggers a variety of immunological processes including phagocytosis, cytotoxicity, inflammation, and immunosuppression.1,2 The structure of the Fc domain is stabilized by the N-linked glycan, attached to Asn297 of each IgG heavy chain.3−6 This glycan is critical for Fc function: genetic or enzymatic removal leads to an almost complete loss of antibody effector functions.7,8 Moreover, composition of the glycan is a key parameter in determining the balance between pro-inflammatory or anti-inflammatory effects. For example, removal of the core α1,6-linked fucose residue of the IgG1 Fc glycan enhances binding for FcγRIIIA,9,10,12,14 while elevation of the levels of terminal α2,6-sialylation leads to decreased natural killer cell activation11 and other potent cell-mediated immunosuppressive effects.13 In addition to natural variations in Fc glycosylation, a growing number of enriched and engineered Fc glycoforms are finding application in therapeutic monoclonal antibodies when a particular balance of effector functions is desirable.15−17 The three-dimensional structure of the Fc glycoforms has been investigated3,18−23 as have the independent effects of glycan and protein engineering on receptor binding.9,10,24−27 By comparison, however, relatively little is known about the interdependence of glycan composition and protein structure on receptor binding.28,29
The N-linked glycans in IgG1 Fc are complex, mostly core-fucosylated, biantennary-type structures with varying amounts of bisecting GlcNAc, terminal galactose, and sialic acid residues.30 Levels of sialylation are low with <10% of total Fc glycans from serum IgG being sialylated.30 Tri- or tetra-antennary glycans are generally not found in serum IgG Fc. The absence of larger, branched, and/or sialylated structures is notable, especially when compared to the glycosylation of other serum or cell-surface glycoproteins.30,31 X-ray crystallographic3,4,20,21 and NMR studies32−34 of the IgG Fc domain have defined the conformation of the N-linked glycans at Asn297. In the complex-type IgG Fc glycoforms, the conformation of the oligosaccharide is well conserved and contacts over 500 Å2 of the surface of each Cγ2 domains.3,4 The six arms of the glycan chain makes several stable interactions with hydrophobic amino acid residues of the Cγ2 domain. The terminal Gal6′ (see legend to Figure 1 for terminology) on the six arm has been shown to restrict glycan flexibility through interaction with the Fc protein backbone.32 Key amino acid residues that interact with the 6-arm glycans through hydrogen bonds and hydrophobic interactions include: Phe241, Phe243, Val262, Val264, Asp265, Lys246, and Arg301. Aromatic rings of Phe241 and Phe243 form CH−π interactions with the GlcNAc2 and GlcNAc5′ residues of the Fc glycan and contribute to the stability of the Fc domain.28,29,35,36 The three arm on the other hand makes fewer contacts with the protein backbone with hydrophobic interactions between Man4 and Lys334 being the only observable protein–glycan interaction.3,4 While the presence of extensive protein–glycan interactions suggests a relatively immobile carbohydrate conformation and reduced enzymatic processing, recent NMR spectroscopic studies indicate a more dynamic and mobile role for the Fc glycans.33,34 Nonetheless, the relatively limited processing of the Fc glycan indicates a reduced accessibility to glycan reactive enzymes in the Golgi apparatus.
Figure 1.

Mass spectrometric analysis of N-glycans released from IgG Fc-F241A. (A) Negative ion ESI spectrum. (B) The data from panel A were processed with the maximum entropy 3 function of MassLynx to convert multiply charged ions to singly charged ions. The position of the fucose residue in the triantennary glycans was not determined. The ion at m/z 3169 gave a composition corresponding to the tetrasialylated triantennary glycan, but this was not confirmed by fragmentation. (C) An example of negative ion collision-induced dissociation spectrum of the monosialylated, fucosylated biantennary glycan. Fragment ions are labeled according to the scheme devised by Domon and Costello.39 (D) Spectra showing trigalactosylated structures with three (triply charged) and four (quadruply charged) sialic acids attached, respectively. Key: Integrated oligosaccharide nomenclature follows that of Bowden et al.20 Residue labeling follows that of Vliegenhart et al.40 with the additional modifications of 7 for sialic acid, 1′ for α1→6-linked core fucose.41 These residue labels are in bold face throughout the manuscript. The symbolic representation of glycans follows that of Harvey et al.42 with residues in both the schematic diagrams and molecular graphics following the color scheme of the Consortium for Functional Glycomics.
The influence of hydrophobic residues in the protein–glycan interface of the Fc on glycan processing was first observed in mutational studies on mouse–human chimeric IgG3 antibodies, where replacement of Phe241, Phe243 or Val264 with Ala resulted in elevated levels of mono- and disialylated glycans and decreased binding to C1q and FcγR activity.28 In addition, ribosomal display has been used to discover site-specific Fc mutants with enhanced FcγRIIIA binding.29 This approach identified the F243L mutant which, in addition to enhanced FcγRIIIA binding, also exhibited altered glycan processing including decreased fucosylation.29 Two possible explanations have been proposed to explain these observations:28 either the alteration of the protein–carbohydrate interaction directly affected the Fc protein conformation or the increased glycan processing affected FcγR binding. To discriminate between these possibilities, we isolated and characterized a series of Fc mutants with chemically defined glycosylation.
These glycoform-controlled mutants still exhibited similar reduced FcγR binding indicating that the conformation of the Cγ2 domain is modulated by glycan–protein interaction independently of glycan type. Furthermore we show that the effects of these glycan-destabilizing mutations on FcγR binding are independent of mutations which directly affect the Fc–FcγR interface. Thus, combinatorial mutagenesis can yield antibodies with novel effector properties. For example, we combine Fc–FcγR interface mutations known to selectively enhance FcγRIIB binding, with a glycan–protein interface mutation that decreases binding to all FcγRs, to provide an IgG which more broadly eliminates activatory (but not inhibitory) receptor binding. This study therefore clarifies the role of the Fc glycan in maintaining antibody structure and provides routes to the development of antibody therapeutics with bespoke effector functions.
Results and Discussion
Crystallographic Analysis of IgG1 Fc F241A Mutant
To investigate the structural impact of mutations at the IgG1 Fc protein–glycan interface, we determined the crystal structure of recombinant IgG1 Fc F241A (Table 1). This mutant has been previously reported by Lund et al. to reduce FcγR binding and increase glycan processing.28 Prior to crystallization, we performed electrospray ionization (ESI) mass spectrometric analysis of the N-linked glycans which confirmed extensive branching and terminal sialylation (Figure 1).
