

Abstract
Axon injury is a central determinant of irreversible neurological deficit and disease progression in patients with multiple sclerosis (MS). CD8+ lymphocytes (CTLs) within inflammatory demyelinated MS lesions correlate with acute axon injury and neurological deficits. The mechanisms of these correlations are unknown. We interrogated CTL-mediated axon injury using the transgenic OT-I antigen-specific CTL model system in conjunction with a chambered cortical neuron culture platform that permits the isolated manipulation of axons. Interferon gamma upregulated the axonal expression of major histocompatibility complex class I molecules presenting immunologically-relevant peptides processed from endogenous proteins. Antigen-specific CTLs formed immune synapses with and directly injured axons in a mechanism dependent upon antigen presentation, T cell receptor specificity and granzyme B activity. These findings present a novel model of immune-mediated axon injury and offer anti-axonal CTLs and granzyme B as targets for the therapeutic protection of axons and prevention of neurological deficits in MS patients.
INTRODUCTION
Multiple sclerosis (MS) is the leading cause of non-traumatic neurological disability for young people in Western countries1. Central nervous system (CNS) axon injury and loss are histopathological correlates of irreversible neurological deficits in MS patients2-4. Axon injury is most prominent within active, inflammatory demyelinated MS lesions enriched in cytotoxic CD8+ T lymphocytes (CTLs) of an effector, memory phenotype5-11. Acute axon injury and loss of neurologic function independently correlate with intra-lesion CTL burden and CTL populations within the lesions, blood and cerebrospinal fluid (CSF) of MS patients are clonally expanded12-18. Demyelinated axons within active, inflammatory lesions express major histocompatibility complex class I (MHC-I) molecules and are therefore potential CTL targets19-21. The cytotoxic mechanisms used by CTLs include Fas-L induction of target cell apoptosis, cytokine-mediated toxicity and target cell killing induced by the perforin-granzyme cascade22. CSF levels of granzyme B (GzB) correlate with relapse rate in a sub-population of MS patients23 and animal models of MS link CTL effector functions to axon loss and development of neurological deficits. In the Theiler’s murine encephalomyelitis model, genetic deletion of perforin (PFP) or the disruption of MHC-I surface expression by genetic deletion of β2-microglobulin result in protection of axons and preservation of neurologic function, despite extensive CNS demyelination24-27.
The mechanism(s) of antigen-specific CTL-mediated axon injury is currently unknown. Mature axons are typically, although incorrectly, considered incapable of expressing surface levels of functional, antigen-presenting MHC-I molecules sufficient to trigger CTL recognition and concomitant injury28-33. Accordingly, most hypotheses examining CTL function in the context of MS have viewed axon injury as secondary to CTL-mediated cytotoxicity of glia. Critically, studies that have examined direct CTL-mediated axon injury have failed to differentiate neuron cell body, neuritic and axonal injury mechanisms, although the subcellular structure and functions of these compartments are vastly different and spatially distinct both in vitro and in vivo34.
In this study, we explicitly tested the hypothesis that antigen-specific CTLs directly injure axons through a MHC-I- and GzB-dependent mechanism. To do so, we employed the antigen-specific OT-I CTL model system in conjunction with a chambered cortical neuron culture platform that microfluidically isolates axons from cell bodies. We determined that sustained exposure to interferon gamma (IFNγ), an inflammatory cytokine produced by cells within active MS lesions, was sufficient to upregulate the axolemmal expression of MHC-I H-2Kb molecules competent to present exogenously or endogenously derived SIINFEKL peptides. We discovered that OT-I CTLs formed cytotoxic immune synapses with and injured MHC-I expressing axons. Finally, we characterized the mechanism of OT-I CTL-mediated axon injury as dependent upon peptide-loaded MHC-I expression on axons, antigen-specific CTL T cell receptor (TCR) recognition of axonal MHC-I, and GzB expression by the effector CTLs. These findings provide a novel model of direct, immune-mediated axon injury and suggest discrete strategies for targeted, early therapeutic intervention aimed at protecting axons and preventing the development of irreversible loss of neurological function in MS patients.
RESULTS
IFNy upregulates MHC-I expression in primary neuron cultures.
We incubated cortical neuron-enriched C57BL/6 (WT, MHC-I H-2b haplotype) gang cultures with IFNγ (500 U mL−1) and SIINFEKL peptide (1 μg mL−1), a MHC-I H-2Kb-restricted antigenic fragment of chicken ovalbumin for 72 hr to assess the influence of sustained inflammatory cytokine exposure on MHC-I expression and antigen presentation by neural cells. RT-PCR demonstrated that total MHC-I H-2b mRNA was elevated 39.3±14.4 fold (n=6, P<0.001) in SIINFEKL- and IFNγ-(SIINFEKL+IFNγ) treated cultures as compared to untreated cultures. Similarly, the MHC-I subtypes H-2Kb and H-2Db were induced 6.4±3.6 (n=6, P=0.002) and 29.6±11.8 (n=6, P<0.001) fold respectively, by SIINFEKL+IFNγ treatment (Fig. 1a, Supplementary Table 1). Total MHC-I H-2Kb protein (H-2Kb) was increased on glia, indentified by morphology, and neurons, indentified by neurofilament medium chain (NFM) immunoreactivity, following SIINFEKL+IFNγ treatment. Neuronal H-2Kb was distributed across both cell bodies and neurites (Fig. 1b). Analysis of SIINFEKL-loaded H-2Kb complexes using the 25-D1.16 antibody demonstrated H-2Kb as functionally competent to present exogenous SIINFEKL antigen at the cell surface (Fig. 1c).
Figure 1.

IFNγ upregulates MHC-I expression in primary neuron cultures. (a) RT-PCR demonstrated that levels of total MHC-I H-2b (39.3±14.4, n=6, P<0.001) and MHC-I subtypes H-2Kb (6.4±3.6, n=6, P=0.002) and H-2Db (29.6±11.8, n=6, P<0.001) were elevated in IFNγ-treated, cortical neuron-enriched gang cultures as compared to untreated cultures. Increased total MHC-I H-2Kb (b) and SIINFEKL-loaded H-2Kb (c) expression was localized to glia, neuronal cell bodies and neurites as detected by immunofluorescence. Scale bar (b,c)=100 μm.
Microfluidic device-based cortical neuron culture platform.
Gang cultures do not permit the isolated experimental manipulation of axons. Therefore we employed a two-chamber microfluidic device-based cortical neuron culture platform (MFC, Supplementary Fig. 1a). Each MFC was comprised of a cell body compartment into which neurons were plated, microgroove channels (500 × 10 × 3 μm, length × width × height) through which axonal processes extended, and an axon compartment containing terminal axon projections (Supplementary Fig. 1a,c-f). The MFC system generated highly reproducible populations of pure cortical axons physically and fluidically isolated from dendrites, neuron cell bodies and glia.
IFNy upregulates axonal MHC-I competent to present exogenously and endogenously derived antigens
We first used the MFC platform to examine axon-localized MHC-I expression and antigen presentation following sustained exposure to IFNγ. We treated WT cortical neurons cultured in MFCs (WT MFC) with either SIINFEKL+IFNγ or RAHYNIVTF peptide (E7, H-2Db-restricted peptide control, 1 μg mL−1) and IFNγ (E7+IFNγ) for 72 hr. Increased levels of axonal H-2Kb (defined as H-2Kb colocalized with NFM) were observed in both SIINFEKL+IFNγ- and E7+IFNγ-treated WT MFC axon fields as compared to untreated WT MFC axon fields (SIINFEKL+IFNγ: 0.69±0.07, P<0.001 vs. Untxt: 0.36±0.04, H-2Kb colocalized with NFM normalized to total NFM; Fig. 2c). High resolution imaging with associated single axon fiber analysis revealed H-2Kb distributed in discrete puncta across the axolemma (Fig. 2a). This H-2Kb was competent to present exogenous antigen peptide, as SIINFEKL+IFNγ-treated WT MFC axon fields demonstrated strong SIINFEKL-loaded H-2Kb complex immunoreactivity (25-D1.16 staining) whereas E7+IFNγ-treated and untreated MFC axon fields did not (SIINFEKL+IFNγ: 0.59±0.09, P<0.001 vs. E7+IFNγ: 0.25±0.09, and P<0.001 vs. Untxt: 0.22±0.05, SIINFEKL-bound H-2Kb colocalized with NFM normalized to total NFM; Fig. 2d). SIINFEKL-loaded H-2Kb complexes were localized in discrete puncta across the axolemma in a distribution similar to that of total H-2Kb (Fig. 2b).
