

Abstract
The catalytic (C) subunit of protein kinase A is regulated in part by cotranslational N-myristylation and ligand binding. Using a combination of time-resolved fluorescence anisotropy and molecular dynamics (MD) simulations, we characterized the effect of N-myristylation and ligand binding on C-subunit dynamics. Five single-site cysteine-substitution mutants of the C-subunit were engineered with and without N-terminal myristylation and labeled with fluorescein maleimide, and time-resolved fluorescence anisotropy decays were measured to assess the flexibility of the labeled regions in the presence and absence of ligands. A parallel set of in silico experiments were performed to complement the experimental findings. These experiments showed that myristylation produces both local and global effects on C-subunit dynamics. The local effects include stabilization of the N-terminus and myristate pocket, and the global effects include small increases in mobility along the C-tail at residue C343. Additionally, ligand binding was associated with an increase in mobility of the myristate binding pocket for both the myristylated and nonmyristylated enzyme on the basis of both the experimental and MD results. Also, MD simulations suggest that the myristylated protein exhibits increased dynamics when bound to ligands compared to the nonmyristylated protein.
The catalytic (C) subunit of cAMP-dependent protein kinase (PKA) is a Ser/Thr kinase that mediates many cAMP signaling functions.1 At low concentrations of cAMP, PKA exists as a heterotetrameric holoenzyme composed of an R-subunit homodimer that binds and inactivates two C-subunit monomers.2 At high concentrations, cAMP binds to the R-subunit dimer to induce a conformational change that reduces the affinity of the R-subunit toward the C-subunit and leads to C-subunit activation.3 The C-subunit has served as an important model for the protein kinase family not only because it was the first kinase whose structure was solved4 but also because its regulatory component is separate from its catalytic component, and therefore, it can be studied independently.
Protein kinase structures include a bilobal structure comprised of the N-lobe (or small lobe) and C-lobe (or large lobe).4 ATP binds in the cleft formed between these lobes, thereby coupling them.5,6 Crystal structures of the C-subunit of PKA reveal multiple conformations of the enzyme termed “open”, “closed”, and “intermediate” on the basis of the relative orientations of the small and large lobes with respect to each other.7 The open conformation is typically observed with the apo C-subunit.8,9 The closed conformation is typically observed when nucleotide and IP20 are crystallized with the protein,5,10 and the intermediate conformation is observed when the C-subunit is crystallized with adenosine alone.11 The C-subunit is regulated by noncovalent ligand and protein binding as well as covalent co- and post-translational modifications. For example, the N-terminus undergoes cotranslational N-myristylation of Gly1, phosphorylation of Ser10, and deamidation of Asn2,12 which may regulate C-subunit interaction with membranes or subcellular sites.12−14 Additionally, N-myristylation increases the structural stability globally15 and locally near the myristylation site.16−18
To further assess how myristylation and ligand binding influence C-subunit dynamics, a combination of time-resolved fluorescence anisotropy and molecular dynamics (MD) simulations was utilized. Specifically, five single-site cysteine-substitution C-subunit mutants were engineered and conjugated with fluorescein maleimide (FM). The cysteine mutations were placed along the A-helix at the N-terminus of the protein (K7C and K16C) near the myristate pocket (N99C), and at the C-tail (S325C and native C343) (Figure 1A–C). Additionally, the mutants were prepared in both myristylated and nonmyristylated configurations, and the time-resolved fluorescence anisotropy measurements were performed in the absence of ligands (apo/open state) and in the presence of ATP plus IP20 (ternary/closed state). Parallel molecular dynamics (MD) simulations were performed on the myristylated and nonmyristylated C-subunit in the presence and absence of ligands with a recent crystal structure of the N-myristylated protein.16 These fluorescence and MD experiments identified a stabilizing effect of myristylation at the N-terminus and myristate pocket and identified long-range allosteric effects of this modification and ligand binding.
Figure 1.

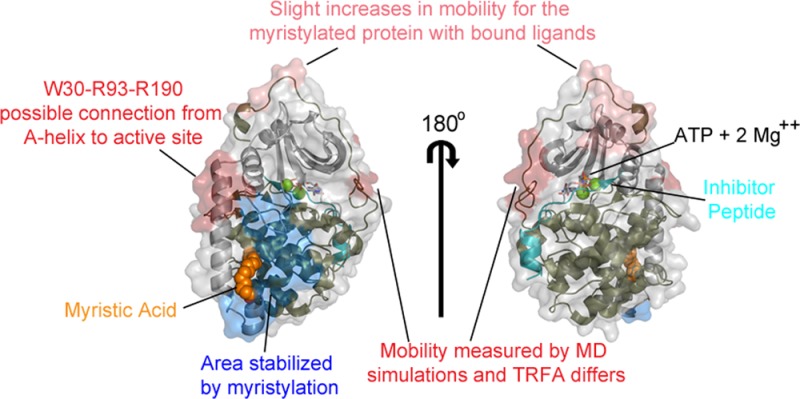
Sites of FM conjugation in the C-subunit. (A) The structure of the N-myristylated C-subunit of PKA (PDB: 4DFX(16)) is displayed in cartoon representation with the small lobe (1–126) in gray, large lobe (127–300) in olive, C-tail (301–350) in red, inhibitor peptide in cyan, myristic acid in orange as a stick representation, and sites of mutation in yellow as a stick and surface representation. (B) A stereoview of the A-helix and myristate pocket is shown in cartoon representation with the regions of the protein and sites of FM conjugation depicted and colored as in (A). Several other residues within or near the myristate pocket are also shown in stick representation, and myristic acid is shown in sphere representation. (C) A stereoview of regions of the protein near the other sites of labeling is shown in cartoon representation with the regions of the protein and sites of fluorescent labeling depicted and colored as in (A).
