

Abstract
Extracellular vesicles are membrane micro/nanovesicles secreted by many cell types into the circulation and the extracellular milieu in physiological and pathological conditions. Evidence suggests that extracellular vesicles, known as matrix vesicles, play a role in the mineralization of skeletal tissue, but emerging ultrastructural and in vitro studies have demonstrated their contribution to cardiovascular calcification as well. Cells involved in the progression of cardiovascular calcification release active vesicles capable of nucleating hydroxyapatite on their membranes. This review discusses the role of extracellular vesicles in cardiovascular calcification, and elaborates on this additional mechanism of calcification as an alternative pathway to the currently accepted mechanism of biomineralization via osteogenic differentiation.
Introduction
Cardiovascular calcification, a growing burden in Westernized countries, is not only a risk factor for cardiovascular events, but may itself contribute to cardiovascular risk (1). A growing number of studies have demonstrated that microcalcification in vulnerable plaques contribute to plaque destabilization and fatal plaque rupture (2–5). Calcification of the cardiovascular system — including the coronary arteries and heart valves — follows an active process in which smooth-muscle cells (SMCs) or valve interstitial cells undergo osteogenic transformation (6–8). In addition, it is now evident that calcification progresses through, and indeed may be initiated by, the release of calcifying extracellular vesicles by cells residing in the calcification niche (2, 9).
Extracellular vesicles possess a metabolically active outer membrane that protects the internal cargo — consisting of proteins, miRNA, and other components from the parental cell. They can be found throughout the body in various tissues and fluids, and they participate in both physiological and pathological processes. Their involvement in a broad range of pathological pathways has made them attractive diagnostic biomarkers (10), while their therapeutic use is an emerging field (11, 12). Extracellular vesicles appear to have advantages over existing drug delivery systems due to their size, lack of toxicity, and target specificity. A growing number of studies have contributed to the concept that cells implicated in the progression of cardiovascular calcification release active extracellular vesicles capable of nucleating hydroxyapatite (13–15). This emerging subset of the field provides additional mechanisms by which to therapeutically target cardiovascular calcification.
Discovery of Calcifying Extracellular Vesicles
The discovery of cell-derived extracellular vesicles followed the introduction of transmission electron microscopy in the mid-20th century. The groups of Anderson and Bonucci discovered that extracellular vesicles associate with the earliest sites of mineral formation in bone and cartilage mineralization (16, 17). These extracellular membrane-bound structures were later termed matrix vesicles (MVs). Physiological mineralization is now widely believed to be initiated in bone, dentin, and cartilage by vesicles released from specific regions of the outer membranes of osteoblasts, odontoblasts, and osteoblasts (18). Using ultrastructural, histological, and cytochemical techniques, Anderson and colleagues showed the presence of matrix vesicle-like structures that were believed to originate from smooth muscle cells (9, 19).
Classification Controversy
Much controversy exists in this field regarding the classification and nomenclature used for extracellular vesicles. Depending on size and type, extracellular vesicles are broadly classified as ectosomes (or shedding microvesicles), exosomes, and apoptotic bodies (20). Ectosomes, also known as microparticles, are large extracellular vesicles ranging from 50–1000 nm in diameter; exosomes are small membranous vesicles of endocytic origin ranging from 40–100 nm in diameter; and apoptotic bodies are released from fragmented apoptotic cells and are 50–5000 nm in diameter. MVs (the main focus of this review) are another category that should be added to this classification. MVs are small membranous structures (30–300 nm in diameter) surrounded by a lipid biolayer, are produced by blebbing of plasma membrane, and can calcify. The current criteria for the classification of extracellular vesicles includes size, density, morphology, lipid/protein composition, and subcellular origin (Table) (21). Several limitations exist in our current understanding of the field. The recently formed International Society of Extracellular Vesicles hopes to overcome these issues by producing guidelines to standardize the field (http://www.isevmeeting.org).
Table.