Figure 2.

IFNγ upregulates axonal MHC-I competent to present exogenously and endogenously derived antigenic peptides. (a) Increased axonal H-2Kb protein expression was detected by immunofluorescence of WT MFC cultures treated with exogenous SIINFEKL peptide and IFNγ or exogenous E7 peptide and IFNγ as compared to untreated MFC cultures. Increased axonal H-2Kb was also observed when OVA MFC cultures were treated with IFNγ. (b) Elevated H-2Kb presenting SIINFEKL antigen was detected by 25-D1.16 immunofluorescence, in both SIINFEKL+IFNγ-treated WT and IFNγ-treated OVA MFC cultures as compared to E7+IFNγ-treated WT, untreated WT or untreated OVA MFC cultures (not shown). Scale bars (a,b; 20x)=100 μm and (a,b; 100x)=20 μm. Total H-2Kb (c) and SIINFEKL-loaded H-2Kb (d) colocalized with the axon marker NFM was quantified and normalized to total NFM. Full statistical measurements are presented in Supplementary Table 1. Key data and statistical measurements: Total H-2Kb: Untreated_WT, 0.36±0.04, n=6; SIINFEKL+INFγ_WT, 0.69±0.07, n=8; E7+INFγ_WT, 0.8±0.2, n=4; Untreated_OVA, 0.4±0.1, n=6; INFγ_OVA, 0.7±0.1, n=7 (Untreated_WT vs. SIINFEKL+INFγ_WT, P<0.001; SIINFEKL+INFγ_WT vs. E7+INFγ_WT, P=0.23; Untreated_OVA vs. INFγ_OVA, P<0.001; SIINFEKL+INFγ_WT vs. INFγ_OVA, P=0.957). SIINFEKL-loaded H-2Kb: Untreated_WT, 0.22±0.04, n=8; SIINFEKL+INFγ_WT, 0.59±0.08, n=11; E7+INFγ_WT, 0.2±0.1, n=6; Untreated_OVA, 0.2±0.1, n=6; INFγ_OVA, 0.6±0.1, n=7 (Untreated_WT vs. SIINFEKL+INFγ_WT, P<0.001; SIINFEKL+INFγ_WT vs. E7+INFγ_WT, P<0.001; Untreated_OVA vs. INFγ_OVA, P<0.001; SIINFEKL+INFγ_WT vs. INFγ_OVA, P=0.811). (e) RT-PCR demonstrated that levels of total MHC-I H-2b (101±13-fold, n=6, P<0.001) and MHC-I subtypes H-2Kb (11±2-fold, n=6, P<0.001) and H-2Db (77±14-fold, n=6, P<0.001) were elevated in IFNγ-treated, WT MFC. Error bars are 95% CI.
C57BL/6-Tg(CAG-OVA)916Jen/J (OVA) mice express full-length ovalbumin protein in all cells. Transgenically expressed ovalbumin is cleaved via antigen processing pathways and peptide derivates such as SIIMFEKL are loaded and presented on MHC-I35. We cultured OVA cortical neurons in MFCs to examine the effect of sustained IFNγ exposure on endogenous antigen presentation by axons. Axonal FI-2Kb was markedly increased, at levels similar to SIINFEKL+IFNγ-treated WT MFC cultures, in OVA MFC cultures exposed to IFNγ alone (500 U mL−1) as compared to untreated OVA cultures (IFNγ OVA: 0.7±0.1, P=0.957 vs. WT SIINFEKL+IFNγ: 0.70±0.07; P<0.001 vs. Untxt OVA: 0.4±0.1; Fig. 2c). IFNγ-induced H-2Kb was axolemmal and punctate in distribution (Fig. 2a). IFNγ-treated OVA MFC axon fields also demonstrated elevated levels of axonal, SIINFEKL-loaded H-2Kb as detected by 25-D1.16 antibody reactivity, similar to those observed in WT MFC axon fields following SIINFEKL+IFNγ treatment, as compared to untreated OVA, untreated WT or E7+IFNγ-treated WT MFC cultures (IFNγ OVA: 0.57±0.09, P=0.811 vs. WT SIINFEKL+IFNγ: 0.59±0.09; P<0.001 vs. OVA Untxt: 0.25±0.09; Fig. 2d). The axonal distribution of endogenously processed SIINFEKL antigen presented on H-2Kb was similar to that of exogenously loaded SIINFEKL antigen presented on H-2Kb (Fig. 2b).
We also examined the influence of sustained IFNγ exposure, independent of peptide-mediated effects, on MHC-I mRNA expression in WT MFC cultures by RT-PCR. Total MHC-I H-2b was elevated 101±13 fold (n=6, P<0.001) in IFNγ-treated (72 hr, 500 U mL−1) MFC cultures as compared to untreated MFC cultures. Similarly, H-2Kb and H-2Db were induced 11±2 (n=6, P=0.004) and 77±14 (n=6, P=0.002) fold respectively, by IFNγ treatment (Fig. 2e).
CTLs form cytotoxic immune synapses with MHC-I expressing axons.
CTLs isolated from C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-I) mice express a transgenic TCR engineered to recognize H-2Kb presenting SIINFEKL peptide. To interrogate CTL interactions with MHC-I expressing axons, we exposed WT MFC axon fields, pretreated for 72 hr with SIINFEKL+IFNγ to 105 activated OT-I, OT-Ipfp1−/− or OT-IGzAB−/− CTLs for one hour. Actin cytoskeleton and cytotoxic granule polarization occurred in all OT-I effector types as assessed by high resolution phalloidin (actin) and PFP and GzB (cytotoxic granule) immunofluorescence (Fig 3a-c). PFP and GzB immunoreactivity were absent in OT-Ipfp1−/− and OT-IGzAB−/− CTLs, respectively (Fig. 3b,c). Scanning (SEM) and transmission (TEM) electron microscopy demonstrated axon fibers intercalated within membrane folds of OT-I CTLs (Fig. 3d,f) and discrete regions of fusion between CTL membranes and axolemma consistent with sustained immune synapse formation (Fig. 3e-g,i, red arrows). Foci of axon fiber scission (Fig. 3d,e yellow arrows), axolemmal perforations (Fig. 3h red arrows) and axoplasmic multivesicluar bodies, morphologically consistent with autophagosomes or lysosomes, were identified near putative cytotoxic immune synapses (Fig. 3f,i,j,j inset yellow arrows). TEM also demonstrated axon spheroids containing similar autophagosome or lysosome structures (Fig. 3k).
Figure 3.
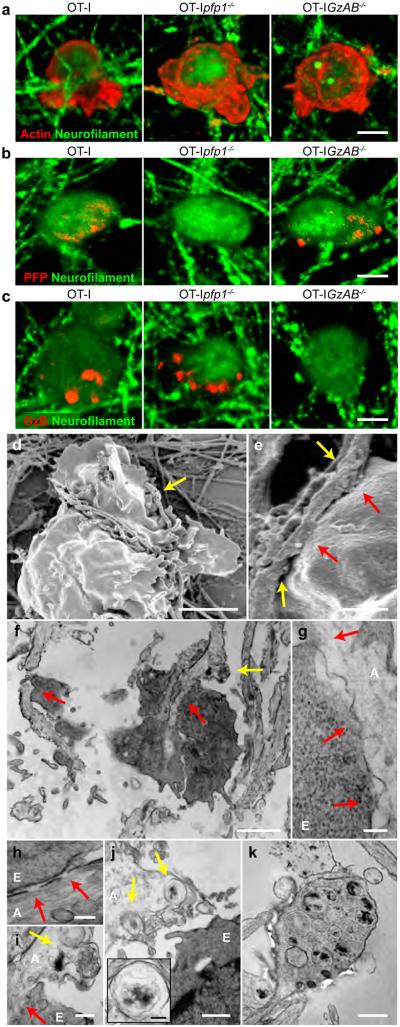
CTLs form cytotoxic immune synapses with MHC-I expressing axons. Immunofluorescence and electron microscopy demonstrated that OT-I CTLs polarized actin (a), PFP(b) and GzB (c) proteins, intercalated with IFNγ-treated, SIINFEKL-loaded WT axon fibers (d, SEM; f, TEM) and formed discrete regions of fusion between CTL membranes and the axolemma (red arrows; e, SEM; f,g,i, TEM) consistent with cytotoxic immune synapse formation. Axon fiber scission (yellow arrows; d,e, SEM), loss of axolemmal integrity (red arrows; h, TEM) and axoplasmic multivesicular bodies (yellow arrows; f,i,j,j inset, TEM) were observed in regions adjacent to OT-I CTL-axon immune synapses. (k) Axon spheroids containing similar axoplasmic multivesicular bodies were also noted. A: Axon, E: OT-I CTL effector. Scale bars (a-c)=20 μm, (d)=5 μm, (e,j)=1 μm, (f)=2 μm, (g, j inset)=200 nm, (h)=100 nm, (i)=400 nm and (k)=500 nm.