Experimental Procedures
Purification of the C-Subunit Proteins
All mutations were generated using a Quikchange mutagenesis kit (Stratagene). The K7C and K16C mutations were introduced into the WT C-subunit vector. The N99C and S325C mutations were introduced into vectors containing a C199A mutation. Finally, the C199A mutation was introduced into the WT vector so that the endogenous C343 could be selectively labeled. The nonmyristylated C-subunit was expressed and purified as described previously.19 The myristylated C-subunit was prepared by coexpression with yeast NMT as described previously20 and purified using a protocol described previously.16 Three of these mutations, K7C, K16C, and C199A were characterized previously and shown not to negatively affect C-subunit activity,16,21 and the activity of the other mutants, N99C and S325C, were tested with a spectrophotometric assay as described by Cook et al. with the soluble substrate kemptide (LRRASLG), which showed similar levels of activity as the WT protein (data not shown), suggesting that the mutations did not alter normal C-subunit function.22
Fluorescein Maleimide Labeling
The C-subunit samples (12.5–25 nmol) were initially buffer exchanged by elution through a PD10 column (GE Healthcare Bio-Sciences Corp., Piscataway, NJ) equilibrated with 20 mM MOPS and 150 mM KCl, pH 7.0, at 4 °C. The protein fractions were pooled, and the concentrations of the pooled samples were determined by spectrophotometry using an A280 extinction coefficient of 45 000 M–1 cm–1 and ranged between 3 and 6 μM. With the exception of the C199A mutant labeling, the reaction mixtures were made with 4 mM ATP and 8 mM MgCl2 to protect from labeling the endogenous cysteine residues (C199 and C343) because it was previously shown that the presence of MgATP prevents labeling of these native cysteine residues in PKA.23 For the N-terminal mutants, the stoichiometry of FM to protein was 2:1, and for the C199A mutants, it was 3:1. The reactions were allowed to proceed for 1 h at room temperature, protected from light, and then eluted through a PD10 column equilibrated with 20 mM MOPS, 150 mM KCl, and 5 mM β-ME, pH 7.0, at 4 °C. Absorbance measurements at 280 and 495 nm were made on each column fraction, and the light absorbing fractions with retention times that corresponded to unmodified catalytic subunit were pooled. The concentration of labeled protein was determined by spectrophotometry using an A495 extinction coefficient of 65 000 M–1 cm–1, and the concentration of total protein (labeled and unlabeled) was calculated with the expression
| 1 |
Time-Resolved Fluorescence Anisotropy
Emission anisotropy was determined as previously described.24 Samples were excited by using a 484 nm laser diode from HORIBA Jobin Yvon Inc. (Edison, New Jersey, NJ) and a Semrock Inc. (Rochester, NY) single-band bandpass filter (FF01-482/35-25) in the excitation pathway to enhance its monochromaticity. A Semrock Inc. single-band bandpass filter (FF01-536/40-25) selected the emission and rotatable Glan–Thompson polarizers were placed in the excitation and emission pathways. Unless stated otherwise, emission anisotropy decays were analyzed with the impulse reconvolution method implemented in the DAS6 software package from HORIBA Jobin Yvon Inc. (Edison New Jersey NJ) described elsewhere.25 Briefly and simply, this approach splits the analysis into two steps: analysis of the total emission decay, S(t), from the vertically, I||(t), and orthogonally, I⊥(t), polarized emission components followed by analysis of the vertical/perpendicular difference emission decay, D(t). S(t) is the sum of the three axes of emission and is free of anisotropy effects. It is given by the expression
| 2 |
and is analyzed as a biexponential function. G is a measure of the instrumental polarization bias for each sample, and the values ranged between 0.97 and 0.995 depending on the instrument configuration. D(t), which includes both fluorescence and anisotropy parameters, is given by the expression
| 3 |
D(t) is deconvolved with the results from the S(t) analysis as a constraint yielding
| 4 |
where r(t) is the time-dependent anisotropy.
Here, β1 and β2 are the amplitudes of the anisotropy at time zero for the fast and slow anisotropy decay processes, respectively. Φfast and Φslow are the fast and slow rotational correlation times of the anisotropy decay, respectively. Φslow usually yields an estimate of the whole-body rotational correlation time when it is less than about five times the emission lifetime, and when the rate of the segmental motions around the site of reporter group conjugation differs greatly from the rate of the whole-body diffusion. The accuracy of this estimate increases with the proportion of the total decay that is associated with the slow decay processes. A nonassociative model was assumed, where the emission relaxation times are common to all the rotational correlation times. Goodness of fit was evaluated from the values of the reduced χ2r and by visual inspection of the weighted-residual plots. All time-resolved anisotropy measurements were performed with samples suspended in 20 mM MOPS, 150 mM KCl, and 5 mM β-ME, pH 7.0, at 22 °C.
Molecular Dynamics Simulations
System Preparation
Four unique systems were set up based on the X-ray refined structure with PDB ID 4DFX.16 The four systems are defined to have the following configurations: a myristylated N-termini with ATP and SP20 bound, nonmyristylated N-termini with ATP and SP20 bound, myristylated N-termini without ATP and SP20 bound, and nonmyristylated N-termini without ATP and SP20 bound. The myristylated glycine was prepared by first geometry optimizing with Gaussian (g03),26 with B3LYP and the 6-31G(d) basis set. Electrostatic potential fits were obtained from Gaussian using the geometry optimized structure and Hartree–Fock level of theory with the 6-31G* basis set. The Restrained Electrostatic Potential (RESP) module within the Antechamber program in AmberTools27,28 was used to assign charges to the atoms using the Gaussian output. The H++ web-server29 was used to predict the protonation state of the residues in the structure at a pH of 7. Only one histidine residue had a particularly low pKa in PKA and was modified to be a charged HIP residue. Other histidines were designated as either HIE or HID (with the ϵ or δ nitrogen protonated, respectively) by evaluating the local environments of each side chain in the crystal structure.
Molecular Dynamics Details
Solvation and Equilibration
The systems were prepared using the AMBER ff99SB30 force field, with the ildn modification for Ile, Leu, Asp, and Asn residues.31 Each system was solvated in a 10.0 Å TIP3P box, and sodium ions were added to neutralize the charge. Systems were heated to 300 K at constant volume (NVT) with restraints on the protein that were gradually reduced from 200 to 0 kcal/(mol Å2) over a period of 150 ps. The langevin thermostat was used with a collision frequency of 1.0 ps–1. The SHAKE algorithm32 was used to constrain bonds to nonpolar hydrogens, and a 2.0 fs time-step was used during dynamics. A 10.0 Å cutoff was used for nonbonded interactions and the Particle Mesh Ewald method33 was used for calculation of long-range electrostatics. Equilibration runs (100 ps) were done in the NPT ensemble using isotropic pressure scaling and a pressure relaxation time of 2.0 ps.
Production Simulations
For each system, six independent simulations were started from different snapshots from the equilibration simulations (taken at arbitrary intervals after the density of the water box was equilibrated) with randomized velocities. All production runs were conducted in the same NVT conditions as equilibration. All simulations were performed using the PMEMD module within the Amber11 and 12 simulation packages.27,28,34 Production runs, each 150 ns in length, were performed on GTX580 GPUs using the pmemd.CUDA engine.35−37
Results
Myristylation Stabilizes the Most N-Terminal Residues of the C-Subunit
Focusing first on the N-terminal dynamics of the C-subunit, the anisotropy decay results with the FM-K7C and FM-K16C conjugates, with and without N-terminal myristylation, are illustrated in Figure 2 panels A and B, and the decay parameters are summarized in Table 1. Myristylation was only associated with large effects on the anisotropy decay of FM-K7C, where myristylation increased the β2 parameter (amplitude of slower decay processes) from 0.092 to 0.142 in the apo state and from 0.103 to 0.157 in the ternary state (Table 1, Figure 2A), suggesting a significant reduction in the angular excursion of the reporter, which is consistent with the myristyl moiety binding to the hydrophobic pocket in the core of the C-subunit observed in the crystal structure. Myristylation produced a small increase in the anisotropy decay of FM-K16C, as evidenced by a reduction of the β2 parameter from 0.201 to 0.185 in the apo state and from 0.185 to 0.174 in the ternary state (Figure 2B, Table 1). There were modest changes in the decay parameters at the K7C and K16C sites upon formation of a ternary complex that are likely not significant. Still, ligand binding and closed state formation caused a slight decrease in mobility near the K7C site on the basis of increases in the β2 parameter from 0.092 to 0.103 and from 0.142 to 0.157 for the nonmyristylated and myristylated protein, respectively. For K16C, ligand binding showed slight increases in mobility on the basis of the β2 parameter decreasing from 0.201 to 0.174 and from 0.185 to 0.169 going from apo to ternary states for the nonmyristylated and myristylated protein, respectively.
