

Abstract
The lysosomal degradation pathway of autophagy has a crucial role in defence against infection, neurodegenerative disorders, cancer and ageing. Accordingly, agents that induce autophagy may have broad therapeutic applications. One approach to developing such agents is to exploit autophagy manipulation strategies used by microbial virulence factors. Here we show that a peptide, Tat–beclin 1—derived from a region of the autophagy protein, beclin 1, which binds human immunodeficiency virus (HIV)-1 Nef—is a potent inducer of autophagy, and interacts with a newly identified negative regulator of autophagy, GAPR-1 (also called GLIPR2). Tat–beclin 1 decreases the accumulation of polyglutamine expansion protein aggregates and the replication of several pathogens (including HIV-1) in vitro, and reduces mortality in mice infected with chikungunya or West Nile virus. Thus, through the characterization of a domain of beclin 1 that interacts with HIV-1 Nef, we have developed an autophagy-inducing peptide that has potential efficacy in the treatment of human diseases.
Autophagy functions in metazoans in cellular and tissue homeostasis, physiology, development, and protection against disease, and abnormalities in autophagy may contribute to many different pathophysiological conditions1,2. Thus, strategies that augment autophagy may prevent or treat human disease3. Although some drugs in clinical use are capable of augmenting autophagy, these compounds exert pleiotropic effects, revealing an unmet need to develop specific inducers of autophagy.
We sought to develop a specific autophagy-inducing agent with a potentially wide range of therapeutic effects. As viruses often provide key insights into the functionally important domains of host proteins, we investigated the molecular determinants governing the interaction between beclin 1, an essential autophagy protein in the class III phosphatidylinositol-3-OH kinase (PI(3)K) complex involved in autophagic vesicle nucleation4, and the HIV-1 virulence factor, Nef5. These investigations led us to identify a Nef-interacting sequence of beclin 1 that is necessary and sufficient for autophagy induction and which provided the basis for the development of an autophagy-inducing peptide drug that has benefits in the clearance of polyglutamine expansion protein aggregates and the treatment of infectious diseases.
The HIV-1 Nef-interacting domain of beclin 1
HIV-1 Nef acts as an antiautophagic maturation factor through interaction with beclin 1 (ref. 5) and is required for efficient viral replication and AIDS pathogenicity. To map the Nef-interacting domain of beclin 1, we co-transfected Flag-tagged beclin 1 deletion mutants (amino acids 1–377, 141–450 and 257–450) with haemagglutinin (HA)-tagged Nef. These Flag–beclin 1 mutants immunoprecipitated with Nef–HA (Supplementary Fig. 1a, b), indicating that amino acids 257–337, a region in the beclin 1 evolutionarily conserved domain (ECD)6,7, are involved in Nef binding. Studies with beclin 1 mutants containing deletions in the ECD revealed that amino acids 267–299 were required for binding to Nef (Supplementary Fig. 1c). Deletion of amino acids 267–284 weakened binding to Nef more than deletion of amino acids 285–299 (Fig. 1a) but had no effect on binding to Vps34 (also called PIK3C3) (Supplementary Fig. 2a), the class III PI(3)K that interacts with the beclin 1 ECD6. Thus, beclin 1 amino acids 267–284 in the ECD are crucial for beclin 1 binding to Nef. These residues are also required for the autophagy function of beclin 1; Flag–beclin 1, but not Flag–beclin 1(Δ267–284), increased starvation-induced autophagy in MCF7 cells (that express low amounts of endogenous beclin 1 (ref. 8)) as measured by quantification of GFP–LC3 dots, a marker of autophagosomes9 (Fig. 1b).
Figure 1. Tat–beclin 1 peptide induces autophagy in vitro.
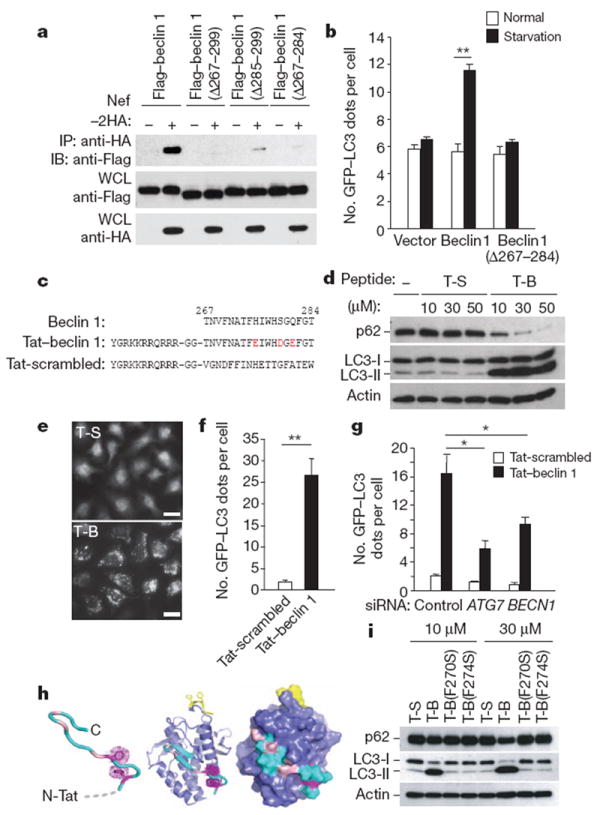
a, Immunoprecipitation of Flag–beclin 1 constructs with Nef–HA in HeLa cells 24 h post-transfection. b, GFP–LC3-positive dots (autophagosomes) in MCF7 cells expressing GFP–LC3 and Flag–beclin 1 constructs grown in either normal medium or starved in EBSS for 2 h. c, Sequences of beclin 1 amino acids 267–284, Tat–beclin 1 (T-B) and Tat-scrambled (T-S) control peptide. Red letters indicate amino acid substitutions to enhance hydrophilicity. d, Biochemical assessment of autophagy (p62 and LC3 immunoblots) in peptide-treated HeLa cells (3 h). e, f, Representative images (e) and quantification of GFP–LC3-positive dots (f) in peptide-treated HeLa/GFP–LC3 cells (30 μM, 3 h). Scale bars, 20 μm. g, GFP–LC3-positive dots in siRNA-transfected peptide-treated HeLa/GFP–LC3 cells (30 μM, 3 h). h, Model of Tat–beclin 1 peptide (left) based on corresponding elements of the beclin 1 evolutionarily conserved domain (ECD) structure (centre). Essential phenylalanine side chains, magenta; positions of solubility mutations, pink; lipid interaction site, yellow. ECD surface representation (right) illustrates exposure of corresponding peptide (cyan). i, p62 and LC3 immunoblots in peptide-treated HeLa cells (3 h). In b, f, g, bars represent mean ± s.e.m. of triplicate samples (50–100 cells per sample). Similar results were observed in three independent experiments. *P < 0.05, **P < 0.01; t-test.
Tat–beclin 1 is an autophagy–inducing peptide
We hypothesized that amino acids 267–284 of beclin 1 may be sufficient to induce autophagy, so we designed a cell-permeable peptide, Tat–beclin 1, composed of the HIV-1 Tat protein transduction domain (PTD)10 attached via a diglycine linker to 18 amino acids derived from amino acids 267–284 of beclin 1 (Fig. 1c). Three substitutions were made (in residues not conserved in all Atg6/beclin 1 orthologues, Supplementary Fig. 3) to enhance peptide solubility. These substitutions had no effect on binding of full-length beclin 1 to Nef (Supplementary Fig. 1d). The 18 amino acids derived from beclin 1 in Tat–beclin 1 were randomly shuffled to generate a control peptide (Tat-scrambled, Fig. 1c). Circular dichroism spectra of Tat–beclin 1 and Tat-scrambled revealed that both peptides are random coils (Supplementary Fig. 4), which is consistent with corresponding structural elements of Tat11 and the beclin 1 ECD7.