Classification of extracellular vesicles.
| Extracellular vesicle | Size (nm) | Density in sucrose (g/ml) | Morphology | Lipid composition | Protein markers | Subcellular location |
|---|---|---|---|---|---|---|
| Exosome | 40–100 | 1.10–1.21 | Cup shape; multivesicular bodies | Enriched in cholesterol, sphingomyelin, ceramide; lipid rafts; exposed phosphatidylserine | Tetraspanins (CD63, CD9), Alix, TSG101 | Endosomes (internal compartments) |
| Ectosome (microparticles) | 50–1000 | Bilamelar round and tubular structures | Enriched in cholesterol and diacylglycerol; exposed phosphatidyserine | Proteolytic enzymes; CR1 | Plasma membrane | |
| Apoptotic body | 50–5000 | 1.16–1.28 | Heterogeneous | Exposed phosphatidylserine | Histones | |
| Matrix vesicle | 30–300 | Double membrane vesicles | Exposed phosphatidylserine | Enriched in annexins | Plasma membrane |
Preparation of Extracellular Vesicles
The isolation of these entities is a major issue of dispute. Different groups use different protocols, which leads to differences between study results. The few groups that study the role of MVs in cardiovascular calcification follow a similar protocol, allowing for more direct comparison of results between groups. The main method for isolating calcifying extracellular vesicles from cell supernatant is differential centrifugation — short time periods of low centrifugal forces to remove cellular fragments (~800–1000 g) and apoptotic bodies (16,500 g), followed by a much higher centrifugal force (~100,000 g) for a longer duration (13, 14). The correctness of this method is debatable; some scientists in the realm of exosomes, for example, prefer to perform a sucrose gradient in which vesicles are separated by density. Future analyses and growth of the field should clarify whether the current protocol of MV isolation needs alteration.
Role of Extracellular Vesicles in Pathogenesis
Circulating microparticles contain large numbers of extracellular vesicles released from platelets, erythrocytes, leukocytes, and endothelial cells — first described as “cell dust”— and are key in the haemostatic response (22, 23). They participate in the pathogenesis of thromboembolic diseases, wherein the number of circulating procoagulant-microparticles increases greatly (23). In the field of cancer research, exosomes — extracellular counterparts of endosomes — may enable invasive growth of tumor cells (24). Circulating extracellular vesicles represent excellent biomarkers for determining the severity of various diseases. Early evidence suggests that increased numbers of endothelial extracellular vesicles in the pulmonary circulation demonstrate advancement of pulmonary arterial hypertension (25). In addition, extracellular vesicles contain varying cargoes of lipids, proteins, and miRNA, which have sparked interest in their potential as diagnostic markers of disease (26).
Pathological Calcification
Cardiovascular calcification is now recognized as regulated biomineralization that follows similar pathways to that of bone development. Nuclear magnetic resonance and X-ray diffraction techniques have shown that the matrix-mineral atomic interface in calcified plaque is similar to that in bone (27). Several studies have indicated that, in addition to bone-like mechanisms, other mechanisms - including cell death - also contribute to cardiovascular calcification (28, 29, 30). A combination of in vitro studies (31, 32) and molecular imaging have formed the concept of an inflammation-dependent calcification paradigm (30, 33), suggesting that macrophage infiltration and inflammation precede calcification, and activated pro-inflammatory pathways induce osteogenic transformation of SMCs or release of MVs.
Calcifying extracellular vesicles also have been identified in calcifying aortic valves (34), atherosclerotic intimal lesions (15), and medial arterial calcification (2, 34, 35) — similar to the MVs involved in physiological bone mineralization (18). Literature is lacking regarding the role of MVs in calcifying aortic valves, which could differ from the role of MVs in arterial calcification — although this concept requires further investigation. MVs may initiate the mineralization processes akin to those in bone, but also may lead to the rupture of vulnerable plaques (2). During the early stages of calcification, MVs and apoptotic bodies released from macrophages and SMCs may contribute to the calcification process (14, 15). Regions of “spotty” calcifications in atherosclerotic lesions that contain calcified vesicles have been predicted, via finite element simulations, to have increased stress levels and to be prone to rupture (5), which can lead to acute thrombosis and fatal myocardial infarction (36). Patients with chronic renal disease (CRD) have advanced atherosclerosis and a high cardiovascular mortality rate (37). The severity and complexity of CRD causes a perfect storm of metabolic dysfunction, leading to accelerated intimal and extensive medial calcification (37, 38). Concentrations of extracellular calcium and phosphate, similar to those found in serum of CRD patients on dialysis, induce the release of calcifying MVs from cells involved in cardiovascular calcification, such as vascular SMCs and macrophages (14, 15). The plaques of non-CRD patients also may contain MVs (39), suggesting that this phenomenon also occurs in an atherosclerotic or diabetic milieu.