Antigen-specific CTLs injure axons expressing MHC-I molecules presenting exogenously or endogenously derived antigen peptides.
To examine the consequences of cytotoxic immune synapse formation, we incubated 5×105 activated OT-I CTLs for three hours with either WT MFC axon fields pretreated with SIINFEKL+IFNγ (elevated exogenous SIINFEKL-loaded H-2Kb, Fig. 2) or with OVA MFC axon fields pretreated with IFNγ alone (elevated endogenous SIINFEKL-loaded H-2Kb, Fig. 2), for three hours. Optimal OT-I CTL number and incubation time was determined by dose response and time course studies (data not shown). Pan-neurofilament (NF) immunofluorescence was used to assess axon integrity and OT-I CTLs were identified within axon chambers by DAPI reactivity. Both SIINFEKL+IFNγ-pretreated WT and IFNγ-pretreated OVA MFC axon fields incubated with OT-I CTLs exhibited robust loss of axon integrity (Fig. 4e-h) as compared to MFC axon fields in the absence of OT-I CTLs (Fig. 4a-d). Axon spheroids containing whorled clusters of NF, indicative of transected axons, were identified by high resolution imaging in both exogenously and endogenously loaded axons (Fig. 4f,h white arrows). Axon integrity index quantification (AIIQ, Supplementary Fig. 1) was performed to assess axon injury. The AIIQ score identified a statistically significant loss of axon integrity in both SIINFEKL+IFNγ-pretreated WT and IFNγ-pretreated OVA MFC axon fields following OT-I CTL incubation as compared to axon fields in the absence of OT-I CTLs. The magnitude of OT-I CTL-mediated axon injury was equivalent for both WT and OVA axons, as assessed by AIIQ score. (Fig. 4i, data and statistical analyses, Supplementary Table 1).
Figure 4.

Antigen-specific CTLs injure axons expressing MHC-I molecules presenting exogenously or endogenously derived antigens. Axon injury was observed when the axon fields of SIINFEKL- and IFNγ-pretreated WT (e,f; exogenous peptide condition) and IFNγ-pretreated OVA (g,h; endogenous peptide condition) MFC cultures were exposed to OT-I CTLs. Axon spheroids, indicative of transected axons, were observed in OT-I CTL-treated axon fields (f,h; white arrows). Axon fields not exposed to OT-I CTLs (a,b; exogenous peptide control; c,d; endogenous peptide control) demonstrated intact axons. Scale bars (a,c,e,g; 20x)=100 μm and (b,d,f,h; 100x)=20 μm. (i) Axon injury was quantified as demonstrated in Supplementary Figure 1. Error bars are 95% CI. Full data and statistical measurements are presented in Supplementary Table 1. Key data and statistical measurements (axon field region, untreated vs. OT-I, P-value): WT MFC Untreated (a, n=9) vs. WT MFC OT-I (e, n=15): Region 4, 298±52 vs. 142 ±43, P<0.001; Region 5, 265±46 vs. 124±40, P<0.001; Region 6, 238±50 vs. 97±31, P<0.001; Region 7, 211±68 vs. 76±26, P<0.001. OVA MFC Untreated (c, n=6) vs. OVA MFC OT-I (g, n=6): Region 4, 256±28 vs. 149±27 P=0.016; Region 5, 218±39 vs. 105±33, P=0.007; Region 6, 192±35 vs. 75±41, P=0.003; Region 7, 169±35 vs. 108±38, P=0.008.
CTL-mediated axon injury is dependent upon antigen-specific TCR recognition of peptide-loaded axonal MHC-I.
The immunological mechanisms underpinning CTL-mediated axon injury were delineated by manipulating components of the MHC-I:antigen:TCR complex. Maximal injury occurred when WT MFC cultures were pretreated with SIINFEKL+IFNγ (Fig. 2) and axon fields were subsequently exposed to 5×105 SIINFEKL:H-2Kb-restricted OT-I CTLs (CD45hiCD8+Vα2+, Supplementary Fig. 2) for three hours (Fig. 5b). Elevated SIINFEKL-loaded H-2Kb expression alone was insufficient to generate axon injury (Fig. 5a). Likewise, WT MFC cultures not pretreated with IFNγ (low SIINFEKL-loaded H-2Kb expression, Fig. 2), with or without OT-I CTL exposure demonstrated no axon injury (Fig. 5c,d). Replacing SIINFEKL antigen with E7 antigen also prevented axon injury (Fig. 5e). Non-transgenic WT CTLs, lacking SIINFEKL:H-2Kb specificity (CD45hiCD8+Vα2−, Supplementary Fig. 2), and conditioned media from OT-I CTL cultures failed to injure SIINFEKL+IFNγ-pretreated WT MFC axon fields (Fig. 5f-g). The AIIQ score demonstrated a statistically significant difference between WT MFC axon fields pretreated with SIINFEKL+IFNγ and exposed to OT-I CTLs and all other treatment or culture conditions. (Fig. 5h, Supplementary Table 1).
Figure 5.