We used several complementary methods9 to demonstrate that the Tat–beclin 1 peptide induces autophagy. Treatment with Tat–beclin 1, but not Tat-scrambled, resulted in a dose-dependent decrease in amounts of p62, a selective autophagy substrate, and a dose-dependent conversion of the non-lipidated form of LC3, LC3-I, to the lipidated, autophagosome-associated form of LC3, LC3-II, in multiple cell lines and primary murine embryonic fibroblasts (MEFs) (Fig. 1d and Supplementary Fig. 5). A peptide consisting of the HIV Tat PTD fused to the wild-type sequence of beclin 1 amino acids 267–284 was not soluble in saline-based solutions; however, when dissolved in H2O, it induced LC3-II conversion and p62 degradation more efficiently than Tat–beclin 1 (Supplementary Fig. 6). The previously reported Tat–vFLIP α2 peptide12, derived from viral-encoded FLICE inhibitory protein, induced autophagy as efficiently as Tat–beclin 1 (Supplementary Fig. 7). In HeLa/GFP–LC3 cells13, Tat–beclin 1 increased GFP–LC3 dot numbers (autophagosomes) 27-fold (Fig. 1e, f). Ultrastructurally, increased numbers of autophagosomes and autolysosomes were observed in Tat–beclin 1-treated HeLa cells (Supplementary Fig. 8a). Bafilomycin A1, an inhibitor of autophagosomal/lysosomal fusion, further increased amounts of LC3-II and p62 in Tat–beclin 1-treated HeLa and COS-7 cells, indicating enhanced autophagic flux (Supplementary Fig. 8b). The magnitude of change in p62 upon bafilomycin A1 treatment was similar in Tat–beclin 1-treated cells to that observed in cells subjected to starvation (Supplementary Fig. 9). Long-lived protein degradation was increased in Tat–beclin 1-treated cells, and partially blocked by 3-methyladenine (3-MA), an autophagy inhibitor that targets class III PI(3)K (ref. 3) (Supplementary Fig. 8c).
Together, these results indicate that the Tat–beclin 1 peptide induces a complete cellular autophagy response. This response involves the canonical autophagy pathway, as short interfering RNA (siRNA) knockdown of two essential autophagy genes, BECN1 (involved in vesicle nucleation) and ATG7 (involved in the protein conjugation system) (Supplementary Fig. 10), decreased the number of Tat–beclin 1-induced autophagosomes (Fig. 1g). The upstream signalling events in Tat–beclin 1-induced autophagy might be partially distinct from those in starvation-induced autophagy, as additive effects were observed in cells simultaneously treated with Tat–beclin 1 and subjected to starvation (Supplementary Fig. 11).
A sequence alignment of Atg6/beclin 1 orthologues revealed two conserved aromatic residues within amino acids 267–284: F270 and F274 (Supplementary Fig. 3). In the beclin 1 ECD structure (a domain that binds to liposomes in vitro7 at the yellow-coloured site in Fig. 1h), these aromatic side chains intercalate in the hydrophobic core in a T-shaped geometry and help to position the intervening residues in a surface-exposed loop. They are predicted to have a similar function in the Tat–beclin 1 peptide, thus contributing to its structural stability (Fig. 1h). Consistent with this prediction, mutation of either of these two residues to serine (F270S and F274S) abrogated the peptide’s autophagy-inducing activity (Fig. 1i). Moreover, the subcellular localization of Tat–beclin 1(F270S) lacked the discrete punctate appearance of Tat–beclin 1 (Supplementary Fig. 12).
Tat–beclin 1 binds the autophagy inhibitor GAPR–1
We performed biochemical analyses to identify cellular proteins that bind to a biotin-conjugated form of the autophagy-inducing Tat–beclin 1 peptide. A protein that migrated at a molecular mass between 15 and 20 kDa bound to biotin–Tat–beclin 1 but not biotin–Tat-scrambled (Supplementary Fig. 13). This band was identified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) as Golgi-associated plant pathogenesis-related protein 1 (GAPR-1) (also known as GLIPR2), a protein that associates with lipid rafts at the cytosolic leaflet of the Golgi membrane14. Endogenous GAPR-1, but not Vps34, immunoprecipitated with the biotin–Tat–beclin 1 peptide (Fig. 2a and Supplementary Fig. 2b). Flag–beclin 1, but not Flag–beclin 1(D267–284), immunoprecipitated with GAPR-1 in HeLa cells expressing GAPR-1–Myc and Flag–beclin 1 (Fig. 2b). Thus, amino acids 267–284 of beclin 1 are necessary and sufficient for beclin 1 to bind GAPR-1. Biotin conjugates of mutant peptides defective in autophagy induction (Fig. 1i) failed to bind to GAPR-1 (Fig. 2a).
Figure 2. Tat–beclin 1 peptide binds to GAPR-1, a beclin 1-interacting protein.
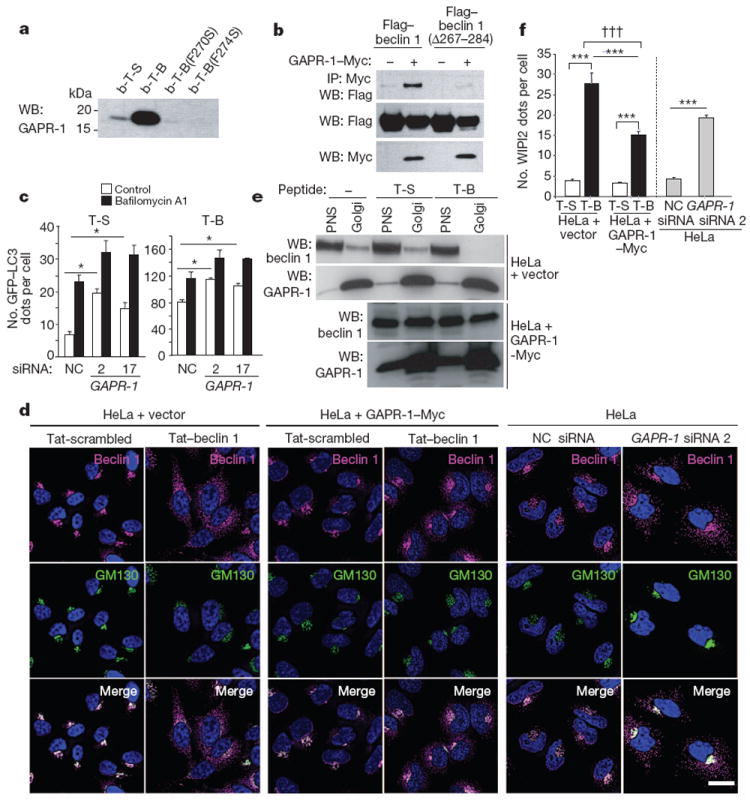
a, HeLa cells were treated with biotin-conjugated peptides (30 μM, 3 h) and proteins bound to peptides were analysed by immunoblot with anti-GAPR-1. b-T-B, biotin–Tat–beclin 1; b-T-S, biotin–Tat-scrambled. b, Immunoprecipitation of Flag–beclin 1 with GAPR-1–Myc in HeLa cells 24 h post-transfection. c, GFP–LC3-positive dots in GAPR-1 siRNA-transfected peptide-treated HeLa/GFP–LC3 cells (20 μM, 3 h) with or without 100nM bafilomycin A1. Bars represent mean ± s.e.m. of triplicate samples (50–100 cells per sample). Similar results were observed in three independent experiments. NC, non-silencing control. d, Localization of beclin 1 and GM130 (a Golgi marker) in HeLa cells stably transduced with empty vector (left panel) or GAPR-1–Myc (middle panel) or transfected with GAPR-1 siRNA (right panel) and treated with peptide (20 μM, 1 h). Scale bar, 20 μm. e, Immunoblot of beclin 1 in post-nuclear supernatant (PNS) and Golgi-enriched fractions in HeLa cells stably transduced with empty vector or GAPR-1–Myc after peptide treatment (20 μM, 2 h). f, WIPI2 dots in cells in the experimental conditions shown in d. Bars represent mean ± s.e.m. for 100–150 cells. *P < 0.05, ***P < 0.001; t-test. †††P < 0.001; two-way ANOVA for comparison of magnitude of changes between groups.