Structure
Ultrastructural studies of components of the cardiovascular system have identified highly heterogeneous mineral-associated vesicles (MVs) ranging from 30–300 nm in diameter and displaying different structural appearances (2, 34, 35). They are typically double-membrane- bound bodies that are round or ovoid in shape (17), often associated with extracellular matrix components, particularly collagen, and displaying evidence of hydroxyapatite crystals on the inner membrane within the lumen, and/or on the outer membrane of the vesicle (Figure 1).
Figure 1.
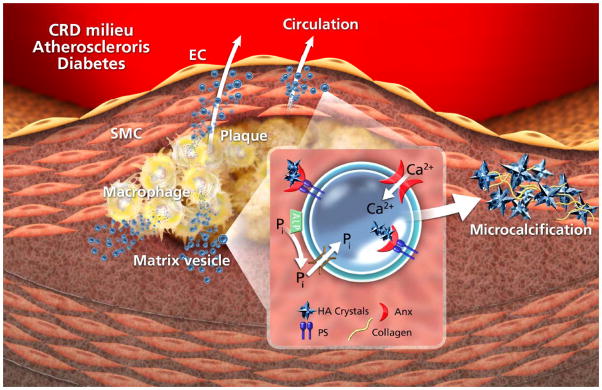
Mechanism of pathological matrix vesicle mineralization: A mineral imbalance leads to an influx of phosphate and calcium into the vesicle via appropriate channels – ALP generates inorganic phosphate in the extravesicular space. Nucleation of hydroxyapatite (HA) is facilitated by an annexin (Anx)-PS complex, enabling generation of microcalcification/calcification.
Biogenesis
MVs arise from cells during physiological mineralization through a “budding” process (40), originating from specialized regions of the cell plasma membrane and released in the same orientation as that of the membrane (41). When the vesicle buds from the cell, it takes a subset of the cell’s “cargo” with it, safely packaged within a region of the membrane. Evidence suggests that the plasma membrane of the parenting cell contains components similar to those of the released vesicle, but in different quantities (42). MVs are rich in annexins and acidic phospholipids, such as phosphatidylserine (PS), compared to the corresponding plasma membranes, which enable them to calcify (14, 15, 42).
Matrix Vesicle Cargo
Extracellular vesicles mediate active communication between cells. They participate in the exchange of functional and genetic information, and in the mediation of adaptive immune responses. The cargo carried by extracellular vesicles dictates their function. Proteomics studies have provided evidence of the cargo contained within osteoblast-derived and chondrocyte-derived MVs (14, 43, 44). Numerous non-calcifying extracellular vesicles also appear within calcifying plaques. Studies by Shananan and colleagues (14, 29) have provided evidence suggesting that non-calcifying vesicles contain inhibitors of calcification such as fetuin- A and matrix Gla protein, which decrease in MVs released by cells in a CRD milieu, enabling them to calcify. These MVs may possess a physiological function under non-pathological conditions. In one study, proteomics analysis of vascular SMC-MVs detected signaling molecules (14). Calcifying vesicles therefore may be merely dysfunctional vesicles, released to act as intercellular communicators. On the other hand, cells may release these particular vesicles as a reaction to stress, as a means of releasing unwanted “cargo” — such as excess calcium.
Nucleation Core
PS forms a complex with calcium at sites of early mineralization in skeletal tissue (45), and inorganic phosphate is required to form the nucleation core (46). We now believe that in both physiological and pathological MVs, annexins also may contribute to the formation of nucleation complex (14, 15). Annexins — specifically, annexin-2, -5, and -6 — are major proteins within MVs (15, 45). They facilitate calcium influx and mineralization by binding to PS and forming ion channels in the MV membrane (45, 47). The exact importance of annexins in MV mineralization, however, is still under investigation.