Table 1. Crystallographic Data and Refinement Statistics of F241A Fc.
| Data Collection | |
| beamline | Diamond I04 |
| resolution range (Å) | 50.0–1.94 (2.01–1.94)a |
| space group | P212121 |
| cell dimensions | |
| a, b, c (Å) | 48.87, 73.38, 135.95 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| wavelength (Å) | 0.917 |
| unique reflections | 36 211 (3559) |
| Completeness (%) | 99.5 (100) |
| Rmerge (%)b | 9.1 (74.9) |
| I/σI | 16.4 (2.0) |
| ave redundancy | 5.8 (6.0) |
| Refinement | |
| resolution range (Å) | 50.0–1.947 (1.997–1.947) |
| number of reflections | 34 319 (2241) |
| Rwork (%)c | 19.8 |
| Rfree (%)d | 23.4 |
| rmsde | |
| bonds (Å) | 0.009 |
| angles (°) | 1.3 |
| homodimers per asu.f | 1 |
| atoms per asu | |
| (protein/carbohydrate/water) | 3160/85/297 |
| average B-factors (Å2) | |
| (protein/carbohydrate/water) | 45.6/73.6/45.2 |
| model quality (Ramachandran plot)g | |
| most favored region (%) | 98.7 |
| allowed region | 1.3 |
Numbers in parentheses refer to the relevant outer resolution shell.
Rmerge = Σhkl Σi|I(hkl;i) – ⟨I(hkl)⟩|/Σhkl ΣiI(hkl;i), where I(hkl;i) is the intensity of an individual measurement and ⟨I(hkl⟩ is the average intensity from multiple observations.
Rwork = Σhkl||Fobs| – k|Fcalc||/Σhkl |Fobs|
Rfree is calculated as for Rwork but using only 5% of the data, which were sequestered prior to refinement.
rmsd: root-mean-square deviation from ideal geometry.
asu: asymmetric unit.
Ramachandran plots were calculated with Molprobity.37
The IgG1 Fc F241A crystallized in the primitive orthorhombic spacegroup, P212121, with one molecule of the Fc homodimer in the asymmetric unit. Data were collected to a resolution of 1.9 Å (Table 1). As observed in many crystal structures of the Fc, there was notable asymmetry between the two chains of the Fc. The protein and glycan moieties of the Cγ2 domain from one chain (chain B) exhibited a high degree of disorder (Figure 2D).
Figure 2.

Packing of N-link glycans in native (A–C) and F241A mutant (D–F) IgG Fc. Glycans are displayed as blue (GlcNAc), red (Fuc), and green (Man) sticks. Protein is displayed as a gray cartoon with four hydrophobic residues at the protein–glycan interface highlighted in pink (sticks). Overall structure of (A) native (PDB ID 3AVE) and (B) F241A mutant IgG1 Fc. The Cγ2 domains from chain A (B and E) are shown with close-ups of the protein-glycan interfaces (C and F). Four hydrophobic residues located on the protein–glycan interface are highlighted in pink (sticks). Electron density corresponding to carbohydrate is depicted as a blue mesh (2Fo–Fc map contoured at 1σ) around the carbohydrate moiety of the mutant Fc reported herein. Integrated oligosaccharide nomenclature follows that of Bowden et al.,20 see legend to Figure 1 for further details. Secondary structure was defined by Ksdssp.43
Interpretable electron density was observable for seven monosaccharide residues on chain A (Figure 2E,F). In the structure of the wild-type Fc, Phe241 packs with the GlcNAc2 and Man3 residues4,28 (Figure 2B,C). In contrast, electron density in our structure of the F241A mutant, revealed the F241A site-directed substitution and a destabilization of the protein–carbohydrate interface (Figure 2E,F). The N-linked glycan rests in the established position along the Cγ2 domain with defined electron density corresponding to the 6-arm, while only diffuse density corresponding to the 3-arm is observed in the 2Fo–Fc map.
This weak density indicates that the 3-arm is either conformationally disordered or glycoforms differing in 3-arm composition are adopting different conformations where the X-ray scattering does not sum to yield consensus electron density. Previous reports had suggested that the intrinsic flexibility of the 3-arm causes the lack of electron density of the galactose in native galactosylated Fc.3,38 Consistent with a loss of electron density of the 3-arm, detailed negative-ion ESI mass spectrometry with fragmentation analysis revealed extensive branching on the 3-arm but not the 6-arm (Figure 1).
The localized induction of disorder at the 3-arm does not fully account for the apparent increase in accessibility of the glycans to Golgi-resident glycosyltransferases (e.g., increase in 6-arm galactosylation) or the reduction in FcγR binding affinity28 (Figure 2). The mutation may influence the dynamics of the glycan–protein interface not sufficiently captured by low-temperature X-ray crystallographic methods.33 For example, the hydrophobic interface mutations may affect the position of equilibrium between the protein ‘bound’ and ‘free’ conformations proposed by NMR studies to yield more accessible glycans and potentially more widely spaced Cγ2 domains.33 Interestingly, NMR studies have indicated that α2,6-sialylation has minimal impact on glycan dynamics in the Fc domain.34 This finding is entirely consistent with our observation of an elevation in 6-arm sialylation by electrospray mass spectrometry and the conserved conformation of the 6-arm observed by X-ray crystallography (Figure 2C,F).
Having established that mutations at the glycan–protein interface can induce at least localized glycan disorder, we next sought to determine to what extent the reduction in FcγRIIIA binding can be attributed to disruption of the glycan–protein interface or to changes in glycan processing. To this end, we used a combination of site-directed mutagenesis and glycan engineering.
HPLC Analysis of IgG1 Glycoforms
A series of human IgG1 Fc mutants was generated containing mutations that modulated the protein–carbohydrate interactions within the Fc: F241A, F243A, V262E, and V264E. The panel of IgG1 b12 mutants was expressed in either human embryonic kidney (HEK) 293T or GlcNAc transferase (GnT) I-deficient HEK 293S cells.44 The glycans were released from the purified antibody via protein N-glycosidase F (PNGase F). The free sugars were fluorescently labeled and resolved via normal-phase high-performance liquid chromatography (HPLC) using a TSK-amide column. The HPLC spectra from IgG1 Fc mutants expressed in HEK 293T (black spectra) or GnT I-deficient HEK 293S cells (blue spectra) are shown in Figure 3.
Figure 3.

HPLC analysis of 2AA-labeled N-linked glycans from monoclonal IgG1 b12 mutants expressed in HEK 293T and HEK 293S cells. Normal-phase HPLC analysis of 2-AA-labeled N-linked glycans released from target antibody glycoforms by in-gel protein PNGase F digestion. Glycan profile of IgG1 b12 expressed in HEK 293T (black) and HEK 293S (blue) for the following variants: (A) wild type; (B) F241A; (C) F243A; (D) V262E; and (E) V264E. The y-axis displays relative fluorescence (RF).
