

Abstract
Thoracic aortic dissection (TAD) is a highly lethal vascular disease. In many patients with TAD, the aorta progressively dilates and ultimately ruptures. Dissection formation, progression, and rupture cannot be reliably prevented pharmacologically because the molecular mechanisms of aortic wall degeneration are poorly understood. The key histopathologic feature of TAD is medial degeneration, a process characterized by smooth muscle cell depletion and extracellular matrix degradation. These structural changes have a profound impact on the functional properties of the aortic wall and can result from excessive protease-mediated destruction of the extracellular matrix, altered signaling pathways, and altered gene expression. Review of the literature reveals differences in the processes that lead to ascending versus descending and sporadic versus hereditary TAD. These differences add to the complexity of this disease. Although tremendous progress has been made in diagnosing and treating TAD, a better understanding of the molecular, cellular, and genetic mechanisms that cause this disease is necessary to developing more effective preventative and therapeutic treatment strategies.
Keywords: aortic dissection, media, degeneration, aneurysm
INTRODUCTION
Thoracic aortic dissection (TAD) is estimated to occur at a rate of 3 cases per 100,000 individuals per year and is a major cause of death [1-4]. There are two phases of TAD. The first is the acute phase, which begins with the sudden initial tear in the aortic intima and media. This tear allows pulsatile blood to enter the media and cause separation of the medial layer along the length of the vessel. This leads to the formation of a second channel, termed the false lumen. Because the acute dissection creates a thin, inflamed, and extremely fragile outer aortic wall and can also profoundly disrupt blood flow in branch arteries, the acute phase is characterized by life-threatening complications. Acute TAD renders the aorta prone to rapid dilation and rupture, which is usually fatal.
In patients who survive the acute phase—which is arbitrarily defined as the first 14 days after onset—the dissection enters the second, chronic phase. Although the aorta partially heals and thus becomes less fragile than it is in the acute phase, the outer aortic wall remains weakened and prone to gradual dilatation and aneurysm formation. In patients with progressive aneurysm expansion, surgical treatment is required to prevent fatal aortic rupture. The transition period between the acute and chronic phases is sometimes described as the subacute phase.
Thoracic aortic dissections are associated with degeneration of the aortic media. The terminology related to medial degeneration has evolved over time. Borst and colleagues [5] have reviewed the different terms and definitions used by researchers through the years. Gsell, in 1928, first described “idiopathic media necrosis” as a loss of structural components in the aortic media. Erdheim later used the term “cystic medial necrosis” to refer to a loss of structural components in the aortic media that was associated with the accumulation of semi-mucoid ground substance in cyst-like arrangements. In some cases, this disease process was also referred to as mucoid degeneration of the media [6]. In more recent publications, both “cystic medial degeneration” and “medial degeneration” are common terms. Cystic medial degeneration is usually defined as the fragmentation or loss of elastic fibers in the media, with or without the presence of pools of glycoproteins, but the term still evokes the presence of cysts [7]. Many authors therefore favor a more specific definition: medial degeneration characterized by smooth muscle cell (SMC) loss, fragmentation and depletion of elastic fibers, and accumulation of proteoglycans and glycosaminoglycans within cell-depleted areas of the aortic media [8].
Although the specific etiology of dissection formation is still unclear, the progression of aortic dilatation in the chronic phase probably results from a combination of hemodynamic stress, aortic injury, chronic inflammation, genetic propensity, and epidemiologic risk factors. The extracellular matrix (ECM) of the aortic wall is a highly dynamic environment. Changes in the structural components of the ECM influence the molecular signaling pathways that regulate aortic wall homeostasis, as well as aortic function. A cohesive understanding of the complex structure and function, regulatory factors, and genetic and developmental origins of the aortic wall is necessary to elucidating the pathogenesis of aortic dissection and, ultimately, developing therapeutic modalities to prevent TAD formation, expansion, and rupture.
Our review is organized into four parts. First, we provide an overview of the key structural and functional properties of the aorta. Second, we discuss the major cellular and molecular regulatory pathways in TAD formation. Third, we review the epidemiologic and genetic risk factors for TAD and their histologic and molecular manifestations in the aortic wall. Fourth, we discuss differences between ascending and descending TAD.
KEY STRUCTURAL COMPONENTS OF THE AORTA AND THEIR DEFECTS IN TAD
As previously mentioned, TAD begins with a tear through the intima that extends into the media of the aortic wall, resulting in the separation of the medial layers. The media of the aortic wall is composed of elastic fibers and vascular smooth muscle cells (VSMCs) interconnected with collagen fibers, proteoglycans, glycosaminoglycans, and various adhesive proteins. All of these elements form the ECM and are important for imparting elasticity and tensile strength, sequestering growth factors, and forming structural interactions between ECM components [9]. In TAD, interlaminar fibers are irregularly shaped and arranged [10]; more importantly, elastic fibers, which normally provide elasticity and compliance to the aortic wall, are fragmented [5]. Reductions in overall elastin content [11] and in elastic fiber cross-links [12] are characteristic of the dissected aortic media.
Elastin is the major protein of the ECM and provides the aorta with its elastic properties. Cross-linking of tropoelastin monomers by lysyl oxidase (LOX) forms elastin molecules, which in turn cross-link with microfibrils to form elastic fibers. In addition to its functional role, elastin is also important for vascular morphogenesis [13]. Binding of elastin to integrin αvβ3[14] and elastin binding protein [15] promotes cell adhesion and regulates cell proliferation. Disruption of the interaction between elastin and other cellular constituents during morphogenesis results in uncontrolled VSMC proliferation, which manifests as supravalvular stenosis in elastinhaploinsufficient mice [16] and as severe occlusive disease in elastin-deficient mice [17]. However, these mice are not reported to develop thoracic aortic aneurysm or dissection (TAAD). This suggests that although elastin is important for aortic recoil, isolated defects in elastin as part of the structural complex of elastic fibers may not be necessary for TAD formation.
Microfibrils comprise 10% of elastic fibers, provide a scaffold for LOX to cross-link tropoelastin monomers to form mature elastic fibers, and provide extensibility to the aortic wall [18]. Microfibrils are composed of fibrillin and several microfibril-associated proteins, such as latent transforming growth factor beta (TGF-β) binding proteins (LTBP 1-4) [19], elastin microfibril interface-located protein 1 (EMILIN-1), microfibril-associated glycoproteins (MAGP-1 and -2), and fibulins [20].
Fibrillin-1 (FBN1) and fibrillin-2 are two well-characterized isoforms that are important for aortic function. FBN1 is a major structural component of microfibrils in the mature aorta and, in addition, it regulates tissue homeostasis by sequestering growth factors such as TGF-β [21]. FBN1 can also influence cell signaling by binding to integrin receptors through Arg-Gly-Asp (RGD)–containing sequences [22], and it can interact with cell-surface heparan sulfate proteoglycans to regulate matrix deposition [23]. Fibrillin-2 is primarily expressed in the embryonic period and is important for aortic morphogenesis [24]. Mice deficient in the FBN1 gene (Fbn1 -/-) recapitulate the human aortic Marfan syndrome phenotype, have thin and fragmented elastic fibers [22], and are prone to aortic aneurysm and dissection. In contrast, mice deficient in the fibrillin-2 gene (Fbn2 -/-) have normal elastic fibers and are not reported to develop aortic aneurysms or dissection during development [25].
Microfibril-associated glycoproteins (MAGP)-1 and -2 are constitutive components of microfibrils and are capable of binding members of the TGF-β growth factor family [26]. However, reports indicate that inactivation of either protein’s associated gene does not result in cardiovascular defects in mice [27, 28] but does cause neurovascular and ophthalmic vascular abnormalities in zebrafish [29]. Furthermore, analysis of aortic tissue from MAGP-1-deficient mice (Mfap2) reveals normal elastic fiber architecture and similar stress-strain patterns, suggesting normal elastic fiber cross-linking [28]. Although this finding indicates that MAGP-1 is dispensable for elastic fiber assembly and that MAGP-1 deficiency may not be critical for dissection formation in large arteries, Mfap2 deficient mice lack the vascular manifestations found in mfap2 deficient zebrafish, suggesting that the manifestation of MAGP-1 deficiency is species specific. More studies are needed to evaluate the functional role of MAGP when elastic fibers are subjected to increased hemodynamic stress in vivo.
The fibulins are a group of seven ECM proteins that are important for aortic wall homeostasis. Fibulin-1 is commonly located within the elastin core [30]. Fibulin-2 and -4 are located at the interface between the elastin core and microfibrils, and fibulin-5 is associated with microfibrils [27]. Mice with deficiency of the fibulin-1 gene (Fbln1) develop abnormal endothelial integrity [31]. However, deficiency of Fbln1, Fbln2, or Fbln3 has no effect on elastic fiber formation [32, 33]. Fibulin-4 is required for LOX recruitment to cross-link elastin molecules [34]. Studies of aortic tissue from humans and mice with FBLN4/Fbln4 deficiency show increased TGF-β signaling, which may result from impaired LOX-mediated TGF-β repression [35, 36]. Fibulin-5 is essential for lamellar unit formation [37], and Fbln5-deficient mice have disrupted elastic fiber assembly [11], as well as less compliant aortas [38]. Fibulin-6 and -7 are the only fibulins that are not found in elastic tissues. These findings suggest that fibulin-4 and -5 have key roles in elastic fiber assembly; deficiency of either one may predispose patients to aortic dissection.
The elastin microfibril interface-located proteins (EMILIN-1, -2, and -3) and the multimerins (multimerin 1 and 2) are glycoproteins of the ECM that have a unique N-terminus cysteine-rich EMI-domain [39]. EMILIN-1 is the most well-studied member of this family of proteins and is commonly found at the interface between microfibrils and amorphous elastin [40]. EMILIN-1 is also capable of binding both tropoelastin and fibulin-5, and deficiency in EMILIN-1 (EMILIN1 -/-) results in abnormal elastic fiber architecture [41]. However, the role of EMILIN-1 in dissection formation is not entirely clear. EMILIN-1 is known to inhibit TGF-β signaling by binding to its precursor, pro-TGF-β, and inhibiting its maturation by furin convertases [42]. Yet, Emilin1 -/- mice, although they have elevated TGF-β levels, have a low incidence of aneurysms and dissection. Instead, Emilin1 -/- mice have abnormal interaction between SMCs and elastic fibers, which may result in narrower blood vessels and systemic hypertension [39, 41, 42]. This suggests that although EMILIN-1 is important for elastogenesis, defects in EMILIN-1 affect the formation and function of elastic lamellae. EMILIN-2 is another member of this family of proteins and, in contrast to EMILIN-1, appears to be important primarily for cardiovascular development [43]. Like Emilin1 -/- mice, Emilin2 -/- mice have hypertensive phenotypes, but EMILIN-2 is not involved in pro-TGF-β processing [39]. The least well characterized member of the EMILIN family is EMILIN-3. Like EMILIN-1, EMILIN-3 serves as a pro-TGF-β antagonist; however, EMILIN-3 is not found in the cardiovascular system [44]. Like the studies of MAGP, these studies did not evaluate the functional role of EMILINs in elastic fiber homeostasis after hemodynamic stress. Future studies should address the role of different isoforms of EMILIN in aortic morphogenesis and elastic fiber assembly.
Collagen is another important component of the ECM and contributes to both the structural and the functional properties of the aorta. From a structural perspective, collagen type 1 and type 3 are the most abundant collagen fibers in the aortic wall and are important for imparting tensile strength. From a functional perspective, collagen is important for sequestering cytokines and mediating cell proliferation by binding to integrins [45]. Intact elastic fibers are necessary for organized collagen deposition and efficient collagen function. In TAD, type I and type III collagen expression is increased [46], as is the level of spiraled collagen [47]. The increased expression and disorderly deposition of collagen may correspond to a slow reparative process triggered by elastic fiber fragmentation and depletion. This fibrosis may also be due to an increase in the expression and release of sequestered growth factors [48]. The increase in collagen could lead to increased arterial stiffness, thus enhancing the aorta’s susceptibility to dissection and rupture [46]. However, in a study of aortic samples taken from patients with acute dissection, de Figueiredo Borges and colleagues [49] found an abnormally low proportion of collagen in the outer half of the dissected aortic wall. Less than 3 days had elapsed between the onset of symptoms and the operation during which these aortic samples were collected. Therefore, it is possible that the collagen content in the media decreases before dissection, thus weakening the aortic wall and making dissection more likely. The observed fibrosis may occur later as the wall remodels. In contrast to type I and type III collagens, the basement membrane-specific type IV collagen, especially its α2- and α5-chains, is underexpressed in TAD [50]. Weakening of the tunica intima may provide another avenue for dissection formation. Therefore, mutations in collagen-encoding genes may lead to dissection formation.
The COL1A1 gene encodes a constituent of α1 chains in type I collagen. Mice with double replacement targeted 1283-bp deletion in the 1462-bp first intron of the Col1a1 gene (Δ/Δ mice) develop age-dependent aortic dissections that often rupture [51, 52]. The age dependency of the onset of aortic dissection may be related to the normal decrease in collagen synthesis with age. The expression of type I collagen is moderately reduced in mice heterozygous for Col1a1 and is substantially reduced in homozygous Δ/Δ mice, which may result in aortic dissection and rupture [53]. Mutations in the Col3a1 gene (i.e., those associated with vascular-type Ehlers-Danlos), which encodes the pro-α(1) chains of type III collagen, result in abnormal collagen fibrillogenesis within the aortic wall and are associated with TAD formation [53].