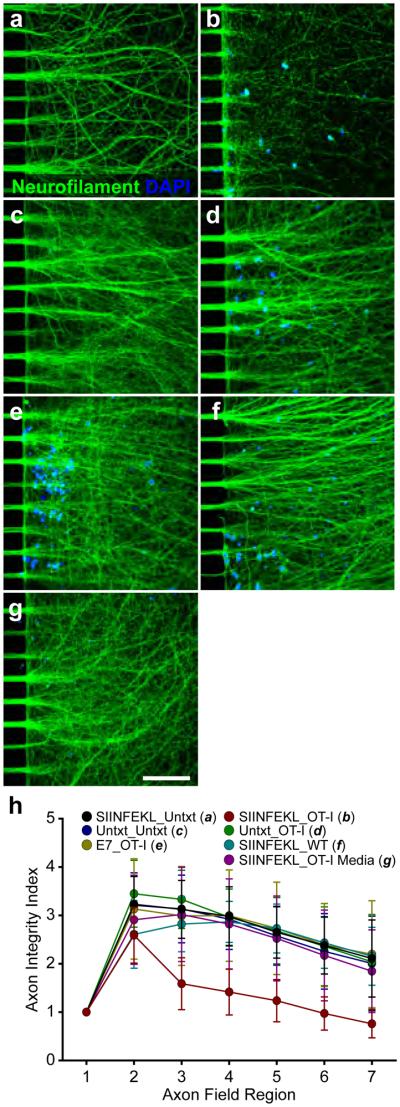
CTL-mediated axon injury is dependent upon antigen-specific TCR recognition of antigen-loaded axonal MHC-I. Axon injury was observed when the axon fields of SIINFEKL+IFNγ-pretreated, WT MFC axon fields were exposed to OT-I CTLs (b). Untreated cultures (no IFNγ, no peptide) demonstrated intact axons in the absence (c) or presence (d) of OT-I CTLs. Cultures pretreated with E7+IFNγ demonstrated intact axons following OT-I CTL exposure (e). SIINFEKL+IFNγ-pretreated cultures not exposed to OT-I CTLs (a), exposed to WT CTLs (f) or exposed to conditioned media from activated OT-I CTL cultures (g) were similarly uninjured. Scale bars (a-g; 20x)=100 μm. (h) Axon injury was quantified as demonstrated in Supplementary Figure 1. Error bars are 95% CI. Full data and statistical measurements are presented in Supplementary Table 1. Key data and statistical measurements: No IFNγ_OT-I (d, n=11) vs. SIINFEKL+IFNγ_OT-I (b, n=15): Region 4, 296±51 vs. 142 ±43, P<0.001; Region 5, 268±52 vs. 124±40, P<0.05; Region 6, 239±61 vs. 97±31, P<0.05; Region 7, 204±86 vs. 76±26, P<0.05. E7+ IFNγ_OT-I (e, n=5) vs. SIINFEKL+IFNγ_OT-I (b, n=15): Region 4, 300±66 vs. 142 ±43, P<0.001; Region 5, 273±67 vs. 124±40, P<0.05; Region 6, 239±61 vs. 97±31, P<0.05; Region 7, 220±78 vs. 76±26, P<0.05. SIINFEKL+IFNγ_WT (f, n=6) vs. SIINFEKL+IFNγ_OT-I (b, n=15): Region 4, 287±32 vs. 142 ±43, P<0.001; Region 5, 273±23 vs. 124±40, P<0.05; Region 6, 243±40 vs. 97±31, P<0.05; Region 7, 215±46 vs. 76±26, P<0.05
CTL-mediated axon injury is GzB-dependent and caspase-independent
We exposed the axon fields of SIINFEKL+IFNγ-pretreated WT MFC cultures to 5×105 OT-I, OT-Ipfp1−/− or OT-IGzAB−/− CTLs for three hours. Axon integrity was compromised in OT-I CTL-treated axon fields (Fig. 6b) as compared to unmanipulated axon fields (Fig. 6a). In contrast, axon fields exposed to OT-Ipfp1−/− (Fig. 6c) or OT-IGzAB−/− CTLs (Fig. 6d) were not damaged, indicating that an intact perforin-granzyme cascade competent to deliver functional granzyme into the axoplasm was required for OT-I CTL-mediated axon injury. SIINFEKL+IFNγ-pretreated WT MFC axon fields were also exposed to OT-I CTLs in the presence of the GzB inhibitor Ac-IEPD-CHO (10 μM, Fig. 6e) or the pan-caspase inhibitor Z-VAD-FMK (10 μM, Fig. 6f). Axon fields were preserved only with GzB inhibition. The AIIQ score indentified a statistically significant loss of axon integrity in both WT MFC axon fields and Z-VAD-FMK-treated axon fields incubated with OT-I CTLs as compared to untreated, OT-Ipfp1−/−, OT-IGzAB−/− or OT-I and Ac-IEPD-CHO CTL-treated axon fields. (Fig. 6g, Supplementary Table 1).
Figure 6.

CTL-mediated axon injury is GzB-dependent and caspase-independent. Axon injury was observed when the axon fields of SIINFEKL- and IFNγ-pretreated WT MFC cultures were exposed to OT-I CTLs (b) or OT-I CTLs in the presence of the pan-caspase inhibitor, Z-VAD-FMK (f). SIINFEKL- and IFNγ-pretreated cultures not exposed to OT-I CTLs (a), or exposed to OT-Ipfp1−/− (c), OT-IGzAB−/− (d) or OT-I CTLs in the presence of the GzB inhibitor, Ac-IEPD-CHO (e) demonstrated intact axons. Scale bars (a-f; 20x)=100 μμm. (g) Axon injury was quantified as demonstrated in Supplementary Figure 1. Error bars are 95% CI. Full data and statistical measurements are presented in Supplementary Table 1. Key data and statistical measurements: OT-Ipfp1−/− (c, n=6) vs. OT-I (b, n=15): Region 4, 288±57 vs. 142 ±43, P<0.001; Region 5, 277±67 vs. 124±40, P<0.001; Region 6, 269±82 vs. 97±31, P<0.001; Region 7, 246±100 vs. 76±26, P<0.001. OT-IGzAB−/− (d, n=7) vs. OT-I (b, n=15): Region 4, 296±42 vs. 142 ±43, P<0.001; Region 5, 272±36 vs. 124±40, P<0.001; Region 6, 248±50 vs. 97±31, P<0.001; Region 7, 221±58 vs. 76±26, P<0.001. Ac-IEPD-CHO & OT-I (e, n=8) vs. OT-I (b, n=15): Region 4, 243±22 vs. 142 ±43, P<0.001; Region 5, 230±28 vs. 124±40, P<0.001; Region 6, 212±47 vs. 97±31, P<0.001; Region 7, 200±66 vs. 76±26, P<0.001. Z-VAD-FMK & OT-I (f, n=9) vs. OT-I (b, n=15): Region 4, 157±37 vs. 142 ±43, P=0.607; Region 5, 124±40 vs. 124±40, P=0.993; Region 6, 93±38 vs. 97±31, P=0.891; Region 7, 66±33 vs. 76±26, P=0.785
DISCUSSION
The mechanisms of MS-related axon injury remain poorly understood despite the established correlation of CTLs and axon loss to irreversible neurological deficits in MS patients. Furthermore, focal axon injury in the context of MS primarily occurs independently and at a distance from the neuron cell body. Nonetheless, axon-specific injury mechanisms remain poorly differentiated from general neurite or cell body neurotoxic mechanisms. In this work, we present a unified model, focused specifically upon the axon, in which antigen-specific CTLs directly injure axons through a mechanism dependent upon formation of the MHC-I:antigen:TCR complex and GzB protease activity. We established that OT-I CTLs directly engage and injure axons expressing MHC-I H-2Kb loaded with SIINFEKL peptide. Sustained exposure to IFNγ initiated the upregulation of axonal MHC-I H-2Kb competent to present either exogenously loaded SIINFEKL or SIINFEKL derived from endogenously expressed OVA (Fig. 2). These observations build upon previously published in vitro hippocampal neuron gang culture and in vivo experiments29,30,36 and demonstrate that cortical axons, distant from cell bodies and in the absence of glia, display complete, functional, antigen-loaded MHC-I complexes. Critically, our experiments are the first to show that axons present immunologically relevant antigens derived from endogenously expressed proteins and highlight the potential relevance of endogenous neuron and axon peptides to anti-axonal CTL recognition in MS lesions21,37.
Our work further demonstrates that antigen-specific CTLs form cytotoxic immune synapses with axons expressing antigen-bound MHC-I complexes (Fig. 3). OT-I CTLs formed cytotoxic immune synapses with axons expressing SIINFEKL-loaded H-2Kb as illustrated by cytoskeleton and cytotoxic granule polarization and fusion of the OT-I CTL membrane with the axolemma38. Axon changes indicative of injury, including axon fiber scission, loss of axolemma integrity, the formation of axoplasmic autophagosomes or lysosomes and axon spheroids, were also noted proximal to immune synapses (Fig. 3). Following three hours of OT-I CTL exposure to axons presenting either exogenous or endogenous SIINFEKL antigen, axon spheroids and a frank loss of axon integrity were clearly detected. These OT-I CTL-mediated axon injury observations unequivocally confirm that antigen-specific CTLs directly injure axons expressing exogenously or endogenously derived antigens loaded on MHC-I (Fig. 4).