Annexin-5 was previously shown to facilitate nucleation and growth of mineral (48). Despite this finding, a recent in vivo study produced evidence that a lack of annexin-5 and/or annexin-6 functionality does not affect skeletal development in mice (49). These results suggest that a compensatory mechanism is at play — another member of the annexin family may compensate for the loss of annexin-5 and/or annexin-6. Annexin-5 is seemingly, if controversially, important in the mineralization potential of macrophage-derived MVs involved in biomineralization (15). In contrast, annexin-6 seems to play a main role in SMC-MV mineralization (14). The nucleation core is required for nucleation — a physiochemical process through which ions accumulate with the correct orientation to mimic a crystal surface — to ensue. We recently demonstrated that pro-inflammatory and pro-thrombotic S100A9 might be involved in nucleation of MVs via formation of PS–annexin5–S100A9 complex (15).
Nucleation, Crystallization, and Microcalcification Formation
Whether crystal formation or transdifferentiation of vascular SMCs occurs first is debatable (50). Whether osteogenic transformation of SMCs is required for MV release and calcification is also unknown. But we do know that in skeletal tissue, the first crystals of hydroxyapatite are formed within extracellular vesicles. This phenomenon can be split into two phases: In conditions of mineral imbalance, the influx of calcium and phosphate into the extracellular vesicles, via their appropriate channels, leads to initial mineral accumulation in the form of amorphous calcium phosphate (51). For mineralization to proceed, the levels of phosphate and pyrophosphate must be in balance (52), achieved via alkaline phosphatase (ALP) within the membranes of calcifying extracellular vesicles in bone (53). Whether MVs involved in cardiovascular calcification contain ALP, however, is controversial — with conflicting reports on ALP within vascular SMC-derived MVs (13, 14). The second phase, mineral propagation, seems to ensue via the release of crystal through the MV membrane, exposing preformed hydroxyapatite nanocrystals to the extracellular fluid. Once exposed to this fluid, the nanocrystals can act as loci or templates for the formation of new crystals via homologous nucleation (9), and perhaps mineralize extracellular matrix (ECM) components. We presume that MVs associated with pathological ectopic calcification generate crystals in a similar way.
MVs bind to ECM components, such as collagens and glycosaminoglycans (GAGs). Vesicles appear to be the initial site of nucleation, and intravesicular mineral crystals seed these ECM components. Vesicle–GAG interaction is enhanced in mineralization-competent/calcifying chondrocyte-derived MVs (54). Atherosclerotic plaque contains a predominance of GAGs associated with mineralization similar to that of bone, further suggesting that GAGs play a direct role in initiating pathological and physiological mineralization (27). Crystalline extracellular vesicles and calcified collagen fibers that abound in Randall’s plaques (found in renal medulla) seem to follow this alternative mechanism of MV mineralization, akin to vascular calcification (55).
Molecular imaging has demonstrated that inflammation and microcalcification evolve within close proximity in intimal arterial calcification, and overlap at border regions that are prone to rupture (33). A positive feedback loop of calcification and inflammation drives disease progression (56). Calcified extracellular vesicles/microcalcification in vulnerable macrophage- rich plaques may contribute to plaque destabilization and rupture, as predicted by Vengrenyuk and colleagues (2–4). Components of calcifying extracellular vesicles, including pro-thrombotic tissue factor and S100A9 may further lead to acute cardiovascular events. A similar inflammation-driven mechanism appears to ensue in calcific aortic valve disease (33, 38). While our electron microscopy observations show that MVs closely associate with microcalcification, suggesting a role of calcifying MVs in the generation of microcalcification (Figure 2), the exact mechanism of pathological microcalcification formation by MVs remains to be elucidated.
Figure 2.