To evaluate the function of GAPR-1 in autophagy, we treated HeLa/GFP–LC3 cells with siRNAs directed against GAPR-1 (Supplementary Fig. 14a). Knockdown of GAPR-1 led to increased numbers of autophagosomes in both Tat-scrambled- and Tat–beclin 1-treated cells (Fig. 2c). This was due to an increase in autophagic flux, rather than a block in autophagosome maturation, as numbers of autophagosomes increased further upon bafilomycin A1 treatment. (We infer that the number of lysosomes available for autophagolysosomal fusion may be rate-limiting, as absolute numbers of autolysosomes did not increase (Supplementary Fig. 15).) Taken together, our data indicate that GAPR-1 is a beclin 1-interacting protein that negatively regulates autophagy.
We examined the subcellular localization of endogenous beclin 1 after Tat–beclin 1 treatment, GAPR-1 overexpression or GAPR-1 knockdown (Fig. 2d and Supplementary Fig. 14). Beclin 1 levels were not affected by GAPR-1 overexpression or GAPR-1 knockdown. In control-transfected HeLa cells treated with control peptide, beclin 1 staining predominantly overlapped with a Golgi apparatus marker (GM130). After Tat–beclin 1 peptide treatment, beclin 1 redistributed to non-Golgi subcellular compartments; beclin 1 demonstrated co-labelling with markers for organelles such as endosomes (EEA1), lysosomes (LAMP1) and endoplasmic reticulum (PD1) (data not shown). In addition, we observed a significant increase in the number of punctate structures containing WIPI2 (Fig. 2f and Supplementary Fig. 16), a mammalian protein that binds to phosphatidylinositol 3-phosphate at the site of early autophagosomal structures15. In HeLa cells expressing GAPR-1–Myc, Tat–beclin 1 treatment resulted in less redistribution of beclin 1 to non-Golgi sites and decreased WIPI2 puncta. Conversely, even in the absence of Tat–beclin 1 peptide, GAPR-1 siRNA knockdown led to an increase in beclin 1 redistribution to non-Golgi sites and in WIPI2 puncta.
To confirm that Tat–beclin 1 induced beclin 1 localization to non-Golgi sites, we measured the amounts of beclin 1 in purified HeLa cell Golgi fractions after peptide treatment (Fig. 2e). In control-transfected cells, treatment with Tat–beclin 1 led to the disappearance of beclin 1 in the Golgi fraction, whereas in cells expressing GAPR-1–Myc, increased amounts of beclin 1 were detected in the Golgi fraction, which were only minimally decreased by treatment with Tat–beclin 1. Thus, GAPR-1 may function to tether beclin 1 in the Golgi apparatus (where it is inactive in autophagy), and the Tat–beclin 1 peptide may promote the release of beclin 1 from the Golgi, resulting in enhanced early autophagosome formation. Other unknown mechanisms may also contribute to the autophagy-inducing activity of Tat–beclin 1.
Tat–beclin 1 has beneficial effects in vitro
Our discovery that Tat–beclin 1 is an inducer of autophagy led us to explore whether it may represent a candidate therapeutic agent for neurodegenerative diseases and infectious diseases. Pharmacological activation of autophagy reduces levels of soluble and aggregated proteins involved in polyglutamine repeat expansion disorders (such as Huntington’s disease, the spinocerebellar ataxias, and synucleinopathies and tauopathies) and reduces their cytotoxicity in vitro and neurotoxicity in mouse or Drosophila models3,16. We therefore investigated whether Tat–beclin 1 peptide treatment decreases aggregates of a model polyglutamine expansion protein (htt103Q) derived from exon 1 of human mutant huntingtin protein17.
In HeLa cells expressing doxycyline-repressible CFP fused to htt103Q (ref. 17), Tat–beclin 1 treatment did not affect the number of large (>1 μm) CFP–htt103Q aggregates (data not shown), consistent with previous reports that autophagy does not clear large protein aggregates16. In contrast, Tat–beclin 1 treatment decreased the number of small (<1 μm) CFP–htt103Q aggregates as efficiently as doxycycline (Fig. 3a and Supplementary Fig. 17). (Similar results were obtained using Torin1, an inhibitor of mTOR that induces autophagy3 (Supplementary Fig. 18).) We confirmed our findings biochemically using a filter trap assay that separates large protein aggregates, small protein aggregates and soluble proteins; Tat–beclin 1 decreased small htt103Q aggregates to a similar degree as doxycycline (Fig. 3b). Tat–beclin 1 peptide also decreased amounts of soluble htt103Q protein, indicating that Tat–beclin 1 can clear the pre-aggregated and small aggregated form of htt103Q protein. Further studies are required to determine whether Tat–beclin 1 peptide is effective in animal models of full-length disease-causing polyglutamine expansion proteins.
Figure 3. Tat–beclin 1 peptide decreases aggregates of a polyglutamine expansion protein and has anti-infective activity.
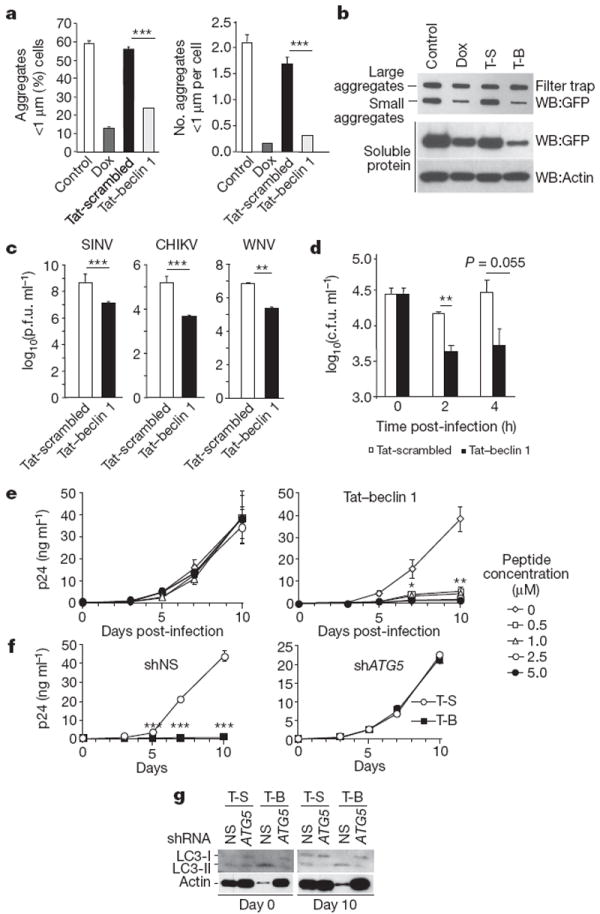
a, Percentage of cells with small htt103Q aggregates (left) and number of aggregates per cell (right) in HeLa cells expressing doxycycline (Dox)-repressible CFP–htt103Q after daily treatment with doxycycline or peptide (20 μM, 4 h per day) for 2 days. Bars represent mean ± s.e.m. of triplicate samples (60–120 cells per sample). Similar results were observed in three independent experiments. b, Filter trap assays for htt103Q large and small aggregates in HeLa/htt103Q cells. c, Viral titres in HeLa cells infected with 0.1 plaque-forming units (p.f.u.) per cell of SINV, CHIKV or WNV (strain TX02) and treated with peptide (10 μM, 4–8 h post-infection). Values represent geometric mean ± s.e.m. for triplicate samples of supernatants collected 18 h post-infection (SINV) or 24 h post-infection (CHIKV and WNV). Similar results were observed in three independent experiments. d, Bacteria colony-forming units (c.f.u.) in primary BMDMs infected with L. monocytogenes ΔactA mutant strain DPL-402927 for 30 min and treated with peptide (10 μM from 0 to 2 h post-infection). Bars represent mean ± s.e.m. of triplicate samples. Similar results were observed in three independent experiments. e, HIV-1 p24 antigen release in primary human MDMs infected with HIV-1 24 h after initiation of daily peptide treatment. Values represent mean ± s.e.m. of triplicate samples. Similar results were observed in MDMs from three independent donors. f, HIV-1 p24 antigen release in MDMs transduced with nonspecific scrambled shRNA (shNS) or ATG5 shRNA (shATG5) and treated daily with peptide (5 μM). Values represent mean ± s.e.m. of triplicate wells. Similar results were observed in MDMs from two independent donors. g, LC3 immunoblot of MDMs transduced with the indicated shRNA at day 0 and day 10 after HIV-1 infection. *P<0.05; **P < 0.01; ***P < 0.001; t-test.