The glycans from IgG b12 expressed in HEK 293T cells are composed of a series of fucosylated, biantennary, complex-type carbohydrates, typical of the protein-directed glycosylation observed for IgG (Figure 3A; black spectrum). The most abundant species observed were agalactosylated structures with smaller amounts of mono and digalactosylated structures. A small population of sialylated material was also present, showing the typical glycan profile for recombinant IgG1 Fc as reported previously.20,35 Consistent with previous analyses of IgG3,28 mutations interrupting the hydrophobic protein–glycan interface led to dramatic increases in terminal β1,4 linked galactose levels, with digalactosylated species representing the most abundant glycan populations for all mutants generated from HEK 293T cells (Figure 3B–E; black spectra). Tri- and tetra-antennary species, not normally observed on Fc, were detected, most notably on V262E and V264E. Additionally, increased bisecting GlcNAc and terminal sialylation were also evident for these mutants. An unusual digalactosylated, trisialylated species was also detected in the HPLC spectra of all of the mutants. Similar unusually sialylated structures have been detected in mouse serum glycoproteins.45,46 This structural assignment was confirmed by electrospray mass spectrometry of recombinant IgG1 Fc (Figure 1) and b12 mutants (Figures S1 and S2). Further matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) (Figure S3) and HPLC analysis of desialylated mutants (Figures S4 and S5) were carried out to validate the HPLC assignments.
In contrast to the heterogeneous complex-type glycan spectra observed when the IgG panel was expressed in HEK 293T cells, expression in GnT I-deficient HEK 293S cells led to a purely oligomannose-type glycan profile composed of Man5GlcNAc2 together with a small population of fucosylated Man5GlcNAc2 (Figure 3; blue spectra). The latter structure has previously been shown to arise via the inefficient GnT I-independent fucosylation pathway.47 Thus mutations that disrupt the glycan–protein interface affected neither oligomannose processing to Man5GlcNAc2 nor the proportion of GnT I-independent fucosylation. In addition to glycan analysis, the N-glycosylation site occupancy at the Asn297 was examined, as previous report showed decreased glycan site occupancy for the hydrophobic mutants.28 A combination of PNGase F treatment and trypsin digestion, followed by MALDI-TOF-MS, showed that the N-glycosylation site on Fc Asn297 is fully occupied for all the mutants (Figures S6 and S7). The discrepancy might be due to the different antibody isotypes used or methods used to examine site occupancy.
To investigate the impact of sialylation on FcγR binding, in vitro glycosyltransferase reactions were used to generate hyper-α2,6-sialylated IgG (Figure 4). As there is a potential for α2,3-linked sialic acid to be present in glycoproteins derived from HEK 293T cells, we first performed a sialidase digestion. Sialylation was not detected by HPLC analysis (Figure 4A,B). The sialidase-treated material was then subjected to sequential β1,4-galactosylation and α2,6-sialylation (Figure 4C,D, respectively). HPLC analysis revealed that a trace population (∼5%) of monogalctosylated glycans remained after the galactosyltransferase reaction (Figure 4C).
Figure 4.
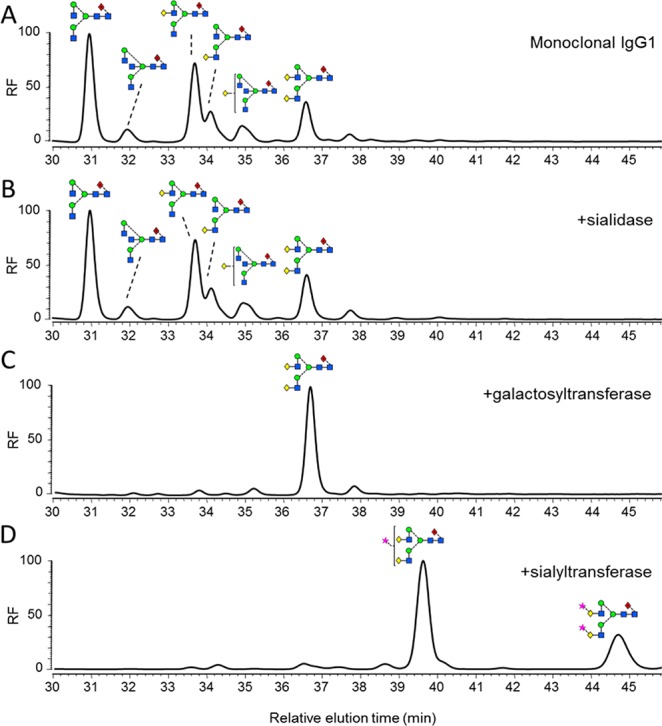
Generation of differentially glycosylated IgG1 Fc. Normal-phase HPLC analysis of 2-AA-labeled N-linked glycans, released from target antibody glycoforms by in-gel PNGase F digestion. (A) Glycan profile of monoclonal IgG1 b12. (B) Glycan profile of IgG1 incubated with 50 U/mL Clostridium perfringens neuraminidase for 48 h at 37 °C. (C) Glycan profile of IgG1 incubated with 25 μg/mL β1,4-galactosyltransferase (B4GALTI) and 80 μM uridine 5′-diphosphogalactose in 50 mM HEPES, 10 mM MnCl2, pH 7.5 for 48 h at 37 °C. (D) Glycan profile of IgG1 sequentially treated with B4GALTI and α2,6-sialyltransferase I (ST6GALI) as described above.
Affinity of IgG1 Fc Mutants for Fc Receptors
We determined the role of the disrupted protein–glycan interactions in IgG Fc binding to Fc–FcγRs by determining the binding of our panel of IgG mutants to the extracellular regions of FcγRIA, FcγRIIA, FcγRIIB, FcγRIIIA, and FcγRIIIB (Figure 5).
Figure 5.

ELISA of monoclonal IgG variants binding human FcγRIA, FcγRIIA, FcγRIIB, FcγRIIIA, and FcγRIIIB. The FcγRs were plated at 5 μg/mL overnight at 4 °C, IgG variants F241A, F243A, V262E, and V264E were incubated for 1.5 h, and binding was detected by HRP-conjugated goat antihuman Fab antibody. Symbolic representation of IgG mutation and glycovariants: solid black square = wild-type native; solid blue square = wild-type Man5GlcNAc2; open black triangle = mutant native; open blue triangle = mutant Man5GlcNAc2; solid red square = wild-type hypergalactosylated and hypersialylated, ELISA binding curves of the four IgG hydrophobic mutants for (A) FcγRIA, IgG variant starting concentration at 10 μg/mL. (B) FcγRIIA, IgG variant starting concentration at 100 μg/mL. (C) FcγRIIB, IgG variant starting concentration at 300 μg/mL. (D) FcγRIIIA, IgG variant starting concentration at 100 μg/mL. (E) FcγRIIIB, IgG variant starting concentration at 300 μg/mL. (F) FcγRIIIA, IgG variant starting concentration at 100 μg/mL. All data points represent the calculated mean of two independent measurements from a total of at least two experiments.