Identification of the specific immunological mechanisms responsible for CTL-mediated axon injury was facilitated by our deliberate selection of the transgenically defined, antigen-specific OT-I model system. By sequentially manipulating axonal MHC-I expression (IFNγ treatment), antigen presentation (SIINFEKL/E7 peptides) and TCR antigen specificity (OT-I/WT CTLs) we demonstrated that antigen-specific CTLs require a fully engaged MHC-I:antigen:TCR complex to injure axons (Fig. 5). Correspondingly, direct contact between CTLs and axons is also required for axon injury as conditioned media from activated OT-I CTL cultures, enriched in CTL-derived cytokines, did not injure axons. These findings directly confirm the early hypotheses of Manning and colleagues30,39 and Medana and colleagues31,33 which implicated a MHC-I-dependent mechanism in the CTL-mediated injury of neurons and neurites. However, our observations contradict the work of Giuliani and colleagues which demonstrated that non-specific populations of activated CD8+ or CD4+ T cells damage human fetal neurons, independent of MHC-I or antigen40. Different intrinsic susceptibilities of human fetal and mouse cortical neurons to immune-mediated injury or non-classical MHC-I ligand engagement may explain the divergence of our findings41. However, it is more likely that these differences highlight the importance of delineating cell body versus axon injury mechanisms. Within gang and organotypic slice cultures, inflammatory cytokines, Fas-Fas-L, CD40-CD40-L and the perforin-granzyme cascade are implicated as possible neuron injury mechanisms32,33,40,42,43. These factors induce changes within neuron cell bodies, including nuclear pyknosis, calcium mobilization and caspase-3 activation, consistent with the induction of apoptotic signaling cascades40,43,44. In the axon, nuclear apoptotic signaling mechanisms do not exist. Accordingly, axon-specific injury mechanisms are likely distinct from cell body-specific mechanisms. We found that CTL-mediated injury, at the level of the axon, is executed directly by GzB, independent of caspase activation (Fig. 6). Axon injury was prevented when GzB activity was blocked by pharmacologic inhibition (Ac-IEPD-CHO) or genetic deletion (OT-IGzAB−/−). Additionally, when GzB was unable to access the axoplasm, due to genetic deletion of perforin (OT-Ipfp1−/−), axon integrity was maintained. PFP activity alone, in contrast with other reports, was insufficient to induce axon injury, as PFP competent but GzB deficient OT-IGzAB−/− CTLs were not found to damage axons44. Axoplasmic GzB activity may also serve as a final axon execution mechanism for the indirect injury of neurons and axons. Bystander axon injury is reported in model systems of myelin antigen-directed and astrocyte-targeted CTLs45-48 and experiments using gang culture neurons demonstrate that purified GzB and exogenously applied lytic granules containing GzB are capable of destabilizing the axon cytoskeleton49,50. Therefore, regardless of the mechanism by which lytic granules encounter the axon, either through direct targeting by axon-specific CTLs, as presented in this work, or by collateral diffusion from glia-targeted immune synapses, the introduction of active GzB into the axoplasm may prove singularly sufficient to injure axons.
Our experimental findings provide a novel model of direct, CTL-mediated axon injury, mechanistically distinct from general neuron-injury models and specifically dependent upon MHC-I:antigen:TCR complex engagement and axoplasmic GzB activity. When modeled in the context of human MS, these finding suggest that demyelinated axons within active, inflammatory lesions are competent to express antigen-loaded MHC-I complexes and are thus susceptible to injury mediated by antigen-specific CTLs. Broadly, the therapeutic targeting of CTLs may prove axon protective and may therefore prevent the irreversible loss of neurological function in MS patients51,52. However, we propose that the refined elimination of pathogenic antigen-specific, anti-axonal CTL populations or specific inhibition of axoplasmic GzB activity may limit MS disease progression without the deleterious complications of immunocompromise.
METHODS
Mouse cortical neuron gang cultures
Cortical neuron-enriched gang cultures were prepared from embroyonic-day-15 C57BL/6 (WT, The Jackson Laboratory, Bar Harbor, Maine) fetuses using methods previously described53. Cortices were isolated in Ca2+- and Mg2+- free Hank’s Buffered Saline Solution containing 1 mg mL−1 glucose (HBSS), incubated in 0.2% papain (HBSS, 15 min, 37 °C, Worthington, Lakewood, New Jersey) and dissociated by trituration. Cells were transferred into plating medium (DMEM, 10% bovine calf serum, 10% Ham’s F12, 1% antimycotic and 1% antibiotic; Life Technologies, Grand Island, New York) and plated onto poly-L-ornithine (Sigma-Aldrich, St. Louis, Missouri) coated, acid-washed cover glasses (Carolina Biological Supply, Burlington, North Carolina). After cell adherence, plating medium was replaced with feed medium (Neurobasal, 10% B27, 50 μM glutamine, 1% antimycotic, 1% antibiotic, 10 ng mL−1 rhIGF-1 and 1 ng mL−1 rhBDNF; Life Technologies; BD Biosciences, San Jose, California; R & D Systems; Minneapolis, Minnesota). SIINFEKL (1 μg mL−1, AnaSpec, Freemont, California) and IFNγ (500 U mL−1, PeproTech, Rocky Hill, New Jersey) treatments were initiated 6-8 days-in-vitro (DIV) and included one medium change 48 hr following treatment initiation.
Microfluidic chamber device neuron culture platform
The microfluidic culture platform (MFC, Supplementary Fig. 1) was produced as previously described54. Chambers were fabricated in polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning, Midland, Michigan) using soft lithography and replica molding. Molds were constructed at the Cornell NonoScale Science and Technology Facility. Chambers were cast of PDMS, cured (100 °C, overnight, 20 psi vacuum), sterilized by autoclaving and adhered to poly-L-ornithine coated, acid-washed 22 × 22 mm cover glasses. Two MFC devices, with microgrooves spaced 100 μm (Fig. 4h) or 50 μm (Fig. 4a) apart, were used interchangeably.
MFC culture and treatment
Dissociated cortical cells from WT and C57BL/6-Tg(CAG-OVA)916Jen/J (OVA, The Jackson Laboratory) fetuses were prepared as described for gang cultures. 1.25×105 cells suspended in 15 μL of plating medium were added to the cell body chamber of pre-prepared MFCs; 20 μL of plating medium without cells was added to the MFC axon chamber. After cell adherence, feed medium was added to cell body (100 μL) and axon (80 μL) chambers. Inter-compartmental fluidic isolation was generated by small volume differences in medium (> 20 μL) between compartments. IFNγ, SIINFEKL and RAHYNIVTF peptide (E7, 1 μg mL−1, Mayo Clinic Peptide Synthesis Core, Rochester, Minnesota) treatments were initiated 5-7 DIV and included one medium change 48 hr following treatment initiation. CTL incubations were performed at 8-10 DIV. Ac-IDEP-CHO (10 μM, EMD Millipore, Billerica, Massachusetts) and Z-AVD-FMK (10 μM, ENZO Life Sciences, Farmingdale, New York) treatments occurred simultaneously with CTL exposure.
H-2Kb and SIINFEKL-bound H-2Kb immunofluorescence
α-H-2Kb (AF6-88.5, BioLegend, San Diego, California) and α-SIINFEKL-bound H-2Kb (25-D1.16, eBioscience) antibodies were diluted 1:50 in feed medium with or with IFNγ and applied to gang or MFC cultures (60 min, 37 °C). Samples were washed in 4 °C dPBS (2 × 30 s) and fixed (20 min, 4% paraformaldehyde, Sigma-Aldrich); MFC chambers were removed following 5 min of fixation. Fixed cells were incubated in immunofluorescence blocking buffer (IF buffer; dPBS, 10% donkey serum, 10% bovine serum albumin, 0.2% Triton X-100 (Sigma-Aldrich); 60 min, 21 °C) and transferred to IF buffer containing biotin-SP-conjugated, donkey α-mouse (1:100, Jackson ImmunoResearch, West Grove, Pennsylvania) and α-neurofilament medium chain (NFM, 1:100, AB1987, Millipore) antibodies (72 hr, 4 °C). Secondary and tertiary labeling was performed with Texas Red-conjugated, donkey α-rabbit and Alexa Fluor 488-conjugated streptavidin (1:100, 60 min, 37 °C, Jackson ImmunoResearch, Life Technologies). Cover glasses were mounted using anti-fade mounting medium (Vector Laboratories, Burlingame, California). Images were collected using a LSM780 laser scanning microscope (Carl Zeiss, Oberkochen, Germany). Images were processed and area measurements completed using ImageJ1.47e (US National Institutes of Health, Bethesda, MD) with colocalization data generated using the RG2B Colocalization plug-in.