Autophagy genes have beneficial effects in animal models of certain viral and intracellular bacterial infections, and autophagy-inducing agents (such as rapamycin and vitamin D) inhibit HIV replication in primary human monocyte-derived macrophages (MDMs)3. We therefore investigated the effects of the Tat–beclin 1 peptide on the replication of three positive-stranded RNA viruses (Sindbis virus (SINV), chikungunya virus (CHIKV), West Nile virus (WNV)), HIV-1, and the intracellular bacterium, Listeria monocytogenes. Tat–beclin 1 treatment (10 μM for 4 h beginning 4 h post-infection) reduced SINV, CHIKV and WNV titres 10–50-fold in HeLa cells (Fig. 3c). Tat–beclin 1 treatment reduced CHIKV replication even if administered 24 h post-infection (Supplementary Fig. 19). Tat–beclin 1-mediated reduction in viral titres was not due to peptide cytotoxicity, as the peptide did not kill uninfected cells (Supplementary Fig. 20a) and there was increased, rather than decreased, cell survival in Tat–beclin 1 peptide-treated virally infected cells (data not shown). Thus, the decreased viral titres probably reflect xenophagic degradation of the viruses or other antiviral effects of increased autophagy.
Tat–beclin 1 peptide decreased the intracellular survival of L. monocytogenes in primary murine bone-marrow-derived macrophages (BMDMs) (Fig. 3d). We used a strain of L. monocytogenes lacking the autophagy evasion protein, ActA18, and a dose of the peptide (10 μM for 2 h) that is non-toxic to uninfected BMDMs (Supplementary Fig. 20b, c). This antibacterial effect was decreased in BMDMs derived from Atg5flox/floxlysozymeM-Cre mice19 that had decreased Atg5 expression and decreased Tat–beclin 1-induced autophagy (Supplementary Fig. 21).
Tat–beclin 1 markedly inhibited HIV-1 replication in primary human MDMs. Using an established pre-treatment model20,21, we observed a dose-dependent inhibition of HIV p24 antigen release in MDMs cultured in the presence of non-toxic concentrations (0.5, 1, 2.5, and 5 μM) of Tat–beclin 1, with nearly undetectable antigen levels in cells treated daily with 5 μM Tat–beclin 1 peptide (Fig. 3e and Supplementary Fig. 22). The magnitude of inhibition of HIV-1 replication was similar to that observed with the mTOR inhibitor rapamycin, which also induces autophagy (Supplementary Fig. 23). Tat–beclin 1 peptide-mediated inhibition of HIV-1 replication appeared to occur via the canonical autophagy pathway, as short hairpin RNA (shRNA) knockdown of the essential autophagy gene, ATG5 (Supplementary Fig. 22c), decreased Tat–beclin 1-induced autophagy (as assessed by LC3 lipidation (Fig. 3g)) and completely abrogated the antiviral effects of Tat–beclin 1 treatment (Fig. 3f) without any decrease in cell survival (Supplementary Fig. 22d).
Tat–beclin 1 induces autophagy in vivo
To show that Tat–beclin 1 induces autophagy in vivo, we examined numbers of autophagosomes in tissues of mice that transgenically express GFP–LC3 (ref. 22) and compared two forms of Tat–beclin 1, including the sequence in Fig. 1c composed of l-amino acids, and a retro-inverso sequence of Tat–beclin 1 composed of d-amino acids, which is predicted to be more resistant to proteolytic degradation23. Significantly more GFP–LC3 dots (autophagosomes) were observed in skeletal muscle, cardiac muscle and pancreas after intraperitoneal (i.p.) treatment with the l- or d-form of Tat–beclin 1 as compared to the l-form of Tat-scrambled or d-form of Tat (Fig. 4a and Supplementary Fig. 24). By imaging tissue stained with streptavidin linked to an infrared dye, we confirmed that biotin-conjugated Tat–beclin 1 enters muscle cells (Supplementary Fig. 25). In 5-day-old suckling mouse brains, treatment with the d-form (but not l-form) of Tat–beclin 1 reduced p62 levels (Fig. 4b). Thus, Tat–beclin 1 can induce autophagy in peripheral tissues in adult mice as well as in the central nervous system of neonatal mice.
Figure 4. Tat–beclin 1 peptide induces autophagy and exerts antiviral activity in vivo.
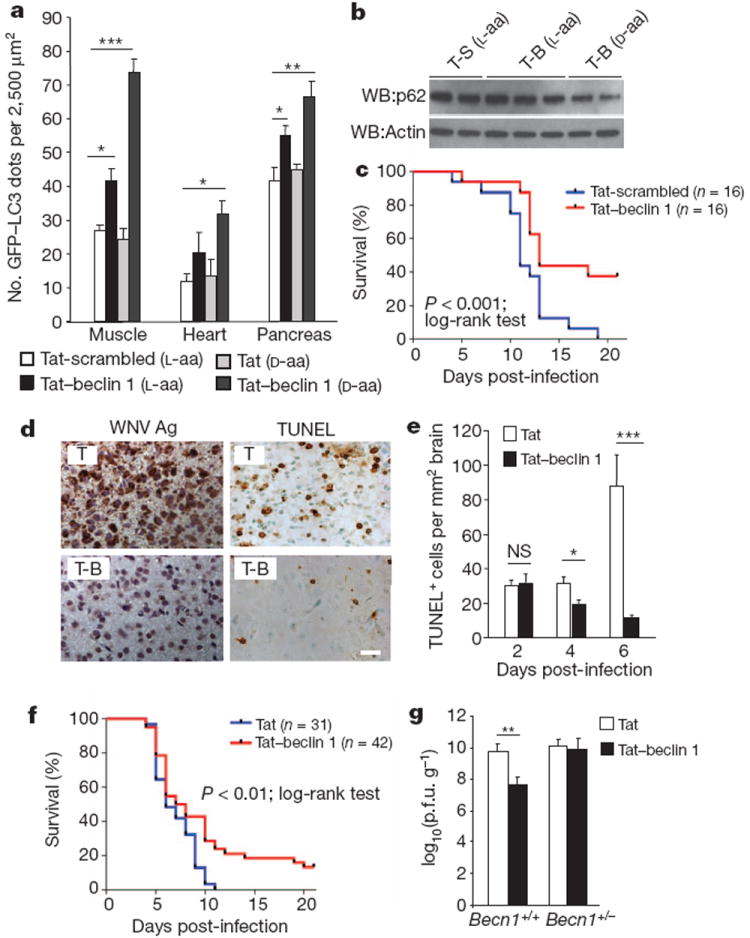
a, GFP–LC3-positive dots in tissues of 6-week-old GFP–LC3 mice treated with the indicated peptide (20 mg kg−1 i.p., 6 h). A minimum of ten fields was counted per tissue section. Bars represent mean ± s.e.m. for three mice. Similar results were observed in three independent experiments. b, p62 immunoblot of brains of 5-day-old GFP–LC3 mice treated with the indicated peptide (20 mg kg−1 i.p., 6 h). c, Survival curves of 5-day-old C57BL/6J mice infected with CHIKV(106 p.f.u. s.c.) and treated daily with peptide (15 mg kg−1 i.p. beginning 1 day post-infection). d–f, Representative images of WNV envelope antigen and TdT-mediated dUTP nick end labelling (TUNEL) staining (d) (T, Tat alone), quantification of cell death in brain (e), and survival curves (f) for 5-day-old C57BL/6J mice infected with WNV (Egypt strain 101, 1 p.f.u. intracerebral (i.c.)) and treated daily with peptide (d-amino acid forms, 20 mg kg−1 i.p. beginning 1 day post-infection). Images in d are from cerebral cortex day 6 post-infection. Similar results were observed in all regions of the brain for three mice per group. Scale bar, 20 μm. Bars in e represent mean ± s.e.m. TUNEL-positive cells per unit area of brain for three mice. g, Geometric mean + s.e.m. viral titres of WNV-infected mouse brains day 6 post-infection. Values represent combined data for 12–20 mice per treatment group from 10 to 12 litters. Data in c and f represent combined survival probabilities for three and four independent litters, respectively, in each group. Similar results were observed in each independent experiment. *P < 0.05; **P < 0.01; ***P < 0.001; NS, not significant; t-test.