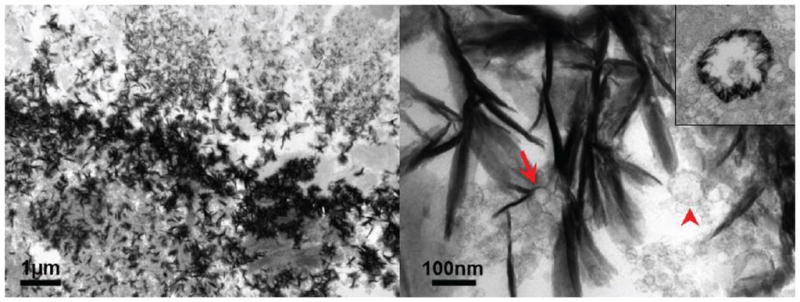
Ultrastructural images of a calcified human tissue: A, defined border of microcalcification; B, higher magnification shows extracellular vesicles (red arrowhead) within regions of microcalcification and nucleation of hydroxyapatite on the outer membrane of MV (red arrow). Inset image shows the nucleation of MV on the inner membrane.
Conclusion
Much of our knowledge of the role of extracellular vesicles in cardiovascular calcification relies heavily on previously established evidence of MVs involved in physiological bone mineralization. Although research in this field dates back to the 1970s, the role of extracellular vesicles in pathological calcification is still largely unknown. Progression of this field has been hindered by the lack of sophisticated modalities to visualize extracellular vesicles in vitro and in vivo. Novel modalities that can detect the size and numbers of extracellular vesicles and visualize them in vitro in real time — such as nanoparticle tracking analysis — are emerging. In addition, molecular imaging has proven useful in visualizing fluorescently probed calcifying vesicular structures in vivo in live animals (30). The development of innovative technologies would further advance this field of research. Further understanding of extracellular vesicle structure and function may pinpoint novel means of treating diseases associated with these entities.
Acknowledgments
The authors would like to thank Dr Erzsébet Ligeti for insightful conversation and Sara Karwacki for excellent editorial assistance.
Sources of Funding
This work was funded by a grant from the National Institutes of Health (R01HL114805, to Dr. Aikawa) and by Kowa Company, Ltd.
Footnotes
Disclosure
None
References
- 1.Johnson RC, Leopold JA, Loscalzo J. Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res. 2006;99:1044–1059. doi: 10.1161/01.RES.0000249379.55535.21. [DOI] [PubMed] [Google Scholar]
- 2.Bobryshev YV, Killingsworth MC, Lord RS, Grabs AJ. Matrix vesicles in the fibrous cap of atherosclerotic plaque: possible contribution to plaque rupture. J Cell Mol Med. 2008;12:2073–2082. doi: 10.1111/j.1582-4934.2008.00230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vengrenyuk Y, Carlier S, Xanthos S, Cardoso L, Ganatos P, Virmani R, Einav S, Gilchrist L, Weinbaum S. A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proc Natl Acad Sci USA. 2006;103:14678–14683. doi: 10.1073/pnas.0606310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vengrenyuk Y, Kaplan TJ, Cardoso L, Randolph GJ, Weinbaum S. Computational stress analysis of atherosclerotic plaques in ApoE knockout mice. Ann Biomed Eng. 2010;38:738–747. doi: 10.1007/s10439-009-9897-5. [DOI] [PubMed] [Google Scholar]
- 5.Wenk JF. Numerical modeling of stress in stenotic arteries with microcalcifications: a parameter sensitivity study. J Biomech Eng. 2011;133:014503. doi: 10.1115/1.4003128. [DOI] [PubMed] [Google Scholar]
- 6.Bostrom K, Watson KE, Stanford WP, Demer LL. Atherosclerotic calcification: relation to developmental osteogenesis. J Am Cardiol. 1995;75:88B–91B. doi: 10.1016/0002-9149(95)80020-s. [DOI] [PubMed] [Google Scholar]
- 7.Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, Schinke T, Karsenty G, Giachelli CM. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–1154. doi: 10.1161/hh2401.101070. [DOI] [PubMed] [Google Scholar]
- 8.Towler DA, Shao JS, Cheng SL, Pingsterhaus JM, Loewy AP. Osteogenic regulation of vascular calcification. Ann NY Acad Sci. 2006;1068:327–333. doi: 10.1196/annals.1346.036. [DOI] [PubMed] [Google Scholar]