Figure 2.

Myristylation stabilizes the N-terminus of the C-subunit, measured via time-resolved fluorescence anisotropy and MD simulations. (A) Anisotropy decay of FM-K7C in myristylated/apo state (blue), nonmyristylated/apo state (red), myristylated/ternary state (orange), and nonmyristylated/ternary state (green). IRF is the instrument response function. (B) Anisotropy decay of FM-K16C with the different states colored as in (A). (C) The root mean squared fluctuation (RMSF) values averaged from six replicate MD simulations of myristylated and nonmyristylated configurations with and without ligands, plotted in angstroms for the A-helix residues (2–30).
Table 1. Time-Resolved Fluorescence Anisotropy Parameters of the FM-Conjugates Free in Solution (Apo) or Bound to MgATP and IP20 (Ternary).
| protein | β1a | β2b | Φfast (ns)c | Φslow (ns)d | χ2re | τf |
|---|---|---|---|---|---|---|
| FM-K7C (apo) | 0.130 ± 0.019 | 0.092 ± 0.004 | 0.6 ± 0.2 | 11.9 ± 4.0 | 1.4 ± 0.2 | 3.8 ± 0.2 |
| FM-Myr-K7C (apo) | 0.097 ± 0.007 | 0.142 ± 0.012 | 0.6 ± 0.1 | 14.1 ± 2.3 | 1.5 ± 0.3 | 3.6 ± 0.1 |
| FM-K7C (ternary) | 0.117 ± 0.011 | 0.103 ± 0.001 | 0.5 ± 0.1 | 12.2 ± 0.2 | 1.8 ± 0.0 | 3.7 ± 0.2 |
| FM-Myr-K7C (ternary) | 0.089 ± 0.003 | 0.157 ± 0.001 | 0.7 ± 0.1 | 17.3 ± 0.3 | 1.5 ± 0.3 | 2.8 ± 1.8 |
| FM-K16C (apo) | 0.069 ± 0.007 | 0.201 ± 0.005 | 0.5 ± 0.2 | 14.2 ± 2.4 | 1.2 ± 0.2 | 3.5 ± 0.2 |
| FM-Myr-K16C (apo) | 0.085 ± 0.009 | 0.185 ± 0.005 | 0.5 ± 0.2 | 14.0 ± 2.4 | 1.5 ± 0.4 | 3.8 ± 0.2 |
| FM-K16C (ternary) | 0.068 ± 0.018 | 0.174 ± 0.020 | 0.7 ± 0.2 | 19.1 ± 0.6 | 1.1 ± 0.0 | 3.9 ± 0.0 |
| FM-Myr-K16C (ternary) | 0.093 ± 0.002 | 0.169 ± 0.000 | 0.5 ± 0.1 | 20.3 ± 0.9 | 1.1 ± 0.1 | 4.3 ± 0.0 |
| FM-N99C (apo) | 0.099 ± 0.011 | 0.158 ± 0.024 | 0.6 ± 0.1 | 18.3 ± 2.6 | 1.1 ± 0.1 | 4.2 ± 0.0 |
| FM-Myr-N99C (apo) | 0.057 ± 0.017 | 0.200 ± 0.022 | 1.6 ± 0.3 | 22.8 ± 1.3 | 1.0 ± 0.1 | 4.3 ± 0.2 |
| FM-N99C (ternary) | 0.135 ± 0.026 | 0.120 ± 0.020 | 0.3 ± 0.2 | 17.9 ± 2.2 | 1.2 ± 0.1 | 4.2 ± 0.0 |
| FM-Myr-N99C (ternary) | 0.075 ± 0.018 | 0.181 ± 0.024 | 0.8 ± 0.3 | 20.0 ± 0.8 | 1.1 ± 0.1 | 4.4 ± 0.1 |
| FM-S325C (apo) | 0.124 ± 0.045 | 0.154 ± 0.022 | 0.4 ± 0.2 | 21.0 ± 0.8 | 1.0 ± 0.0 | 4.1 ± 0.1 |
| FM-Myr-S325C (apo) | 0.095 ± 0.014 | 0.182 ± 0.018 | 0.6 ± 0.4 | 19.6 ± 0.7 | 1.1 ± 0.1 | 4.1 ± 0.0 |
| FM-S325C (ternary) | 0.150 ± 0.045 | 0.094 ± 0.013 | 0.4 ± 0.2 | 14.9 ± 1.4 | 1.1 ± 0.1 | 4.3 ± 0.0 |
| FM-Myr-S325C (ternary) | 0.141 ± 0.022 | 0.106 ± 0.006 | 0.4 ± 0.1 | 13.7 ± 1.1 | 1.1 ± 0.1 | 4.3 ± 0.0 |
| FM-C343 (apo) | 0.079 ± 0.003 | 0.154 ± 0.002 | 0.9 ± 0.1 | 19.5 ± 0.6 | 1.0 ± 0.0 | 4.3 ± 0.0 |
| FM-Myr-C343 (apo) | 0.087 ± 0.011 | 0.150 ± 0.008 | 0.8 ± 0.3 | 19.4 ± 1.1 | 1.1 ± 0.1 | 4.3 ± 0.0 |
| FM-C343 (ternary) | 0.059 ± 0.003 | 0.174 ± 0.000 | 1.3 ± 0.2 | 24.2 ± 0.2 | 1.0 ± 0.1 | 4.3 ± 0.1 |
| FM-Myr-C343 (ternary) | 0.089 ± 0.017 | 0.151 ± 0.015 | 0.6 ± 0.3 | 21.6 ± 1.9 | 1.0 ± 0.1 | 4.3 ± 0.1 |
Amplitude of the fast anisotropy decay processes.
Amplitude of the slow anisotropy decay processes.
Rotational correlation time of fast anisotropy decay processes.
Rotational correlation time of slow anisotropy decay processes.
Reduced ψr2 of the anisotropy decay analysis.
Amplitude weighted averaged emission lifetime (∑αiτi, where ∑αi = 1). The error terms represent the SEM of the results from three replicate sample preparations, each of which was analyzed three times.
The results of MD simulations analysis of N-terminal dynamics are illustrated in Figure 2C and summarized in Table 2. The MD simulations agree with the time-resolved fluorescence anisotropy results because the root mean squared fluctuation (RMSF) values of the main-chain atoms of the nonmyristylated protein are much greater than the myristylated protein in both apo and ternary states. This is illustrated in Figure 2C and in the lower average main-chain atom RMSF values of residues 2–40 for the myristylated compared with the nonmyristylated protein (Table 2), indicating that the N-terminus is more flexible with the nonmyristylated protein, and this agrees with the anisotropy data at both K7C and K16C. However, there are small differences between the MD and time-resolved fluorescence data because the MD simulations showed slightly higher mobility of the K16 site for the nonmyristylated compared to myristylated protein (Figure 2C), and the time-resolved fluorescence anisotropy data showed slightly lower mobility of the K16 site for the nonmyristylated compared to myristylated protein (Table 1). However, the differences in mobility seen from both methods are very small and likely not significant. In general, both MD simulations and time-resolved fluorescence anisotropy suggest similar levels of mobility of the K16 region for both the myristylated and nonmyristylated enzyme.
