

Abstract
TLR9-/-NOD mice develop a significantly reduced incidence of diabetes. This study was to investigate the molecular mechanisms of the protective role of TLR9 deficiency. Through gene screening and confirmation by both mRNA and protein expression, we found a significant increase in CD73-expressing immune cells from peripheral lymphoid tissues in TLR9-/-NOD mice. The elevated frequency of CD73-expressing immune cells seemed to be specific for TLR9 deficiency and was MyD88-independent. Moreover, the increased frequency of CD73 expression was limited to the NOD background. Increased frequency of CD73 expression was also associated with lower levels of pro-inflammatory cytokines and more anti-inflammatory cytokine production in CD4+ T cells in TLR9-/-NOD mice. Purified CD73+CD4+ T cells showed stronger immunosuppressive function in vitro and delayed diabetes development in vivo. The immunosuppression appeared to be mediated by TGFβ. In addition, elevated frequency of CD73 expressing cells was associated with improved β cell function. Our observations were further confirmed by protection from diabetes with similar alterations in CD73 in the NY8.3 T cell receptor (TCR) NOD mouse model crossed with TLR9 deficient mice and by the use of a TLR9 inhibitor in NOD mice. Our novel findings suggest an important immune regulatory role of CD73 in regulation of diabetes development and may offer a new therapeutic strategy for specific intervention to prevent T1D.
Keywords: innate immunity, TLR9, CD73, Type 1 diabetes, Non-obese diabetic mice
Introduction
Type 1 diabetes (T1D) is an organ specific autoimmune disease characterized by T cell-mediated destruction of the insulin-producing pancreatic β cells (1-3). Growing evidence has shown that Toll-like receptors (TLRs), which are pattern-recognition receptors that recognize structurally conserved microbial molecular components, are involved in autoimmune diseases, such as experimental autoimmune encephalomyelitis (EAE), systemic lupus erythematosus (SLE), rheumatoid arthritis (RA) and T1D (4-9). TLR9 recognizes unmethylated CpG DNA that is rich in bacteria and also present in mammalian cells (10, 11) and signaling occurs through the MyD88 pathway (12-14). We recently reported that TLR9-deficient (TLR9-/-) NOD mice are significantly protected from T1D development (15). However, the mechanisms by which the mice are protected from disease have not been fully elucidated. Zhang et al have recently found that TLR9-/-NOD mice expressed lower levels of IFN-α in pancreatic lymph nodes (PLNs) and reduced frequencies of plasmacytoid DCs (pDC) and diabetogenic CD8+ T cells compared with wild type (WT) NOD mice (16). The authors concluded that TLR9 activation contributed to the spontaneous diabetes onset in NOD mice by increasing IFN-α and promoting diabetic CD8+ T cell activation. However, it is likely that other mechanisms are also involved in diabetes protection, in addition to reduction in IFN-α in TLR9-/- NOD mice. The role of IFN-α in T1D is complex; our previous studies showed that over-expression of IFN-α promoted diabetes development in one diabetes model whereas it protected against diabetes development in another (17).
CD73 is a 70-kDa glycosylphosphatidylinositol (GPI)-anchored protein with ecto 5’-nucleotidase enzyme activity that catalyses the dephosphorylation of AMP to generate adenosine (18-20). Adenosine has various immunoregulatory activities mediated through four adenosine receptors (A1AR, A2AAR, A2BAR and A3AR), which are expressed in some lymphocyte subsets and endothelial cells (21, 22). Recent studies showed that CD73 clearly contributes to the generation of extracellular adenosine in a number of physiologically relevant experimental models (23-25). However, there are other functions which include a critical role in host defense (20), involvement in lymphocyte adhesion to endothelium and restriction of lymphocyte migration into draining lymph nodes (26), costimulation of T cell activation and control of B cell-follicular DC interaction (27). CD73 expression can be detected in human and mouse T and B lymphocytes; as well as DCs and macrophages (27). However, it is particularly highly expressed in T regulatory (Treg) cells, which can suppress effector T cells by converting 5’-adenosine monophosphate (AMP) to adenosine (28, 29). In spite of these important immune functions, little is known the role of CD73 in T1D development, especially in relation to innate immunity. In this study, we performed microarray analysis in immune cells from TLR9-/- and WT NOD mice. We found a significant increase in frequency of CD73-expressing T cells in TLR9-/-NOD and TLR9-/-NY8.3NOD mice and the diabetes protected phenotype seen in both models was associated with the up-regulation of CD73 and anti-inflammatory functions of CD73+ T cells.