Consistent with studies with FcγRI, our data show that abolition of the Fc glycan–protein hydrophobic interaction resulted in a decreased Fc affinity for the FcγRIA (Figure 5D).28 A similar pattern, albeit to different extents, was observed in the binding analysis with FcγRIIIA, FcγRIIA, FcγRIIB, and FcγRIIIB (Figure 5). The high affinity, activatory FcγRIA and low-affinity, inhibitory FcγRIIB were least prone to such modulation. Interestingly, the V264E mutant contains the highest level of sialylated structures among mutants tested (Figure 3E). V264 packs directly against the core residue, GlcNAc2 (Figure 2C). The V264E mutation would be predicted to perturb the overall trajectory of the glycan away from the surface of the Cγ2 domain and may account for the extensive glycan terminal processing due to increased steric accessibility.
To investigate the effect of these hydrophobic mutations on FcγR binding independent of the differential Fc glycoforms, we compared FcγR binding to the panel of uniformly glycosylated IgG1 Fc mutants expressed in the GnT I-deficient HEK 293S cells (Figure 3; blue spectra).
Consistent with previous reports, wild-type IgG1 Fc with Man5GlcNAc2 glycosylation exhibits increased affinity for FcγRIIIA (Figure 5D) and decreased affinity for the FcγRIIB (Figure 5E) compared with Fc with native biantennary complex glycans.26,48 We also demonstrate that the Man5GlcNAc2 glycoform exhibits decreased Fc affinity for FcγRIIA, which is highly homologous to FcγRIIB (Figure 5B,C).
Mutations that disrupt the hydrophobic interface significantly decrease the Fc affinity for FcγRIIA, FcγRIIIA, and FcγRIIIB, largely independently of the glycoform. This effect is evident by comparison of the wild-type and mutants expressed in GnTI-deficient HEK 293S cells (Figure 3; blue spectra). Mutations affecting the hydrophobic interface resulted in significantly decreased Fc affinity for FcγRIIA, FcγRIIIA, and FcγRIIIB (Figure 5B,D,E), while interestingly, no significant changes were observed for FcγRIIB binding (Figure 5C).
For the high affinity FcγRIA, only V264E caused a significant decrease in binding (Figure 5A). Together, our results show that the hydrophobic mutations disrupt the Fc binding to the activatory FcγRs independently of the Fc glycan, indicating that the productive engagement of Fc–FcγR requires the protein–glycan interaction at the Fc Cγ2 domain.
Unlike F241A, V262E, and V264E, the F243A mutant expressed as the Man5GlcNAc2 glycoform has a minimal effect on FcγRs binding (Figure 5). This minimal effect can be explained by the different protein–glycan interfaces of oligomannose-, hybrid-, and complex-type antibody glycoforms as revealed by X-ray crystallography.20 In contrast to the other residues, F243 exhibits minimal van der Waals contacts with the 6-arm mannose residues in the predicted structure of the Man5GlcNAc2 glycoform.20 Therefore, in contrast to the significant effect of F243A mutation on the mobility of complex-type glycans, F243A would be predicted to have a minimal impact on the mobility of Man5GlcNAc2 structures. The relative affinity of the single mutants for FcγRs is summarized in Table S2.
Remarkably, the interaction between FcγRIIB and IgG1 Fc is relatively unperturbed by the disruption of Fc protein–glycan interactions (Figure 5C). We hypothesized that further enhancements to the FcγRIIB selectivity could be achieved by combining our glycan–protein interface mutations with previously reported hinge-proximal Cγ2 mutations (S267E/L328F) that exhibit selective FcγRIIB-binding.24,25 We generated two novel IgG1 Fc mutants, V262E/S267E/L328F and V264E/S267E/L328F, which exhibited enhanced affinity for the FcγRIIB and, by comparison to the double mutant alone,24,25 significantly decreased affinity for FcγRIA, FcγRIIA, and FcγRIIIB (Figure 6; Table S3). The double mutant S267E/L328F shows significantly increased affinity for the inhibitory FcγRIIB and decreased affinity for the activatory FcγRIIIA, while the binding for the other FcγRs remains similar to the wild type (Figure 6), consistent with published data.25 The increased levels of glycan terminal processing and bisection of these triple mutants are comparable to those of the single V262E and V264E mutants (Figure S8).
Figure 6.

ELISA of monoclonal IgG variants binding human FcγRIA, FcγRIIA, FcγRIIB, FcγRIIIA and FcγRIIIB. The FcγRs were plated at 5 μg/mL overnight at 4 °C, IgG variants S267E/L328F, V262E/S267E/L328F, and V264E/S267E/L328F were incubated for 1.5 h and binding was detected by HRP-conjugated goat antihuman Fab antibody. Symbolic representation of IgG mutation and glycovariants: solid black square = wild-type native; solid blue square = wild-type Man5GlcNAc2; open black triangle = mutant native; open blue triangle = mutant Man5GlcNAc2, ELISA binding curves of the four IgG hydrophobic mutants for (A) FcγRIA, IgG variant starting concentration at 10 μg/mL. (B) FcγRIIA, IgG variant starting concentration at 100 μg/mL. (C) FcγRIIB, IgG variant starting concentration at 300 μg/mL. (D) FcγRIIIA, IgG variant starting concentration at 100 μg/mL. (E) FcγRIIIB, IgG variant starting concentration at 300 μg/mL. All data points represent the calculated mean of two independent measurements from a total of at least two experiments.
Affinity of Hypersialylated IgG1 Fc Mutants for FcγRIIIA
Our results indicate that destabilization of glycan–protein interactions modify the Fc structure relevant for FcγR binding (Figure 5). However, it has previously been shown that hypersialylation of IgG1 Fc, regardless of the linkage type, decreases Fc binding to the FcγRIA, FcγRIIB, and FcγRIIIA, which also reduces antibody-mediated cytotoxicity both in vivo and in vitro.11,49,50 Therefore, we generated hypergalactosylated and hypersialylated human IgG1 Fc (Figure 4) and examined their affinity for FcγRIIIA, a critical determinant of natural killer cell-mediated ADCC. Usually, the Fc–FcγR affinity strongly correlates with effector functions measured by cellular assays.17,24,25,51−54 However, our data indicate that the hypersialylated Fc binds to the FcγRIIIA with very similar affinity to the wild type, supported by both ELISA (Figure 5F) and surface plasmon resonance (SPR) data (Figure 7). One possible explanation for the reduced cytotoxicity is that terminal sialic acid might interact with inhibitory sialic acid binding Ig-like lectins (Siglec) present on the surface of immune cells including macrophages and natural killer cells.55 This minimal effect of Fc sialylation on FcγRIIIA binding is also supported by the structural analysis presented here, which reveals that the protein–glycan interface, rather than glycan terminal processing, modulates Cγ2 domain conformation (Figure 5).