Table 2. Average Root Mean Squared Fluctuation (RMSF) Values in Angstroms of the Backbone Atoms for Different Regions of the Protein from the MD Simulations.
| region of protein | residue range | Myr apo (Å)a | Myr ternary (Å)a | apo (Å)b | ternary (Å)b |
|---|---|---|---|---|---|
| entire protein | 2–350 | 1.07 | 1.16 | 1.18 | 1.17 |
| N-terminus/A-helix | 2–40 | 1.97 | 2.41 | 2.89 | 3.40 |
| core (N/C-lobes) | 41–300 | 0.89 | 0.95 | 0.92 | 0.83 |
| N-lobe | 41–126 | 0.89 | 0.94 | 0.95 | 0.79 |
| C-lobe | 127–300 | 0.89 | 0.95 | 0.90 | 0.86 |
| C-tail | 301–350 | 1.31 | 1.33 | 1.22 | 1.15 |
| myristate pocket | 13–18; 95–105; 150–160; 300–308 | 0.80 | 1.04 | 0.96 | 1.11 |
| catalytic loop | 164–172 | 0.68 | 0.54 | 0.68 | 0.54 |
| DFG motif | 184–186 | 0.97 | 0.60 | 0.86 | 0.55 |
| ATP site residues | 49–55, 70, 120–127, 327–331 | 1.19 | 0.95 | 1.04 | 0.84 |
| Activation Loop | 192–200 | 1.15 | 1.23 | 1.11 | 0.97 |
| B-helix | 76–81 | 1.18 | 1.16 | 1.39 | 0.98 |
| C-helix | 84–96 | 0.77 | 0.92 | 0.86 | 0.75 |
| β-strand 2 | 56–61 | 0.78 | 0.89 | 0.75 | 0.63 |
| β-strand 3 | 68–75 | 0.76 | 0.78 | 0.78 | 0.64 |
| β-strand 4 | 106–111 | 0.66 | 0.78 | 0.80 | 0.68 |
| β-strand 5 | 115–121 | 0.67 | 0.76 | 0.74 | 0.60 |
These simulations are of the protein when it is myristylated at its N-terminal glycine residue.
These simulations correspond to the protein that is not myristylated. Apo simulations are of the protein in the absence of ligands, and ternary state corresponds to simulations where the protein was bound to ATP and an inhibitory peptide, SP20.
Flexibility of the C-Subunit at the Myristate Pocket
In addition to the N-terminus, the effects of myristylation and ligand binding on the myristate pocket were examined. On the basis of the X-ray structure, we chose to mutate and conjugate Asn99. The Asn99 residue does not directly interact with the myristyl moiety and is directly above the pocket near Phe100, which forms part of the pocket. We reasoned that if this residue was modified, it should not interfere with the conformational activity of the pocket (Figure 1A,B). The results from the FM-N99C mutant are shown in Figure 3A. In the absence of ligands, myristylation causes a dramatic decrease in the mobility of this site on the basis of slower anisotropy decay of Φfast and an increase in the β2 parameter (0.158 to 0.200) (Table 1 and Figure 3A). Thus, myristylation appears to greatly stabilize the N99C site and presumably the residues near the myristate pocket in an apo state. Interestingly, the addition of MgATP/IP20 to form a ternary complex produced an increase in the mobility of the myristate pocket for both the nonmyristylated and myristylated protein. The increased mobility for the nonmyristylated protein is evidenced by a faster anisotropy decay (Φfast) for the ternary nonmyristylated FM-N99C compared to the apo state (Figure 3A) and by the decrease in the β2 parameter (0.158 to 0.120) (Table 1). Similarly, the β2 parameter decreased for the myristylated protein (0.200 to 0.181) along with Φ1 (1.6 to 0.8 ns), suggesting increased probe mobility with the formation of the ternary complex. Together, myristylation appears to stabilize residues in the myristate pocket compared to the stability of nonmyristylated protein in the presence and absence of ligands, but ligand binding and closed state formation increases the mobility of the myristate pocket for both the nonmyristylated and the myristylated enzyme.
Figure 3.

Myristylation and ligand binding produces altered mobility of the myristate pocket. (A) Anisotropy decay of FM-N99C in myristylated/apo state (blue), nonmyristylated/apo state (red), myristylated/ternary state (orange), and nonmyristylated/ternary state (green). IRF is the instrument response function. (B–D) RMSF values of the main-chain atoms from MD simulations for residues 90–105 (B), 150–160 (C), and 300–310 (D).
The results from the MD simulations are in agreement with the fluorescence results and are illustrated in Figure 3B–D and summarized in Table 2. The MD simulations show that Asn99, Glu155, and Tyr306, which are in or near the myristate pocket, exhibit decreased mobility for the myristylated compared to the nonmyristylated protein in an apo state. Also, after forming a ternary complex, the MD simulations show that these regions become more flexible for both the myristylated and nonmyristylated protein (Figure 3B–D). Additionally, the average main-chain atom RMSF values of residues at the myristate pocket are lowest for the apo myristylated protein, and the values increase for both the myristylated and nonmyristylated C-subunit going from an apo state to a ternary complex (Table 2).
Effects of Myristylation and Ligand Binding on the Dynamics of the C-Tail
Two sites in the C-terminal tail, S325C and the native C343, were examined with fluorescence anisotropy. The S325C site is near the “FDDY” motif that is thought to be important for forming part of the nucleotide binding pocket,5 and the C343 site is further along the C-tail and closer to the small lobe and A-helix (Figure 1A,C). The mobility of the FM-S325C site was unaffected by myristylation but exhibited dramatic increases in mobility following MgATP/IP20 binding. This increased mobility is evident from the sharp decrease in the rate of anisotropy decay in the presence of MgATP/IP20 as illustrated in Figure 4A and is based on the large decrease in the β2 parameter (0.154 to 0.094) (Table 1). The MD simulations did not agree with these results; instead, they showed the highest mobility for the nonmyristylated protein in an apo state and similar mobility for the other three states (Figure 4B).
Figure 4.

Effects of myristylation and ligand binding on the mobility of the C-tail. (A) Anisotropy decay of FM-S325C in myristylated/apo state (blue), nonmyristylated/apo state (red), myristylated/ternary state (orange), and nonmyristylated/ternary state (green). IRF is the instrument response function. (B) RMSF values for the backbone atoms from the MD simulations near the S325 residue. (C) Anisotropy decay of FM-C343 with the different states colored as in (A). (D) The RMSF values of the backbone atoms near the C343 residue.
For the myristylated FM-C343, no significant differences in mobility were observed between apo and ternary states (Figure 4C). For the nonmyristylated protein, on the other hand, the probe mobility decreased upon ligand binding. This effect is primarily seen as an increase in the β2 parameter (0.154 to 0.174) (Table 1). Thus, the mobility of this region was greater for the myristylated protein than the nonmyristylated protein in a ternary complex based on a lower β2 parameter for the myristylated (0.151) compared to the nonmyristylated protein (0.174) in a ternary complex. Here, myristylation blocked the stabilizing effect of ligand binding near the C-terminus. The MD simulations provide some agreement with the experimental results because the nonmyristylated protein in a ternary complex is generally the least mobile and the other three states exhibit similar mobility (Figure 4D). Also, the average RMSF values of the entire C-tail suggest a similar result of ligand binding showing minimal effects at the C-tail for the myristylated protein but decreasing mobility for the nonmyristylated protein (Table 2).