MHC-I RT-PCR
Two-step RT-PCR was used to quantify relative MHC-I mRNA expression55. RNA was prepared using the Qiagen RNeasy microRNA kit (Qiagen, Hilden, Germany); concentration and quality was assessed by NanoDrop100 (Thermo Scientific, Wilmington, Delaware). cDNA synthesis was accomplished using the Roche Transcriptor First Strand cDNA synthesis kit with 1 μg (gang) or 100 ng (MFC) of RNA and anchored oligo(dT) primers (Roche Applied Sciences, Mannheim, Germany). Amplification was performed with the Roche LightCycler 480 SYBR Green I Master kit and LightCycler 480 system. The following primers, designed using NCBI Primer-BLAST, were used for Total MHC-I H-2b: forward 5’-CTGAAAACGTGGACGGCGGC-3’, reverse 5’-GGCCCAGCACCTCAGGGTGA-3’, H-2Kb (NM 001001892.2): forward 5’-GCCCGCAGAACTCAGAAGTCGC-3’, reverse 5’-CCTGAATAGTGTGAGAGCCGCCC-3’ and H-2Db (NM 010380.3): forward 5’-AAGCCAAGGGCCAAGAGCAGTGG-3’, reverse 5’-CCTTCATCGGCGAACTGCAG-3’. PCR protocol included polymerase activation (95 °C, 5 min) and 45 cycles of melting (95 °C, 10 s), annealing (60 °C, 10 s) and elongating (72 °C, 20 s). Fluorescence intensity was measured at each elongation step, plotted against cycle number and exported into SigmaPlot-11 (Systat, Chicago, IL) for crossing point analysis. Crossing point values were used to calculate fold induction using the Pfaffl equation56, with reaction efficiency (E) estimated at 1.9 for all targets.
Neurofilament immunofluorescence and axon integrity index quantification.
MFC cultures were processed as described for MHC-I immunofluorescence experiments. Immunolabeling was performed using α-pan-neurofilament antibody (NF, SMI312, 1:100, Millipore) for 72 hr at 4 °C; secondary labeling was completed with FITC-conjugated, donkey α-mouse (1:100, IF buffer, 60 min, 37 °C, Jackson ImmunoResearch) and cover glasses were mounted using anti-fade mounting medium with DAPI (Vector Laboratories). Images were collected and processed as described for MHC-I immunofluorescence experiments. Axon integrity index quantification was performed as described in Supplementary Fig. 1. All image processing was conducted using ImageJ1.47e and the ROI Manager plug-in.
Primary CTL effectors isolation and activation. WT,
C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-I, The Jackson Laboratory), OT-Ipfp1tm1Sdz (J. Trapani, Peter MacCallum Cancer Center) and OT-IGzAtm1LeyGzBtm2.1Ley (J. Trapani) splenocytes were isolated by mechanical disruption and ACK lysis buffer treatment. OT-I, OT-Ipfp−/− and OT-IGzAB−/− splenocytes were cultured for 48 hr at 3×105 cells mL−1 in CML medium (RPMI-1640, 3% fetal calf serum, 25 μM glutamine, 1% sodium pyruvate, 1% nonessential amino acids, 2 mM mercaptoethanol,1% antimycotic and 1% antibiotic, Life Technologies) with SIINFEKL (2 μg mL−1) and rhIL-2 (1 ng mL−1, Millipore). WT splenocytes were cultured in CML with α-CD3 (2C11, 5 μg mL−1, Bio-X-Cell, West Lebanon, New Hampshire) and α-CD28 (371.5, 10 μg mL−1, Bio-X-Cel) soluble antibodies and rhIL-2 (1 ng mL−1). After 48 hr, all cell types were transferred to fresh CML with rhIL-2 (1 ng mL−1) for 24 hr. Prior to experimentation, effectors were purified via Ficoll-Paque (GE Life Sciences, Pittsburgh, Pennsylvania) gradient centrifugation. Isolated splenocytes and post-activation effectors were evaluated by flow cytometry using the following antibodies: α-CD45 (30-F11, PerCp), α-CD4 (RM4-5, APC), α-CD8α (53-6.7, FITC) and α-Vα2 TCR (B20.6, PE) (1:200, BD Biosciences, San Jose, California). Flow cytometry data was gathered using an Accuri C6 cytometer and analyzed with C6 software (BD Biosciences).
51Cr release assay
51Cr release assays were performed according to established methods57. EL-4 cells (H-2b haplotype, ATCC, Manassas, Virginia) were pulsed with SIINFEKL (10 μmL−1), E7 (10 μg mL−1) or no peptide and loaded with Na51Cr (100 μCi, 60 min, 37 °C, Amersham Pharmacia Biotech; Piscataway, New Jersey). CTLs were incubated (30:1) with 51Cr-loaded EL-4 cells (three hours, 37 °C); maximal 51Cr release was obtained using 5% Triton X-100.
OT-I CTL effector immunofluorescence
Samples were processed as described for MHC-I immunofluorescence experiments. Immunolabeling was performed using α-NF (SMI312, 1:100) and Rhodamine-conjugated phalloidin (1:1,000, Life Technologies), α-PFP (CB5.4, 1:100, AbCam; Cambridge, MA) or α-GzB (1:100, AbCam) antibodies (72 hr, 4 °C); secondary labeling was performed with FITC-conjugated, donkey α-mouse (1:100, Jackson ImmunoResearch) and Texas Red-conjugated, donkey α-rat or α-rabbit (1:100, Jackson ImmunoResearch) (60 min, 37 °C). Images were collected and processed as described for MHC-I immunofluorescence experiments.
Scanning and transmission electron microscopy
EM studies were performed at the Mayo Clinic EM Facility. Samples were immersed in 4 °C Trump’s fixative for a minimum of 24 hr; MFC chambers were following 5 min of fixation. SEM samples were stained with 1% osmium tetroxide, dehydrated, dried using CO2, mounted on an aluminum stub and sputter-coated with gold-palladium. SEM images were acquired using a Hitachi 4700 scanning-electron microscope (Hitachi, Schaumburg, Illinois). TEM samples were incubated in 1% osmium tetroxide, dehydrated and embedded in Quetol 651 (Ted Pella, Redding, California). Serial thin sections (0.09–0.1 μm) were cut using a diamond knife (Diatome, Hatfield, Pennsylvania) and Ultracut E microtome (Reichert-Jung, Wien, Austria), collected on copper grids and post-stained with lead citrate; images were collected at ~80 kV with a JEOL 1400 transmission electron microscope (JEOL, Peabody, Massachusetts).
Animal use and care
All animal experiments were approved by the Mayo Clinic Institutional Animal Care and Use Committee in accordance with National Institutes of Health guidelines.
Statistical analysis
All analyses were performed using SigmaStat11. α=0.05 and β=0.2 were established a priori. Normality was determined by the Shapiro-Wilk test and normally distributed data were checked for equal variance. Parametric tests were only applied to data that were both normally distributed and of equal variance. The Holm-Sidak (parametric) or Dunn’s method (nonparametric) pairwise comparison tests were used for all post-hoc sequential comparisons. Curran-Everett guidelines were used for reporting statistical values 58. All non-parametric P-values were reported as unadjusted P-values.
Supplementary Material
Supplementary Figure 1. Microfluidic chamber neuron culture platform. (a) Microfluidic chamber design constructed in PDMS. (b) Fluidic isolation of the cell body and axon chambers occurs by hydrostatic pressure variation across the microgrooves as generated by small difference in fluid volume (> 20 μL) between the two compartments. (c) E15 cortical neurons plated in the cell body chamber (left) of the MFC (shown at 5x) extend long axons through the microgrooves (500 × 10 × 3 μm, length × width × height) into the axon chamber (right). (d-f) NF (axons) and DAPI (cell bodies) immunofluorescence images (20x) of the cell body chamber (d), microgrooves (e) and axon chamber (f). (g) Axon integrity index quantification (AIIQ). 20x NF immunofluorescence images of axon fields were collected, converted to single channel binary images and partitioned into seven bins (g, inset). Bin 1 encompassed the microgroove MFC region 25 μm proximal to the leading edge of the axon chamber; bins 2-7 extended distally from the axon chamber leading edge in 50 μm increments. An axon integrity index for each bin was calculated as the percent NF immunofluorescence signal area of each bin normalized to the percent area of bin 1. The axon chamber image (f), as converted to a single channel binary image is presented (g, inset) and the resultant, normalized data is shown (g, graph) and contextualized with the full unmanipulated axon field data set. Error bars are 95% CI. Scale bars (c)=200 μm and (d-f)=100 μm.
Supplementary Figure 2. CTL effector characterization. Example flow cytometry plots of splenocyte-derived OT-I CTL effectors pre- (a) and post- (b) in vitro activation and expansion. Effector OT-I CTLs are defined as CD45hiCD8+Vα2+. (c) Flow cytometric characterization of pre-activation, splenocyte-derived WT and OT-I cells and post-activation WT, OT-I, OT-Ipfp1−/− and OT-IGzAB−/− CTLs. (d) 51Cr release assay characterizing the cytotoxic capacity of activated WT, OT-I, OT-Ipfp1−/− and OT-IGzAB−/− CTLs; conditions presented reflect the experimental paradigms of Figure 6. (c,d) Full statistical measurements are presented in Supplementary Table 1; error bars are 95% CI.