Daily Tat–beclin 1 peptide treatment was well-tolerated in both neonatal and adult mice. During a 2-week course, neonatal mice had normal weight gain, motor activity, renal function and clinical status as well as normal histology of the heart, kidney, brain and liver (Supplementary Fig. 26 and Supplementary Table 1, and data not shown). These parameters, as well as hepatic and haematological function, were also normal in Tat–beclin 1 peptide-treated 3-month-old mice (Supplementary Table 1 and data not shown).
Tat–beclin 1 improves clinical outcomes
We investigated the effect of the Tat–beclin 1 peptide on the outcome of neonatal infection with CHIKV and WNV. After subcutaneous (s.c.) inoculation with CHIKV, neonatal mice develop lethal infection similar to that observed in human neonates, characterized by flaccid paralysis and abundant viral replication in muscle, skin and joints24. Tat–beclin 1 peptide treatment (15 mg kg−1 i.p. daily, l-form, beginning one day post-infection) reduced viral titres at day 4 and day 6 in the soleus and vastus lateralis muscles (Supplementary Fig. 27a) and decreased clinical paralysis (data not shown). In neonatal GFP–LC3 transgenic mice, autophagosomes were increased in muscle, including in cells that expressed CHIKV E2 envelope glycoprotein (Supplementary Fig. 27b). Importantly, Tat–beclin 1 treatment reduced mortality of neonatal mice infected with CHIKV; 100% of mice treated with control Tat-scrambled peptide died as compared to 62.5% of mice treated with Tat–beclin 1 peptide (Fig. 4c).
Arbovirus infections of the central nervous system (CNS) in human infants are often lethal and have no effective treatment25,26. We evaluated the effects of the Tat–beclin 1 peptide (d-form) on clinical outcome in a neonatal mouse model of WNV CNS infection. Tat–beclin 1 peptide treatment increased brain viral titres at day 2 post-infection, but resulted in more than a 1,000-fold reduction in brain viral titres by day 6 post-infection (Supplementary Fig. 28). The brains of control Tat peptide (d-form)-treated mice had higher levels of viral antigen staining at day 4 and day 6 post-infection, more neuropathology, and more cell death than those of Tat–beclin 1 peptide-treated mice (Fig. 4d, e and data not shown). Tat–beclin 1 peptide treatment also significantly reduced mortality in WNV-infected mice (Fig. 4f). (The antiviral effects of Tat–beclin 1 in vivo may be autophagy-dependent, as CNS viral titres were not reduced in Tat–beclin 1 peptide-treated mice with monoallelic deletion of Becn1 (Fig. 4g).) Taken together, our findings demonstrate that the Tat–beclin 1 peptide improves the clinical outcome of mice with CNS WNV infection.
Discussion
By mapping the domain of beclin 1 targeted by the HIV-1 pathogenicity protein, Nef, we identified a functionally important region of beclin 1, leading to the synthesis of a cell-permeable autophagy-inducing peptide and the discovery of a negative regulator of autophagy, GAPR-1. The autophagy-inducing activity of the peptide was associated with clearance of small polyglutamine expansion protein aggregates, reduced titres of several positive-stranded RNA viruses, decreased intracellular survival of the bacterium, L. monocytogenes, inhibition of HIV-1 replication in human macrophages, and a reduction in the mortality of neonatal mice infected with CHIKV and WNV. Because autophagy dysregulation may underlie the pathogenesis of metabolic, inflammatory and neoplastic diseases (in addition to its role in infections and neurodegenerative disorders), specific autophagy-inducing agents such as the Tat–beclin 1 peptide may have potential for the prevention and treatment of a broad range of human diseases.
METHODS
Cell culture
COS-7, HeLa, A549 and MCF7 cells were cultured in DMEM containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. HBEC30-KT cells were cultured in keratinocyte-SFM with 50 mg m−1 bovine pituitary extract, 5 ng ml−1 recombinant epidermal growth factor (Gibco) and 1% penicillin/streptomycin28. THP-1 and HCC827 cells were cultured in RPMI 1640 containing 10% FBS and 1% penicillin/streptomycin. For monocytic differentiation, THP-1 cells were incubated with 10 nM PMA (Sigma) for 72 h. MEFs were cultured in DMEM containing 15% FBS, 1% penicillin/streptomycin, 118 μM β-mercaptoethanol, and 1× MEM non-essential amino acids. HeLa/GFP-LC3 cells and HeLa-htt103Q cells have been described previously13,17. Murine BMDMs were cultured in DMEM containing 20% heat-inactivated FBS, 20% L-cell supernatant, and 1% penicillin/streptomycin. Atg5flox/flox and Atg5flox/flox-LysM-Cre BMDMs have been described19. Primary human MDMs were prepared from whole blood and cultured in RPMI 1640 containing 10% charcoal/dextran-treated, heat-inactivated FBS (Gemini Bio-Products) and 10 ng ml−1 macrophage CSF (R&D Systems) for 9 days as described21. Venous blood was obtained from HIV-1 seronegative subjects using a protocol approved by the Human Research Protections Program of the University of California, San Diego (Project 08-1613) in accordance with the Code of Federal Regulations on the Protection of Human Subjects (45 CFR part 46 and 21 CFR part 50 and 56). Written informed consent was obtained from blood donors before their participation.
Plasmid construction and transfection
pCDNA3.1 Myc–Vps34 has been described previously6. BECN1 cDNA was subcloned from pCR3.1-beclin 1 (ref. 29) into pBICEP-CMV2 (Sigma). Truncated constructs of Flag–beclin 1 were subcloned as PCR products containing each domain into pBICEP-CMV2. Deletion constructs of Flag–beclin 1 and the Flag–beclin 1(H275E/S279D/Q281E) mutant were generated by PCR mutagenesis and subcloned into pBICEP-CMV2. The HIV-1 SF2 Nef plasmid, pcDNA-Nef30, was provided by J. L. Foster. The Nef protein was fused with a 2× HA sequence at the C terminus by PCR and subcloned into pcDNA3.1. GAPR-1 cDNA from pCMV6-XL4 (Origene) was fused with a Myc sequence at the C terminus by PCR and subcloned into pcDNA3.1. Cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Immunoprecipitation and fluorescent microscopy analysis were performed 20–27 h post-transfection. GFP–LC331 and mRFP-GFP-LC332 plasmids were provided by T. Yoshimori.
Antibodies and reagents
For immunoblot analyses, rat monoclonal anti-HA (1:500 dilution, Roche), mouse monoclonal anti-Flag M2 (1:500 dilution, Sigma), mouse monoclonal anti-Myc (1:500 dilution, Santa Cruz), rabbit anti-LC3 (1:300 dilution, Novus Biologicals), mouse monoclonal anti-p62 (1:500 dilution, BD Biosciences), guinea-pig anti-p62 (1:500 dilution, Progen), mouse monoclonal anti-actin (1:2,000 dilution, Santa Cruz), rabbit anti-beclin 1 (1:1,000 dilution, Santa Cruz), rabbit anti-ATG7 (1:1,000 dilution, (Sigma), anti-ATG5 (1:500 dilution, Novus Biologicals), anti-GFP (1:500 dilution, Invitrogen) and rabbit anti-GAPR-1 (1:10,000 dilution)14 were used. 3-MA (Sigma) was dissolved in H2O and used at a 10 μM concentration. Torin1 (gift from D. Sabatini)33, rapamcyin (Sigma) and bafilomycin A1 (Sigma) were dissolved in DMSO and used at concentrations of 1 μM, 500 nM and 100 nM, respectively.
Immunoblot analyses
Cells were rinsed with Dulbecco’s phosphate buffered saline (PBS(−), (Sigma)) and lysed in lysis buffer (20 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, protease inhibitor cocktail (Roche)) on ice for 1 h. Lysates were centrifuged at 16,100g for 10 min at 4 °C, and resolved by SDS–PAGE and transferred to PVDF membranes. Membranes were blocked with 5% non-fat dry milk (NFDM) in PBST (PBS 0.05% Tween-20), incubated overnight at 4 °C with primary antibodies in PBST containing 5% NFDM, washed with PBST, and incubated for 1 h at room temperature with HRP-conjugated secondary antibodies diluted in PBST containing 5% NFDM. Immunoreactive bands were visualized with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) or Amersham ECL Plus Western Blotting Detection System (GE Healthcare).