Principal Component Analysis of the MD Simulations
To further characterize the dynamics of PKA in myristylated and nonmyristylated forms and in the presence and absence of ligands, we utilized principal component analysis (PCA) of the MD simulations to determine the most dominant modes in the dynamics. Specifically, all simulations were combined and the backbone atoms—C, CA, N, and O—on residues 2–350 were used in the analysis. The two principal components (PC1 and PC2) are illustrated in Figure 5A–C and are attached as movies in the Supporting Information. In this analysis, PC1 and PC2 describe 33% and 15%, respectively, of the total variance of the motions in the simulation. The projections of the PKA simulations are shown in Figure 5A, and the starting crystal structure is shown as a red diamond in the figure for reference. The major motions displayed in PC1 showed that as the N-terminus moved vertically, the Gly-rich loop would raise or lower (Figure 5B,C). In PC2, the major motions were the rotation of the N-terminus coupled to rotation of the Gly-rich loop (Figure 5B,C). The PCA analysis shows a larger range of motion for the nonmyristylated protein than the myristylated protein. Also, the myristylated protein shows mostly PC1 movements unlike the nonmyristylated protein, which has a large range of motions.
Figure 5.

PCA analysis of the MD simulations of the C-subunit. The first two principal components were calculated from the backbone atoms of all MD simulations. (A) Structural ensembles of the backbone atoms for the nonmyristylated C-subunit in apo and ternary states and myristylated C-subunit in apo and ternary states are shown. (B) Major movements associated with PC1 and PC2 are depicted on the C-subunit. PC1 is associated with a vertical movement of the N-terminus and Gly-rich loop, and PC2 involves a rotation of the N-terminus and Gly-rich loop. (C) Changes in the Gly-rich loop are shown for PC1 (gray) and PC2 (olive) through the projections aligned with the crystal structure 4DFX (black).
Discussion
The conformational dynamics of the C-subunit are known to be important in the opening and closing of the active-site cleft of the enzyme and potentially in product turnover and allostery.38 We utilized time-resolved fluorescence anisotropy and MD simulations to study the mobility of the C-subunit in myristylated and nonmyristylated configurations and in the presence and absence of ligands. We found that myristylation greatly stabilizes the most N-terminal residues of the A-helix and stabilizes the myristate pocket residues, which is consistent with recent crystal structures of the myristylated C-subunit that display an ordered N-terminus with myristic acid stably bound within its hydrophobic pocket.16 We also found that the myristylated protein exhibits increased dynamics of some regions of the protein in the presence of ligands compared to the dynamics of nonmyristylated protein.
We investigated the mobility of residues at the N-terminus and near the myristyl binding pocket for effects of N-myristylation and ligand binding. N-myristylation greatly stabilized the first 10–12 residues at the N-terminus but exhibited less of an effect further along the A-helix near K16 (Figure 2A–C). Also, ligand binding did not greatly alter the mobility of the N-terminus. To test for mobility of the myristate pocket, we labeled N99C in the αC-β4 loop. The αC-β4 loop contains many highly conserved residues among AGC kinases including Phe100, Pro101, Phe102, Val104, and L106.39 Also, this region of the protein serves as a key link between the N- and C-lobes, and it is the only segment of the N-lobe that tracks with the C-lobe when the rigid-body movements of the C-subunit were analyzed.40 The myristylated FM-N99C protein and N99 site showed much lower mobility than the nonmyristylated protein, but the flexibility of the pocket increased for both the myristylated protein and nonmyristylated protein upon ligand binding (Figure 3A–D). This increase in dynamics at the myristate pocket in a ternary complex observed experimentally and with MD simulations suggests that the myristate pocket and active site may exhibit crosstalk consistent with recent X-ray crystallography,16 NMR,18 and MD simulation studies.17
The increased mobility near N99C and at the myristate pocket upon ligand binding may seem surprising because Val104 in this loop helps to form the binding pocket for the adenine ring of ATP (Figure 6A). However, we believe this result may be explained on the basis of which regions are likely to be stabilized by ATP binding. Val104 forms part of the adenine binding pocket, and its dynamics are likely quenched by ATP binding. Additionally, the C-helix is also likely anchored by ATP binding because of Glu91 in the helix that helps to position ATP (Figure 6A). The N99C site is centered between these two regions that likely become anchored by nucleotide binding. We believe that anchoring of both the N- and C-terminal portions of this loop may be compensated for by increased movements of the loop itself. This is one possible explanation for how ATP binding could increase movements of the N99C site and of the myristate pocket in general.
Figure 6.
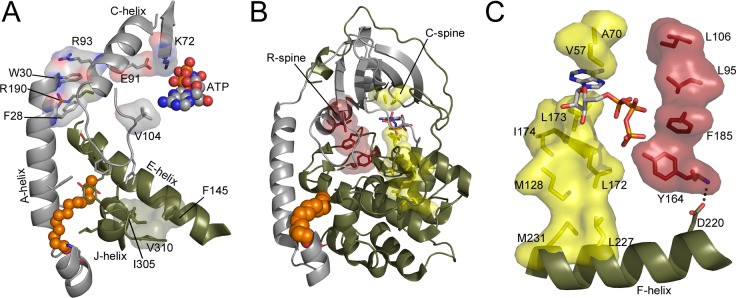
Possible modes of crosstalk between the myristate pocket and active site. (A) The N-lobe is gray, C-lobe and C-tail are olive, the myristic acid group is shown in sphere representation in orange, and ATP is shown in sphere representation and colored by element. Several residues that may mediate crosstalk between the myristate pocket and active site are shown in stick representation or stick and surface representation. Some residues in the myristic acid pocket are not labeled for viewing clarity but are labeled in Figure 1B. (B) The protein is colored as in (A). ATP is shown in stick representation. The regulatory and catalytic spines (R-spine and C-spine) are shown in stick and surface representation in red and yellow, respectively. (C) The C-spine and R-spine are colored and depicted as in (B), and one connection between the F-helix and R-spine is shown.
There is mounting evidence that N-myristylation may influence the mobility of different regions within the C-subunit, including the active site, and could be involved in allosteric regulation of the C-subunit. NMR experiments displayed altered chemical shifts at the active site with the myristylated protein.18 X-ray crystal structures showed that the structure of the N-terminus is influenced by ligand binding,16 and previous MD simulations showed that the conformation of the N-terminus is coupled to dynamics at the active site of the enzyme.17 It was hypothesized, on the basis of the X-ray crystal structures, that effects of N-myristylation could be transmitted to the active site via interactions between Trp30 on the A-helix, Arg190 on the activation loop, and Arg93 on the C-helix (Figure 6A),16 and the previous MD simulations of the N-myristylated protein supported this hypothesis.17 The MD simulations analyzed here also support this finding. There is increased dynamics of the C-helix, activation loop, and many of the β-strands in the N-lobe of the C-subunit for the myristylated protein in a ternary state, whereas the flexibility of these regions decreases for the nonmyristylated C-subunit (Table 2). Thus, these MD simulations also support increased dynamics of regions near the active site and support the hypothesis that Arg190 and Arg93 could help to mediate these effects on the basis of increases in flexibility of the activation loop and C-helix for the myristylated protein in a ternary complex. Additionally, other important regions at the active site of the C-subunit including the catalytic loop, the DFG motif, and residues involved in ATP binding show similar decreases in mobility going from apo to ternary states for the myristylated and nonmyristylated protein (Table 2). Therefore, this result suggests that the C-helix, activation loop, and β-strands are unique in displaying increased movements at the active site with the myristylated protein, and it is not simply increased dynamics for the entire active site with the myristylated protein.