The lysyl oxidases comprise a family of five enzymes (LOX and four LOX-like proteins) critical for collagen and elastin cross-linking. Although the specific function of the four LOX-like proteins is not fully characterized, LOX is an important regulator of the ECM [54]. LOX and LOX-like protein 1 interact with fibulin-4 [36], fibulin-5 [55], and fibrillin-2 [53] during elastogenesis. In addition to its role in forming lysine- and hydroxylysine-derived cross-links in collagen and elastin, LOX also regulates SMC signaling [56], represses TGF-β [35], and regulates the expression of both elastin (through TGF-β signaling) [57] and COL3A (possibly through the Ku antigen) [58]. Lox-deficient mice have highly fragmented elastic fibers and are prone to aneurysm formation [36]. Therefore, deficiencies in LOX weaken the aortic wall, alter ECM homeostasis, and may predispose patients to TAD.
An increase in proteoglycan and glycosaminoglycan content is also associated with degeneration of the aortic media in aortic dissection [59]. These proteins resist compressive forces, sequester growth factors, and form structural interactions between ECM components [9]. Proteoglycans are the most abundant glycoproteins found in the vessel wall and can be categorized into two different classes: large proteoglycans, such as versican and aggrecan, and small, leucine-rich proteoglycans, such as decorin, biglycan, fibromodulin, osteoglycin, and lumican. Versican is important for creating the pericellular matrix that is required for the proliferation and migration of VSMCs [60, 61]. Injury to the vascular wall is known to increase versican degradation and downregulate elastin levels [60, 61]. In aneurysmal aortas, there is an increase in versican degradation products and downregulation of the versican V0 isoform [62]. In normal aortas, most of the versican is seen in the intima and media, whereas in aortic dissection, versican degradation is more prominent in the external half of the media where the dissection occurs [63]. Therefore, degradation of versican may predispose the aorta to dissection. The role of aggrecan in the vessel wall is unclear.
Small, leucine-rich proteoglycans bind to key ECM molecules such as collagen, tropoelastin, fibronectin, and microfibrils and may also have a role in TAD formation. Biglycan and decorin are the most well-studied proteoglycans of this family. Decorin is found only in the adventitia, whereas biglycan is found throughout the aortic wall. Both biglycan and decorin regulate collagen fibrillogenesis and are capable of binding to TGF-β [64]. Mice null for decorin or biglycan have decreased collagen fiber diameter and dysregulated collagen organization [65]. Analysis of tissue from aortic dissection patients shows downregulation of decorin [66], and biglycan-deficient mice often die from spontaneous aortic dissection and rupture [65]. Proteoglycans can be degraded by a disintegrin and metalloproteinase with thrombospondin motifs) [67], production of which has been shown be to significantly increased in patients with TAAD [68, 69].
Several secreted and transmembrane glycoproteins involved in cellular adhesion are also downregulated in TAD. More specifically, decreased expression has been reported for fibronectin [47] and the microfibril-associated protein 4 (MFAP4) in TAD tissue [70]. Fibronectin is a glycoprotein that binds to various ECM components, and MFAP4 is a collagen-binding glycoprotein. Also downregulated are the polycystin-1 and -2 genes (PC1 and PC2), which are important for the manufacture of cell-cell adhesion proteins [51]. In hypomorphic polycystic kidney disease 1 (Pkd1nl/nl) mice, no alterations have been noted in the overall structure of elastic lamellae or components involved in elastin organization (e.g., FBN1) [71]; however, aortic dissections occur in the descending segment of the thoracic aorta. SMCs in Pkd1nl/nl mice are characterized by an increased expression of α-actin, proteoglycans, and fibronectin and by a widened space between the elastic lamellae, which may reflect a loss of contact between elastic fibers and SMCs. These findings suggest that that the structural integrity of the ECM is as important as the assembly of elastic fibers in determining the risk of TAD.
Depletion of SMCs is also a characteristic feature of medial degeneration [72, 73]. In TAD, SMCs show signs of apoptosis, which appear even stronger in areas rich in ground substance and in the vasa vasorum [8, 74]. SMC apoptosis—evidenced by p53 accumulation in the nuclei of SMCs, increased expression of the proapoptotic BAX gene in their cytoplasm, and terminal deoxynucleotidyl transferase–mediated dUTP-biotin nick end labeling (TUNEL)-positive VSMCs—may be mediated by the altered expression of angiotensin II type 2 receptors and result in abnormal matrix deposition in the aortic media and adventitia [49, 75, 76]. Using a mouse model that overexpresses diphtheria toxin in SMCs in ApoE-/- mice, Clarke et al [76] showed that SMC apoptosis is associated with medial expansion, increased elastic lamina breaks, and abnormal matrix deposition, suggesting that SMC apoptosis is important for degenerative medial remodeling and may contribute to AAD formation.
Additionally, the phenotype of SMCs is changed from contractile to synthetic [77]. Evidence of this transition lies in the loss of myofilaments by SMCs, the downregulation of genes encoding for cytoskeletal and myofibrillar proteins (e.g., α-actinin and several members of the myosin gene family), and the formation of an extensive rough endoplasmic reticulum and a large Golgi complex [51, 71, 77]. Reduced expression of cytoskeletal and myofibrillar proteins disrupts cell mobility, while the extensive rough endoplasmic reticulum and large Golgi complex contribute to the overproduction of substances involved in the remodeling of the aortic wall, including various ECM structural components, connective tissue growth factors, and proteases. Cytoskeletal protein degradation decreases tensile-stress–induced phenotypic modulation of SMCs in an organ culture module [78]. Despite the characteristic observation of SMC apoptosis in TAD, we also observe regions of SMC proliferation and compensational hyperplasia in our TAD patients, reflective of regenerative processes in the aortic media [75].
Altered Expression of Multiple Proteins
Degeneration of the aortic wall in TAD could also result from impaired aortic remodeling. Evidence of the altered expression of multiple proteins involved in aortic remodeling corroborates this proposition. More specifically, the expression of smooth muscle 22-α, destrin, α-actinin-2-associated LIM protein, skeletal muscle LIM-protein FHL1, and heat shock protein 27 is altered in TAD [79]. Smooth muscle 22-α is an SMC-specific actin-binding protein, as well as a contractile marker of differentiated VSMCs. Destrin regulates the actin cytoskeleton. α-Actinin-2-associated LIM protein and skeletal muscle LIM-protein FHL1 are both involved in cytoskeletal integrity and reorganization. Heat shock protein 27 may have a role in actin filament remodeling during SMC migration and contractions. Deficiencies of any one of these proteins may alter the organizational, synthetic, and contractile capabilities of SMCs and promote TAD formation.
CELLULAR AND BIOCHEMICAL REGULATORY PATHWAYS IN TAD
Inflammation
SMC apoptosis and ECM destruction in the aortic wall are accompanied by an increased degree of inflammation, as evidenced by the presence of T-lymphocytes, macrophages, mast cells, and neutrophils. This suggests that inflammation participates in the pathogenesis of aortic dissection formation by regulating aortic wall homeostasis. T-lymphocytes and macrophages can be found diffusely throughout the media or in focal accumulations between SMC layers and inside the wall of vasa vasorum, suggesting their possible migration from the adventitia to the media of the aortic wall [8]. Several cytokines and chemokines that promote the recruitment of inflammatory cells to the aortic wall, such as TNF-α, interferon (IFN)-γ, interleukin (IL)-1, IL-2, IL-6, and IL-8, are upregulated in TAD [80]. This increased expression of cytokines is accompanied by an increased expression of other chemokines, such as monocyte chemoattractant protein 1 (MCP-1), and of integrins αM and α2, all of which are linked to inflammatory processes. These inflammatory cells are potential sources of proteases that can degrade the ECM and weaken the aortic wall. For example, MMP-9, elastase, and collagenase secreted from macrophages and neutrophils are capable of directly degrading the ECM and may also contribute to the detachment of SMCs from the ECM, leading to cell death [81]. Furthermore, pathways protective of the action of inflammatory proteases appear to be dysfunctional in TAD. Schachner and colleagues [82] observed decreased levels of α1-antitrypsin in the aortic wall of patients with acute dissection. This serine protease inhibitor normally protects tissues from enzymes produced by inflammatory cells and also regulates the expression of proinflammatory cytokines. An imbalance between proteases and their inhibitors—in favor of proteases—may result in tissue damage in TAD.
Histologic analysis of TAD tissue consistently reveals a large number of adaptive immune cells within the outer media and adventitia. High levels of CD3+, CD4+, CD8+, and CD45+ T cells and CD68+ monocytes have been identified in the aortic wall of TAD patients [8]. High levels of NK, B, and CD8+CD28 in the peripheral blood of patients with acute Stanford type A TAD suggest that innate and cytotoxic cells are involved in aortic wall rupture [80]. These changes are not accompanied by an increase in Th2 cytokines, suggesting that CD4+ T cell– mediated response is not critical to TAD formation. This finding contrasts with reports of attenuated aneurysm formation in CD4 -/- mice and IFN-γ -/- mice and of a Th2-predominant immune response in patients with abdominal aortic aneurysm, suggesting that the pathogenesis of aneurysm and dissection formation may be time dependent [80, 83, 84]. Because CD8+ T cells regulate CD4+ cells, patients with acute dissection may not have significant changes in CD4+ population. CD8+CD28- cells are a subset of CD8+ T cells that have a triple role in inhibiting the activation of CD4+ T cells, promoting CD4+ T cell apoptosis, and promoting regulatory T cell differentiation. Therefore, acute aortic dissection may result from a cytotoxic T cell–mediated response, whereas the T cell population and pathogenesis may shift with continued injury and dissection propagation. Whether a Th1 or Th2 cytokine response dominates in the pathogenesis of TAD remains unknown.
The chronic inflammatory state found in TAD tissue suggests that the ECM has an immunomodulatory role [85, 86]. Increasing evidence indicates that ECM fragments can act as chemoattractants for immune cells. Although data are limited regarding the effect of altered ECM on immune cell behavior in TAD, we can extrapolate from studies of other tissues. For instance, after lipopolysaccharide is administered to murine lung, selective cleavage of collagen type 1 by MMP-8 and MMP-9 produces an acetyl-PRP tripeptide that closely mimics the chemoattractant effects of CXC-chemokine ligand 8 (CXCL8) [87] and shares homology with bacterial peptides capable of activating CXC-chemokine receptor (CXCR1 and CXCR2) to activate neutrophils [88]. Elastin fragments from MMP-9 cleavage are also strong monocyte chemoattractants and can enhance the expression of Th1 type cytokines [89]. In the renal inflammation model, biglycan binds to TLR2 and TLR4, leading to upregulation of TNF expression by macrophages [90]. Hyaluronan fragments derived from intratracheal bleomycin infusion have also been shown to perpetuate the inflammatory response by binding to TLR2 and TLR4 [91]. Extracellular matrix remodeling can also affect the genetic expression of inflammatory cells. For instance, integrin-mediated interactions of monocytes and macrophages with the ECM alter the expression of inflammatory and immune response genes, affecting overall monocyte and macrophage function [92]. These findings indicate that damage to the ECM, when it results in elastic fiber fragmentation or the generation of proteoglycan degradation products, can perpetuate an inflammatory process by recruiting, activating, and inducing the differentiation of immune cells.
The distribution of inflammatory cells varies within the layers of the aortic wall (unpublished data) and may contribute to different inflammatory mechanisms of vascular degeneration [93]. For instance, blockade of neutrophil-derived MMP-9 in the intima significantly attenuated AAD development in Ang II-infused WT mice treated with β-aminopropionitrile monofumarate [94]. At the adventitia-media border, activated dendritic cells can produce chemokines that trigger the recruitment of macrophages and CD4+ T cells [95, 96]. Once activated, these cells undergo clonal expansion and secrete cytokines, including IFN-γ, involved in the regulation of macrophage differentiation and function [95]. The ultimate consequence of these inflammatory activities is elastic fiber fragmentation, as well as activation of repair mechanisms such as cell proliferation and angiogenesis [97, 98].
Hypoxia and Increased Oxidative Stress
Hypoxia and increased oxidative stress also probably contribute to medial degeneration. In support of this hypothesis, research conducted in TAD patients shows increased levels of the oxidative stress marker malondialdehyde and decreased levels of the extracellular superoxide dismutase, an antioxidative enzyme [79]. Increased oxidative stress from vasa vasorum that is impaired (e.g., by hypertension) alters the elastic and collagen fibers in the outer media, stiffens the aortic wall, increases circumferential wall stress, and may predispose patients to TAD [99]. Hypoxia also promotes MMP-2 and MMP-9 upregulation through the HIF-1 α/Ets-1 pathway in aortic aneurysms [100]. In addition, mice deficient in superoxide-generating reduced nicotinamide-adenine dinucleotide phosphate oxidase NOX1 have an increased tissue inhibitor of metalloproteinase 1 (TIMP-1) level and a decreased susceptibility to aortic dissection after angiotensin II infusion [101]. However, it has not yet been determined whether these phenomena actually contribute to TAD through a protease/inhibitor imbalance or result from the inflammatory processes that are triggered when or after the aortic wall dissects.
SMC apoptosis, increased degrees of inflammation and oxidative stress, imbalance between proteases and their inhibitors resulting in a shift toward proteolysis, and impaired remodeling in the aortic wall may accelerate the rate of SMC loss and tissue destruction, possibly before the onset of dissection. An ongoing, disorganized regenerative process leads to the formation of irregularly arranged fibers in the aortic media. This irregular arrangement may contribute to the reduced functional properties of the aortic wall and could promote aortic dissection formation and progression [75].