Figure 7.
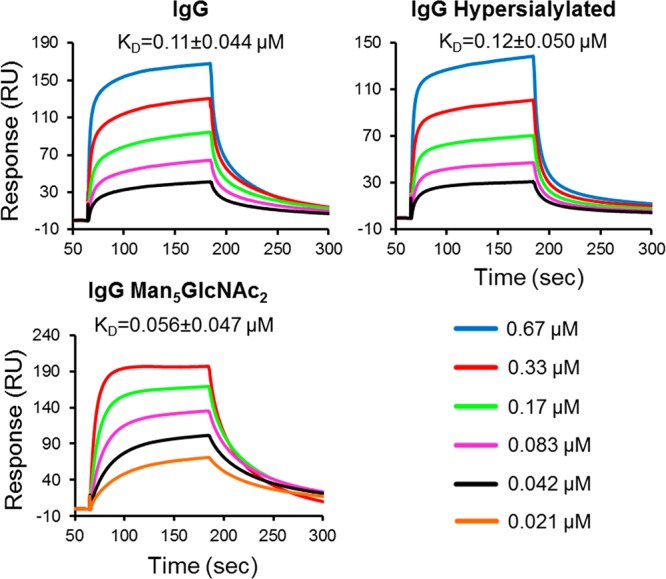
SPR analysis of monoclonal IgG variants binding to human FcγRIIIA. The human FcγRIIIA was immobilized on the CM5 sensorchip by amine coupling. The IgG variants were injected at 5 different concentrations at a flow rate of 30 μL/min: IgG and IgG hypersialylated (0.67, 0.33, 0.17, 0.083, and 0.042 μM); IgG Man5GlcNAc2 (0.33, 0.17, 0.083, 0.042, and 0.021 μM). The association time was 2 min, and dissociation time was 3 min. The chip was regenerated with 10 mM glycine-HCl, pH 1.7. Sensorgrams were fitted with a global 1:1 interaction, and the ka, kd, and KD were calculated, all using BIA evaluation software 2.0.3. KD values are reported as mean ± SD, and sensorgrams are representative a total of three independent experiments.
Conclusions
Modulation of the multiple hydrophobic interactions between the protein surface and the glycan within the IgG Fc strongly influences FcγR binding, presumably by altering the dynamics of the Fc and affecting the adoption of receptor binding conformations. While these mutations also modulate glycan processing, expression of Fc domains with homogenized glycoforms revealed that the interaction between the glycan and the protein independently influences FcγR binding. By combining interface mutations with glycan engineering, new portfolios of effector activities can be generated.
Experimental Procedures
Molecular Cloning and Mutagenesis
The pFUSE vector with the human IgG1 Fc insert was obtained from Invivogen, U.K. The vectors encoding IgG1 b12 light and heavy chains were kindly provided by Prof. Ian A. Wilson (The Scripps Research Institute, CA, U.S.A.).56 Protein mutagenesis was performed using the QuikChange kit (Agilent Technology, U.K.) to generate the IgG1 Fc mutant F241A and the full-length IgG1 b12 mutants F241A, F243A, V262E, and V264E. The mutated Fc (residues 225–447, SWISS-PROT accession number P01857.1) encompassing hinge, Cγ2 and Cγ3 domains was cloned into the mammalian expression vector, pHLSec. The vectors containing full-length FcγRIA, FcγRIIA (His131 variant), FcγRIIB, FcγRIIIA (Val158 variant), FcγRIIIB, and mouse β-1,4-galactosyltransferase I (B4GALTI) were all purchased from Open Biosystems, U.K. The vector containing full-length rat α2,6-sialyltransferase I (ST6GALI) was a gift from Prof. Karen Colley (University of Illinois, IL, U.S.A.). The soluble extracellular regions of each FcγR, B4GALTI and ST6GALI were cloned into the pHLSec vector as described for the Fc: FcγRIA (residues 16–288; SWISS-PROT accession number BC152383); FcγRIIA (residues 34–217; SWISS-PROT accession number BC020823); FcγRIIB (residues 42–225; SWISS-PROT accession number NM_001190828) FcγRIIIA (residues 16–288; SWISS-PROT accession number BC033678); FcγRIIIB (residues 17–200; SWISS-PROT accession number BC128562); ST6GALI (residues 89–403; SWISS-PROT accession number NP_001106815); and B4GALTI (residues 127–399; SWISS-PROT accession number BC053006). For B4GALTI, the residue Cys339 was mutated to Thr to minimize the potential for aggregation.57 The pHLSec vector encodes a hexahistidine tag at the C-terminus.58
Protein Expression
The Fc, FcγRs, B4GALTI, and ST6GALI were expressed in HEK 293T cells as previously described.58 Briefly, HEK 293T cells (ATCC number CRL-1573) were grown to 90% confluence and transiently transfected with polyethyleneimine (PEI),59 using a transfection mix with DNA and PEI in ratio of 1:1.5. Following transfection, cells were grown in DMEM/1% fetal bovine serum at 37 °C and 5% CO2 for 5 days. Protein was purified from cell supernatant by immobilized metal affinity chromatography using chelating sepharose fast flow Ni2+-agarose beads (GE Healthcare, U.K.) followed by size exclusion chromatography using a Superdex S-200 column equilibrated in phosphate buffered saline (PBS) (for FcγRs) or 10 mM HEPES pH 7.4, 150 mM NaCl (for Fc).
Full-length IgG1 b12 was transiently expressed in HEK 293T or GnT I-deficient HEK 293S cells.44,60 Prior to transfection, light and heavy chain plasmids were mixed in a mass ratio of 4:1, and the total DNA was mixed with PEI in a mass ratio of 1:1.5. After incubation for 4 days at 37 °C, cell culture supernatant was harvested and IgG1 b12 was purified using Protein A Sepharose (GE Healthcare, U.K.) according to the manufacturer’s directions.
Enzymatic Release of N-Linked Glycans
Oligosaccharides were released from Coomassie blue-stained reducing SDS-PAGE gel bands containing ∼40 μg of IgG Fc as previously reported.61 Gel bands were excised, washed with acetonitrile and water, and dried. Gel bands were rehydrated with 30 μL of 30 mM NaHCO3 pH 7.0 containing 100 units/mL PNGase F (New England Biolabs, U.K.) and incubated for 12 h at 37 °C.61 The enzymatically released N-linked glycans were eluted with water. Desialylation was carried out using linkage nonspecific neuraminidase from Clostridium perfringens (New England Biolabs, U.K.) for 48 h at 37 °C.