The Trp30, Arg190, or Arg93 interaction is one way that the active site and N-myristylation could influence each other. Another way that myristylation and the active site could communicate is via the regulatory spine (R-spine). Leu106 is another residue that is near the N99C site, and it is a key hydrophobic contact to the R-spine. The R-spine is a stack of four hydrophobic residues that are assembled in every active kinase, typically as a consequence of phosphorylation of the activation loop.41−43 The adenine ring of ATP completes the parallel C-spine (Figure 6B,C).44 It may be possible that myristylation could influence these spines, especially the regulatory spine, because of its closer proximity to the myristyl moiety. If myristylation influences either or both spines, then it could influence mobility of the entire protein because these residues span both lobes of the kinase.
To further address how myristylation can influence the dynamics of the C-subunit and how N-myristylation may impact the active site, we performed principal component analysis (PCA) of the MD simulations. The movements associated with the two principal components PC1 and PC2 are shown in Figure 5B,C and attached as movies in the Supporting Information. PC1 and PC2 illustrate that the movements of the N-terminus are coupled with movements at the active site and with the global conformation of the protein. PC1 shows that as the N-terminus moves vertically away from the protein, the Gly-rich loop rises and the C-subunit adopts a more open conformation. PC2 shows that a rotation of the N-terminus away from the core of the protein is coupled to rotation of the Gly-rich loop and N-lobe. Also, projecting the motions of the myristylated and nonmyristylated protein onto the PC space shows that the nonmyristylated protein adopts a much wider range of motions than the myristylated protein (Figure 5A). Furthermore, the myristylated protein adopts more PC1 movements than the nonmyristylated protein. These results suggest that, by stabilizing the N-terminus and its possible conformations, N-myristylated stabilizes the amount and types of global conformations and movements of the protein. Also, the global dynamics and active site dynamics that are experienced by the myristylated protein are largely opening and closing of the Gly-rich loop and not rotations of the active site.
The implications of the PCA results are interesting to consider in terms of what they suggest about the possible role of N-myristylation on C-subunit activity or interactions. The results suggest that N-myristylation may prevent or limit drastic conformational changes of the enzyme, possibly due to stabilization of the N-terminus. This is supported by the relatively small range of motions adopted by the myristylated protein in apo and ternary states on the basis of PCA analysis (Figure 5A). Therefore, N-myristylation may keep the C-subunit in a relatively stable conformation, which could be important for ligand binding and activity. This possibility is exemplified in the PCA plots (Figure 5A) because the range of motions exhibited by the nonmyristylated protein in an apo and ternary state are very different, but for the myristylated protein, on the other hand, the motions adopted in apo or ternary states are very similar. Therefore, N-myristylation may help to lock the enzyme in conformations that are more representative of the active state of enzyme, because the ternary complex is more likely to adopt conformations suitable for catalysis. Also, because the PCA analysis shows that the motions adopted by the N-myristylated protein are mostly opening and closing of the Gly-rich loop, this result suggests that N-myristylation could influence the ability for substrates to bind and be turned over by the enzyme by modulating opening and closing of the active site. Finally, in general, the PCA results further imply that the active site and the myristyl moiety as well as its binding pocket may influence each other.
In addition to the myristate pocket, N-terminus, and possibly the active site, the flexibility of other regions may be influenced by N-myristylation. For instance, another site that we labeled for time-resolved fluorescence anisotropy was the native C343. This region is more mobile in a ternary complex for the myristylated C-subunit than the nonmyristylated enzyme (Figure 4C). The mobility of this region is not greatly influenced by ligand binding for the myristylated protein but becomes less mobile upon ligand binding for the nonmyristylated protein on the basis of time-resolved fluorescence anisotropy, and this result is also in agreement with the MD simulations based on the average RMSF values of the C-tail, which are very similar for the myristylated protein in apo and ternary states but decrease from apo to ternary states for the nonmyristylated protein (Table 2). Myristylation, thus, prevents the stabilizing effect caused by ligand binding that occurs with the nonmyristylated protein and, effectively, results in increased mobility of this region in a ternary state relative to the nonmyristylated protein.
Additionally, the average main-chain atom RMSF values from several regions throughout the C-subunit suggest that ligand binding increases flexibility in the myristylated enzyme but decreases dynamics for the nonmyristylated C-subunit (Table 2). For instance, the N-lobe, the C-lobe, and the C-tail all show increases in average RMSF values, indicating increased flexibility, for the myristylated protein going from an apo to ternary state (Table 2). The nonmyristylated protein, on the other hand, shows decreases in average RMSF values for these same regions, indicating decreased mobility, going from apo to ternary state (Table 2). Thus, the flexibility of the myristylated protein is generally increased upon ligand binding and generally decreased upon ligand binding for the nonmyristylated protein. These results suggest that myristylation can influence other regions within the C-subunit following ligand binding including, potentially, the active site.
We also wanted to test the mobility of the C-tail region near the FDDY motif that forms part of the ATP binding site (Figure 1C). To do this, we investigated the S325 site. This is an important region because the S325C site is near the PxxP motif in the C-tail (residues 313–316 in PKA), which is a known protein binding site in another AGC kinase, PKC, and may be a binding site in PKA as well.45 Furthermore, 3-phosphoinositide dependent protein kinase 1 (PDK1), which phosphorylates the activation loop of different kinases, binds to the C-tail of PKA near this region.46 With time-resolved fluorescence anisotropy, the FM-S325C site showed large changes in mobility upon ligand binding for both the myristylated and nonmyristylated protein (Figure 4A), but the MD simulations displayed increased mobility for the apo myristylated protein with similar mobility for the other states (Figure 4B). Obviously, the discrepancy between the MD and fluorescence data at the S325C site is disappointing. There are some possible explanations for the different results between the two methods. For example, it is possible that the time-resolved fluorescence anisotropy results are distorted by the fluorophore, fluorescein-5-maleimide, altering the natural mobility of the C-tail. It is possible that the fluorophore could interact with other residues in the C-tail or near the active site, and this could cause the increased mobility upon formation of a ternary complex that may not represent natural mobility of the site. Alternatively, there could be problems with the MD simulation results. One possible explanation for discrepancies in the MD simulation results is problems with sampling. It is possible that these simulations may represent outliers in possible motions of the C-tail, and if more simulations were performed or longer simulations were performed, the final RMSF values may change and could be closer to the experimental findings. Additionally, one should keep in mind that the MD simulations are not “measuring” the same things as time-resolved fluorescence anisotropy (TRFA). The MD simulations are estimating spatial displacement of the main-chain atom over time, whereas TRFA monitors motions of a reporter at the end of a cysteine side chain. (The motions of the reporter group are influenced by (1) the torsional motions of the side chain and linking tether arm, (2) the backbone motions of the peptidyl chain around the conjugated cysteine, and (3) the whole-body rotation diffusion of the protein.) Clearly, caution should be used in interpreting the results from either method.