- 9.Anderson HC. Mechanisms of pathologic calcification. Rheum Dis Clin North Am. 1988;14:303–319. [PubMed] [Google Scholar]
- 10.Zhou H, Pisitkun T, Aponte A, Yuen PS, Hoffert JD, Yasuda H, Hu X, Chawla L, Shen RF, Knepper MA, Star RA. Exosomal Fetuin-A identified by proteomics: a novel urinary biomarker for detecting acute kidney injury. Kidney Int. 2006;70:1847–1857. doi: 10.1038/sj.ki.5001874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delcayre A, Le Pecq JB. Exosomes as novel therapeutic nanodevices. Curr Opin Mol Ther. 2006;8:31–38. [PubMed] [Google Scholar]
- 12.Lee Y, EL Andaloussi S, Wood MJA. Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Hum Mol Genet. 2012;21:R125. doi: 10.1093/hmg/dds317. [DOI] [PubMed] [Google Scholar]
- 13.Chen NX, O’Neill KD, Chen X, Moe SM. Annexin-mediated matrix vesicle calcification in vascular smooth muscle cells. J Bone MinerRes. 2008;23:1798–1805. doi: 10.1359/JBMR.080604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M, Shanahan CM. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res. 2011;109:e1–12. doi: 10.1161/CIRCRESAHA.110.238808. [DOI] [PubMed] [Google Scholar]
- 15.New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, Libby P, Shanahan CM, Croce K, Aikawa E. Macrophage-Derived Matrix Vesicles: An Alternative Novel Mechanism for Microcalcification in Atherosclerotic Plaques. Circ Res. 2013 doi: 10.1161/CIRCRESAHA.113.301036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson HC. Electron microscopic studies of induced cartilage development and calcification. J Cell Biol. 1967;35:81–101. doi: 10.1083/jcb.35.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonucci E. Fine structure of early cartilage calcification. J Ultrastruct Res. 1967;20:33–50. doi: 10.1016/s0022-5320(67)80034-0. [DOI] [PubMed] [Google Scholar]
- 18.Anderson HC. Matrix vesicles and calcification. Curr Rheumatol Rep. 2003;5:222–226. doi: 10.1007/s11926-003-0071-z. [DOI] [PubMed] [Google Scholar]
- 19.Anderson HC, Mulhall D, Garimella R. Role of extracellular membrane vesicles in the pathogenesis of various diseases, including cancer, renal diseases, atherosclerosis, and arthritis. Lab Invest. 2010;90:1549–1557. doi: 10.1038/labinvest.2010.152. [DOI] [PubMed] [Google Scholar]
- 20.Kalra H, Simpson RJ, Ji H, et al. Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol. 2012;10:e1001450. doi: 10.1371/journal.pbio.1001450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van der Pol E, Boing AN, Harrison P, Sturk A, Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmaco Rev. 2012;64:676–705. doi: 10.1124/pr.112.005983. [DOI] [PubMed] [Google Scholar]
- 22.Chargaff E, West R. The biological significance of the thromboplastic protein of blood. J Biol Chem. 1946;166:189–197. [PubMed] [Google Scholar]
- 23.Furie B, Furie BC. Mechanisms of thrombus formation. NEJM. 2008;359(9):938–949. doi: 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- 24.Hendrix A, Westbroek W, Bracke M, De Wever O. An ex(o)citing machinery for invasive tumor growth. Cancer Res. 2010;70:9533–9537. doi: 10.1158/0008-5472.CAN-10-3248. [DOI] [PubMed] [Google Scholar]
- 25.Amabile N, Heiss C, Real WM, Minasi P, McGlothlin D, Rame EJ, Grossman W, De Marco T, Yeghiazarians Y. Circulating endothelial microparticle levels predict hemodynamic severity of pulmonary hypertension. Am J Respir Crit Care Med. 2008;177:1268–1275. doi: 10.1164/rccm.200710-1458OC. [DOI] [PubMed] [Google Scholar]