Supplementary Table 1. Complete data and statistical methods and analyses. Abbreviations: Confidence Interval (CI), Normality Test (Normality), Equal Variance Test (Variance), P-value (P), Unadjusted P-value (Unadj. P), Critical Level P-value (Critical P)
ACKNOWLEDGEMENTS
This work was supported by a generous gift from Donald and Frances Herdrich to C.L.H., grant RG3636 from the United States National Multiple Sclerosis Society to C.L.H., the Mayo Foundation for Medical Research and United States National Institutes of Health Medical Scientist Training Program grant GM065841 to B.M.S.. We thank Rachel Bergstrom for providing technical assistance with the microfluidic neuron culture platform, Joseph Trapani and Vivien Sutton of the Peter MacCallum Cancer Center, Melbourne, Australia for providing the OT-IGzAB−/− and OT-Ipfp−/− mouse strains and Trace Christensen and Scott Gamb for technical assistance with the electron microscopy experiments. We also thank Joshua Burda and Joshua Piotrowski for assistance with manuscript preparation.
Footnotes
CONTRIBUTIONS
B.M.S. and C.L.H. conceived, designed, and analyzed all experiments and wrote the manuscript. B.M.S. performed all experiments. W.F.S. assisted with RT-PCR, 51Cr release assays and manuscript preparation.
REFERNCES
- 1.Conway D, Cohen JA. Combination therapy in multiple sclerosis. Lancet neurology. 2010;9:299–308. doi: 10.1016/S1474-4422(10)70007-7. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain: a journal of neurology. 1997;120:393–399. doi: 10.1093/brain/120.3.393. Pt 3. [DOI] [PubMed] [Google Scholar]
- 3.Dutta R, Trapp BD. Pathogenesis of axonal and neuronal damage in multiple sclerosis. Neurology. 2007;68:S22–31. doi: 10.1212/01.wnl.0000275229.13012.32. discussion S43-54. [DOI] [PubMed] [Google Scholar]
- 4.Tallantyre EC, et al. Clinico-pathological evidence that axonal loss underlies disability in progressive multiple sclerosis. Multiple sclerosis. 2010;16:406–411. doi: 10.1177/1352458510364992. [DOI] [PubMed] [Google Scholar]
- 5.Trapp BD, et al. Axonal transection in the lesions of multiple sclerosis. The New England journal of medicine. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 6.Kornek B, et al. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. The American journal of pathology. 2000;157:267–276. doi: 10.1016/S0002-9440(10)64537-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bjartmar C, Trapp BD. Axonal and neuronal degeneration in multiple sclerosis: mechanisms and functional consequences. Current opinion in neurology. 2001;14:271–278. doi: 10.1097/00019052-200106000-00003. [DOI] [PubMed] [Google Scholar]
- 8.Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Annals of neurology. 2001;50:389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- 9.Traugott U, Reinherz EL, Raine CS. Science. Vol. 219. New York, N.Y: 1983. Multiple sclerosis: distribution of T cell subsets within active chronic lesions; pp. 308–310. [DOI] [PubMed] [Google Scholar]
- 10.Booss J, Esiri MM, Tourtellotte WW, Mason DY. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. Journal of the neurological sciences. 1983;62:219–232. doi: 10.1016/0022-510x(83)90201-0. [DOI] [PubMed] [Google Scholar]
- 11.Hauser SL, et al. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Annals of neurology. 1986;19:578–587. doi: 10.1002/ana.410190610. [DOI] [PubMed] [Google Scholar]
- 12.Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Bruck W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain. 2000;123:1174–1183. doi: 10.1093/brain/123.6.1174. Pt 6. [DOI] [PubMed] [Google Scholar]
- 13.Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, Bruck W. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain. 2002;125:2202–2212. doi: 10.1093/brain/awf235. [DOI] [PubMed] [Google Scholar]
- 14.Frischer JM, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain: a journal of neurology. 2009;132:1175–1189. doi: 10.1093/brain/awp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Babbe H, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. The Journal of experimental medicine. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skulina C, et al. Multiple sclerosis: brain-infiltrating CD8+ T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2428–2433. doi: 10.1073/pnas.0308689100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacobsen M, et al. Oligoclonal expansion of memory CD8+ T cells in cerebrospinal fluid from multiple sclerosis patients. Brain: a journal of neurology. 2002;125:538–550. doi: 10.1093/brain/awf059. [DOI] [PubMed] [Google Scholar]
- 18.Junker A, et al. Multiple sclerosis: T-cell receptor expression in distinct brain regions. Brain: a journal of neurology. 2007;130:2789–2799. doi: 10.1093/brain/awm214. [DOI] [PubMed] [Google Scholar]
- 19.Hoftberger R, et al. Brain pathology. Vol. 14. Zurich, Switzerland: 2004. Expression of major histocompatibility complex class I molecules on the different cell types in multiple sclerosis lesions; pp. 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liblau RS, Gonzalez-Dunia D, Wiendl H, Zipp F. Neurons as targets for T cells in the nervous system. Trends in neurosciences. 2013 doi: 10.1016/j.tins.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 21.Neumann H, Medana IM, Bauer J, Lassmann H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends in neurosciences. 2002;25:313–319. doi: 10.1016/s0166-2236(02)02154-9. [DOI] [PubMed] [Google Scholar]
- 22.Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. 2009;9:393–407. doi: 10.1038/nri2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malmestrom C, et al. Relapses in multiple sclerosis are associated with increased CD8+ T-cell mediated cytotoxicity in CSF. Journal of neuroimmunology. 2008;196:159–165. doi: 10.1016/j.jneuroim.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 24.Rivera-Quinones C, et al. Absence of neurological deficits following extensive demyelination in a class I-deficient murine model of multiple sclerosis. Nature medicine. 1998;4:187–193. doi: 10.1038/nm0298-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howe CL, Adelson JD, Rodriguez M. Absence of perforin expression confers axonal protection despite demyelination. Neurobiology of disease. 2007;25:354–359. doi: 10.1016/j.nbd.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deb C, et al. Demyelinated axons and motor function are protected by genetic deletion of perforin in a mouse model of multiple sclerosis. Journal of neuropathology and experimental neurology. 2009;68:1037–1048. doi: 10.1097/NEN.0b013e3181b5417e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deb C, et al. CD8+ T cells cause disability and axon loss in a mouse model of multiple sclerosis. PLoS ONE. 2010;5:e12478. doi: 10.1371/journal.pone.0012478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joly E, Oldstone MB. Neuronal cells are deficient in loading peptides onto MHC class I molecules. Neuron. 1992;8:1185–1190. doi: 10.1016/0896-6273(92)90138-4. [DOI] [PubMed] [Google Scholar]
- 29.Neumann H, Cavalie A, Jenne DE, Wekerle H. Science. Vol. 269. New York, N.Y: 1995. Induction of MHC class I genes in neurons; pp. 549–552. [DOI] [PubMed] [Google Scholar]
- 30.Neumann H, Schmidt H, Cavalie A, Jenne D, Wekerle H. Major histocompatibility complex (MHC) class I gene expression in single neurons of the central nervous system: differential regulation by interferon (IFN)-gamma and tumor necrosis factor (TNF)-alpha. The Journal of experimental medicine. 1997;185:305–316. doi: 10.1084/jem.185.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Medana I, Martinic MA, Wekerle H, Neumann H. Transection of major histocompatibility complex class I-induced neurites by cytotoxic T lymphocytes. The American journal of pathology. 2001;159:809–815. doi: 10.1016/S0002-9440(10)61755-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rensing-Ehl A, et al. Neurons induced to express major histocompatibility complex class I antigen are killed via the perforin and not the Fas (APO-1/CD95) pathway. European journal of immunology. 1996;26:2271–2274. doi: 10.1002/eji.1830260945. [DOI] [PubMed] [Google Scholar]