Beyond adding insight into the conformational dynamics of the C-subunit, the general but not complete consistency between the fluorescence anisotropy and MD simulations and previous NMR and X-ray crystallographic results adds credence to the use of fluorescence anisotropy to measure protein conformational dynamics. Clearly, there are limitations to its use. One such limitation is the possibility that the mutation and conjugation significantly perturb local dynamics and/or function. For instance, if the native C199A site in PKA is labeled, then PKA loses catalytic activity.23 Fortunately, with PKA, labeling of the native cysteine residues is prevented with the addition of MgATP, but with other proteins, the native cysteine residues may need to be mutated in order to perform time-resolved fluorescence anisotropy. Also, if mutation of the cysteine residues negatively impacts the protein’s natural function or interactions, then time-resolved fluorescence anisotropy experiments become impractical. Additionally, MD simulations also have limitations and should ideally be verified with experimental findings because artifacts from MD simulations are possible due, for example, to short simulation times or very few simulations. When only a small number of simulations and/or short simulation times are utilized, then it is possible to see artifacts in the simulations. Like other techniques, it is ideal to perform replications to ensure that results are representative of actual motions and do not represent outliers in possible dynamics. Additionally, MD simulations are reliant on a solved crystal structure. Therefore, it is possible for MD simulations to be biased by the starting crystal structure especially if the structure does not represent a typical conformation of the protein in solution. Therefore, there are some limitations to both MD simulations and time-resolved fluorescence anisotropy, so some caution should be used in the interpretation of results from these approaches. Ideally, cross validation should be utilized for either method, and the general agreement in these studies between the MD simulations and fluorescence anisotropy results supports the use and findings obtained from each method.
In summary, the results of these studies provide new insights into the motions adopted by the C-subunit in response to N-myristylation and ligand binding. As several studies showed previously,16−18 we also find that N-myristylation stabilizes the N-terminus and myristate pocket, and N-myristylation also may exhibit crosstalk with the active site of the enzyme. We found that ligand binding increases movements at the myristate pocket, and myristylation in combination with ligand binding showed modest increases in dynamics near the active site. Together this suggests that N-myristylation may impart allosteric regulation of the C-subunit.
Acknowledgments
We thank J. Andrew McCammon for advice and guidance, and we thank Alexandr Kornev for helpful input on sites for mutation.
Glossary
ABBREVIATIONS:
- PKA
protein kinase A
- C-subunit
catalytic subunit of PKA
- R-subunit
regulatory subunit of PKA
- myr
myristylated
- PKI
protein kinase inhibitor
- IP20
peptide corresponding to residues 5–24 from PKI
- SP20
peptide of residues 5–24 from PKI with N20A, A21S mutations
- FM
fluorescein maleimide
- MD
molecular dynamics
Supporting Information Available
Movies highlighting the movements of the first two principal components, PC1 and PC2, in the range from −150 Å to +150 Å. This material is available free of charge via the Internet at http://pubs.acs.org.
The project described was supported by grant number GM19301 (S.S.T.) and F31GM099415 (A.C.B.) from the National Institute of General Medical Sciences. Additionally, A.C.B was funded by the Ford Foundation Diversity Fellowship. R.C.W and L.C.P. acknowledge funding from the National Science Foundation through the Scientific Software Innovations Institutes program (NSF SI2-SSE, grants NSF1047875 and NSF1148276). R.C.W. additionally acknowledges funding through the NSF XSEDE program and through a fellowship from NVIDIA Inc. Computer time was provided by the San Diego Supercomputer Center and Georgia Institute of Technology through National Science Foundation award TG-CHE130010 to R.C.W.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Taylor S. S.; Keshwani M. M.; Steichen J. M.; Kornev A. P. (2012) Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos. Trans. R. Soc., B 367, 2517–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C.; Cheng C. Y.; Saldanha S. A.; Taylor S. S. (2007) PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell 130, 1032–1043. [DOI] [PubMed] [Google Scholar]
- Taylor S. S.; Kim C.; Cheng C. Y.; Brown S. H.; Wu J.; Kannan N. (2008) Signaling through cAMP and cAMP-dependent protein kinase: diverse strategies for drug design. Biochim. Biophys. Acta 1784, 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knighton D. R.; Zheng J. H.; Ten Eyck L. F.; Xuong N. H.; Taylor S. S.; Sowadski J. M. (1991) Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253, 414–420. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Knighton D. R.; ten Eyck L. F.; Karlsson R.; Xuong N.; Taylor S. S.; Sowadski J. M. (1993) Crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MgATP and peptide inhibitor. Biochemistry 32, 2154–2161. [DOI] [PubMed] [Google Scholar]
- Masterson L. R.; Mascioni A.; Traaseth N. J.; Taylor S. S.; Veglia G. (2008) Allosteric cooperativity in protein kinase A. Proc. Natl. Acad. Sci. U. S. A. 105, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D. A.; Akamine P.; Radzio-Andzelm E.; Madhusudan M.; Taylor S. S. (2001) Dynamics of cAMP-dependent protein kinase. Chem. Rev. 101, 2243–2270. [DOI] [PubMed] [Google Scholar]
- Akamine P.; Madhusudan Wu, J.; Xuong N. H.; Ten Eyck L. F.; Taylor S. S. (2003) Dynamic features of cAMP-dependent protein kinase revealed by apoenzyme crystal structure. J. Mol. Biol. 327, 159–171. [DOI] [PubMed] [Google Scholar]
- Wu J.; Yang J.; Kannan N.; Madhusudan, Xuong N. H.; Ten Eyck L. F.; Taylor S. S. (2005) Crystal structure of the E230Q mutant of cAMP-dependent protein kinase reveals an unexpected apoenzyme conformation and an extended N-terminal A helix. Protein Sci. 14, 2871–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossemeyer D.; Engh R. A.; Kinzel V.; Ponstingl H.; Huber R. (1993) Phosphotransferase and substrate binding mechanism of the cAMP-dependent protein kinase catalytic subunit from porcine heart as deduced from the 2.0 A structure of the complex with Mn2+ adenylyl imidodiphosphate and inhibitor peptide PKI(5–24). EMBO J. 12, 849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayana N.; Cox S.; Nguyen-huu X.; Ten Eyck L. F.; Taylor S. S. (1997) A binary complex of the catalytic subunit of cAMP-dependent protein kinase and adenosine further defines conformational flexibility. Structure 5, 921–935. [DOI] [PubMed] [Google Scholar]
- Tholey A.; Pipkorn R.; Bossemeyer D.; Kinzel V.; Reed J. (2001) Influence of myristoylation, phosphorylation, and deamidation on the structural behavior of the N-terminus of the catalytic subunit of cAMP-dependent protein kinase. Biochemistry 40, 225–231. [DOI] [PubMed] [Google Scholar]