Structural Determination of N-Glycans
Glycans were labeled with anthranilic acid (2-AA) as previously described62 and separated using TSK amide column (Sigma-Aldrich, U.K.). The released N-glycans, dissolved in water and 2-AA labeling buffer (3/8, v/v) were mixed with 2-AA and sodium cyanoborohydride and incubated for 1 h at 80 °C; excess 2-AA dye was removed using a Spe-ed Amide-2 column (Systematic Systems, U.K.). HPLC was carried out in a linear gradient of solvents at room temperature. Solvent A was acetonitrile, solvent B was Milli-Q water, and solvent C was 800 mM ammonium hydroxide adjusted to pH 3.85 using acetic acid. Solvent C was in a constant gradient of 2.5% throughout the run. The gradient was a constant 71.6% A for 6 min at a flow rate of 0.8 mL/min, followed by a linear gradient of 71.6–35% A over 80 min at 0.8 mL/min. Afterward, the gradient was a linear 35–71.6% A for 1 min at a flow rate of 0.8 mL/min; then at the same gradient, the flow rate increased from 0.8 mL/min to 1.2 mL/min over 1 min and followed by the same gradient and flow rate for 13 min. The run finished by returning the flow rate to 0.8 mL/min over 1 min. Fluorescence was detected at 425 nm, and the excitation wavelength was 360 nm. Chromatography data were processed by the Empower software (all instruments and software from Interlink Scientific Services Limited, U.K.). Assignments were consistent with previously reported serum IgG N-linked glycan profiles30 and were confirmed by MALDI-TOF MS and exoglycosidase analysis.
In vitro Modulation of IgG Glycosylation
Hyper-α2,6-sialylated IgG was generated by incubating with B4GALTI in the presence of 80 μM uridine 5′-diphosphogalactose (Sigma-Aldrich, U.K.) in 50 mM HEPES, 10 mM MnCl2, pH 7.5 for 48 h at 37 °C. The hyper-β1,4-galactosylated IgG was treated with ST6GALI in the presence of 70 μM cytidine-5′-monophospho-N-acetylneuraminic acid (Sigma-Aldrich, U.K.) in 50 mM HEPES, 10 mM MnCl2, pH 6.5 for 48 h at 37 °C. The composition of the glycoform was verified by HPLC analysis after each enzymatic treatment. IgG deglycosylation was confirmed by a protein band shift in SDS-PAGE.
Crystallization and Structure Determination
Recombinantly expressed mutant IgG1 Fc (F241A) was concentrated to 7.0 mg/mL and crystallized after 10 days using the sitting drop vapor diffusion method using 100 nL protein plus 100 nL precipitant equilibrated against 95 μL reservoirs. Crystals of F241A IgG1 Fc grew at room temperature in a precipitant containing 28% polyethylene glycol monomethyl ether 2000 in 0.1 M BIS-TRIS buffer at pH 6.5. Crystals were flash frozen by immersion in a cryoprotectant containing the mother liquor diluted in 30% polyethylene glycol and then rapidly transferred to a gaseous nitrogen stream. Crystallographic data were collected to 1.9 Å resolution at beamline I04 at the Diamond Light Source (Oxfordshire, U.K.). Images were indexed, integrated, and scaled using HKL2000.63 The structure was solved using molecular replacement with the program PHASER64 using native Fc (PDB accession no. 3AVE) as a search model. Model building was performed with COOT65 and iteratively refined using restrained refinement in the CCP4 supported program, Refmac5, with the incorporation of translation-libration-screw (TLS) parametrization and automatically generated local noncrystallographic symmetry restraints.66Model quality was validated with Molprobity.67 Data processing and refinement statistics are presented in Table 1.
FcγR Binding Assays
Recombinant FcγRs at 5 μg/mL in PBS were coated on high-binding microtiter plates (3690, Corning, NY, U.S.A.) overnight at 4 °C. Coated plates were washed with PBS containing 0.05% Tween 20 (Sigma-Aldrich, U.S.A.) and blocked for 1 h at room temperature with 5% bovine serum albumin (BSA) in PBS. Recombinant IgG1 b12 expressed from HEK 293T cells or from GnT I-deficient HEK 293S cells were then added and allowed to bind for 1.5 h at room temperature. Plates were washed five times with PBS containing 0.05% Tween and binding was detected using a horse radish peroxidase (HRP)-conjugated Fab fragment specific for Human IgG Fab (Abcam, U.K.). The 3,3′,5,5′-tetramethylbenzidine substrate (TMB; Thermo Scientific, U.S.A.) was used for development according to the manufacturer’s directions and was stopped by the addition of 2 M H2SO4. Absorbance was measured at 450 nm on a Spectramax M5 (Molecular Devices, California, U.S.A.) multiwell plate reader. Apparent affinity was calculated as the concentration of IgG1 b12 corresponding to half-maximal binding on the ELISA binding curve.
Surface Plasmon Resonance
SPR experiments were carried out using the BIAcore T100 instrument (GE Healthcare, U.K.). Briefly, the FcγRIIIA (Val158 variant) was immobilized onto the surface of CM5 sensor chip (GE Healthcare, U.K.) to about 1000 RU during each independent experiment. All experiments were carried out in the HBS-EP running buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA and 0.005% surfactant P20), at a flow rate of 30 μL/min. The monoclonal IgG1 b12 protein and the hypersialylated and high-mannose glycol variants were injected at 5 different concentrations, allowing 2 min for association and 3 min for dissociation. After each run, the sensor chip was regenerated using 10 mM glycine-HCl, pH1.7. The sensorgrams were fitted to a global 1:1 interaction, and the ka, kd, and KD were calculated, all using BIAevaluation software 2.0.3 (GE Healthcare, U.K.).
Acknowledgments
We thank Prof. James Scrivens, Biological Sciences, University of Warwick for use of the Synapt G2 electrospray mass spectrometer, Dr. Charlotte Scarff for advice on MS data acquisition, Prof. Elspeth Garman and the staff of beamline I04 for assistance with crystallographic data collection, and Dr. Mark Wormald and Prof. Raymond Dwek for helpful discussions. T.A.B. is a Sir Henry Wellcome Postdoctoral Fellow (grant no. 089026/Z/09/Z) and Junior Research Fellow at University College, Oxford. We thank the Wellcome Trust, International AIDS Vaccine Initiative, and the Scripps Korea Antibody Institute for funding. X.Y. gratefully acknowledges the financial support of Mr. Qijun Yu and Ms. Peiqing Hu and a scholarship awarded by the Oxford Glycobiology Institute. The atomic coordinates and crystallographic structure factors of the F241A IgG1 Fc have been deposited in the Protein Data Bank (PDB) with accession code 4BM7.