Matrix Metalloproteinases in TAD
The dissected aortic wall is characterized by an excess of proteases, including cathepsins, chymase, tryptase, neutrophil-derived serine elastase, tissue and urokinase plasminogen activators, plasmin, and matrix metalloproteinases (MMPs). MMPs are proteases that can act on a variety of extracellular protein substrates, activating some and degrading others [102]. In the context of TAD, however, MMPs mainly act as proteases on ECM components. Several studies confirm the increased expression of MMPs in TAD cases, especially MMP-1, -2, -7, and -9 [48, 50, 103, 104]. MMP-1 degrades collagen types I, II, and III. It is actually thought to be responsible for the initial cleavage of collagen, which would be further degraded by other MMPs, particularly MMP-2 and MMP-9 [48]. MMP-2 is also capable of degrading type IV collagen, elastin, and various basement membrane proteins of SMCs. MMP-2 expression seems to increase immediately after the onset of aortic dissection and to peak about 1 week later [105]. Its expression decreases in the chronic phase, possibly reflecting a decrease in the ability of the media to heal. MMP-7 expression is increased in the mucoid areas of the dissected aortic wall and appears to be co-localized with MMP-3. MMP-7 degrades elastin, fibronectin, laminin, and some types of collagen and proteoglycans. For example, MMP-7 can cleave versican, resulting in the accumulation of versican fragments in the aortic media. In addition to being able to directly degrade elastin, MMP-7 may contribute to elastolysis through the inactivation of serine protease inhibitors. MMP-9 degrades type IV collagen, elastin, and various basement membrane proteins of SMCs. Its expression increases within 3 hours of the onset of dissection, peaks about 2 weeks after onset, and remains high throughout the subacute phase, at least in patients with Stanford type B aortic dissection [104].
MMP-9 probably plays a part in the remodeling of the aortic wall. It is expressed by medial SMCs and macrophages, suggesting that macrophages may also have a role in the healing process of the aortic wall. Furthermore, in a genetic association study, the single nucleotide polymorphism MMP9-8202A/G was found to be associated with TAD [48]; however, the functional consequences of the SNP were not evaluated, and the association has not been replicated. In addition to MMP-1, -2, -7, and -9, other MMPs are also upregulated, notably MMP-11, -14, and -19 [50]. MMP-11 degrades fibronectin and proteoglycans; MMP-14 breaks down these substances plus collagen types I, II, and III; and MMP-19 degrades membrane proteins of SMCs.
Like the variety of substrates that MMPs can act on, these proteases can be regulated through various mechanisms [102, 106]. These include the cysteine switch; allosteric activation; furin activation; activation by other MMPs, plasmin, other serine proteases, or reactive oxygen species; and inhibition by TIMPs. In the dissected aortic media, the expression of TIMPs would be expected to decrease; however, upregulation has been observed in several studies [77, 103, 105]. Theoretically, this upregulation should reduce MMP expression, yet the expression of several MMPs is also upregulated. Therefore, it has been suggested that an imbalance between MMP and TIMP expression is responsible for this shift toward a proteolytic state. This imbalance may in part be due to an increase in the concentration of medin oligomers, which activate the production of MMP-2 [107]. Medin oligomers may also directly contribute to SMC apoptosis, whereas medin monomers and mature fibrils do not seem to be toxic to SMCs.
Pathologic Signaling in TAD
TGF-β signaling
TGF-β is commonly known for its role in matrix synthesis. However, TGF-β signaling is also involved in the degradation of the ECM through the upregulation of MMP-2 and -9 and may contribute to TAD formation [108]. TGF-β upregulates multiple factors involved in ECM synthesis, including fibronectin, collagens I, III, V, and VI, proteoglycans, tenascin, and more [109]. Additionally, TGF-β contributes to cell-matrix adhesion by upregulating integrin receptors for collagen, fibronectin, laminin, and vitronectin [110]. In the ECM, TGF-β is inactive when bound to the large latent complex, which comprises latent TGF-β binding protein, latency-associated protein, and FBN1. Activation of TGF-β depends on either proteolytic cleavage to release TGF-β or direct cell binding. FBN1 fragmentation can lead to improper sequestration of TGF-β and, subsequently, increased TGF-β availability [21]. However, conflicting evidence exists regarding the pathologic nature of the increased TGF-β signaling that leads to aneurysm or dissection formation. One study found that mice deficient in Emilin1 had increased TGF-β activity; however, these mice had a low incidence of aneurysms and no dissection [42]. Another study showed that embryonic ablation of TGF-β signaling through lineage-specific mutation of the type II TGF-β receptor gene (Tgfbr2) impaired elastogenesis and resulted in aneurysm formation [111]. Nonetheless, pathologic TGF-β activity has been reported in several heritable connective tissue disorders associated with a high incidence of aneurysm and dissection formation, such as Marfan syndrome, Loeys-Dietz syndrome, and Shprintzen-Goldberg syndrome [112].
TGF-β (TGF-β1, TGF-β2, or TGF-β3) normally binds to the TGFBR2 homodimer, activating it, which in turn activates the TGFBR1 homodimer. In the canonical pathway, the active complex proceeds to phosphorylate Smad 2 and Smad 3 proteins, which recruit Smad 4 to form a Smad2/3-Smad4 complex. This complex translocates to the nucleus, activating the transcription of TGF-β target genes that affect cell proliferation, growth, development, and death [113]. The regulation of the canonical pathways is key to normal biological function. When the Smad2/3-Smad4 complex activates the transcription of TGF-β target genes, it also promotes the transcription of Smad 6 and Smad 7 proteins. Both are inhibitory Smads that antagonize TGF-β signaling; the Smad 7 protein specifically binds to TGFBR1 and inhibits the phosphorylation of Smad 2 and 3 proteins [114, 115]. In addition to the inhibitory Smads, the proto-oncoproteins Ski and SnoN are also important negative regulators of the canonical TGF-β signaling pathway. Ski and SnoN interact with the Smad2/3-Smad4 complex, as well as the Smad-binding element of the Smad 7 promoter [116]. In addition, Ski functions as a direct antagonist of TGFBR1, which leads to accumulation of a non-functional R-smad/smad4 complex [117]. The interaction of Ski and SnoN represses the Smad2/3-Smad4 complex’s ability to activate the transcription of TGF-β target genes [118-120]. In addition to corepressing gene transcription, SnoN acts to further sequester Smad proteins in the nucleus [121]. Furthermore, Ski and SnoN compete with transcriptional coactivators p300/CREB binding protein for SMAD binding and actively recruit a transcriptional co-repressor complex containing N-COR and HDAC [118, 119, 122-124]. Collectively, Ski and SnoN regulate TGF-β signaling on many levels, and alterations of these proto-oncoproteins may be critical to the development of TAD [112].
An increase in TGF-β signaling could also be the result of non-canonical signaling pathways such as the Rho-associated protein kinase cascade and the mitogen-activated protein kinase (MAPK) cascade [125, 126]. These pathways have been shown to contribute to the progression of aortic aneurysms in a mouse model of Marfan syndrome and may also have some relevance for TAD formation [127]. If the TGFBR 1/2 signaling cascade has separate canonical and non-canonical activities, compensatory upregulation occurs when Ski, SnoN, and Smad 7 provide feedback regulation to canonical pathways [128]. Although Ski and SnoN are degraded upon initial exposure to TGF-β, the expression of SnoN appears to be induced after extended exposure to TGF-β treatment, which would occur with the less regulated non-canonical TGF-β signaling [121]. These alternative signaling pathways may explain the formation of dissection in patients with increased TGF-β activity.
Angiotensin II signaling
Angiotensin II (Ang II) is another signaling pathway that is interrelated with TGF-β signaling and may have a role in TAD formation. Angiotensin II can affect the vascular wall through direct vasoconstriction, resulting in hypertension, stimulation of sodium reabsorption, aldosterone synthesis, and the modulation of the function of multiple adhesion molecules, cytokines, chemokines, and growth factors that may be responsible for cell proliferation, hypertrophy, fibrosis, and inflammation [129, 130]. Through Ang II receptor 1 (AT1), Ang II can enhance TGF-β signaling, as well as activate Smad and MAPK signaling in a TGF-β–independent manner [131]. Infusing Ang II into apolipoprotein E–deficient (ApoE -/-) mice results in aneurysm and dissection formation [132]. Histologically, these mouse aortas show elastin degradation, upregulation of MCP-1 and IL-6, and macrophage accumulation at the site of dissection in the media [133]. Mice deficient in the MCP-1 receptor gene Ccr2 are resistant to Ang II-induced dissection, and treating Fbn1 -/- mice with losartan attenuates and reverses ECM changes [134]. These findings suggest that blockade of Ang II signaling may attenuate TAD formation and progression.
However, the role of the Ang II receptor 2 (AT2) in dissection formation remains less clear. AT2 is known to inhibit AT1-induced TGF-β signaling and to attenuate both canonical and non-canonical TGF-β signaling [131]. AT2 is also known to induce VSMC apoptosis [135]. Thus, AT2 may have a role in inducing or inhibiting dissection formation. In the β-aminopropionitrile fumarate (BAPN)-treated rat model of aortic dissection formation, there is increased expression of AT2 and increased VSMC apoptosis [136]. Treatment with an angiotensin-converting enzyme (ACE) inhibitor, but not a selective AT1 inhibitor, attenuates aneurysm and dissection formation [137]. Histologically, there is a decrease in interlaminar elastic fibers, suggesting a loss of connections among elastic lamina and between elastin and SMCs. The BAPN treatment also reduced elastin cross-linking by inhibiting LOX. Whether dissection formation or its attenuation results from changes in AT2 signaling or can be attributed to other defects, such as defects in LOX, is unclear.
RISK FACTORS FOR TAD
Many risk factors have been associated with aortic dissection, including male gender, older age, and hypertension [138-140]. In addition, aortic root motion may contribute to the onset of TAD: downward displacement of the aortic root, enhanced by aortic valve regurgitation, may cause tears in the aortic wall, especially in the context of increased aortic tissue stiffness [141]. The degree of ascending aortic curvature has also been suggested as an important cause of aortic dissection and may be a more important factor than aortic diameter, blood pressure, or cardiac output in aortic wall stress [142]. It may even help predict the location of the tear. Concurrent aortic aneurysms are also a risk factor for TAD, as are smoking, illegal drug use, dyslipidemia, atherosclerosis, and inflammatory diseases that affect the aorta, such as giant cell arteritis, Takayasu aortoarteritis, and syphilis [140, 143]. TAD may also have other external causes, including strenuous exercise, chest trauma, iatrogenic injury (notably during catheterization procedures), and even pregnancy, although most of the pregnancy-related cases seem to be associated with some kind of connective tissue disorder. Additionally, individuals with congenital vascular diseases such as coarctation and bicuspid aortic valve may have a higher risk for TAD [7, 144].
Researchers are gaining a better understanding of how some of these risk factors manifest themselves at the histologic level. For example, male sex is a risk factor for TAD, and aortic dissections in general affect more men than women [7, 144]. In familial cases of TAAD that involve TGFBR1 mutations, men appear more likely to develop aortic aneurysms and dissections than women, and men tend to die younger than women [145]. Heegard and colleagues [65] recently discovered that male Bgn-deficient mice have collagen fibrils of smaller diameter than female Bgn-deficient and wild-type mice. This may be true in humans, as well, and would in part explain the higher incidence of aortic dissection among men.
Normal aging of the aorta also appears to contribute to TAD [146, 147]. As the aorta ages, its well-defined structures shift to unorganized deposits. For instance, elastin fragmentation and fibrosis, which mostly occur in the inner layers of the media, are more severe in the ascending aorta and aortic arch than in the descending or abdominal aorta. These changes reduce the elasticity of the aortic wall and increase its stiffness, making the aortic wall more prone to dissection. Morrison and colleagues [148] found that the thoracic aorta enlarges circumferentially as it ages, but its cyclic ability to deform decreases. Additionally, the aging process of the aorta is characterized by an increase in mucopolysaccharides and by the focal loss of SMC nuclei, although some signs of SMC proliferation have been observed.
Another major risk factor for TAD is hypertension [10]. Although hypertension has been linked to medial degeneration in many studies, some studies have found no significant relationship between the two [147]. Nevertheless, several researchers report hypertension to be associated with elastolysis and hyalinization of collagen in the aortic wall, fibrosis, cell apoptosis, extracellular fatty acid deposition, and even calcification [149]. In combination with hemodynamic stress, particularly in the ascending aorta, hypertension may lead to changes in medial SMCs, increased expression of MMPs, and alteration of the ECM structure [47]. Rather than causing medial degeneration, hypertension may accelerate the onset of dissection in an already disturbed aortic wall, as observed in the BAPN rat model [136].
Attention has also been brought to the increased risk of aortic dissection, especially in young patients, after the use of drugs like methamphetamine and cocaine. Cocaine can upregulate endothelin production, stop nitric oxide release, and block norepinephrine and dopamine reuptake at the presynaptic level, resulting in the release of catecholamines [150]. This catecholamine release causes hypertension and increases vasoconstriction, which ultimately lead to increased shear and circumferential stress on the aortic wall, making it more susceptible to dissection. Additionally, there is evidence that cocaine can trigger SMC apoptosis in a concentration-dependent manner in rats [151].
Advanced atherosclerosis that causes inflammation, destruction, and calcification of the aortic wall may result in decreased aortic elasticity and increased hemodynamic stress on the wall, possibly leading to dissection of the aorta. The calcification that takes place in the aortic wall could be the consequence of improper vitamin K–dependent γ-carboxylation due to single nucleotide polymorphisms in VKORC1[152]. The VKORC1 gene encodes for the vitamin K epoxide reductase complex subunit 1 involved in the mediation of the γ-carboxylation process. Variants in this gene could result in γ-carboxylation dysfunction and increased calcification. Of note, atherosclerosis has been found to be more severe in DeBakey type III aortic dissections, which originate in the descending thoracic aorta, than in dissections that originate in the ascending aorta [153]. Other mechanisms such as chronic inflammation and MMP production may link atherosclerosis with aortic destruction.
Inflammatory disorders, such as giant cell arteritis, have also been associated with TAD. Some cases of giant cell arteritis are characterized by infiltrates of lymphocytes, macrophages, and multinuclear giant cells in the vascular wall, whereas others involve lymphomononuclear cell, neutrophil, and eosinophil accumulation [154].