For detection of beclin 1 and GAPR-1 in Golgi fractions of HeLa cells and HeLa/GAPR-1–Myc cells, Golgi-enriched fractions were isolated as described34,35 from cells treated with peptide (20 μM, 1 h), then trypsinized and homogenized with a Balch homogenizer (gap size 9 μm). Post-nuclear supernatant (PNS) was obtained after centrifugation of the cell homogenate (1,700 r.p.m., 10 min, 4 °C). The PNS was fractionated by sucrose density gradient ultracentrifugation (SV40 rotor at 100,000g, 2 h 40 min) and approximately 1 ml of a Golgi-enriched fraction was collected at the 35–29% sucrose interface. Membranes were pelleted by ultracentrifugation (100,000g, 30 min) after the addition of three volumes of PBS to one volume of Golgi-enriched fraction.
Immunoprecipitation
Cell lysates were centrifuged at 16,100g for 10 min at 4 °C and the supernatants were subjected to immunoprecipitation with an anti-HA antibody (1:100 dilution, Roche) and protein G plus-agarose (Santa Cruz) or with anti-Myc agarose (1:20 dilution, Santa Cruz) and subjected to immunoblot analyses.
Cell viability assays
Cytotoxicity was measured in HeLa cells using an LDH cytotoxicity kit (Cayman Chemical). Cytotoxicity in primary human MDMs was measured using WST-1 (Roche). Cell survival in primary murine BMDMs was measured by trypan blue exclusion or a CellTiter-Glo Luminescent Cell Viability assay kit (Promega).
siRNA and shRNA suppression of gene expression
siRNAs were purchased from Thermo Scientific Dharmacon. siRNAs specific to BECN1 (5′-GCACUCAAGUUCAUGCUUA-3′), ATG7 (5′-GGGUUAUUACUACAAUGGUGUU-3′), C9orf19 (GAPR-1) (no. 2, 5′-GCAAGAACCUCAACCGGGA-3′ and no. 17, 5′-GUUACCUAGACCACGAUUA-3′) and a non-silencing control siRNA (NC-2) were transfected using Lipofectamine 2000. Immunoblot and fluorescent microscopic analyses were performed 48–72 h post-transfection. MISSION short-hairpin (shRNA) lentiviral particles were from Sigma: ATG5 (SHCLNV-NM_004849/TRCN0000150940), or scrambled non-targeting negative control (Scr, SHC002V).
Fluorescence microscopy and image analysis
For measurement of autophagy in HeLa/GFP–LC3 cells or MCF7 cells, cells were fixed with 2% paraformaldehyde (PFA) in PBS, and GFP–LC3-positive dots per cell was quantified as described29. To measure autophagic flux, HeLa cells were co-transfected with an mRFP–GFP–LC3 plasmid32 and siRNA, treated with peptide (25 μM, 3 h) 72 h post-transfection, and then fixed as above. GFP-positive autophagosomes and RFP-positive autolysosomes per cell were quantified. For the htt103Q aggregate assay, CFP-positive aggregates <1 μm were counted.
For immunofluorescence (IFA) analysis of cultured cells, HeLa cells were fixed as above, permeabilized in 0.5% Triton X-100/PBS, blocked with 0.5% BSA in PBS, stained with anti-WIPI215 (mouse, 1:500), anti-beclin 1 (rabbit, 1: 250, Santa Cruz), anti-GM130 (mouse, 1:250, Novus Biologicals), and labelled with goat-anti-rabbit AlexaFluor 647 and goat-anti-mouse AlexaFluor 488 secondary antibodies (1:500) or stained with anti-streptavidin-AlexaFluor 488 (1:500) (after treatment with Tat–biotin-conjugated peptides). For the beclin 1 primary antibody, specificity was confirmed by siRNA knockdown. Z-stacks were acquired with a Zeiss AxioImager M2 microscope equipped with a Photometrics CoolSnap HQ2 camera using a Zeiss PLAN APO 20X/0.8 NA wide-field objective with the same acquisition times for samples stained with identical primary antibody pairs, then deconvolved with AutoDeBlur (Biotplane). Imaris version 7.4.0 (Bitplane) was used for analysis. Thresholding for background secondary antibody staining was performed in all experiments. Representative images were chosen after examining 60–120 cells from triplicate samples for each condition. Nuclear staining (besides DAPI) was masked using the DAPI channel and setting non-DAPI signal within the nuclear surface to zero. Quantification of WIPI2-positive dots per cell was performed by an observer blinded to experimental condition.
For IFA of mouse tissues, 5-day-old GFP–LC3 transgenic mice infected with CHIKV were treated with peptide (20 mg kg−1, 6 h). Muscle sections were prepared as described in the section below, stained with an anti-CHIKV E2 antibody13,36, and images were acquired as described for cultured cells above. For infrared (IR) dye images, muscle tissue was collected from peptide-treated GFP–LC3 mice (25 mg kg−1 i.p., 2 h), and sections were stained with streptavidin-conjugated IRDye 800CW (1:500 dilution; Licor). Images were acquired with a Zeiss AxioObserver Z1 microscope equipped with a Hamamatsu OrcaII-ER camera using a Zeiss PLAN APO 63×/1.4 NA oil objective and analysed by an observer blinded to experimental condition.
Electron microscopy
HeLa cells were fixed overnight with 2.5% glutaraldehyde in 0.1 M (pH 7.4) cacodylate buffer and samples were prepared and analysed as described8.
Immunohistochemistry
Mouse brain sections were fixed in 4% PFA. Paraffin-embedded sagittal sections were TUNEL-stained to detect apoptotic nuclei (Apoptag peroxidase in situ Apoptosis Detection Kit; Chemicon International). Immunoperoxidase staining was performed to detect WNV antigen (rat polyclonal anti-WNV antibody; 1:100 dilution; provided by M. Diamond).
Peptide design
An evolutionary trace (ET) analysis37 revealed regions within the 267–299 domain of beclin 1 likely to be of evolutionary importance. To enhance the solubility of the Tat PTD (amino acids 47–57)10 fused to the beclin 1 267–284 fragment, ET analysis of beclin 1 further identified residue positions unlikely to be functionally important occupied by charged residues in beclin 1 orthologues. These cognate residues were then substituted into the peptide (H275E, S79D and Q281E) to increase its solubility without disrupting its mimicry of beclin 1 function.
Peptide synthesis
l-amino acid peptides were synthesized by the University of Texas Southwestern Medical Center (UTSW) Protein Chemistry Technology Core and purified to >95% by HPLC (confirmed by mass spectrometry). The Tat–beclin 1 peptide sequence, YGRKKRRQRRRGGTNVFNATFEIWHDGEFGT, consisted of 11 amino acids from the Tat PTD at the N terminus, a GG linker to increase flexibility, and at the C terminus, 18 amino acids derived from beclin 1 267–284 containing three substitutions, including H275E, S279D, Q281E. Wild-type Tat–beclin 1 peptide consisted of the Tat PTD, a GG linker, and 18 amino acids derived from the natural beclin 1 sequence (267–284) (YGRKKRRQRRRGGTNVFNATFHIWHSGQFGT). Control peptide, Tat-scrambled, consisted of the Tat protein transduction domain, a GG linker, and a scrambled version of the C-terminal 18 amino acids from Tat–beclin 1 (YGRKKRRQRRRGGVGNDFFINHETTGFATEW). For experiments comparing Tat–beclin 1 and Tat-scrambled, peptides were dissolved in PBS(−), whereas in the experiment comparing Tat–beclin 1 and wild-type Tat–beclin 1, peptides were dissolved in H2O. Peptides were stored at −80 °C. For peptide treatment, cells were washed with PBS(−) and treated with peptides (10–50 μM, 1–4 h) dissolved in OPTI-MEM (Gibco) acidified with 0.15% (v/v) 6N HCl. For treatment of primary human MDMs, cells were washed with PBS and pre-treated with peptides (0.5–5 μM, 24 h) in 500 μl macrophage-SFM (serum-free media) (Gibco) before infection with HIV-1.
d-amino acid peptides were synthesized at the HHMI Mass Spectrometry Laboratory at UC-Berkeley. The retro-inverso Tat–beclin 1 d-amino acid sequence was RRRQRRKKRGYGGTGFEGDHWIEFTANFVNT. Peptides were synthesized by solid-phase methodology on Wang resin of 0.44 meq g−1 substitution using an ABI 431A synthesizer. Appropriate N-FMOC amino acid derivatives were coupled via dicyclohexylcarbodiimide activation in dichloro-methane/N-methylpyrrolidone using user-devised extended activation, coupling and piperidine deprotection cycles. Dried resin-peptide was deprotected in reagent K 4 h at room temperature. Peptide was extracted with warm acetonitrile/water, lyophilized, and purified by RPLC. Crude peptide purity was roughly 75%; after purification, 95%. d-amino acid peptides were dissolved in H2O and stored at −80 °C until use. Peptide purity was assessed by FTICR mass spectrometry.