Materials and methods
Mice
NOD/Caj mice were originally obtained from the Jackson Laboratory and have been maintained at Yale University for many years. TLR2, TLR4, TLR9, MyD88 and TRIF deficient mice were generated as previously described (10, 30-34). We backcrossed these innate immune deficient mice on to the NOD/Caj genetic background for over 10 generations. NY8.3 NOD mice, obtained from the Jackson Laboratory, have been reported previously (35) and we bred NY8.3 NOD mice with TLR9-/-NOD mice to obtain TLR9-/-NY8.3NOD mice. TLR9-/-B6 mice were obtained by breeding TLR9 deficient mice to C57BL/6 mice. All the mice were kept in specific pathogen-free conditions in a 12-hour dark/light cycle and housed in individually ventilated filter cages with autoclaved food. The use of the animals and the procedures applied in this study were approved by the Institutional Animal Care and Use Committee of Yale University.
Natural history of diabetes development
Incidence of diabetes was observed in NOD, TLR9-/-NOD, NY8.3NOD and TLR9-/-NY8.3NOD mice by weekly screening for urine glucose. When glycosuria was observed, diabetes was then confirmed by blood glucose ≥250 mg/dl (13.9 mmol/l).
Microsatellite analysis of Idd markers
TLR9-/-NOD mice were examined for 32 known Idd markers by PCR using specific primers for the Idd markers (http://type1diabetes.jax.org), controlled using DNA samples from WT NOD and C57BL/6 mice.
Antibodies and Reagents
All the fluorochrome-conjugated mAbs used in this study were purchased from eBioscience or Biolegend unless otherwise stated. Hybridoma supernatants containing mAbs, used for cell purification or stimulation, were generously provided by the late Charles Janeway Jr. (Yale University). Magnetic beads conjugated with goat anti-mouse IgG, goat anti-mouse IgM, or goat anti-rat IgG were purchased from Qiagen. RPMI-1640 medium and heat-inactivated FCS were purchased from Invitrogen and Gemini, respectively.
Immunization
NOD or TLR9-/-NOD mice (2-months old) were injected subcutaneously (s.c.) with keyhole limpet hemocyanin (KLH, Sigma), as a foreign antigen, emulsified in Alum (Pierce). Mice were sacrificed 7 days after immunization and lymphocytes from draining lymph nodes and spleens were tested for recall immune responses to the immunized antigen. Two separate experiments were performed (n=3-4 mice/group/experiment).
Intracellular cytokine (ICC) or cytotoxic protein detection assay
ICC was performed according to the protocol provided with kits from eBioscience. Briefly, cells were stimulated with anti-CD3 (clone 2C-11) and anti-CD28 (clone 37N51) antibodies overnight followed by further stimulation with PMA (50 ng/ml, Sigma) and ionomycin (500 ng/ml, Sigma) in the presence of Golgi-plug (eBioscience) for an additional four hours. The cells were then stained with surface markers before fixation and permeabilization. Fc receptors were blocked with 2.4G2 Fc-blocking antibody before staining with the recommended amount of fluorochrome-labeled antibody for the detection of intracellular cytokines or cytotoxic protein (granzyme B and perforin). The live lymphocytes were first gated according to the parameters of forward scatter and side scatter. The expression of cytokine was then analyzed in gated CD4 or CD8 T cells.
Cell proliferation assay
MACS bead-purified splenic CD4+ T cells (105 cells/well) from BDC2.5 TCR-transgenic NOD mice were cultured in the presence or absence of BDC2.5 mimotope (10 ng/ml) with FACS sorted splenic CD73+CD4+ or CD73-CD4+ T cells (105 cells/well) from WT NOD or TLR9-/-NOD mice (7-8 week-old, sex-matched). Irradiated (3000 rads) total splenocytes (105 cells/well) from NOD mice were used as antigen presentation cells and 3H-thymidine was added during the last 18 hours of a 4-day culture. Proliferation was measured by 3H-thymidine incorporation. Neutralizing antibody, anti-TGF-β (clone 1D11.16.8; BioXcell) or anti-IL-10 (JES5-2A5; BioXcell) was added in some proliferation assays, as indicated, to test for regulatory cytokine mediated immune suppression.
Adoptive transfer
Irradiated (650 rads) 6-7-week-old female NOD mice were used as recipients in adoptive transfer experiments. Splenocytes (8×106) from diabetic NOD mice with or without sorted splenic CD73+CD4+ T cells (1.7×106) from 6-7 week-old NOD or TLR9-/-NOD mice were injected (i.v.) into age and sex-matched recipients (all females). All the recipients were monitored for glycosuria weekly, and the experiments were terminated 3 months after the cell transfer unless the mice developed diabetes, confirmed by blood glucose greater than 250 mg/dL (13.9 mmol/l).