Most of the previously described risk factors for TAD correspond to acquired conditions. However, congenital abnormalities and inherited connective tissue disorders also are major contributors to TAD. In patients with bicuspid aortic valve, aortic dilation and dissection primarily affect the ascending segment of the aorta. The aortic wall in these patients is characterized by FBN1 deficiency, SMC apoptosis, and, possibly, elevated MMP levels [1]. Increased activity of phosphorylated Smad 2, a marker of TGF-β, has also been observed and may reflect an upregulation of TGF-β in patients with bicuspid aortic valve [155].
Patients with Marfan syndrome are at particularly high risk of aortic dissection. Marfan syndrome is characterized by autosomal dominant inheritance and variable penetrance. The changes in the ascending aortic tissue of Marfan patients with TAD appear to be qualitatively similar to the changes in the aortic tissue of non-Marfan patients with aortic dissection [156]. However, there are some quantitative differences. For example, the aortic wall of Marfan patients with TAD has a higher proportion of smooth elastic lamellae. Smooth elastic lamella is defined by the loss of microfibrils associated with elastic extensions, the loss of contact with SMCs, and the formation of short and compact elastic extensions. The development of TAD in Marfan patients is mostly attributed to the production of defective FBN1, which leads to a decrease in FBN1-containing microfibrils [157]. This results in the inappropriate release of bioactive TGF-β, which is surmised to drive aneurysm and dissection formation. The aortic wall of Marfan patients is also characterized by defective type I collagen synthesis, derangement of medial collagen fibers, increased collagen deposition, and decreased concentrations of elastin and the elastin-specific cross-link compound desmosine. It has also been noted that the expression of MMPs and peroxisome proliferator-activated receptor γ in SMCs are upregulated, and this upregulation is accompanied by SMC apoptosis in Marfan patients [138]. Genetic studies have linked Marfan syndrome to more than 600 mutations in FBN1, mapped to chromosome 15q15.3 [157, 158]. Most of these mutations are missense mutations. FBN1 mutations at 15q21.1 may also be responsible for sporadic cases of TAD [159].
The direct consequence of FBN1 mutations may be either a defect in the assembly of microfibrils, resulting in decreased levels of FBN1-containing microfibrils in the aortic wall of Marfan patients, or a progressive loss of FBN1-containing microfibrils due to the integration of the mutant fibrillin monomers into microfibrils. The effects of FBN1 mutations downstream are many and may include 1) excessive release of TGF-β because of the low affinity of the defective FBN-1 for latent TGF-β1 [21], 2) decreased attachment of VSMCs to elastic fibers, which leads to phenotypic alteration of VSMCs [160], 3) overexpression of MMPs [161-163], 4) increased recruitment of macrophages into the aortic wall [8, 164, 165], and 5) increased proteolysis of the mutant FBN [162], which leads to a cycle of MMP upregulation and macrophage recruitment. Each of these mechanisms has the potential to result in TAD formation or progression.
Loeys-Dietz syndrome (LDS) is another connective tissue disorder associated with TAD. This disorder is linked to missense mutations in TGFBR1 and TGFBR2, which encode for the TGF-β type 1 and type 2 receptors, respectively [128, 166]. A diffuse form of medial degeneration is characteristic of the aortic tissue of patients with LDS [155]. This diffuse medial degeneration is defined as intralamellar elastin fragmentation with ECM deposition and differs from the cystic medial degeneration commonly observed in the aortic wall of Marfan patients, where the elastic fiber fragmentation is interlamellar and accompanied by mucopolysaccharide deposition. Increased collagen deposition and increased activity of phosphorylated Smad 2, reflecting increased TGF-β expression, is found in LDS. The overexpression of TGF-β might be responsible for the increased levels of collagen type I and III observed in LDS.
However, the precise role of TGF-β–induced aortic vasculopathy is more complex than simple upregulation. SMAD3 mutations, which would intuitively abrogate TGF-β signaling or induce a functional haploinsufficiency, cause aneurysm osteoarthritis syndrome. Affected patients have a high incidence of early death due to aortic dissection of a mildly dilated aorta (4-4.5 cm) [167]. Furthermore, haploinsufficiency of TGF-β2 or loss of function mutations in TGF-β2 lead to aortic disease with evidence of increased TGF-β signaling [168, 169]. This apparent paradox is postulated to result from compensatory autocrine or paracrine events [168, 169].
Ehlers-Danlos syndrome is also associated with TAD [170]. There are several types of Ehlers-Danlos syndrome, all of autosomal dominant inheritance. Vascular-type Ehlers-Danlos syndrome especially is responsible for aortic defects and translates into a defective procollagen synthesis [5]. Mutations in the gene encoding for COL3A1 have been found in patients with this specific type of Ehlers-Danlos syndrome, whereas Ehlers-Danlos syndrome with kyphoscoliosis (Type VI) would appear to deregulate the synthesis of procollagen lysyl hydroxylase through mutations in its encoding gene on 1p36.2-36.3 [157, 171]. The Ehlers-Danlos variant of periventricular heterotopia is caused by mutations in the filamin A gene (FLNA) and is associated with hyperflexibility and aortic dilatation [172].
Additionally, cases of TAD have been reported in association with autosomal dominant polycystic kidney disease, mutations in FLNA, Turner syndrome, Noonan syndrome, and patent ductus arteriosus. Autosomal dominant polycystic kidney disease has been linked to mutations in the genes that encode for the plasma protein polycystin-1 and the channel-forming protein polycystin-2 [157, 171]. Mutations in the FLNA gene are characterized by X-linked inheritance and result in the disruption of cross-linking between cortical actin filaments [173].
Patients with Turner syndrome, who have a 45, X karyotype or an abnormal second chromosome X, may develop aortic dissection at a relatively young age [174]. Although a reliable medical profile for these patients does not yet exist, they tend to present with other established risk factors for aortic dissection (e.g., hypertension, bicuspid aortic valve), and their aortic wall shows signs of medial degeneration and thickening, possibly from upregulation of TGF-β signaling. Patients with Turner syndrome have been suggested to be at an even greater risk of developing TAD during pregnancy [174] or if treated with growth hormone [171].
Noonan syndrome is a disorder with an autosomal dominant inheritance pattern that has been associated with mutations in PTPN11, the gene encoding for the protein tyrosine phosphatase 11 [157]. This protein is responsible for transducing signals from growth factors, cytokines, and hormones.
Familial aortic dissections (FADs) have become the focus of some research groups. Often characterized by autosomal dominant inheritance, marked variability in the age at onset, and decreased penetrance, FADs are defined as aortic dissection cases in which the proband’s first-degree relatives are at a higher risk of developing TAD and dying from TAD rupture than is the general population [173, 175, 176]. The incidence of TAAD in these familial cases appears to increase with age, and TAAD is sometimes associated with bicuspid aortic valve or patent ductus arteriosus [173]. Loss of elastic fibers, deposits of mucopolysaccharide-like materials, and defective procollagen synthesis have been observed in patients with familial aortic dissection [138]. Cases of heritable, concomitant TAD and patent ductus arteriosus have been found in one American family and one French family [157]. In the affected individuals, the aortic wall was characterized by a focal loss of SMCs, disruption of elastic fibers and collagen, and accumulation of mucopolysaccharides. Patent ductus arteriosus has been linked to mutations in the MYH11 gene encoding for the contractile protein smooth muscle myosin heavy chain (MYH) [177]. This mutation may cause a conformational change in MYH, leading to an early and severe decrease in aortic wall elasticity.
Because FAD is genetically inherited, much of the research has been directed at identifying the loci linked to familial TAD. So far, identified loci include the TAAD1 locus mapped to 5q13-14, the (rare) FAA1 locus mapped to 11q23.3-24, the TAAD2 locus mapped to 3p24-25 (with TGFBR2 being the mutant gene), the TAAD3 locus mapped to 15q24-26, and the TAAD4 locus mapped to 10q23-24 (with ACTA2 being the mutant gene) [173, 178-183]. Mutations in TGFBR2 are responsible for about 5% of familial TAAD. Some families with TGFBR2 mutations have members who exhibit non-cardiovascular features of Marfan syndrome [149,159]. Mutations in ACTA2 at the TAAD4 locus are associated with medial degeneration, disrupted actin filament assembly and integrity, and the proliferation of medial SMCs [179]. ACTA2 mutations may also be responsible for sporadic cases of TAD. Additional mutations identified in familial TAD include mutations in the MYH11 gene [173], located on 16p12-13, and mutations in the FBN1 gene that are not associated with Marfan syndrome. More specifically, the Gly1127Ser mutation, a single nucleotide substitution in the exon 27 of the FBN1 gene, leads to a normal level of fibrillin synthesis but decreased deposition into the ECM in cultured fibroblasts, possibly because of an abnormal folding of the EGF-like domain encoded by exon 27 [184].
In their review of the genetic basis of TAAD, Milewicz and colleagues [166] suggest that many of the previously described mutations can result in decreased SMC contractile function. One of the possible underlying mechanisms of SMC contractile dysfunction is the disruption of some of the proteins involved in the mechanotransduction complex in SMCs. This mechanotransduction complex is composed of α-actin/β-myosin contractile and cytoskeletal filaments inside the SMCs that interact with transmembrane integrin receptors through vinculin, talin, and filamin A. These integrin receptors link the SMC filaments to the FBN1-containing microfibrils in the ECM. Of note, a comparative microarray study of gene expression profiles in ascending aortic dissection showed a decrease in the expression of actin α-2, SM-MYH11, filamin A, and some integrins, among other genes [50]. In the context of the SMC mechanotransduction complex, FBN1 mutations that disrupt the integration of FBN1 monomers into microfibrils are believed to impair the proper function of this complex by preventing SMC contractile filaments from binding to the FBN1-containing microfibrils [22]. Thus, FBN1 mutations can affect SMC contractility. Similarly, COL3A1, FLNA, ACTA2, and MYH11 mutations may disrupt the SMC mechanotransduction complex and impair SMC contractility. COL3A1 mutations can favor binding of SMCs to type III collagen in the ECM through the transmembrane integrin receptors [185-187]; FLNA mutations can limit connections between the SMC contractile filaments and the integrin receptors [188-190]; ACTA2 mutations can disrupt actin-actin interaction sites or actin polymerization [166, 179], thus affecting actin fiber assembly or stability; and MYH11 can alter the structure and function of myosin in the SMC contractile filaments [166].
Additionally, SMC contractile dysfunction may result from improper SMC differentiation triggered by TGFBR1 or TGFBR2 mutations. Differentiation of the neural crest and of mesenchymal SMCs into VSMCs requires activation by TGF-β. This activation is impaired in the context of TGFBR1 and TGFBR2 mutations because the kinase activity of the TGF-β receptors is altered. Undermined SMC contractility may, in turn, lead to upregulation of stress and repair pathways, including the upregulation of trophic factors, such as insulin growth factor-1 (IGF-1), macrophage inflammatory protein-1 α (MIP-1α), MIP-β, and ACE, which would trigger increased Ang II production by SMCs. Increased production of MMPs and proteoglycans by SMCs may occur concomitantly [81].
Many risk factors, whether acquired or inherited, are associated with TAD. Although researchers have made great progress in identifying these risk factors and linking them to specific histologic changes in the aortic wall, the specific processes and pathways responsible for TAD remain speculative.
ASCENDING AND DESCENDING TAD
It remains unknown whether different mechanisms lead to dissection arising in the ascending aorta (i.e., Stanford type A or DeBakey I or II) and those arising in the descending thoracic aorta (i.e., Stanford type B or DeBakey III). However, some differences in the characteristics of both the patients and their dissected aortic media have already been identified [6]. Ascending TAD usually occurs in younger patients than descending TAD does and is more often associated with a connective tissue disorder. The ascending aortic media in these patients is usually characterized by elastic tissue degeneration and, more specifically, by fragile and irregular elastic fibers interconnecting the lamellar units, particularly in the outer media. In contrast, dissection arising in the descending thoracic aorta tends to occur in older patients, who often have a history of hypertension. The descending thoracic aortic media in these patients shows a laminar degeneration of the SMCs, which may mostly reflect the normal aging process of the aorta.
The developmental biology of the aorta may provide an explanation for the differences between these two types of dissection [128]. VSMCs in the root arise from cardiogenic mesoderm cells, whereas the ascending aorta is composed of both cardiogenic mesodermal cells and cardiac neural crest cells [191]. The descending thoracic aorta is composed of somite-derived SMCs, and at the diaphragm, there is an abrupt transition from somatic mesoderm-derived cells to splanchnic mesoderm cells [192]. The transition points between different VSMCs may represent focal points of weakness between lamellar units where dissection occurs or propagates.
Specific molecular pathways also regulate the development of each VSMC lineage, and each lineage has a different ability to respond to stimuli [192]. For instance, transcriptional responses to TGF-β signaling are lineage dependent [193]. In general, mesoderm-derived VSMCs show little promoter activity and growth inhibition in response to TGF-β stimulation compared to ectoderm-derived VSMCs. Therefore, the heterogeneous vascular microenvironment can activate destructive enzymes such as MMPs, amplify signaling through increased ligand binding, and lead to an anisotropic vascular response that predisposes patients to different types of dissection formation. This mismatch may represent a defective intrinsic regulation of signaling pathways and expression of ECM proteins or interactions, and, hence, the alteration of mechanical properties that leads to ascending or descending aortic dissection formation.
Conclusion
Most cases of TAD are characterized by medial degeneration in the aortic wall. This medial degeneration is associated with the fragmentation and loss of elastic fibers, SMC depletion, fibrotic remodeling, and the accumulation of mucopolysaccharides in the aortic media. Although the genetic and molecular pathways responsible for medial degeneration are not fully understood, it is apparent that the process is rather complex. Inflammatory reactions and increased oxidative stress are cellular responses to aortic injury, and they may induce protease-mediated destruction of the aortic wall. Changes in the expression of molecules that regulate MMP activity (i.e., TIMPs, medin oligomers, cytokines, chemokines, and integrins) could also ultimately lead to medial degeneration. Although the destruction of the components of the ECM certainly contributes to TAD, the ongoing but imbalanced and disorganized healing process in the aortic wall probably also plays a role. These molecular pathways may be either triggered or reinforced by environmental and genetic factors. Many other risk factors, acquired or inherited, are associated with TAD and add to the complexity of the disease.