Supporting Information Available
Additional data and methods. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Status
⊥ Deceased.
Supplementary Material
References
- Nimmerjahn F.; Ravetch J. V. Annu. Rev. Immunol. 2008, 26, 513. [DOI] [PubMed] [Google Scholar]
- Ravetch J. V.; Bolland S. Annu. Rev. Immunol. 2001, 19, 275. [DOI] [PubMed] [Google Scholar]
- Krapp S.; Mimura Y.; Jefferis R.; Huber R.; Sondermann P. J. Mol. Biol. 2003, 325, 979. [DOI] [PubMed] [Google Scholar]
- Deisenhofer J. Biochemistry 1981, 20, 2361. [PubMed] [Google Scholar]
- Borrok M. J.; Jung S. T.; Kang T. H.; Monzingo A. F.; Georgiou G. ACS Chem. Biol. 2012, 7, 1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardi E.; Holdom M. D.; Davies A. M.; Sutton B. J.; Beavil A. J. Biochem. J. 2009, 417, 77. [DOI] [PubMed] [Google Scholar]
- Nose M.; Wigzell H. Proc. Natl. Acad. Sci. U.S.A. 1983, 80, 6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazinsky S. L.; Ott R. G.; Silver N. W.; Tidor B.; Ravetch J. V.; Wittrup K. D. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 20167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa R.; Shoji-Hosaka E.; Sakurada M.; Shinkawa T.; Uchida K.; Nakamura K.; Matsushima K.; Ueda R.; Hanai N.; Shitara K. Cancer Res. 2004, 64, 2127. [DOI] [PubMed] [Google Scholar]
- Mori K.; Iida S.; Yamane-Ohnuki N.; Kanda Y.; Kuni-Kamochi R.; Nakano R.; Imai-Nishiya H.; Okazaki A.; Shinkawa T.; Natsume A.; Niwa R.; Shitara K.; Satoh M. Cytotechnology 2007, 55, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scallon B. J.; Tam S. H.; McCarthy S. G.; Cai A. N.; Raju T. S. Mol. Immunol. 2007, 44, 1524. [DOI] [PubMed] [Google Scholar]
- Ferrara C.; Grau S.; Jäger C.; Sondermann P.; Brünker P.; Waldhauer I.; Hennig M.; Ruf A.; Rufer A. C.; Stihle M.; Umaña P.; Benz J. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 12669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony R. M.; Ravetch J. V. J. Clin. Immunol. 2010, 30Suppl 1S9. [DOI] [PubMed] [Google Scholar]
- Ferrara C.; Stuart F.; Sondermann P.; Brunker P.; Umana P. J. Biol. Chem. 2006, 281, 5032. [DOI] [PubMed] [Google Scholar]
- Jefferis R. Nat. Rev. Drug discovery 2009, 8, 226. [DOI] [PubMed] [Google Scholar]
- Niwa R.; Natsume A.; Uehara A.; Wakitani M.; Iida S.; Uchida K.; Satoh M.; Shitara K. J. Immunol. Methods 2005, 306, 151. [DOI] [PubMed] [Google Scholar]
- Forthal D. N.; Gach J. S.; Landucci G.; Jez J.; Strasser R.; Kunert R.; Steinkellner H. J. Immunol. 2010, 185, 6876. [DOI] [PubMed] [Google Scholar]
- Matsumiya S.; Yamaguchi Y.; Saito J.; Nagano M.; Sasakawa H.; Otaki S.; Satoh M.; Shitara K.; Kato K. J. Mol. Biol. 2007, 368, 767. [DOI] [PubMed] [Google Scholar]
- Radaev S.; Motyka S.; Fridman W. H.; Sautes-Fridman C.; Sun P. D. J. Biol. Chem. 2001, 276, 16469. [DOI] [PubMed] [Google Scholar]
- Bowden T. A.; Baruah K.; Coles C. H.; Harvey D. J.; Yu X.; Song B. D.; Stuart D. I.; Aricescu A. R.; Scanlan C. N.; Jones E. Y.; Crispin M. J. Am. Chem. Soc. 2012, 134, 17554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crispin M.; Bowden T. A.; Coles C. H.; Harlos K.; Aricescu A. R.; Harvey D. J.; Stuart D. I.; Jones E. Y. J. Mol. Biol. 2009, 387, 1061. [DOI] [PubMed] [Google Scholar]
- Feige M. J.; Nath S.; Catharino S. R.; Weinfurtner D.; Steinbacher S.; Buchner J. J. Mol. Biol. 2009, 391, 599. [DOI] [PubMed] [Google Scholar]
- Baruah K.; Bowden T. A.; Krishna B. A.; Dwek R. A.; Crispin M.; Scanlan C. N. J. Mol. Biol. 2012, 420, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields R. L.; Namenuk A. K.; Hong K.; Meng Y. G.; Rae J.; Briggs J.; Xie D.; Lai J.; Stadlen A.; Li B.; Fox J. A.; Presta L. G. J. Biol. Chem. 2001, 276, 6591. [DOI] [PubMed] [Google Scholar]
- Chu S. Y.; Vostiar I.; Karki S.; Moore G. L.; Lazar G. A.; Pong E.; Joyce P. F.; Szymkowski D. E.; Desjarlais J. R. Mol. Immunol. 2008, 45, 3926. [DOI] [PubMed] [Google Scholar]
- Zou G.; Ochiai H.; Huang W.; Yang Q.; Li C.; Wang L. X. J. Am. Chem. Soc. 2011, 133, 18975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda Y.; Yamada T.; Mori K.; Okazaki A.; Inoue M.; Kitajima-Miyama K.; Kuni-Kamochi R.; Nakano R.; Yano K.; Kakita S.; Shitara K.; Satoh M. Glycobiology 2007, 17, 104. [DOI] [PubMed] [Google Scholar]
- Lund J.; Takahashi N.; Pound J. D.; Goodall M.; Jefferis R. J. Immunol. 1996, 157, 4963. [PubMed] [Google Scholar]
- Stewart R.; Thom G.; Levens M.; Guler-Gane G.; Holgate R.; Rudd P. M.; Webster C.; Jermutus L.; Lund J. Protein Eng. Des. Sel. 2011, 24, 671. [DOI] [PubMed] [Google Scholar]
- Arnold J. N.; Wormald M. R.; Sim R. B.; Rudd P. M.; Dwek R. A. Annu. Rev. Immunol. 2007, 25, 21. [DOI] [PubMed] [Google Scholar]
- Rudd P. M.; Dwek R. A. Crit. Rev. Biochem. Mol. Biol. 1997, 32, 1. [DOI] [PubMed] [Google Scholar]
- Wormald M. R.; Rudd P. M.; Harvey D. J.; Chang S. C.; Scragg I. G.; Dwek R. A. Biochemistry 1997, 36, 1370. [DOI] [PubMed] [Google Scholar]