Oral glucose tolerance test (OGTT)
Mice were fasted overnight (free water access) before giving glucose (2 mg/g body weight) by oral gavage and blood glucose was measured at different time points.
Quantitative real-time PCR (qPCR)
Total RNA was isolated from MACS bead-purified splenic CD4+ and CD8+ T cells or FACS sorted splenic CD73+CD4+ and CD73-CD4+ T cells from NOD or TLR9-/- NOD mice (7-8 week-old, sex-matched, both females and males) using RNeasy Mini kit (Qiagen) or TRIzol (Invitrogen) and then reverse transcribed to cDNA using SuperScript III First-strand synthesis kit with random hexamers (Invitrogen). Quantitative real-time PCR (qPCR) was performed using Bio-Rad iQ5 qPCR detection system according to the manufacturer’s instructions. The relative mRNA levels of CD73, TGFβ, IRF1, IRF5, IRF7, IRF8, CXCR4, HIF-α, SOCS3 and IGF-1 were determined using the 2-ΔΔCt method by normalization with the house-keeping gene GAPDH.
Chloroquine administration test
One-month-old NOD mice were injected with chloroquine (Sigma, 20 μg/g body weight) or PBS (i.p.) daily for 5 days and twice/wk thereafter for an additional 3 weeks. OGTT was performed one month after the treatment. CD73 expression in lymphocytes from different peripheral lymphoid tissue was also evaluated by flow cytometry. Pancreata were taken from the mice after three-month treatment with chloroquine (20 μg/g body weight), fixed with formalin and embedded in paraffin. The tissue blocks were cut (5-6 μm), mounted on microscope slides and stained with hematoxylin and eosin (H+E). Insulitis was scored by an individual in a blinded fashion.
Adenosine deaminase (ADA) activity
ADA activity was measured in serum and spleen cell lysates of 7-8 week-old NOD or TLR9-/- NOD mice (age and sex-matched, males and females) according to the method by Mishra et al, (36) with modification. Briefly, splenocytes (10×106 cells) were lysed with PTNG buffer (50 mM Hepes, pH 7.5, 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1.5 mM MgCl2, 1 mM EGTA) at 4°C for 1 hr. The protein concentration was determined by Bio-Rad Protein Assay Kit (Cat # 500-001). The enzyme assay mixture (250 μl) contained 0.1 mM adenosine, 15 mM potassium phosphate buffer (pH 7.4), 1.25% glycerol and 25 μl of sera or 50 μg of protein. The rate of disappearance of adenosine was taken as an index of ADA activity and was followed by measuring the rate of decrease in optical density at 265 nm. The ADA activity was represented by the converted adenosine (nmol/min/ml or nmol/min/mg).
Immunohistochemical staining
Pancreata taken from 6-7 week-old female NOD or TLR9-/-NOD mice were fixed overnight in periodate-lysine-paraformaldehyde fixative buffer (2% paraformaldehyde, 0.075 M lysine, 0.037 M sodium phosphate, 0.01 M periodate). The tissue was then embedded in Tissue-Tek OCT compound and was snap frozen. Cryosections (10 μM) were rehydrated with 1×PBS followed by blocking with 2% donkey serum. The primary antibodies were biotin labeled anti-mouse CD73 (BioLegend) and guinea pig anti-mouse insulin (Zymed). FITC-conjugated streptavidin (Invitrogen) and PE-conjugated goat anti-guinea pig IgG (H+L) (Santa Cruz) were used as secondary antibodies. Pancreatic sections were examined and photographed using an Olympus fluorescent microscope BX50.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software. Diabetes incidence was compared using log-rank test. In vitro assays were analyzed with Student’s t test or ANOVA and P<0.05 was considered significant.