- Gangal M.; Clifford T.; Deich J.; Cheng X.; Taylor S. S.; Johnson D. A. (1999) Mobilization of the A-kinase N-myristate through an isoform-specific intermolecular switch. Proc. Natl. Acad. Sci. U. S. A. 96, 12394–12399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepperkok R.; Hotz-Wagenblatt A.; Konig N.; Girod A.; Bossemeyer D.; Kinzel V. (2000) Intracellular distribution of mammalian protein kinase A catalytic subunit altered by conserved Asn2 deamidation. J. Cell Biol. 148, 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemoto W.; McGlone M. L.; Taylor S. S. (1993) N-myristylation of the catalytic subunit of cAMP-dependent protein kinase conveys structural stability. J. Biol. Chem. 268, 2348–2352. [PubMed] [Google Scholar]
- Bastidas A. C.; Deal M. S.; Steichen J. M.; Keshwani M. M.; Guo Y.; Taylor S. S. (2012) Role of N-terminal myristylation in the structure and regulation of cAMP-dependent protein kinase. J. Mol. Biol. 422, 215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cembran A.; Masterson L. R.; McClendon C. L.; Taylor S. S.; Gao J.; Veglia G. (2012) Conformational Equilibrium of N-Myristoylated cAMP-Dependent Protein Kinase A by Molecular Dynamics Simulations. Biochemistry 51, 10186–10196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffarogullari E. C.; Masterson L. R.; Metcalfe E. E.; Traaseth N. J.; Balatri E.; Musa M. M.; Mullen D.; Distefano M. D.; Veglia G. (2011) A myristoyl/phosphoserine switch controls cAMP-dependent protein kinase association to membranes. J. Mol. Biol. 411, 823–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herberg F. W.; Bell S. M.; Taylor S. S. (1993) Expression of the catalytic subunit of cAMP-dependent protein kinase in Escherichia coli: multiple isozymes reflect different phosphorylation states. Protein Eng. 6, 771–777. [DOI] [PubMed] [Google Scholar]
- Duronio R. J.; Jackson-Machelski E.; Heuckeroth R. O.; Olins P. O.; Devine C. S.; Yonemoto W.; Slice L. W.; Taylor S. S.; Gordon J. I. (1990) Protein N-myristoylation in Escherichia coli: reconstitution of a eukaryotic protein modification in bacteria. Proc. Natl. Acad. Sci. U. S. A. 87, 1506–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangal M.; Cox S.; Lew J.; Clifford T.; Garrod S. M.; Aschbaher M.; Taylor S. S.; Johnson D. A. (1998) Backbone flexibility of five sites on the catalytic subunit of cAMP-dependent protein kinase in the open and closed conformations. Biochemistry 37, 13728–13735. [DOI] [PubMed] [Google Scholar]
- Cook P. F.; Neville M. E. Jr.; Vrana K. E.; Hartl F. T.; Roskoski R. Jr. (1982) Adenosine cyclic 3′,5′-monophosphate dependent protein kinase: kinetic mechanism for the bovine skeletal muscle catalytic subunit. Biochemistry 21, 5794–5799. [DOI] [PubMed] [Google Scholar]
- Nelson N. C.; Taylor S. S. (1981) Differential labeling and identification of the cysteine-containing tryptic peptides of catalytic subunit from porcine heart cAMP-dependent protein kinase. J. Biol. Chem. 256, 3743–3750. [PubMed] [Google Scholar]
- Hibbs R. E.; Radic Z.; Taylor P.; Johnson D. A. (2006) Influence of agonists and antagonists on the segmental motion of residues near the agonist binding pocket of the acetylcholine-binding protein. J. Biol. Chem. 281, 39708–39718. [DOI] [PubMed] [Google Scholar]
- Birch D. J. S., and Imhof R. E. (1991) in Topics in Fluorescence Spectroscopy: Techniques Vol. 1 (Lakowicz J. R., Ed.) Plenum, New York, NY. [Google Scholar]
- Frisch M. J., . et al. (2004) Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford CT. [Google Scholar]
- Case D. A., Darden T. A., Cheatham T. E. III, Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., and Merz K. M. (2010) Amber 11, University of California, San Francisco. [Google Scholar]
- Salomon-Ferrer R.; Case D. A.; Walker R. C. (2013) An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 3, 198–210. [Google Scholar]
- Anandakrishnan R.; Aguilar B.; Onufriev A. V. (2012) H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 40, W537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Palmo K.; Maragakis P.; Klepeis J. L.; Dror R. O.; Shaw D. E. (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. C. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 327–341. [Google Scholar]
- Darden T.; York D.; Pedersen L. (1993) Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 98, 10089–10092. [Google Scholar]
- Case D. A., Darden T. A., Cheatham T. E. III, Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., and Merz K. M. (2012) Amber12, University of California, San Francisco. [Google Scholar]
- Gotz A. W.; Williamson M. J.; Xu D.; Poole D.; Le Grand S.; Walker R. C. (2012) Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grand S.; Gotz A. W.; Walker R. C. (2013) SPFP: Speed without compromise-A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 184, 374–380. [Google Scholar]
- Salomon-Ferrer R.; Goetz A. W.; Poole D.; Le Grand S.; Walker R. C. (2013) Routine microsecond molecular dynamics simulations with AMBER - Part II: Particle Mesh Ewald. J. Chem. Theory Comput. 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- Masterson L. R.; Shi L.; Metcalfe E.; Gao J.; Taylor S. S.; Veglia G. (2011) Dynamically committed, uncommitted, and quenched states encoded in protein kinase A revealed by NMR spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 108, 6969–6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan N.; Haste N.; Taylor S. S.; Neuwald A. F. (2007) The hallmark of AGC kinase functional divergence is its C-terminal tail, a cis-acting regulatory module. Proc. Natl. Acad. Sci. U. S. A. 104, 1272–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsigelny I.; Greenberg J. P.; Cox S.; Nichols W. L.; Taylor S. S.; Ten Eyck L. F. (1999) 600 ps molecular dynamics reveals stable substructures and flexible hinge points in cAMP dependent protein kinase. Biopolymers 50, 513–524. [DOI] [PubMed] [Google Scholar]
- Kornev A. P.; Haste N. M.; Taylor S. S.; Eyck L. F. (2006) Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. U. S. A. 103, 17783–17788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S. S.; Kornev A. P. (2011) Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem. Sci. 36, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steichen J. M.; Kuchinskas M.; Keshwani M. M.; Yang J.; Adams J. A.; Taylor S. S. (2012) Structural basis for the regulation of protein kinase A by activation loop phosphorylation. J. Biol. Chem. 287, 14672–14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornev A. P.; Taylor S. S.; Ten Eyck L. F. (2008) A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. U. S. A. 105, 14377–14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould C. M.; Kannan N.; Taylor S. S.; Newton A. C. (2009) The chaperones Hsp90 and Cdc37 mediate the maturation and stabilization of protein kinase C through a conserved PXXP motif in the C-terminal tail. J. Biol. Chem. 284, 4921–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano R. A.; Kannan N.; Kornev A. P.; Allison C. J.; Taylor S. S. (2009) A chimeric mechanism for polyvalent trans-phosphorylation of PKA by PDK1. Protein Sci. 18, 1486–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.