- 33.Medana IM, et al. MHC class I-restricted killing of neurons by virus-specific CD8+ T lymphocytes is effected through the Fas/FasL, but not the perforin pathway. European journal of immunology. 2000;30:3623–3633. doi: 10.1002/1521-4141(200012)30:12<3623::AID-IMMU3623>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 34.Bradke F, Dotti CG. Establishment of neuronal polarity: lessons from cultured hippocampal neurons. Current opinion in neurobiology. 2000;10:574–581. doi: 10.1016/s0959-4388(00)00124-0. [DOI] [PubMed] [Google Scholar]
- 35.Ehst BD, Ingulli E, Jenkins MK. Development of a novel transgenic mouse for the study of interactions between CD4 and CD8 T cells during graft rejection. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2003;3:1355–1362. doi: 10.1046/j.1600-6135.2003.00246.x. [DOI] [PubMed] [Google Scholar]
- 36.Vass K, Lassmann H. Intrathecal application of interferon gamma. Progressive appearance of MHC antigens within the rat nervous system. The American journal of pathology. 1990;137:789–800. [PMC free article] [PubMed] [Google Scholar]
- 37.Forooghian F, Cheung RK, Smith WC, O'Connor P, Dosch HM. Enolase and arrestin are novel nonmyelin autoantigens in multiple sclerosis. Journal of clinical immunology. 2007;27:388–396. doi: 10.1007/s10875-007-9091-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dustin ML, Chakraborty AK, Shaw AS. Understanding the structure and function of the immunological synapse. Cold Spring Harbor perspectives in biology. 2010;2:a002311. doi: 10.1101/cshperspect.a002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manning PT, Johnson EM, Jr., Wilcox CL, Palmatier MA, Russell JH. MHC-specific cytotoxic T lymphocyte killing of dissociated sympathetic neuronal cultures. The American journal of pathology. 1987;128:395–409. [PMC free article] [PubMed] [Google Scholar]
- 40.Giuliani F, Goodyer CG, Antel JP, Yong VW. Vulnerability of human neurons to T cell-mediated cytotoxicity. Journal of immunology. 2003;171:368–379. doi: 10.4049/jimmunol.171.1.368. [DOI] [PubMed] [Google Scholar]
- 41.Saikali P, et al. NKG2D-mediated cytotoxicity toward oligodendrocytes suggests a mechanism for tissue injury in multiple sclerosis. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:1220–1228. doi: 10.1523/JNEUROSCI.4402-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Downen M, Amaral TD, Hua LL, Zhao ML, Lee SC. Neuronal death in cytokine-activated primary human brain cell culture: role of tumor necrosis factor-alpha. Glia. 1999;28:114–127. [PubMed] [Google Scholar]
- 43.Bien CG, et al. Destruction of neurons by cytotoxic T cells: a new pathogenic mechanism in Rasmussen's encephalitis. Annals of neurology. 2002;51:311–318. doi: 10.1002/ana.10100. [DOI] [PubMed] [Google Scholar]
- 44.Meuth SG, et al. Cytotoxic CD8+ T cell-neuron interactions: perforin-dependent electrical silencing precedes but is not causally linked to neuronal cell death. The Journal of neuroscience: the official journal of the. Society for Neuroscience. 2009;29:15397–15409. doi: 10.1523/JNEUROSCI.4339-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McPherson SW, Heuss ND, Roehrich H, Gregerson DS. Bystander killing of neurons by cytotoxic T cells specific for a glial antigen. Glia. 2006;53:457–466. doi: 10.1002/glia.20298. [DOI] [PubMed] [Google Scholar]
- 46.Tsuchida T, et al. Autoreactive CD8+ T-cell responses to human myelin protein-derived peptides. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:10859–10863. doi: 10.1073/pnas.91.23.10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sobottka B, et al. Collateral bystander damage by myelin-directed CD8+ T cells causes axonal loss. The American journal of pathology. 2009;175:1160–1166. doi: 10.2353/ajpath.2009.090340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bahbouhi B, et al. T cell recognition of self-antigen presenting cells by protein transfer assay reveals a high frequency of anti-myelin T cells in multiple sclerosis. Brain: a journal of neurology. 2010;133:1622–1636. doi: 10.1093/brain/awq074. [DOI] [PubMed] [Google Scholar]
- 49.Miller NM, et al. Lymphocytes with cytotoxic activity induce rapid microtubule axonal destabilization independently and before signs of neuronal death. ASN Neuro. 2012;5 doi: 10.1042/AN20120087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haile Y, et al. Granule-derived granzyme B mediates the vulnerability of human neurons to T cell-induced neurotoxicity. Journal of immunology. 2011;187:4861–4872. doi: 10.4049/jimmunol.1100943. [DOI] [PubMed] [Google Scholar]
- 51.Polman CH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. The New England journal of medicine. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 52.Havrdova E, et al. Effect of natalizumab on clinical and radiological disease activity in multiple sclerosis: a retrospective analysis of the Natalizumab Safety and Efficacy in Relapsing-Remitting Multiple Sclerosis (AFFIRM) study. Lancet neurology. 2009;8:254–260. doi: 10.1016/S1474-4422(09)70021-3. [DOI] [PubMed] [Google Scholar]
- 53.Hilgenberg LG, Smith MA. Preparation of dissociated mouse cortical neuron cultures. Journal of visualized experiments: JoVE. 2007;562 doi: 10.3791/562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taylor AM, et al. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nature methods. 2005;2:599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deb C, Howe CL. Functional characterization of mouse spinal cord infiltrating CD8+ lymphocytes. Journal of neuroimmunology. 2009 doi: 10.1016/j.jneuroim.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wallace D, Hildesheim A, Pinto LA. Comparison of benchtop microplate beta counters with the traditional gamma counting method for measurement of chromium-51 release in cytotoxic assays. Clin Diagn Lab Immunol. 2004;11:255–260. doi: 10.1128/CDLI.11.2.255-260.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Curran-Everett D, Benos DJ. Guidelines for reporting statistics in journals published by the American Physiological Society: the sequel. Adv Physiol Educ. 2007;31:295–298. doi: 10.1152/advan.00022.2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Microfluidic chamber neuron culture platform. (a) Microfluidic chamber design constructed in PDMS. (b) Fluidic isolation of the cell body and axon chambers occurs by hydrostatic pressure variation across the microgrooves as generated by small difference in fluid volume (> 20 μL) between the two compartments. (c) E15 cortical neurons plated in the cell body chamber (left) of the MFC (shown at 5x) extend long axons through the microgrooves (500 × 10 × 3 μm, length × width × height) into the axon chamber (right). (d-f) NF (axons) and DAPI (cell bodies) immunofluorescence images (20x) of the cell body chamber (d), microgrooves (e) and axon chamber (f). (g) Axon integrity index quantification (AIIQ). 20x NF immunofluorescence images of axon fields were collected, converted to single channel binary images and partitioned into seven bins (g, inset). Bin 1 encompassed the microgroove MFC region 25 μm proximal to the leading edge of the axon chamber; bins 2-7 extended distally from the axon chamber leading edge in 50 μm increments. An axon integrity index for each bin was calculated as the percent NF immunofluorescence signal area of each bin normalized to the percent area of bin 1. The axon chamber image (f), as converted to a single channel binary image is presented (g, inset) and the resultant, normalized data is shown (g, graph) and contextualized with the full unmanipulated axon field data set. Error bars are 95% CI. Scale bars (c)=200 μm and (d-f)=100 μm.
Supplementary Figure 2. CTL effector characterization. Example flow cytometry plots of splenocyte-derived OT-I CTL effectors pre- (a) and post- (b) in vitro activation and expansion. Effector OT-I CTLs are defined as CD45hiCD8+Vα2+. (c) Flow cytometric characterization of pre-activation, splenocyte-derived WT and OT-I cells and post-activation WT, OT-I, OT-Ipfp1−/− and OT-IGzAB−/− CTLs. (d) 51Cr release assay characterizing the cytotoxic capacity of activated WT, OT-I, OT-Ipfp1−/− and OT-IGzAB−/− CTLs; conditions presented reflect the experimental paradigms of Figure 6. (c,d) Full statistical measurements are presented in Supplementary Table 1; error bars are 95% CI.
Supplementary Table 1. Complete data and statistical methods and analyses. Abbreviations: Confidence Interval (CI), Normality Test (Normality), Equal Variance Test (Variance), P-value (P), Unadjusted P-value (Unadj. P), Critical Level P-value (Critical P)