Results
CD73 is uniquely up-regulated in TLR9-deficient NOD mice
Consistent with our previous observation that TLR9-/-NOD mice were protected from diabetes development (15), the observation for diabetes in our recent cohorts also showed that TLR9-/-NOD mice were significantly protected from diabetes development in both genders compared to WT NOD counterparts (Fig. 1A). To exclude potential effects of carry-over genes from the original TLR9 targeted ES cells, we examined 32 known Idd markers (http://type1diabetes.jax.org) by microsatellite analysis and all the markers were shown to be of NOD origin (data not shown) including Idd2 that is on Chr.9 and ~27cM away from the TLR9 gene (Fig. 1B). To investigate potentially novel genes in T cells that contribute to diabetes reduction in TLR9-/-NOD mice we performed Illumina mRNA microarray analysis using FACS purified T cells from spleens of WT NOD and TLR9-/-NOD mice. CD73 expression was significantly increased in T cells from TLR9-/-NOD mice compared to WT NOD mice (Accession number GSE48781, http://www.ncbi.nlm.nih.gov/geo/info/linking.html). We confirmed CD73 gene up-regulation by qPCR not only in purified CD4 but also in purified CD8+ T cells (Fig. 1C). Using flow cytometry, we have also demonstrated that CD73-expressing splenocytes were significantly increased in frequency in TLR9-/- NOD mice compared to their WT counterparts (Fig. 1D). Moreover, the frequency of CD73-expressing splenic DCs and macrophages were also increased in TLR9-/-NOD compared to the equivalent cells from WT NOD mice (data not shown). Similar results were found in peripheral lymph nodes as well (data not shown). To determine whether the difference in CD73 expression is part of a general phenotype of TLR9 deficiency, we examined TLR9-/-B6 mice and found that the frequency of CD73-expressing B cells, but not T cells, were modestly increased in TLR9-/-B6 mice compared to B6 WT mice (Fig. 1E). This suggests that the up-regulation of CD73 in the immune cells examined in the absence of TLR9 is related to the NOD genetic background. To investigate whether the enhanced CD73 expression is unique to TLR9 deficiency, we then tested CD73 expression in TLR2-/-, TLR4-/-, MyD88-/- and TRIF-/- NOD mice. It is interesting that elevated frequency of CD73-expressing immune cells was unique to TLR9-/-NOD mice and was MyD88 independent (Fig. 1F) as none of the tested NOD strains deficient with the innate immune receptors or adaptors showed the elevation (Fig. 1F). Furthermore, the elevated frequency of CD73 expressing cells in TLR9-/-NOD mice was not age dependent as both young (~2-month-old) and aged (~6-month-old) mice showed the same elevation (data not shown).
Figure 1.
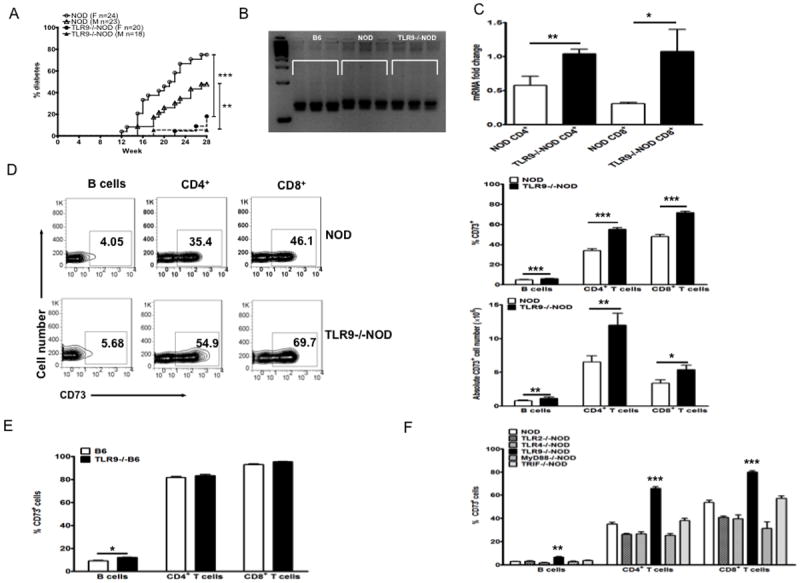
Elevated CD73 expression in immune cells of TLR9-/-NOD mice.