- 26.Goettsch C, Hutcheson JD, Aikawa E. MicroRNA in Cardiovascular Calcification: Focus on Targets and Extracellular Vesicle Delivery Mechanisms. Circ Res. 2013;112:1073–84. doi: 10.1161/CIRCRESAHA.113.300937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duer MJ, Friscic T, Proudfoot D, Reid DG, Schoppet M, Shanahan CM, Skepper JN, Wise ER. Mineral surface in calcified plaque is like that of bone: further evidence for regulated mineralization. Arterioscler Thromb Vasc Biol. 2008;28:2030–2034. doi: 10.1161/ATVBAHA.108.172387. [DOI] [PubMed] [Google Scholar]
- 28.Proudfoot D, Skepper JN, Hegyi L, Farzaneh-Far A, Shanahan CM, Weissberg PL. The role of apoptosis in the initiation of vascular calcification. Z Kardiol. 2001;90 (Suppl 3):43–46. doi: 10.1007/s003920170041. [DOI] [PubMed] [Google Scholar]
- 29.Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, Jahnen-Dechent W, Weissberg PL, Shanahan CM. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15:2857–2867. doi: 10.1097/01.ASN.0000141960.01035.28. [DOI] [PubMed] [Google Scholar]
- 30.New SE, Aikawa E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ Res. 2011;108:1381–91. doi: 10.1161/CIRCRESAHA.110.234146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abedin M, Tintut Y, Demer LL. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol. 2004;24:1161–70. doi: 10.1161/01.ATV.0000133194.94939.42. [DOI] [PubMed] [Google Scholar]
- 32.Tintut Y, Patel J, Parhami F, Demer LL. Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation. 2000;102:2636–2642. doi: 10.1161/01.cir.102.21.2636. [DOI] [PubMed] [Google Scholar]
- 33.Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–2850. doi: 10.1161/CIRCULATIONAHA.107.732867. [DOI] [PubMed] [Google Scholar]
- 34.Kim KM. Calcification of matrix vesicles in human aortic valve and aortic media. Fed Proc. 1976;35:156–162. [PubMed] [Google Scholar]
- 35.Tanimura A, McGregor DH, Anderson HC. Matrix vesicles in atherosclerotic calcification. Proc Soc Exp Biol Med. 1983;172:173–177. doi: 10.3181/00379727-172-41542. [DOI] [PubMed] [Google Scholar]
- 36.Aikawa M, Libby P. The vulnerable atherosclerotic plaque: pathogenesis and therapeutic approach. Cardiovasc Pathol. 2004;13:125–138. doi: 10.1016/S1054-8807(04)00004-3. [DOI] [PubMed] [Google Scholar]
- 37.Goodman WG, London G, Amann K, Block GA, Giachelli C, Hruska KA, Ketteler M, Levin A, Massy Z, McCarron DA, Raggi P, Shanahan CM, Yorioka N. Vascular calcification in chronic kidney disease. Am J Kidney Dis. 2004;43:572–579. doi: 10.1053/j.ajkd.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 38.Aikawa E, Aikawa M, Libby P, Figueiredo JL, Rusanescu G, Iwamoto Y, Fukuda D, Kohler RH, Shi GP, Jaffer FA, Weissleder R. Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation. 2009;119:1785–1794. doi: 10.1161/CIRCULATIONAHA.108.827972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bobryshev YV, Killingsworth MC, Huynh TG, Lord RS, Grabs AJ, Valenzuela SM. Are calcifying matrix vesicles in atherosclerotic lesions of cellular origin? Basic Res Cardiol. 2007;102:133–143. doi: 10.1007/s00395-006-0637-9. [DOI] [PubMed] [Google Scholar]
- 40.Golub EE. Role of matrix vesicles in biomineralization. Biochim Biophys Acta. 2009;1790:1592–1598. doi: 10.1016/j.bbagen.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fedde KN. Human osteosarcoma cells spontaneously release matrix-vesicle-like structures with the capacity to mineralize. Bone Miner. 1992;17:145–151. doi: 10.1016/0169-6009(92)90726-t. [DOI] [PubMed] [Google Scholar]
- 42.Majeska RJ, Wuthier RE. Studies on matrix vesicles isolated from chick epiphyseal cartilage. Association of pyrophosphatase and ATPase activities with alkaline phosphatase. Biochim Biophys Acta. 1975;391:51–60. doi: 10.1016/0005-2744(75)90151-5. [DOI] [PubMed] [Google Scholar]