Although many genetic and molecular pathways involved in the pathogenesis of AAD have been elucidated, studying the molecular mechanisms of aortic wall failure remains a significant challenge for investigators. The spectrum of AAD in patients with genetic abnormalities spans both disease severity and time, suggesting multiple converging and compensatory pathways for disease development. An analysis of gene abnormalities and end-stage human aortic tissue represents only two snapshots of AAD’s natural history. Research regarding the development of the aorta will provide great insight into how circumferential and longitudinal forces, such as those exerted when the body grows, result in normal or abnormal growth. Delineation of the different phases of the disease process is also necessary to understand when to intervene or when to bolster natural physiologic processes. This will allow movement away from characterizing processes as good or bad; instead, cellular, signaling, and molecular processes will be characterized in terms of sufficient or insufficient aortic remodeling. Also, often ignored in any study involving the pathogenesis of AAD is the relation of mechanical forces to disease development. Intuitively, excessive circumferential stress or strain will damage the aorta. However, like the heart, the ultimate function of the aorta is mechanical, and the long-term contribution of hemodynamic forces to disease development remains unknown. Future studies elucidating how biomechanical forces shape aortic development and identifying signaling pathways that lead to AAD development are necessary to develop more effective treatments and, possibly, preventive therapies. Given that TAD ranks among the most lethal vascular diseases, it is important that efforts continue to focus on improving our understanding of this disease and its underlying mechanisms.
Fig 1.
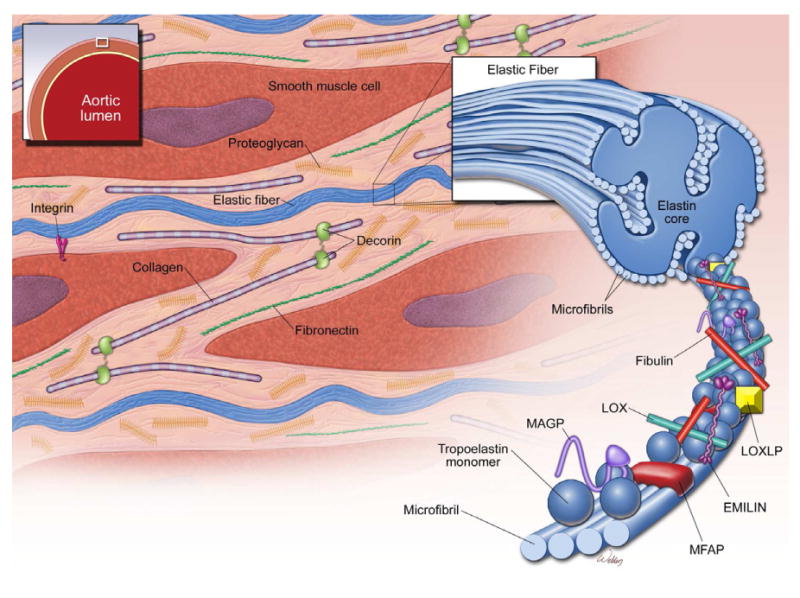
Key structural components of the aorta. The media of the aortic wall is composed of vascular smooth muscle cells (SMCs) and an extracellular matrix (ECM) of elastic fibers, collagen fibers, and proteoglycans. Elastic fiber is the major ECM component and provides extensibility to the aortic wall. Cross-linking of tropoelastin monomers by lysyl oxidase (LOX) forms elastin molecules, which in turn cross-link with microfibrils to form elastic fibers. Microfibrils provide a scaffold for tropoelastin cross-linking. Microfibrils are composed of fibrillin and several microfibril-associated proteins (MFAPs), such as elastin microfibril interface-located protein 1 (EMILIN-1), microfibril-associated glycoproteins (MAGP-1 and -2), and fibulins.
Fig 2.
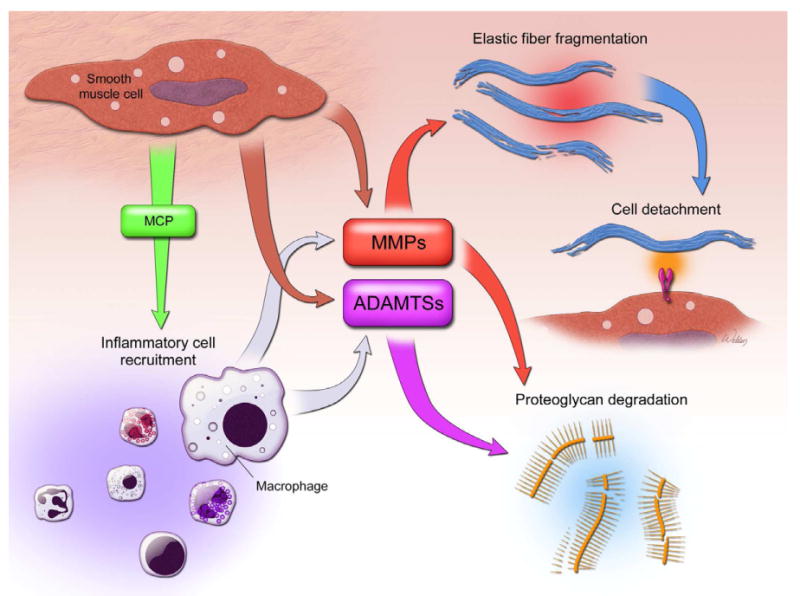
Aortic inflammation. Injured or stressed SMCs produce chemokines such as monocyte chemoattractant protein 1 (MCP-1) to promote the recruitment of inflammatory cells to the aortic wall. Elastin fragments are also strong monocyte chemoattractants. Extracellular matrix proteases such as MMPs (matrix metalloproteinase) and ADAMTS (A disintegrin and metalloproteinase with thrombospondin motifs) produced from SMCs and macrophages are capable of directly degrading the elastic fibers and proteoglycan, respectively. The ECM degradation contributes to the SMC detachment from the ECM. Inflammation also causes SMC injury, dedifferentiation, dysfunction, and, ultimately, death.
Fig 3.
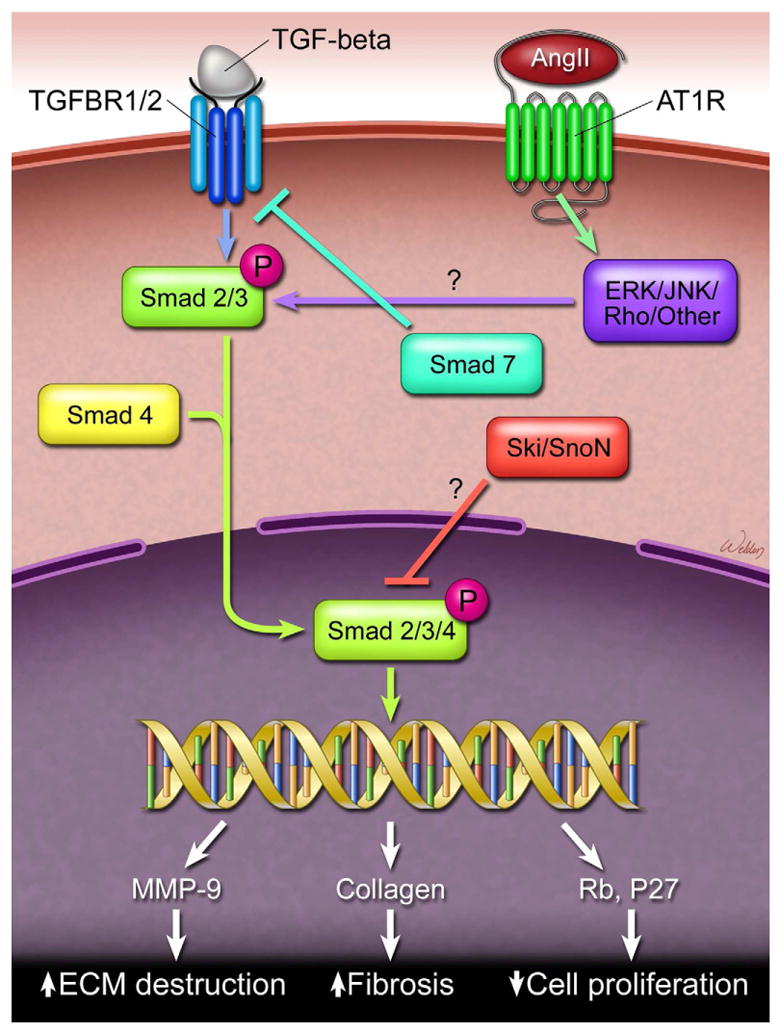
TGF-β signaling. TGF-β binds and activates its receptor (TGFBR). In the canonical pathway, the active complex proceeds to phosphorylate Smad 2 and Smad 3 proteins, which recruit Smad 4 to form a Smad2/3-Smad4 complex. This complex translocates to the nucleus, activating the transcription of TGF-β target genes that affect cell proliferation, cell death, and extracellular matrix (ECM) destruction. TGF-β signaling can be antagonized at the receptor level by Smad 7. The proto-oncoproteins Ski and SnoN also negatively regulate the TGF-β signaling by directly interacting with the Smad2/3-Smad4 complex.
Acknowledgments
Darrell Wu was supported by a training grant (NIH T32 HL007676) through the Department of Molecular Physiology and Biophysics at Baylor College of Medicine. We gratefully acknowledge Stephen N. Palmer, PhD, ELS, of the Texas Heart Institute at St. Luke’s Episcopal Hospital, for providing editorial support, and we thank Scott A. Weldon, MA, CMI, for creating the illustrations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Golledge J, Eagle KA. Acute aortic dissection. Lancet. 2008;372:55. doi: 10.1016/S0140-6736(08)60994-0. [DOI] [PubMed] [Google Scholar]
- 2.Clouse WD, Hallett JW, Jr, Schaff HV, et al. Acute aortic dissection: population-based incidence compared with degenerative aortic aneurysm rupture. Mayo Clin Proc. 2004;79:176. doi: 10.4065/79.2.176. [DOI] [PubMed] [Google Scholar]
- 3.Meszaros I, Morocz J, Szlavi J, et al. Epidemiology and clinicopathology of aortic dissection. Chest. 2000;117:1271. doi: 10.1378/chest.117.5.1271. [DOI] [PubMed] [Google Scholar]
- 4.Kochanek KD, Xu JQ, Murphy SL, et al. Deaths: Final data for 2009. Natl Vital Stat Rep. 2011:60. [PubMed] [Google Scholar]
- 5.Borst HG, Heinemann MK, Stone CD. Histology and ultrastructure. In: Borst HG, Heinemann MK, Stone CD, editors. Surgical Treatment of Aortic Dissection. New York, NY: Churchill Livingstone; 1996. pp. 37–46. [Google Scholar]
- 6.Coady MA, Rizzo JA, Goldstein LJ, et al. Natural history, pathogenesis, and etiology of thoracic aortic aneurysms and dissections. Cardiol Clin. 1999;17:615. doi: 10.1016/s0733-8651(05)70105-3. [DOI] [PubMed] [Google Scholar]
- 7.Homme JL, Aubry MC, Edwards WD, et al. Surgical pathology of the ascending aorta: a clinicopathologic study of 513 cases. Am J Surg Pathol. 2006;30:1159. doi: 10.1097/01.pas.0000213270.38091.69. [DOI] [PubMed] [Google Scholar]
- 8.He R, Guo DC, Estrera AL, et al. Characterization of the inflammatory and apoptotic cells in the aortas of patients with ascending thoracic aortic aneurysms and dissections. J Thorac Cardiovasc Surg. 2006;131:671. doi: 10.1016/j.jtcvs.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 9.Lander AD. Proteoglycans. In: Kreis T, Vale R, editors. Guidebook to the Extracellular Matrix, Anchor, and Adhesion Proteins. New York: Oxford University Press; 1999. pp. 351–356. [Google Scholar]
- 10.Nakashima Y, Shiokawa Y, Sueishi K. Alterations of elastic architecture in human aortic dissecting aneurysm. Lab Invest. 1990;62:751. [PubMed] [Google Scholar]
- 11.Wang X, LeMaire SA, Chen L, et al. Decreased expression of fibulin-5 correlates with reduced elastin in thoracic aortic dissection. Surgery. 2005;138:352. doi: 10.1016/j.surg.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe M, Sawai T. Alteration of cross-linking amino acids of elastin in human aorta in association with dissecting aneurysm: analysis using high performance liquid chromatography. Tohoku J Exp Med. 1999;187:291. doi: 10.1620/tjem.187.291. [DOI] [PubMed] [Google Scholar]
- 13.Karnik SK, Brooke BS, Bayes-Genis A, et al. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003;130:411. doi: 10.1242/dev.00223. [DOI] [PubMed] [Google Scholar]
- 14.Rodgers UR, Weiss AS. Integrin αvβ3 binds a unique non-RGD site near the C-terminus of human tropoelastin. Biochimie. 2004;86:173. doi: 10.1016/j.biochi.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Mochizuki S, Brassart B, Hinek A. Signaling pathways transduced through the elastin receptor facilitate proliferation of arterial smooth muscle cells. J Biol Chem. 2002;277:44854. doi: 10.1074/jbc.M205630200. [DOI] [PubMed] [Google Scholar]
- 16.Wagenseil JE, Nerurkar NL, Knutsen RH, et al. Effects of elastin haploinsufficiency on the mechanical behavior of mouse arteries. Am J Physiol Heart Circ Physiol. 2005;289:H1209. doi: 10.1152/ajpheart.00046.2005. [DOI] [PubMed] [Google Scholar]
- 17.Li DY, Faury G, Taylor DG, et al. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest. 1998;102:1783. doi: 10.1172/JCI4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kielty CM, Baldock C, Lee D, et al. Fibrillin: From microfibril assembly to biomechanical function. Philos Trans R Soc Lond B Biol Sci. 2002;357:207. doi: 10.1098/rstb.2001.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Isogai Z, Ono RN, Ushiro S, et al. Latent transforming growth factor β-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J Biol Chem. 2003;278:2750. doi: 10.1074/jbc.M209256200. [DOI] [PubMed] [Google Scholar]
- 20.Yurchenco PD, Birk DE, Mecham RP. Extracellular matrix assembly and structure. San Diego: Academic Press; 1994. [Google Scholar]
- 21.Neptune ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-β activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 22.Bax DV, Bernard SE, Lomas A, et al. Cell adhesion to fibrillin-1 molecules and microfibrils is mediated by α5β1 and αvβ3 integrins. J Biol Chem. 2003;278:34605. doi: 10.1074/jbc.M303159200. [DOI] [PubMed] [Google Scholar]
- 23.Ritty TM, Broekelmann TJ, Werneck CC, et al. Fibrillin-1 and -2 contain heparin-binding sites important for matrix deposition and that support cell attachment. Biochem J. 2003;375:425. doi: 10.1042/BJ20030649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelleher CM, McLean SE, Mecham RP. Vascular extracellular matrix and aortic development. Curr Top Dev Biol. 2004;62:153. doi: 10.1016/S0070-2153(04)62006-0. [DOI] [PubMed] [Google Scholar]
- 25.Carta L, Pereira L, Arteaga-Solis E, et al. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J Biol Chem. 2006;281:8016. doi: 10.1074/jbc.M511599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Massam-Wu T, Chiu M, Choudhury R, et al. Assembly of fibrillin microfibrils governs extracellular deposition of latent TGF β. J Cell Sci. 2010;123:3006. doi: 10.1242/jcs.073437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev. 2009;89:957. doi: 10.1152/physrev.00041.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weinbaum JS, Broekelmann TJ, Pierce RA, et al. Deficiency in microfibril-associated glycoprotein-1 leads to complex phenotypes in multiple organ systems. J Biol Chem. 2008;283:25533. doi: 10.1074/jbc.M709962200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen E, Larson JD, Ekker SC. Functional analysis of zebrafish microfibril-associated glycoprotein-1 (Magp1) in vivo reveals roles for microfibrils in vascular development and function. Blood. 2006;107:4364. doi: 10.1182/blood-2005-02-0789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roark EF, Keene DR, Haudenschild CC, et al. The association of human fibulin-1 with elastic fibers: an immunohistological, ultrastructural, and RNA study. J Histochem Cytochem. 1995;43:401. doi: 10.1177/43.4.7534784. [DOI] [PubMed] [Google Scholar]
- 31.Kostka G, Giltay R, Bloch W, et al. Perinatal lethality and endothelial cell abnormalities in several vessel compartments of fibulin-1-deficient mice. Mol Cell Biol. 2001;21:7025. doi: 10.1128/MCB.21.20.7025-7034.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McLaughlin PJ, Bakall B, Choi J, et al. Lack of fibulin-3 causes early aging and herniation, but not macular degeneration in mice. Hum Mol Genet. 2007;16:3059. doi: 10.1093/hmg/ddm264. [DOI] [PubMed] [Google Scholar]
- 33.Sicot FX, Tsuda T, Markova D, et al. Fibulin-2 is dispensable for mouse development and elastic fiber formation. Mol Cell Biol. 2008;28:1061. doi: 10.1128/MCB.01876-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horiguchi M, Inoue T, Ohbayashi T, et al. Fibulin-4 conducts proper elastogenesis via interaction with cross-linking enzyme lysyl oxidase. Proc Natl Acad Sci U S A. 2009;106:19029. doi: 10.1073/pnas.0908268106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Atsawasuwan P, Mochida Y, Katafuchi M, et al. Lysyl oxidase binds transforming growth factor-β and regulates its signaling via amine oxidase activity. J Biol Chem. 2008;283:34229. doi: 10.1074/jbc.M803142200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maki JM, Rasanen J, Tikkanen H, et al. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation. 2002;106:2503. doi: 10.1161/01.cir.0000038109.84500.1e. [DOI] [PubMed] [Google Scholar]