(A) Natural history of diabetes development of NOD and TLR9-/NOD mice. The incidence of diabetes in TLR9-/-NOD mice was compared to WT NOD mice. Log-rank test for survival was used for statistical analysis.**, p<0.01, ***, p<0.001. (B) Microsatellite analysis of Idd2. Idd2 marker (D9Mit25) was analyzed by PCR. The size of PCR product for B6 and NOD mice is 130 and 136bp, respectively. As shown in the gel, the size of the Idd2 marker in TLR9-/-NOD mice is the same as WT NOD mice. (C) The level of CD73 mRNA expression was determined by real time PCR in purified splenic CD4+ and CD8+ T cells from 7-8 week-old mice (both male and females). The expression level of CD73 mRNA was normalized to the house-keeping gene GAPDH. The experiment was repeated twice and the mean ± SEM is presented. Student’s t test was used for statistical analysis, * p<0.05; ** p<0.01. (D) Splenocytes from TLR9-/-NOD and WT NOD mice were stained with fluorochrome-conjugated anti-CD73, anti-CD4, anti-CD8 and anti-B220 antibodies. Representative FACS plots are shown on the left, a summary of the percentage of CD73 expressing T and B cells is shown on the upper right (n≥16, sex-matched 7-8 week-old, males and females) and the absolute numbers of CD73+ cells are shown on the lower right (n=5). Student’s t test was used for statistical analysis, *, p<0.05; **, p<0.01; ***, p<0.001. (E) CD73 expression in both B and T lymphocytes of TLR9-/-B6 and wild type B6 mice (n≥4, 7-8 week-old, both males and females). Student’s t test was used for statistical analysis, *, p<0.05. (F) CD73 expression levels of peripheral blood mononuclear cells from different innate immune-deficient NOD mice and wild type NOD mice were examined by flow cytometry after staining with anti-CD73, anti-CD4, anti-CD8 and anti-B220 antibodies. The graph illustrates the values obtained from 7-8 week-old mice (n≥ 8 mice each strain, 7-8 week-old, sex-matched, males and females). Each value represents mean ± SEM. One-way ANOVA was used for statistical analysis, **, p<0.01; ***, p<0.001. Similar patterns were observed in other peripheral lymphoid tissues (data not shown).
Reduced activation of CD73-positive T cells and suppressed immune response to foreign antigen in TLR9-/-NOD mice
To determine the effect of up-regulation of CD73 expression on T cell function, we first examined T cell activation and memory markers. The frequency of naïve CD4+ T cells (CD44lowCD62L+) in TLR9-/-NOD splenic CD4+ T cells was higher than in WT NOD CD4+ T cells whereas the frequency of memory cells (CD44highCD62L-) was lower (data not shown). This was more obvious in CD73-positive CD4+ T cells (Fig. 2A), but also found in the CD8+ T cells (data not shown). Furthermore, the expression of the early activation marker, CD69, was significantly lower in CD73+CD4+cells in TLR9-/-NOD compared to WT NOD mice (Fig. 2B). However, there was no difference in the expression of CD69 in CD73-negative CD4+ T cells (Fig. 2B) or CD8+ cells (data not shown). These results suggest that up-regulation of CD73 expression in TLR9-/-NOD mice leads to less CD4+ T cell activation and fewer memory CD4+ T cells.
Figure 2.
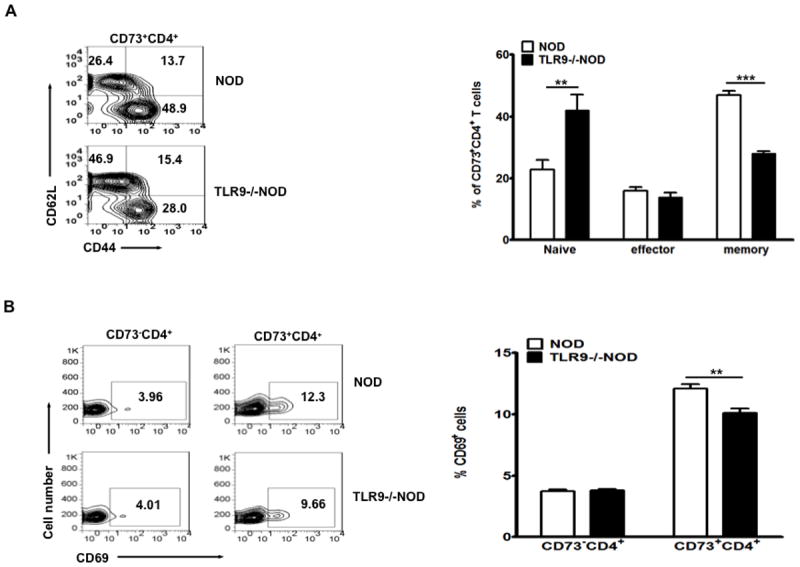
Reduced numbers of activated and memory T cells in TLR9-/-NOD mice.