Circular dichroism spectroscopy
CD spectra of the Tat–beclin 1 and Tat-scrambled peptides were recorded on an AVIV 62DS spectropolarimeter.
Model of beclin 1 peptide
The beclin 1 peptide model was adopted from the corresponding elements of the ECD structure7 (Protein Data Bank accession 4DDP). The first two residues after the diglycine linker are disordered in the ECD structure and were omitted. The side chains of the residues mutated to increase solubility were modelled using the mutagenesis wizard of PyMOL, keeping rotamers with no steric clashes.
Measurement of long-lived protein degradation
COS-7 cells cultured in 6-well plates were incubated in 10% FBS/DMEM containing L-3H leucine (1 μCi ml−1) (Perkin Elmer) and 65 μM of unlabelled leucine for 3 days. Cells were washed with 10% FBS-DMEM three times and incubated with 10% FBS/DMEM containing unlabelled leucine (2 μM) for 1 day. After washing three times with normal medium, cells were treated with indicated peptide or EBSS for 2 h with or without 3-MA. The medium was precipitated with 10% TCA and TCA-soluble radioactivity was measured. Cells were fixed by 10% TCA, lysed in 0.2N NaOH and total cell radioactivity was measured. l-3H-leucine release was estimated as a percentage of the radioactivity in the TCA-soluble material relative to the total cell radioactivity.
Protein purification and mass spectrometry
Tat-scrambled and Tat–beclin 1 peptides were conjugated at their N termini to biotin via a PEG(3) linkage by the UTSW Protein Chemistry Technology Core. Approximately 8 × 106 HeLa cells were treated with biotinylated peptides (30 μM, 3 h), lysed in lysis buffer (20 mM HEPES, 137 mM NaCl, 1 mM EDTA, 0.2% Triton X-100, 10% glycerol and protease inhibitor cocktail) and homogenized using a 25G syringe. Lysates were mixed with M-280 streptavidin Dynabeads (Invitrogen) overnight and 20 μl of laemmli sample buffer was added. After separation by SDS–PAGE, bands were visualized by GelCode blue stain reagent (Thermo Scientific), excised and analysed by LC-MS/MS at the UTSW Protein Chemistry Technology Core. All MS/MS spectra were searched against the protein sequence database at the NCBI using Mascot software.
Filter trap assay
A filter trap assay was performed to measure amounts of soluble htt103Q protein, small htt103Q aggregates (P2 supernatant) and large htt103Q aggregates (P1 supernatant) in HeLa-htt103Q cells as described17,38.
Virus strains and infection
SINV strain SV1A (ATCC) has been described previously39. WNV strain TX0240 was provided by M. Gale and WNV strain Egypt 10141 was provided by M. Diamond. CHIKV strain CHIKV-21 was propagated as described42. All in vitro virus infections were performed at a multiplicity of infection (MOI) of 0.1 in reduced serum (1% heat-inactivated FBS) containing media for 1 h. For titration of viruses in mouse tissues, 10% homogenates of freeze-thawed tissue were used. Viral stocks, cell culture supernatants, and tissue homogenates were titred by plaque assay titration using BHK-21 cells for SINV and CHIKV and Vero cells for WNV.
For HIV-1 experiments, 5 × 105 MDMs were treated with peptide at the indicated dose for 24 h, infected with HIV-1Ba-L (provided by S. Gartner and R. Gallo)43 at an MOI of 0.01 for 3 h, washed 3× with PBS, and then incubated with macrophage-SFM containing the peptide for 10 days. At days 3, 5, 7 and 10, 250 μl of cell supernatant was collected and fresh 250 μl macrophage-SFM containing the peptides was added. Extracellular release of HIV-1 p24 antigen into the supernatants was measured using the Alliance HIV-1 p24 antigen enzyme-linked immunosorbent assay kit (ELISA; PerkinElmer).
Listeria infection
Primary BMDMs were collected from C57/BL6J mice and cultured for 6 days. On day 7, BMDMs were plated in DMEM containing 20% FBS and 20% L-cell supernatant and no antibiotics, and infected 24 h later with the Listeria ΔactA mutant strain DPL-402927 (provided by M. Shiloh) at an MOI of 5 c.f.u. per cell for 30 min at 37 °C and 5% CO2, then gently washed three times with PBS and treated with either 10 μM Tat-scrambled or Tat–beclin 1 peptide for 2 h. After 2 h, cells were washed three times with PBS to remove extracellular bacteria and peptide and the medium was replaced with non-antibiotic-containing medium. BMDMs were lysed in 0.3% Triton X-100 at serial time points and serial dilutions were plated on LB agar overnight at 37 °C for determination of c.f.u.
Animal experiments
To measure autophagy in mouse tissues, 6-week-old GFP–LC3 transgenic mice22 were injected i.p. with Tat-scrambled (l-amino acid), Tat–beclin 1 (l-amino acid) and Tat–beclin 1 (d-amino acid) peptide at 20 mg kg−1 (5.3 μmol kg−1) or with Tat (d-amino acid) peptide at 5.3 μmol kg−1. After 6 h, mice were killed and fixed by perfusion with 4% PFA in PBS. Tissues were fixed in 4% PFA overnight, 15% sucrose for 4 h, and 30% sucrose overnight before frozen sections were prepared and used for fluorescence microscopy analysis as described44. For immunoblot analysis, 5-day-old GFP–LC3 mice were injected i.p. with Tat-scrambled (l-amino acid), Tat–beclin 1 (l-amino acid) and Tat–beclin 1 (d-amino acid) peptide at 20 mg kg−1 (5.3 μmol kg−1) or with Tat (d-amino acid) peptide at 5.3 μmol kg−1. Six hours later, mice were killed and frozen brain tissue homogenates were used. For virus infections, CHIKV and WNV Egypt strain were diluted in HBSS and used to infect 5-day-old C57BL/6J mice with 106 p.f.u. s.c or 1 p.f.u. i.c., respectively. For assessment of peptide toxicity, 6-day-old C57BL/6J mice were injected i.p. with Tat-scrambled (l-amino acid) or Tat–beclin 1 (l-amino acid) at 15 mg kg−1 and with Tat (d-amino acid) at 5.3 mmol kg−1 or Tat–beclin 1 (d-amino acid) at 20 mg kg−1 daily for 2 weeks. Body weight, neurological status and general clinical status was monitored daily, and blood, liver, spleen, heart and brain samples were collected upon death after 2 weeks of peptide treatment. For serum biochemistry and haematological analyses in adult mice, 3-month-old C57BL/6J mice were injected i.p. with Tat-scrambled (l-amino acid) or Tat–beclin 1 (l-amino acid) at 20 mg kg−1 daily for 2 weeks before blood collection. Animal experiments were approved by the UTSW Institutional Animal Care Use Committee and performed in accordance with institutional guidelines.