- 37.Yanagisawa H, Davis EC, Starcher BC, et al. Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature. 2002;415:168. doi: 10.1038/415168a. [DOI] [PubMed] [Google Scholar]
- 38.Spencer JA, Hacker SL, Davis EC, et al. Altered vascular remodeling in fibulin-5-deficient mice reveals a role of fibulin-5 in smooth muscle cell proliferation and migration. Proc Natl Acad Sci U S A. 2005;102:2946. doi: 10.1073/pnas.0500058102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Colombatti A, Spessotto P, Doliana R, et al. The EMILIN/Multimerin family. Front Immunol. 2011;2:93. doi: 10.3389/fimmu.2011.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bressan GM, Daga-Gordini D, Colombatti A, et al. Emilin, a component of elastic fibers preferentially located at the elastin-microfibrils interface. J Cell Biol. 1993;121:201. doi: 10.1083/jcb.121.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zanetti M, Braghetta P, Sabatelli P, et al. EMILIN-1 deficiency induces elastogenesis and vascular cell defects. Mol Cell Biol. 2004;24:638. doi: 10.1128/MCB.24.2.638-650.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zacchigna L, Vecchione C, Notte A, et al. Emilin1 links TGF-β maturation to blood pressure homeostasis. Cell. 2006;124:929. doi: 10.1016/j.cell.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 43.Doliana R, Bot S, Mungiguerra G, et al. Isolation and characterization of EMILIN-2, a new component of the growing EMILINs family and a member of the EMI domain-containing superfamily. J Biol Chem. 2001;276:12003. doi: 10.1074/jbc.M011591200. [DOI] [PubMed] [Google Scholar]
- 44.Schiavinato A, Becker AK, Zanetti M, et al. EMILIN-3, peculiar member of elastin microfibril interface-located protein (EMILIN) family, has distinct expression pattern, forms oligomeric assemblies, and serves as transforming growth factor β (TGF-β) antagonist. J Biol Chem. 2012;287:11498. doi: 10.1074/jbc.M111.303578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pozzi A, Wary KK, Giancotti FG, et al. Integrin α1β1 mediates a unique collagen-dependent proliferation pathway in vivo. J Cell Biol. 1998;142:587. doi: 10.1083/jcb.142.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sariola H, Viljanen T, Luosto R. Histological pattern and changes in extracellular matrix in aortic dissections. J Clin Pathol. 1986;39:1074. doi: 10.1136/jcp.39.10.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ishii T, Asuwa N. Collagen and elastin degradation by matrix metalloproteinases and tissue inhibitors of matrix metalloproteinase in aortic dissection. Hum Pathol. 2000;31:640. doi: 10.1053/hupa.2000.7642. [DOI] [PubMed] [Google Scholar]
- 48.Wang X, LeMaire SA, Chen L, et al. Increased collagen deposition and elevated expression of connective tissue growth factor in human thoracic aortic dissection. Circulation. 2006;114:I200. doi: 10.1161/CIRCULATIONAHA.105.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Figueiredo Borges L, Jaldin RG, Dias RR, et al. Collagen is reduced and disrupted in human aneurysms and dissections of ascending aorta. Hum Pathol. 2008;39:437. doi: 10.1016/j.humpath.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 50.Weis-Muller BT, Modlich O, Drobinskaya I, et al. Gene expression in acute Stanford type A dissection: a comparative microarray study. J Transl Med. 2006;4:29. doi: 10.1186/1479-5876-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rahkonen O, Su M, Hakovirta H, et al. Mice with a deletion in the first intron of the Col1a1 gene develop age-dependent aortic dissection and rupture. Circ Res. 2004;94:83. doi: 10.1161/01.RES.0000108263.74520.15. [DOI] [PubMed] [Google Scholar]
- 52.Hormuzdi SG, Penttinen R, Jaenisch R, et al. A gene-targeting approach identifies a function for the first intron in expression of the alpha1(I) collagen gene. Mol Cell Biol. 1998;18:3368. doi: 10.1128/mcb.18.6.3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith LB, Hadoke PW, Dyer E, et al. Haploinsufficiency of the murine Col3a1 locus causes aortic dissection: a novel model of the vascular type of Ehlers-Danlos syndrome. Cardiovasc Res. 2011;90:182. doi: 10.1093/cvr/cvq356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lucero HA, Kagan HM. Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell Mol Life Sci. 2006;63:2304. doi: 10.1007/s00018-006-6149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu X, Zhao Y, Gao J, et al. Elastic fiber homeostasis requires lysyl oxidase-like 1 protein. Nat Genet. 2004;36:178. doi: 10.1038/ng1297. [DOI] [PubMed] [Google Scholar]
- 56.Hurtado PA, Vora S, Sume SS, et al. Lysyl oxidase propeptide inhibits smooth muscle cell signaling and proliferation. Biochem Biophys Res Commun. 2008;366:156. doi: 10.1016/j.bbrc.2007.11.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oleggini R, Gastaldo N, Di Donato A. Regulation of elastin promoter by lysyl oxidase and growth factors: cross control of lysyl oxidase on TGF-β1 effects. Matrix Biol. 2007;26:494. doi: 10.1016/j.matbio.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 58.Giampuzzi M, Botti G, Di Duca M, et al. Lysyl oxidase activates the transcription activity of human collagene III promoter: possible involvement of Ku antigen. J Biol Chem. 2000;275:36341. doi: 10.1074/jbc.M003362200. [DOI] [PubMed] [Google Scholar]
- 59.Manley G. Histology of the aortic media in dissecting aneurysms. J Clin Pathol. 1964;17:220. doi: 10.1136/jcp.17.3.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Evanko SP, Angello JC, Wight TN. Formation of hyaluronan- and versican-rich pericellular matrix is required for proliferation and migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:1004. doi: 10.1161/01.atv.19.4.1004. [DOI] [PubMed] [Google Scholar]
- 61.Wight TN. Arterial remodeling in vascular disease: a key role for hyaluronan and versican. Front Biosci. 2008;13:4933. doi: 10.2741/3052. [DOI] [PubMed] [Google Scholar]
- 62.Theocharis AD, Tsolakis I, Hjerpe A, et al. Human abdominal aortic aneurysm is characterized by decreased versican concentration and specific downregulation of versican isoform V(0) Atherosclerosis. 2001;154:367. doi: 10.1016/s0021-9150(00)00504-9. [DOI] [PubMed] [Google Scholar]
- 63.Gutierrez PS, Reis MM, Higuchi ML, et al. Distribution of hyaluronan and dermatan/chondroitin sulfate proteoglycans in human aortic dissection. Connect Tissue Res. 1998;37:151. doi: 10.3109/03008209809002435. [DOI] [PubMed] [Google Scholar]
- 64.Hocking AM, Shinomura T, McQuillan DJ. Leucine-rich repeat glycoproteins of the extracellular matrix. Matrix Biol. 1998;17:1. doi: 10.1016/s0945-053x(98)90121-4. [DOI] [PubMed] [Google Scholar]
- 65.Heegaard AM, Corsi A, Danielsen CC, et al. Biglycan deficiency causes spontaneous aortic dissection and rupture in mice. Circulation. 2007;115:2731. doi: 10.1161/CIRCULATIONAHA.106.653980. [DOI] [PubMed] [Google Scholar]
- 66.Mohamed SA, Sievers HH, Hanke T, et al. Pathway analysis of differentially expressed genes in patients with acute aortic dissection. Biomark Insights. 2009;4:81. doi: 10.4137/bmi.s2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Apte SS. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J Biol Chem. 2009;284:31493. doi: 10.1074/jbc.R109.052340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Taketani T, Imai Y, Morota T, et al. Altered patterns of gene expression specific to thoracic aortic aneurysms: microarray analysis of surgically resected specimens. Int Heart J. 2005;46:265. doi: 10.1536/ihj.46.265. [DOI] [PubMed] [Google Scholar]
- 69.Ren P, Zhang L, Xu G, et al. ADAMTS-1 and ADAMTS-4 levels are elevated in thoracic aortic aneurysms and dissections. Ann Thorac Surg. 2013;95:570. doi: 10.1016/j.athoracsur.2012.10.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Muller BT, Modlich O, Prisack HB, et al. Gene expression profiles in the acutely dissected human aorta. Eur J Vasc Endovasc Surg. 2002;24:356. doi: 10.1053/ejvs.2002.1731. [DOI] [PubMed] [Google Scholar]
- 71.Hassane S, Claij N, Lantinga-van Leeuwen IS, et al. Pathogenic sequence for dissecting aneurysm formation in a hypomorphic polycystic kidney disease 1 mouse model. Arterioscler Thromb Vasc Biol. 2007;27:2177. doi: 10.1161/ATVBAHA.107.149252. [DOI] [PubMed] [Google Scholar]
- 72.Henderson EL, Geng YJ, Sukhova GK, et al. Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms. Circulation. 1999;99:96. doi: 10.1161/01.cir.99.1.96. [DOI] [PubMed] [Google Scholar]
- 73.Lopez-Candales A, Holmes DR, Liao S, et al. Decreased vascular smooth muscle cell density in medial degeneration of human abdominal aortic aneurysms. Am J Pathol. 1997;150:993. [PMC free article] [PubMed] [Google Scholar]
- 74.Ihling C, Szombathy T, Nampoothiri K, et al. Cystic medial degeneration of the aorta is associated with p53 accumulation, Bax upregulation, apoptotic cell death, and cell proliferation. Heart. 1999;82:286. doi: 10.1136/hrt.82.3.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nagashima H, Sakomura Y, Aoka Y, et al. Angiotensin II type 2 receptor mediates vascular smooth muscle cell apoptosis in cystic medial degeneration associated with Marfan’s syndrome. Circulation. 2001;104:I282. doi: 10.1161/hc37t1.094856. [DOI] [PubMed] [Google Scholar]
- 76.Clarke MC, Littlewood TD, Figg N, et al. Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ Res. 2008;102:1529. doi: 10.1161/CIRCRESAHA.108.175976. [DOI] [PubMed] [Google Scholar]
- 77.Lesauskaite V, Tanganelli P, Sassi C, et al. Smooth muscle cells of the media in the dilatative pathology of ascending thoracic aorta: morphology, immunoreactivity for osteopontin, matrix metalloproteinases, and their inhibitors. Hum Pathol. 2001;32:1003. doi: 10.1053/hupa.2001.27107. [DOI] [PubMed] [Google Scholar]
- 78.Zheng JP, Ju D, Shen J, et al. Disruption of actin cytoskeleton mediates loss of tensile stress induced early phenotypic modulation of vascular smooth muscle cells in organ culture. Exp Mol Pathol. 2010;88:52. doi: 10.1016/j.yexmp.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liao M, Liu Z, Bao J, et al. A proteomic study of the aortic media in human thoracic aortic dissection: implication for oxidative stress. J Thorac Cardiovasc Surg. 2008;136:65. doi: 10.1016/j.jtcvs.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 80.del Porto F, Proietta M, Tritapepe L, et al. Inflammation and immune response in acute aortic dissection. Ann Med. 2010;42:622. doi: 10.3109/07853890.2010.518156. [DOI] [PubMed] [Google Scholar]
- 81.Allaire E, Schneider F, Saucy F, et al. New insight in aetiopathogenesis of aortic diseases. Eur J Vasc Endovasc Surg. 2009;37:531. doi: 10.1016/j.ejvs.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 82.Schachner T, Golderer G, Sarg B, et al. The amounts of alpha 1 antitrypsin protein are reduced in the vascular wall of the acutely dissected human ascending aorta. Eur J Cardiothorac Surg. 2010;37:684. doi: 10.1016/j.ejcts.2009.07.025. [DOI] [PubMed] [Google Scholar]
- 83.Schönbeck U, Sukhova GK, Gerdes N, et al. TH2 predominant immune responses prevail in human abdominal aortic aneurysm. Am J Pathol. 2002;161:499. doi: 10.1016/S0002-9440(10)64206-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xiong W, Zhao Y, Prall A, et al. Key roles of CD4+ T cells and IFN-γ in the development of abdominal aortic aneurysms in a murine model. J Immunol. 2004;172:2607. doi: 10.4049/jimmunol.172.4.2607. [DOI] [PubMed] [Google Scholar]