(A) Splenocytes from TLR9-/-NOD and WT NOD mice were stained with anti-CD4, anti-CD73, anti-CD44 and anti-CD62L antibodies. A representative set of FACS plots is illustrated on the left and the summary of naïve (CD44lowCD62Lhigh), effector (CD44highCD62Lhigh) and memory (CD44highCD62Llow) subsets (gated on CD73+CD4+ cells) from TLR9-/-NOD and WT NOD is shown on the right (n=8, 7-8 week-old, sex-matched, males and females). (B) A representative set of FACS plots showing expression of the early activation marker CD69 on gated CD73-CD4+ and CD73+CD4+ splenic T cells is shown on the left; the summary of CD69 expression on gated CD73-CD4+ and CD73+CD4+ splenic T cells is illustrated on the right (n=12, 7-8 week-old, sex-matched, males and females). Each bar represents mean ± SEM. Student’s t test was used for statistical analysis, ** p<0.01; ***, p<0.001.
We then tested the effect of enhanced CD73 expression on inflammatory cytokine production in CD4+ T cells after TCR (anti-CD3) stimulation. We found that CD73+CD4+ T cells from TLR9-/-NOD mice produced lower levels of the pro-inflammatory cytokines IFN-γ and TNF-α, compared to WT NOD mice (Fig. 3A and 3B). IFN-γ and TNF-α levels in CD73-CD4+ T cells followed the same pattern, namely, fewer CD73-CD4+ T cells from TLR9-/- NOD mice produced pro-inflammatory cytokines compared with WT NOD mice after T cell stimulation (Fig. 3C).
Figure 3.
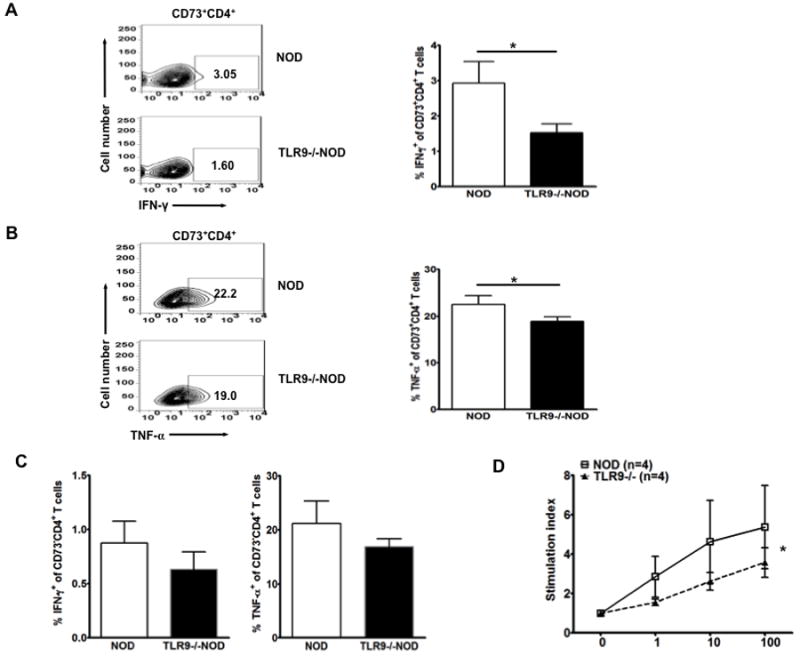
TLR9-deficient NOD mice express reduced anti-inflammatory cytokines in splenic CD73+CD4+ T cells and immune responses to KLH immunization are reduced.
Intracellular cytokines were measured using splenocytes from 7-8 week-old TLR9-/-NOD or WT NOD mice (sex-matched males and females). The cells were co-stained with anti-CD4 and anti-CD73 antibodies. (A) Representative FACS plots for the production of IFN-γ are shown on the left (gated on CD73+CD4+ cells) and the mean percentages of IFN-γ producing CD73+CD4+ cells are presented in a bar chart on the right (n=6). (B) Representative FACS plots of TNF-α producing CD73+CD4+ cells are shown on the left and the mean percentages of TNF-α producing CD73+CD4+ cells are presented in a bar chart on the right (n=5-6). (C) Inflammatory cytokine expression in CD73-CD4+ T cells. Splenocytes from NOD and TLR9-/-NOD mice were stimulated with anti-CD3 (1:100) and anti-CD28 (1:300) overnight and then further stimulated with PMA/ionomycin in the presence of Golgi Plug for an additional 5 hrs followed by surface (CD4 and CD73) and intracellular cytokine IFN-γ (left) and TNF-α (right) staining. Student t-test (two tailed) was used for statistical analysis. *, p<0.05. (D) NOD mice deficient or sufficient in TLR9 were immunized with KLH as described in Materials and Methods. Lymphocytes from draining popliteal lymph nodes cells or spleen were isolated 7 days after immunization and KLH-specific recall immune responses were examined by 3H-thymidine assays in triplicate. The data are presented as stimulation index (SI), which is the mean cpm in the presence of antigen/the mean cpm in the absence of antigen. Background cpm were ~3,000 to 7,000 cpm. 3-4 mice/group (7-8 week-old, sex-matched males) were used in each experiment and the experiment was performed twice. KLH-specific proliferation from splenocytes in one experiment is shown. Student’s t test was used for statistical analysis, * p<0.05.