- 43.Balcerzak M, Malinowska A, Thouverey C, Sekrecka A, Dadlez M, Buchet R, Pikula S. Proteome analysis of matrix vesicles isolated from femurs of chicken embryo. Proteomics. 2008;8:192–205. doi: 10.1002/pmic.200700612. [DOI] [PubMed] [Google Scholar]
- 44.Wuthier RE, Lipscomb GF. Matrix vesicles: structure, composition, formation and function in calcification. Front Biosci. 2012;17:2812–2902. doi: 10.2741/3887. [DOI] [PubMed] [Google Scholar]
- 45.Genge BR, Wu LN, Wuthier RE. Identification of phospholipid-dependent calcium-binding proteins as constituents of matrix vesicles. J Biol Chem. 1989;264(18):10917–10921. [PubMed] [Google Scholar]
- 46.Cotmore JM, Nichols G, Jr, Wuthier RE. Phospholipid-calcium phosphate complex: enhanced calcium migration in the presence of phosphate. Science. 1971:1721339–1341. doi: 10.1126/science.172.3990.1339. [DOI] [PubMed] [Google Scholar]
- 47.Balcerzak M, Hamade E, Zhang L, Pikula S, Azzar G, Radisson J, Bandorowicz-Pikula J, Buchet R. The roles of annexins and alkaline phosphatase in mineralization process. Acta Biochim Pol. 2003;50:1019–1038. [PubMed] [Google Scholar]
- 48.Genge BR, Wu LN, Wuthier RE. Kinetic analysis of mineral formation during in vitro modeling of matrix vesicle mineralization: effect of annexin A5, phosphatidylserine, and type II collagen. AnalBiochem. 2007;367:159–166. doi: 10.1016/j.ab.2007.04.029. [DOI] [PubMed] [Google Scholar]
- 49.Grskovic I, Kutsch A, Frie C, et al. Depletion of annexin A5, annexin A6 and collagen X causes no gross changes in matrix vesicle mediated mineralization, but lack of collagen X affects hematopoiesis and the Th1/Th2 response. J Bone Miner Res. 2012;27:2399–2412. doi: 10.1002/jbmr.1682. [DOI] [PubMed] [Google Scholar]
- 50.Neven E, De Schutter TM, De Broe ME, D’Haese PC. Cell biological and physicochemical aspects of arterial calcification. Kidney Int. 2011;79:1166–1177. doi: 10.1038/ki.2011.59. [DOI] [PubMed] [Google Scholar]
- 51.Warner GP, Hubbard HL, Lloyd GC, Wuthier RE. 32Pi- and 45Ca-metabolism by matrix vesicle-enriched microsomes prepared from chicken epiphyseal cartilage by isosmotic Percoll density-gradient fractionation. Calcif Tissue Int. 1983;35:327–338. doi: 10.1007/BF02405054. [DOI] [PubMed] [Google Scholar]
- 52.Anderson HC, Garimella R, Tague SE. The role of matrix vesicles in growth plate development and biomineralization. Front Biosci. 2005;10:822–837. doi: 10.2741/1576. [DOI] [PubMed] [Google Scholar]
- 53.Anderson HC, Sipe JB, Hessle L, Dhanyamraju R, Atti E, Camacho NP, Millan JL. Impaired calcification around matrix vesicles of growth plate and bone in alkaline phosphatase-deficient mice. J Am Pathol. 2004;164:841–847. doi: 10.1016/s0002-9440(10)63172-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu LN, Genge BR, Lloyd GC, Wuthier RE. Collagen-binding proteins in collagenase-released matrix vesicles from cartilage. Interaction between matrix vesicle proteins and different types of collagen. The J Bioll Chem. 1991;266:1195–1203. [PubMed] [Google Scholar]
- 55.Khan SR, Rodriguez DE, Gower LB, Monga M. Association of Randall plaque with collagen fibers and membrane vesicles. J Urol. 2012;187:1094–1100. doi: 10.1016/j.juro.2011.10.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nadra I, Mason JC, Philippidis P, Florey O, Smythe CD, McCarthy GM, Landis RC, Haskard DO. Proinflammatory activation of macrophages by basic calcium phosphate crystals via protein kinase C and MAP kinase pathways: a vicious cycle of inflammation and arterial calcification? Circ Res. 2005;96:1248–1256. doi: 10.1161/01.RES.0000171451.88616.c2. [DOI] [PubMed] [Google Scholar]