- 85.Adair-Kirk TL, Senior RM. Fragments of extracellular matrix as mediators of inflammation. Int J Biochem Cell Biol. 2008;40:1101. doi: 10.1016/j.biocel.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol. 2010;10:712. doi: 10.1038/nri2852. [DOI] [PubMed] [Google Scholar]
- 87.Weathington NM, van Houwelingen AH, Noerager BD, et al. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med. 2006;12:317. doi: 10.1038/nm1361. [DOI] [PubMed] [Google Scholar]
- 88.Gaggar A, Jackson PL, Noerager BD, et al. A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. J Immunol. 2008;180(5662) doi: 10.4049/jimmunol.180.8.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Debret R, Antonicelli F, Theill A, et al. Elastin-derived peptides induce a T-helper type 1 polarization of human blood lymphocytes. Arterioscler Thromb Vasc Biol. 2005;25:1353. doi: 10.1161/01.ATV.0000168412.50855.9f. [DOI] [PubMed] [Google Scholar]
- 90.Babelova A, Moreth K, Tsalastra-Greul W, et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J Biol Chem. 2009;284:24035. doi: 10.1074/jbc.M109.014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jiang D, Liang J, Fan J, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 92.de Fougerolles AR, Koteliansky VE. Regulation of monocyte gene expression by the extracellular matrix and its functional implications. Immunol Rev. 2002;186:208. doi: 10.1034/j.1600-065x.2002.18617.x. [DOI] [PubMed] [Google Scholar]
- 93.Zhao L, Pratico D, Rader DJ, et al. 12/15-Lipoxygenase gene disruption and vitamin E administration diminish atherosclerosis and oxidative stress in apolipoprotein E deficient mice through a final common pathway. Prostaglandins Other Lipid Mediat. 2005;78:185. doi: 10.1016/j.prostaglandins.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 94.Kurihara T, Shimizu-Hirota R, Shimoda M, et al. Neutrophil-derived matrix metalloproteinase 9 triggers acute aortic dissection. Circulation. 2012;126:3070. doi: 10.1161/CIRCULATIONAHA.112.097097. [DOI] [PubMed] [Google Scholar]
- 95.Xiong W, Zhao Y, Prall A, et al. Key roles of CD4+ T cells and IFN-gamma in the development of abdominal aortic aneurysms in a murine model. J Immunol. 2004;172:2607. doi: 10.4049/jimmunol.172.4.2607. [DOI] [PubMed] [Google Scholar]
- 96.Galle C, Schandene L, Stordeur P, et al. Predominance of type 1 CD4+ T cells in human abdominal aortic aneurysm. Clin Exp Immunol. 2005;142:519. doi: 10.1111/j.1365-2249.2005.02938.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rizas KD, Ippagunta N, Tilson MD., III Immune cells and molecular mediators in the pathogenesis of the abdominal aortic aneurysm. Cardiol Rev. 2009;17:201. doi: 10.1097/CRD.0b013e3181b04698. [DOI] [PubMed] [Google Scholar]
- 98.Shimizu K, Mitchell RN, Libby P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2006;26:987. doi: 10.1161/01.ATV.0000214999.12921.4f. [DOI] [PubMed] [Google Scholar]
- 99.Angouras D, Sokolis DP, Dosios T, et al. Effect of impaired vasa vasorum flow on the structure and mechanics of the thoracic aorta: Implications for the pathogenesis of aortic dissection. Eur J Cardiothorac Surg. 2000;17:468. doi: 10.1016/s1010-7940(00)00382-1. [DOI] [PubMed] [Google Scholar]
- 100.Erdozain OJ, Pegrum S, Winrow VR, et al. Hypoxia in abdominal aortic aneurysm supports a role for HIF-1α and Ets-1 as drivers of matrix metalloproteinase upregulation in human aortic smooth muscle cells. J Vasc Res. 2011:48–163. doi: 10.1159/000318806. [DOI] [PubMed] [Google Scholar]
- 101.Gavazzi G, Deffert C, Trocme C, et al. NOX1 deficiency protects from aortic dissection in response to angiotensin II. Hypertension. 2007;50:189. doi: 10.1161/HYPERTENSIONAHA.107.089706. [DOI] [PubMed] [Google Scholar]
- 102.Ra HJ, Parks WC. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 2007;26:587. doi: 10.1016/j.matbio.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Koullias GJ, Ravichandran P, Korkolis DP, et al. Increased tissue microarray matrix metalloproteinase expression favors proteolysis in thoracic aortic aneurysms and dissections. Ann Thorac Surg. 2004;78:2106. doi: 10.1016/j.athoracsur.2004.05.088. [DOI] [PubMed] [Google Scholar]
- 104.Sangiorgi G, Trimarchi S, Mauriello A, et al. Plasma levels of metalloproteinases-9 and -2 in the acute and subacute phases of type A and type B aortic dissection. J Cardiovasc Med (Hagerstown) 2006;7:307. doi: 10.2459/01.JCM.0000223251.26988.c5. [DOI] [PubMed] [Google Scholar]
- 105.Manabe T, Imoto K, Uchida K, et al. Decreased tissue inhibitor of metalloproteinase-2/matrix metalloproteinase ratio in the acute phase of aortic dissection. Surg Today. 2004;34:220. doi: 10.1007/s00595-003-2683-3. [DOI] [PubMed] [Google Scholar]
- 106.Zhang X, Shen YH, LeMaire SA. Thoracic aortic dissection: are matrix metalloproteinases involved? Vascular. 2009;17:147. doi: 10.2310/6670.2008.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Peng S, Larsson A, Wassberg E, et al. Role of aggregated medin in the pathogenesis of thoracic aortic aneurysm and dissection. Lab Invest. 2007;87:1195. doi: 10.1038/labinvest.3700679. [DOI] [PubMed] [Google Scholar]
- 108.Chung AW, Yang HH, Radomski MW, et al. Long-term doxycycline is more effective than atenolol to prevent thoracic aortic aneurysm in Marfan syndrome through the inhibition of matrix metalloproteinase-2 and -9. Circ Res. 2008;102:e73. doi: 10.1161/CIRCRESAHA.108.174367. [DOI] [PubMed] [Google Scholar]
- 109.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor β in human disease. N Engl J Med. 2000;342:1350. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 110.Massague J. The transforming growth factor-β family. Annu Rev Cell Biol. 1990;6:597. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- 111.Choudhary B, Zhou J, Li P, et al. Absence of TGFβ signaling in embryonic vascular smooth muscle leads to reduced lysyl oxidase expression, impaired elastogenesis, and aneurysm. Genesis. 2009;47:115. doi: 10.1002/dvg.20466. [DOI] [PubMed] [Google Scholar]
- 112.Doyle AJ, Doyle JJ, Bessling SL, et al. Mutations in the TGF-β repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat Genet. 2012;44:1249. doi: 10.1038/ng.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kang JS, Liu C, Derynck R. New regulatory mechanisms of TGF-β receptor function. Trends Cell Biol. 2009;19:385. doi: 10.1016/j.tcb.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 114.Hayashi H, Abdollah S, Qiu Y, et al. The MAD-related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell. 1997;89:1165. doi: 10.1016/s0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- 115.Nakao A, Afrakhte M, Moren A, et al. Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature. 1997;389:631. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 116.Briones-Orta MA, Sosa-Garrocho M, Moreno-Alvarez P, et al. SnoN co-repressor binds and represses smad7 gene promoter. Biochem Biophys Res Commun. 2006;341:889. doi: 10.1016/j.bbrc.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 117.Ferrand N, Atfi A, Prunier C. The oncoprotein c-ski functions as a direct antagonist of the transforming growth factor-β type I receptor. Cancer Res. 2010;70:8457. doi: 10.1158/0008-5472.CAN-09-4088. [DOI] [PubMed] [Google Scholar]
- 118.Luo K. Ski and SnoN: negative regulators of TGF-β signaling. Curr Opin Genet Dev. 2004;14:65. doi: 10.1016/j.gde.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 119.Luo K, Stroschein SL, Wang W, et al. The Ski oncoprotein interacts with the Smad proteins to repress TGFβ signaling. Genes Dev. 1999;13:2196. doi: 10.1101/gad.13.17.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stroschein SL, Wang W, Zhou S, et al. Negative feedback regulation of TGF-β signaling by the SnoN oncoprotein. Science. 1999;286:771. doi: 10.1126/science.286.5440.771. [DOI] [PubMed] [Google Scholar]
- 121.Krakowski AR, Laboureau J, Mauviel A, et al. Cytoplasmic SnoN in normal tissues and nonmalignant cells antagonizes TGF-β signaling by sequestration of the Smad proteins. Proc Natl Acad Sci U S A. 2005;102:12437. doi: 10.1073/pnas.0504107102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Akiyoshi S, Inoue H, Hanai J, et al. c-Ski acts as a transcriptional co-repressor in transforming growth factor-β signaling through interaction with smads. J Biol Chem. 1999;274:35269. doi: 10.1074/jbc.274.49.35269. [DOI] [PubMed] [Google Scholar]
- 123.Chen W, Lam SS, Srinath H, et al. Competition between Ski and CREB-binding protein for binding to Smad proteins in transforming growth factor-β signaling. J Biol Chem. 2007;282:11365. doi: 10.1074/jbc.M700186200. [DOI] [PubMed] [Google Scholar]
- 124.Deheuninck J, Luo K. Ski and SnoN, potent negative regulators of TGF-β signaling. Cell Res. 2009;19:47. doi: 10.1038/cr.2008.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lee MK, Pardoux C, Hall MC, et al. TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007;26:3957. doi: 10.1038/sj.emboj.7601818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang YE. Non-Smad pathways in TGF-β signaling. Cell Res. 2009;19:128. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Holm TM, Habashi JP, Doyle JJ, et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332:358. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature. 2011;473:308. doi: 10.1038/nature10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Qin Z. Newly developed angiotensin II-infused experimental models in vascular biology. Regul Pept. 2008;150:1. doi: 10.1016/j.regpep.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 130.Daugherty A, Cassis L. Angiotensin II-mediated development of vascular diseases. Trends Cardiovasc Med. 2004;14:117. doi: 10.1016/j.tcm.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 131.Habashi JP, Doyle JJ, Holm TM, et al. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. 2011;332:361. doi: 10.1126/science.1192152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Saraff K, Babamusta F, Cassis LA, et al. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:1621. doi: 10.1161/01.ATV.0000085631.76095.64. [DOI] [PubMed] [Google Scholar]
- 134.Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tan NY, Li JM, Stocker R, et al. Angiotensin II-inducible smooth muscle cell apoptosis involves the angiotensin II type 2 receptor, GATA-6 activation, and FasL-Fas engagement. Circ Res. 2009;105:422. doi: 10.1161/CIRCRESAHA.109.203323. [DOI] [PubMed] [Google Scholar]
- 136.Nakashima Y, Sueishi K. Alteration of elastic architecture in the lathyritic rat aorta implies the pathogenesis of aortic dissecting aneurysm. Am J Pathol. 1992;140:959. [PMC free article] [PubMed] [Google Scholar]
- 137.Nagashima H, Uto K, Sakomura Y, et al. An angiotensin-converting enzyme inhibitor, not an angiotensin II type-1 receptor blocker, prevents β-aminopropionitrile monofumarate-induced aortic dissection in rats. J Vasc Surg. 2002;36:818. [PubMed] [Google Scholar]
- 138.Nienaber CA, Eagle KA. Aortic dissection: New frontiers in diagnosis and management. Part I: From etiology to diagnostic strategies. Circulation. 2003;108:628. doi: 10.1161/01.CIR.0000087009.16755.E4. [DOI] [PubMed] [Google Scholar]
- 139.Siegal EM. Acute aortic dissection. J Hosp Med. 2006;1:94. doi: 10.1002/jhm.69. [DOI] [PubMed] [Google Scholar]
- 140.LeMaire SA, Russell L. Epidemiology of thoracic aortic dissection. Nat Rev Cardiol. 2011;8:103. doi: 10.1038/nrcardio.2010.187. [DOI] [PubMed] [Google Scholar]
- 141.Beller CJ, Labrosse MR, Thubrikar MJ, et al. Role of aortic root motion in the pathogenesis of aortic dissection. Circulation. 2004;109:763. doi: 10.1161/01.CIR.0000112569.27151.F7. [DOI] [PubMed] [Google Scholar]