Furthermore, to investigate whether the immune response to foreign antigen was affected by the enhanced expression of CD73, we immunized NOD and TLR9-/-NOD mice with Keyhole Limpet Hemocyanin (KLH) emulsified with Alum. Lymphocytes from draining popliteal LN or spleens of immunized mice were tested for in vitro recall responses. As shown in Figure 3D, the responses to KLH in TLR9-/-NOD mice were significantly reduced compared to WT NOD mice. This result suggests that elevated CD73 expression in immune cells in TLR9-/- NOD mice possibly promoted general immune tolerance.
CD73+CD4+ cells exhibit increased immunosuppressive function
To test whether expression of CD73 was associated with altered numbers or function of Tregs, we examined the number of Foxp3+ Tregs (CD4+CD25+Foxp3+ T cells) and their CD73 expression. Although the frequency of CD4+ Tregs was similar in splenocytes of TLR9-/-NOD mice compared to NOD mice (Fig. 4A, far right), the percentage of CD73 expression in Foxp3+CD4+ T cells was significantly higher in TLR9-/-NOD mice compared with WT NOD mice (Fig. 4A, left and middle). We also found the same results in pancreatic draining lymph nodes (PLN; Supplementary figure 1). To test the suppressive function of CD73+CD4+ cells, we examined the response of diabetogenic BDC2.5 CD4+ T cells to BDC2.5 mimotope in the presence or absence of FACS purified CD73+CD4+ and CD73-CD4+ T cells from splenocytes of TLR9-/-NOD or WT NOD mice. CD73+CD4+ cells from both TLR9-/-NOD and WT NOD mice inhibited the proliferation of effector BDC 2.5 CD4+ T cells whereas CD73-CD4+ T cells were not inhibitory (Figure 4B). To investigate the role of CD73 in immune suppression mediated by Foxp3+ Tregs, we tested the suppressive function of CD73+ Tregs (CD73+CD4+CD25+Foxp3+) and CD73- Tregs (CD73- CD4+CD25+Foxp3+) in proliferation of BDC2.5 CD4 T cells as described above. Not surprisingly, CD73+ Tregs showed a stronger inhibition of BDC2.5 T cell proliferation (Supplementary figure 2). In addition to the inhibition of BDC2.5 T cell proliferation, we also tested the Th1 cytokines (IFN-γ and TNF-α) in the supernatants of the inhibition assay. As shown in Figure 4C, higher levels of IFN-γ were detected in the supernatants where BDC CD4+ T cells were co-cultured with CD73-CD4+ T cells compared with those co-cultured with CD73+CD4+ T cells. CD73-CD4+ T cells from NOD mice appeared to produce the highest amount of IFN-γ. The secretion of TNF-α was low under all the culture conditions (data not shown). To test which anti-inflammatory cytokines (TGF-β or IL-10) were involved in the suppression, we examined the response of diabetogenic BDC2.5 CD4+ T cells to BDC2.5 mimotope in the presence of FACS-sorted splenic CD73+CD4+ T cells from TLR9-/-NOD mice with or without TGF-β or IL-10 neutralizing antibody. As shown in Fig. 4D, the suppression of CD73+CD4+ T cells of the proliferation of effector BDC 2.5 CD4+ T cells was diminished by neutralizing TGF-β while no effect was seen by neutralizing IL-10. These data indicate that immunosuppressive function of CD73+CD4+ T cells is TGF-β dependent and IL-10 independent.
Figure 4.