Retroviral expression system
GAPR-1 (also known as GLIPR2) cDNA (Origene) was subcloned into pBABE-puro (Addgene) by PCR. To generate pBabe-GAPR-1-Myc, GAPR-1 cDNA was fused with a Myc sequence at the C terminus by PCR and subcloned into pBABE-puro. To produce retroviruses, 4 μg of pBABE-GAPR-1 was transfected into 293T cells with 3.6 μg of pUMVC (Addgene) and 0.4 μg of pCMV-VSV-G (Addgene). At 2 and 3 days after transfection, the viral supernatants were collected and filtered through a 0.45-μm membrane filter and the viral supernatants were used to infect HeLa cells in the presence of 6 μg ml−1 of polybrene. At 24 h post-infection, fresh viral supernatants with polybrene were added to the cells for a second round of infection. After an additional 24 h, cells were cultured with 10% FBS/DMEM with 4 μg ml−1 puromycin.
Supplementary Material
Acknowledgments
We thank M. Diamond, J. L. Foster, M. Gale, N. Mizushima, D. Sabatini, M. Shiloh and T. Yoshimori for supplying critical reagents; and H. Ball, A. Bugde and E.-L. Eskelinen for assistance with peptide synthesis, infrared imaging and EM interpretation, respectively. This work was supported by NIH grants U54AI057156 (B.L.), K08 AI099150 (R.S.), ROI NS077874 (S.A.S), RO1 GM094575 (N.V.G.), ROI GM066099 (O.L.), ROI GM079656 (O.L.), ROI NS063973 (A.Y.), ROI NS050199 (A.Y.), U54AI057160 (H.W.V., D.J.L.), ROI DK083756 (R.X.), ROI DK086502 (R.X.), and T32 GM008297 (C.H.); NSF CCF-0905536 (O.L.); an NWO-ALW Open Program Grant 817.02.023 (J.B.H.); Cancer Research UK (S.A.T.); and a Welch Foundation Award I-15-5 (N.V.G.).
Footnotes
Author Contributions B.L., S.S.-K. and O.L. generated the original hypothesis. S.S.-K., R.S., M.L., G.R.C., Z.Z., Q.S., K.P., D.M., C.H., R.E., D.K. and D.V.K. performed experiments. L.K., A.D.W., R.X., O.L. and N.V.G. performed bioinformatics analyses. B.L., S.S.-K., R.S., M.L., L.K., H.W.V., J.B.H., S.A.T., R.X., D.J.L., A.Y., O.L., N.V.G., S.A.S. and D.V.K. provided intellectual contributions throughout the project. B.L., S.S.-K. and R.S. took primary responsibility for writing the manuscript. All authors edited the manuscript.
Supplementary Information is available in the online version of the paper.
The authors declare no competing financial interests.
References
- 1.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nature Rev Drug Discov. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001;2:330–335. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kyei GB, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. 2009;186:255–268. doi: 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Furuya N, Yu J, Byfield M, Pattingre S, Levine B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy. 2005;1:46–52. doi: 10.4161/auto.1.1.1542. [DOI] [PubMed] [Google Scholar]
- 7.Huang W, et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 2012;22:473–489. doi: 10.1038/cr.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang XH, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 9.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van den Berg A, Dowdy SF. Protein transduction domain delivery of therapeutic macromolecules. Curr Opin Biotechnol. 2011;22:888–893. doi: 10.1016/j.copbio.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 11.Bayer P, et al. Structural studies of HIV-1 Tat protein. J Mol Biol. 1995;247:529–535. doi: 10.1006/jmbi.1995.0158. [DOI] [PubMed] [Google Scholar]
- 12.Lee JS, et al. FLIP-mediated autophagy regulation in cell death control. Nature Cell Biol. 2009;11:1355–1362. doi: 10.1038/ncb1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orvedahl AO, et al. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7:115–127. doi: 10.1016/j.chom.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eberle HB, et al. Identification and characterization of a novel human plant pathogenesis-related protein that localizes to lipid-enriched microdomains in the Golgi complex. J Cell Sci. 2002;115:827–838. doi: 10.1242/jcs.115.4.827. [DOI] [PubMed] [Google Scholar]
- 15.Polson HE, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6:506–522. doi: 10.4161/auto.6.4.11863. [DOI] [PubMed] [Google Scholar]
- 16.Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nature Rev Neurol. 2012;8:108–117. doi: 10.1038/nrneurol.2011.200. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto A, Cremona ML, Rothman JE. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol. 2006;172:719–731. doi: 10.1083/jcb.200510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshikawa Y, et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nature Cell Biol. 2009;11:1233–1240. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- 19.Zhao Z, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4:458–469. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campbell GR, Spector SA. Hormonally active vitamin D3 (1α,25-dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV-1 infection. J Biol Chem. 2011;286:18890–18902. doi: 10.1074/jbc.M110.206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campbell GR, Spector SA. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 2012;8:e1002689. doi: 10.1371/journal.ppat.1002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer PM. The design, synthesis and application of stereochemical and directional peptide isomers: a critical review. Curr Protein Pept Sci. 2003;4:339–356. doi: 10.2174/1389203033487054. [DOI] [PubMed] [Google Scholar]
- 24.Couderc T, et al. A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 2008;4:e29. doi: 10.1371/journal.ppat.0040029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson RT. Acute encephalitis. Clin Infect Dis. 1996;23:219–226. doi: 10.1093/clinids/23.2.219. [DOI] [PubMed] [Google Scholar]
- 26.Griffin DE. Emergence and re-emergence of viral diseases of the central nervous system. Prog Neurobiol. 2010;91:95–101. doi: 10.1016/j.pneurobio.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lauer P, Chow MY, Loessner MJ, Portnoy DA, Calendar R. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J Bacteriol. 2002;184:4177–4186. doi: 10.1128/JB.184.15.4177-4186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato M, et al. Multiple oncogenic changes (K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res. 2006;66:2116–2128. doi: 10.1158/0008-5472.CAN-05-2521. [DOI] [PubMed] [Google Scholar]
- 29.Pattingre S, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 30.O’Neill E, et al. Dynamic evolution of the human immunodeficiency virus type 1 pathogenic factor. Nef J Virol. 2006;80:1311–1320. doi: 10.1128/JVI.80.3.1311-1320.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kabeya Y, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 33.Thoreen CC, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balch WE, Dunphy WG, Braell WA, Rothman JE. Reconstitution of the transport of protein between successive compartments of the Golgi measured by the coupled incorporation of N-acetylglucosamine. Cell. 1984;39:405–416. doi: 10.1016/0092-8674(84)90019-9. [DOI] [PubMed] [Google Scholar]
- 35.Vogels MW, et al. Quantitative proteomic identification of host factors involved in the Salmonella typhimurium infection cycle. Proteomics. 2011;11:4477–4491. doi: 10.1002/pmic.201100224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bréhin AC, et al. Production and characterization of mouse monoclonal antibodies reactive to Chikungunya envelope E2 glycoprotein. Virology. 2008;371:185–195. doi: 10.1016/j.virol.2007.09.028. [DOI] [PubMed] [Google Scholar]
- 37.Mihalek I, Res I, Lichtarge O. A family of evolution-entropy hybrid methods for ranking protein residues by importance. J Mol Biol. 2004;336:1265–1282. doi: 10.1016/j.jmb.2003.12.078. [DOI] [PubMed] [Google Scholar]
- 38.Bailey CK, Andriola IF, Kampinga HH, Merry DE. Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2002;11:515–523. doi: 10.1093/hmg/11.5.515. [DOI] [PubMed] [Google Scholar]
- 39.Taylor RM, Hurlbut HS, Work TH, Kingston JR, Frothingham TE. Sindbis virus: a newly recognized arthropod-transmitted virus. Am J Trop Med Hyg. 1955;4:844–862. doi: 10.4269/ajtmh.1955.4.844. [DOI] [PubMed] [Google Scholar]
- 40.Keller BC, et al. Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence. J Virol. 2006;80:9424–9434. doi: 10.1128/JVI.00768-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weiner LP, Cole GA, Nathanson N. Experimental encephalitis following peripheral inoculation of West Nile virus in mice of different ages. J Hyg (Lond) 1970;68:435–446. doi: 10.1017/s0022172400042339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schuffenecker I, et al. Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med. 2006;3:e263. doi: 10.1371/journal.pmed.0030263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gartner S, et al. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science. 1986;233:215–219. doi: 10.1126/science.3014648. [DOI] [PubMed] [Google Scholar]
- 44.Qu X, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.