- 142.Poullis MP, Warwick R, Oo A, et al. Ascending aortic curvature as an independent risk factor for type A dissection, and ascending aortic aneurysm formation: a mathematical model. Eur J Cardiothorac Surg. 2008;33:995. doi: 10.1016/j.ejcts.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 143.Lesauskaite V, Epistolato MC, Castagnini M, et al. Expression of matrix metalloproteinases, their tissue inhibitors, and osteopontin in the wall of thoracic and abdominal aortas with dilatative pathology. Hum Pathol. 2006;37:1076. doi: 10.1016/j.humpath.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 144.Larson EW, Edwards WD. Risk factors for aortic dissection: a necropsy study of 161 cases. Am J Cardiol. 1984;53:849. doi: 10.1016/0002-9149(84)90418-1. [DOI] [PubMed] [Google Scholar]
- 145.Tran-Fadulu V, Chen JH, Lemuth D, et al. Familial thoracic aortic aneurysms and dissections: three families with early-onset ascending and descending aortic dissections in women. Am J Med Genet A. 2006;140:1196. doi: 10.1002/ajmg.a.31236. [DOI] [PubMed] [Google Scholar]
- 146.Dingemans KP, Teeling P, Lagendijk JH, et al. Extracellular matrix of the human aortic media: an ultrastructural histochemical and immunohistochemical study of the adult aortic media. Anat Rec. 2000;258:1. doi: 10.1002/(SICI)1097-0185(20000101)258:1<1::AID-AR1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 147.Schlatmann TJ, Becker AE. Histologic changes in the normal aging aorta: implications for dissecting aortic aneurysm. Am J Cardiol. 1977;39:13. doi: 10.1016/s0002-9149(77)80004-0. [DOI] [PubMed] [Google Scholar]
- 148.Morrison TM, Choi G, Zarins CK, et al. Circumferential and longitudinal cyclic strain of the human thoracic aorta: age-related changes. J Vasc Surg. 2009;49:1029. doi: 10.1016/j.jvs.2008.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nienaber CA, Eagle KA. Aortic dissection: New frontiers in diagnosis and management. Part II: Therapeutic management and follow-up. Circulation. 2003;108:772. doi: 10.1161/01.CIR.0000087400.48663.19. [DOI] [PubMed] [Google Scholar]
- 150.Daniel JC, Huynh TT, Zhou W, et al. Acute aortic dissection associated with use of cocaine. J Vasc Surg. 2007;46:427. doi: 10.1016/j.jvs.2007.05.040. [DOI] [PubMed] [Google Scholar]
- 151.Su J, Li J, Li W, et al. Cocaine induces apoptosis in primary cultured rat aortic vascular smooth muscle cells: possible relationship to aortic dissection, atherosclerosis, and hypertension. Int J Toxicol. 2004;23:233. doi: 10.1080/10915810490471361. [DOI] [PubMed] [Google Scholar]
- 152.Wang Y, Zhang W, Zhang Y, et al. VKORC1 haplotypes are associated with arterial vascular diseases (stroke, coronary heart disease, and aortic dissection) Circulation. 2006;113:1615. doi: 10.1161/CIRCULATIONAHA.105.580167. [DOI] [PubMed] [Google Scholar]
- 153.Nakashima Y, Kurozumi T, Sueishi K, et al. Dissecting aneurysm: a clinicopathologic and histopathologic study of 111 autopsied cases. Hum Pathol. 1990;21:291. doi: 10.1016/0046-8177(90)90229-x. [DOI] [PubMed] [Google Scholar]
- 154.Salvarani C, Cantini F, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. Lancet. 2008;372:234. doi: 10.1016/S0140-6736(08)61077-6. [DOI] [PubMed] [Google Scholar]
- 155.Maleszewski JJ, Miller DV, Lu J, et al. Histopathologic findings in ascending aortas from individuals with Loeys-Dietz syndrome (LDS) Am J Surg Pathol. 2009;33:194. doi: 10.1097/PAS.0b013e31817f3661. [DOI] [PubMed] [Google Scholar]
- 156.Dingemans KP, Teeling P, van der Wal AC, et al. Ultrastructural pathology of aortic dissections in patients with Marfan syndrome: comparison with dissections in patients without Marfan syndrome. Cardiovasc Pathol. 2006;15:203. doi: 10.1016/j.carpath.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 157.Pannu H, Tran-Fadulu V, Milewicz DM. Genetic basis of thoracic aortic aneurysms and aortic dissections. Am J Med Genet C Semin Med Genet. 2005;139:10. doi: 10.1002/ajmg.c.30069. [DOI] [PubMed] [Google Scholar]
- 158.Waldmuller S, Muller M, Warnecke H, et al. Genetic testing in patients with aortic aneurysms/dissections: a novel genotype/phenotype correlation? Eur J Cardiothorac Surg. 2007;31:970. doi: 10.1016/j.ejcts.2007.02.027. [DOI] [PubMed] [Google Scholar]
- 159.LeMaire SA, McDonald ML, Guo DC, et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat Genet. 2011;43:996. doi: 10.1038/ng.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bunton TE, Biery NJ, Myers L, et al. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res. 2001;88:37. doi: 10.1161/01.res.88.1.37. [DOI] [PubMed] [Google Scholar]
- 161.Booms P, Ney A, Barthel F, et al. A fibrillin-1-fragment containing the elastin-binding-protein GxxPG consensus sequence upregulates matrix metalloproteinase-1: biochemical and computational analysis. J Mol Cell Cardiol. 2006;40:234. doi: 10.1016/j.yjmcc.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 162.Charbonneau NL, Carlson EJ, Tufa S, et al. In vivo studies of mutant fibrillin-1 microfibrils. J Biol Chem. 2010;285:24943. doi: 10.1074/jbc.M110.130021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chung AW, Au Yeung K, Sandor GG, et al. Loss of elastic fiber integrity and reduction of vascular smooth muscle contraction resulting from the upregulated activities of matrix metalloproteinase-2 and -9 in the thoracic aortic aneurysm in Marfan syndrome. Circ Res. 2007;101:512. doi: 10.1161/CIRCRESAHA.107.157776. [DOI] [PubMed] [Google Scholar]
- 164.Guo G, Booms P, Halushka M, et al. Induction of macrophage chemotaxis by aortic extracts of the mgR Marfan mouse model and a GxxPG-containing fibrillin-1 fragment. Circulation. 2006;114:1855. doi: 10.1161/CIRCULATIONAHA.105.601674. [DOI] [PubMed] [Google Scholar]
- 165.Guo G, Gehle P, Doelken S, et al. Induction of macrophage chemotaxis by aortic extracts from patients with Marfan syndrome is related to elastin binding protein. PLoS One. 2011;6:e20138. doi: 10.1371/journal.pone.0020138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Milewicz DM, Guo DC, Tran-Fadulu V, et al. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- 167.van de Laar IM, Oldenburg RA, Pals G, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43:121. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 168.Boileau C, Guo DC, Hanna N, et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet. 2012;44:916. doi: 10.1038/ng.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lindsay ME, Schepers D, Bolar NA, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44:922. doi: 10.1038/ng.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Oderich GS, Panneton JM, Bower TC, et al. The spectrum, management and clinical outcome of Ehlers-Danlos syndrome type IV: a 30-year experience. J Vasc Surg. 2005;42:98. doi: 10.1016/j.jvs.2005.03.053. [DOI] [PubMed] [Google Scholar]
- 171.Hasham SN, Guo DC, Milewicz DM. Genetic basis of thoracic aortic aneurysms and dissections. Curr Opin Cardiol. 2002;17:677. doi: 10.1097/00001573-200211000-00015. [DOI] [PubMed] [Google Scholar]
- 172.Sheen VL, Jansen A, Chen MH, et al. Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology. 2005;64:254. doi: 10.1212/01.WNL.0000149512.79621.DF. [DOI] [PubMed] [Google Scholar]
- 173.Milewicz DM, Chen H, Park ES, et al. Reduced penetrance and variable expressivity of familial thoracic aortic aneurysms/dissections. Am J Cardiol. 1998;82:474. doi: 10.1016/s0002-9149(98)00364-6. [DOI] [PubMed] [Google Scholar]
- 174.Carlson M, Silberbach M. Dissection of the aorta in Turner syndrome: Two cases and review of 85 cases in the literature. J Med Genet. 2007;44:745. doi: 10.1136/jmg.2007.052019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Albornoz G, Coady MA, Roberts M, et al. Familial thoracic aortic aneurysms and dissections—incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg. 2006;82:1400. doi: 10.1016/j.athoracsur.2006.04.098. [DOI] [PubMed] [Google Scholar]
- 176.Biddinger A, Rocklin M, Coselli J, et al. Familial thoracic aortic dilatations and dissections: A case control study. J Vasc Surg. 1997;25:506. doi: 10.1016/s0741-5214(97)70261-1. [DOI] [PubMed] [Google Scholar]
- 177.Zhu L, Vranckx R, Khau Van Kien P, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38:343. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 178.Guo D, Hasham S, Kuang SQ, et al. Familial thoracic aortic aneurysms and dissections: genetic heterogeneity with a major locus mapping to 5q13-14. Circulation. 2001;103:2461. doi: 10.1161/01.cir.103.20.2461. [DOI] [PubMed] [Google Scholar]
- 179.Guo DC, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle α-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 180.Hasham SN, Willing MC, Guo DC, et al. Mapping a locus for familial thoracic aortic aneurysms and dissections (TAAD2) to 3p24-25. Circulation. 2003;107:3184. doi: 10.1161/01.CIR.0000078634.33124.95. [DOI] [PubMed] [Google Scholar]
- 181.Kakko S, Raisanen T, Tamminen M, et al. Candidate locus analysis of familial ascending aortic aneurysms and dissections confirms the linkage to the chromosome 5q13-14 in Finnish families. J Thorac Cardiovasc Surg. 2003;126:106. doi: 10.1016/s0022-5223(03)00037-0. [DOI] [PubMed] [Google Scholar]
- 182.Pannu H, Fadulu VT, Chang J, et al. Mutations in transforming growth factor-β receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005;112:513. doi: 10.1161/CIRCULATIONAHA.105.537340. [DOI] [PubMed] [Google Scholar]
- 183.Vaughan CJ, Casey M, He J, et al. Identification of a chromosome 11q23.2-q24 locus for familial aortic aneurysm disease, a genetically heterogeneous disorder. Circulation. 2001;103:2469. doi: 10.1161/01.cir.103.20.2469. [DOI] [PubMed] [Google Scholar]
- 184.Francke U, Berg MA, Tynan K, et al. A Gly1127Ser mutation in an EGF-like domain of the fibrillin-1 gene is a risk factor for ascending aortic aneurysm and dissection. Am J Hum Genet. 1995;56:1287. [PMC free article] [PubMed] [Google Scholar]
- 185.Kim JK, Xu Y, Xu X, et al. A novel binding site in collagen type III for integrins α1β1 and α2β1. J Biol Chem. 2005;280:32512. doi: 10.1074/jbc.M502431200. [DOI] [PubMed] [Google Scholar]
- 186.Raynal N, Hamaia SW, Siljander PR, et al. Use of synthetic peptides to locate novel integrin α2β1-binding motifs in human collagen III. J Biol Chem. 2006;281:3821. doi: 10.1074/jbc.M509818200. [DOI] [PubMed] [Google Scholar]
- 187.Zoppi N, Ritelli M, Colombi M. Type III and V collagens modulate the expression and assembly of EDA+ fibronectin in the extracellular matrix of defective Ehlers-Danlos syndrome fibroblasts. Biochim Biophys Acta. 2012;1820:1576. doi: 10.1016/j.bbagen.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 188.Labeit S, Lahmers S, Burkart C, et al. Expression of distinct classes of titin isoforms in striated and smooth muscles by alternative splicing, and their conserved interaction with filamins. J Mol Biol. 2006;362:664. doi: 10.1016/j.jmb.2006.07.077. [DOI] [PubMed] [Google Scholar]
- 189.Lebart MC, Mejean C, Casanova D, et al. Characterization of the actin binding site on smooth muscle filamin. J Biol Chem. 1994;269:4279. [PubMed] [Google Scholar]
- 190.Yu N, Erb L, Shivaji R, et al. Binding of the P2Y2 nucleotide receptor to filamin A regulates migration of vascular smooth muscle cells. Circ Res. 2008;102:581. doi: 10.1161/CIRCRESAHA.107.162271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Waldo KL, Hutson MR, Ward CC, et al. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev Biol. 2005;281:78. doi: 10.1016/j.ydbio.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 192.Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. 2007;27:1248. doi: 10.1161/ATVBAHA.107.141069. [DOI] [PubMed] [Google Scholar]
- 193.Topouzis S, Majesky MW. Smooth muscle lineage diversity in the chick embryo: two types of aortic smooth muscle cell differ in growth and receptor-mediated transcriptional responses to transforming growth factor-β. Dev Biol. 1996;178:430. doi: 10.1006/dbio.1996.0229. [DOI] [PubMed] [Google Scholar]