- Barb A. W.; Prestegard J. H. Nat. Chem. Biol. 2011, 7, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barb A. W.; Meng L.; Gao Z.; Johnson R. W.; Moremen K. W.; Prestegard J. H. Biochemistry 2012, 51, 4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voynov V.; Chennamsetty N.; Kayser V.; Helk B.; Forrer K.; Zhang H.; Fritsch C.; Heine H.; Trout B. L. PLoS One 2009, 4, e8425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jassal R.; Jenkins N.; Charlwood J.; Camilleri P.; Jefferis R.; Lund J. Biochem. Biophys. Res. Commun. 2001, 286, 243. [DOI] [PubMed] [Google Scholar]
- Davis I. W.; Leaver-Fay A.; Chen V. B.; Block J. N.; Kapral G. J.; Wang X.; Murray L. W.; Arendall W. B. 3rd; Snoeyink J.; Richardson J. S.; Richardson D. C. Nucleic Acids Res. 2007, 35, W375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju T. S. Curr. Opin. Immunol. 2008, 20, 471. [DOI] [PubMed] [Google Scholar]
- Domon B.; Costello C. E. Glycoconj. J. 1988, 5, 397. [Google Scholar]
- Vliegenthart J. F. G.; Dorland L.; van Halbeek H. Adv. Carbohydr. Chem. Biochem. 1983, 41, 209. [Google Scholar]
- Dwek R. A.; Lellouch A. C.; Wormald M. R. J. Anat. 1995, 187Pt 2279. [PMC free article] [PubMed] [Google Scholar]
- Harvey D. J.; Merry A. H.; Royle L.; Campbell M. P.; Dwek R. A.; Rudd P. M. Proteomics 2009, 9, 3796. [DOI] [PubMed] [Google Scholar]
- Kabsch W.; Sander C. Biopolymers 1983, 22, 2577. [DOI] [PubMed] [Google Scholar]
- Reeves P. J.; Callewaert N.; Contreras R.; Khorana H. G. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua S.; Jeong H. N.; Dimapasoc L. M.; Kang I.; Han C.; Choi J. S.; Lebrilla C. B.; An H. J. Anal. Chem. 2013, 85, 4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S. Y.; Chen Y. Y.; Fan Y. Y.; Lin C. W.; Chen S. T.; Wang A. H.; Khoo K. H. J. Proteome Res. 2008, 7, 3293. [DOI] [PubMed] [Google Scholar]
- Crispin M.; Harvey D. J.; Chang V. T.; Yu C.; Aricescu A. R.; Jones E. Y.; Davis S. J.; Dwek R. A.; Rudd P. M. Glycobiology 2006, 16, 748. [DOI] [PubMed] [Google Scholar]
- Ferrara C.; Grau S.; Jager C.; Sondermann P.; Brunker P.; Waldhauer I.; Hennig M.; Ruf A.; Rufer A. C.; Stihle M.; Umana P.; Benz J. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 12669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko Y.; Nimmerjahn F.; Ravetch J. V. Science 2006, 313, 670. [DOI] [PubMed] [Google Scholar]
- Lu J.; Ellsworth J. L.; Hamacher N.; Oak S. W.; Sun P. D. J. Biol. Chem. 2011, 286, 40608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida S.; Misaka H.; Inoue M.; Shibata M.; Nakano R.; Yamane-Ohnuki N.; Wakitani M.; Yano K.; Shitara K.; Satoh M. Clin. Cancer Res. 2006, 12, 2879. [DOI] [PubMed] [Google Scholar]
- Shields R. L.; Lai J.; Keck R.; O’Connell L. Y.; Hong K.; Meng Y. G.; Weikert S. H.; Presta L. G. J. Biol. Chem. 2002, 277, 26733. [DOI] [PubMed] [Google Scholar]
- Dall’Acqua W. F.; Cook K. E.; Damschroder M. M.; Woods R. M.; Wu H. J. Immunol. 2006, 177, 1129. [DOI] [PubMed] [Google Scholar]
- Horton H. M.; Bernett M. J.; Pong E.; Peipp M.; Karki S.; Chu S. Y.; Richards J. O.; Vostiar I.; Joyce P. F.; Repp R.; Desjarlais J. R.; Zhukovsky E. A. Cancer Res. 2008, 68, 8049. [DOI] [PubMed] [Google Scholar]
- Crocker P. R. Curr. Opin. Pharmacol. 2005, 5, 431. [DOI] [PubMed] [Google Scholar]
- Roben P.; Moore J. P.; Thali M.; Sodroski J.; Barbas C. F. 3rd; Burton D. R. J. Virol. 1994, 68, 4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasamy V.; Ramakrishnan B.; Boeggeman E.; Ratner D. M.; Seeberger P. H.; Qasba P. K. J. Mol. Biol. 2005, 353, 53. [DOI] [PubMed] [Google Scholar]
- Aricescu A. R.; Lu W.; Jones E. Y. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 1243. [DOI] [PubMed] [Google Scholar]
- Durocher Y.; Perret S.; Kamen A. Nucleic Acids Res. 2002, 30, E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang V. T.; Crispin M.; Aricescu A. R.; Harvey D. J.; Nettleship J. E.; Fennelly J. A.; Yu C.; Boles K. S.; Evans E. J.; Stuart D. I.; Dwek R. A.; Jones E. Y.; Owens R. J.; Davis S. J. Structure 2007, 15, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küster B.; Wheeler S. F.; Hunter A. P.; Dwek R. A.; Harvey D. J. Anal. Biochem. 1997, 250, 82. [DOI] [PubMed] [Google Scholar]
- Neville D. C.; Coquard V.; Priestman D. A.; te Vruchte D. J.; Sillence D. J.; Dwek R. A.; Platt F. M.; Butters T. D. Anal. Biochem. 2004, 331, 275. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z M. W.Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology; Carter C. W. Jr, Sweet R. M., Eds.; Academic Press: Waltham, MA, 1997; Vol. 276, p 306. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Gross-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. J. Appl. Crystallogr. 2007, 40, 658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4.Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994, 50, 760.15299374 [Google Scholar]
- Davis I. W.; Leaver-Fay A.; Chen V. B.; Block J. N.; Kapral G. J.; Wang X.; Murray L. W.; Arendall W. B. 3rd; Snoeyink J.; Richardson J. S.; Richardson D. C. Nucleic Acids Res. 2007, 35, W375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.