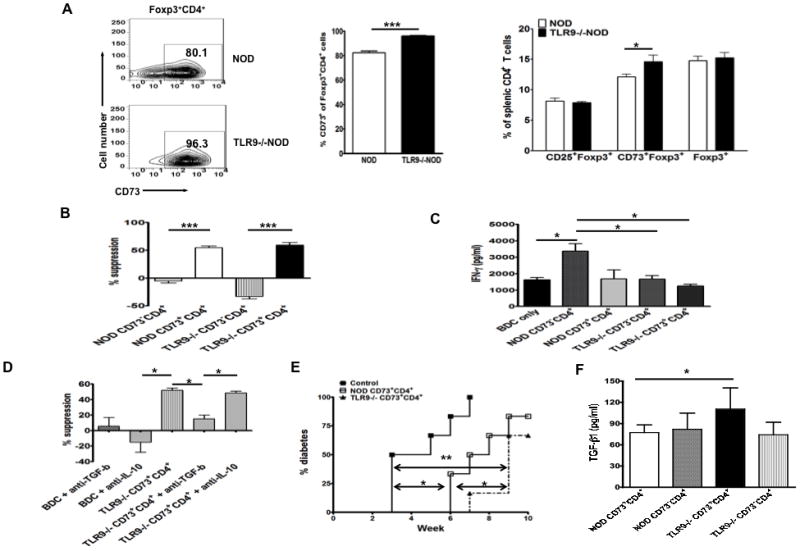
Phenotype and function of Treg cells in the absence of TLR9.
(A) Up-regulation of CD73 expression in Foxp3+ Treg cells in the absence of TLR9. CD73 expression in gated splenic Foxp3+CD4+ Treg cells (left). The mean percentage of CD73+ cells gated on CD4+Foxp3+ cells is demonstrated in the middle and the mean percentage of CD73+Foxp3+, CD25+Foxp3+ or Foxp3+ cells in the gated splenic CD4+ T cell population is presented on the right (6 mice/group, 7-8 week-old, sex-matched males and females). Similar results were found in peripheral lymph nodes (data not shown). (B) Suppression assay. The function of CD73+CD4+ cells was tested in a suppression assay, in which FACS sorted splenic CD73+CD4+ and CD73-CD4+ T cells from 7-8 week-old NOD or TLR9-/-NOD mice were co-cultured (105 cells/well) with purified BDC2.5 CD4+ T effector cells (105 cells/well) and antigen presenting cells (irradiated NOD splenocytes, 3000 rads; 105 cells/well) in the presence or absence of BDC mimotope (10ng/ml). Antigen specific response of BDC2.5 CD4+ T cells was measured by 3H-thymidine incorporation in the last 16-18 hours of the 4-day culture. Results are presented as a percentage of suppression of cell proliferation (stimulation index) in the presence of CD73+CD4+ or CD73-CD4+ T cells compared to BDC2.5 CD4+ T cells alone. The data from one of the four separate experiments are presented. ***, p<0.001. (C) IFNγ production in the culture supernatants of experiments (B) was measured by Luminex. Student’s t-test (two tailed) was used for statistical analysis. *, p<0.05. (D) Suppression of CD73+CD4+ T cells depends on TGF-β. FACS sorted splenic CD73+CD4+ T cells from 7-8 week-old TLR9-/-NOD mice were co-cultured (105 cells/well) with purified BDC2.5 CD4+ T effector cells (105 cells/well) and antigen presenting cells (irradiated NOD splenocytes, 3000 rads; 105 cells/well) in the presence or absence of neutralizing antibody (anti-TGFβ or anti-IL-10, 10 μg/ml) and BDC mimotope (10 ng/ml). Antigen-specific response of BDC2.5 CD4+ T cells was measured by 3H-thymidine incorporation in the last 16-18 hours of the 4-day culture. Results are presented as a percentage of suppression of cell proliferation in the presence or absence of CD73+CD4+ T cells with or without neutralizing antibody compared to BDC2.5 CD4+ T cells alone. *, p<0.05. (E) Delayed diabetes development in adoptive transfer experiments. Splenocytes (8×106 cells/mouse) from new-onset diabetic NOD mice were intravenously injected into irradiated (650 rads) young female NOD mice (6~7 wk) with or without sorted CD73+CD4+ cells (1.7×106 cells/mouse) from female NOD or TLR9-/-NOD mice of similar age. Diabetes was monitored by testing for glycosuria twice a week and diabetes was confirmed by blood glucose measurement (>250mg/dl), 6 mice/group. Log-rank test for survival curve was used for statistical analysis. (F) TGFβ production. Sorted CD73 positive or negative CD4 T cells (106/ml) from spleen of WT or TLR9-/- NOD mice were stimulated with anti-CD3 (2C11, 1:100) and anti-CD28 (37.51, 1:100) for 3 days. Secreted TGFβ in the culture supernatants was measured by ELISA (R&D Systems).
To further test the immunosuppressive effect of CD73+CD4+ in vivo on diabetes development, we adoptively transferred splenocytes from diabetic NOD mice with or without FACS purified CD73+CD4+ cells from either TLR9-/-NOD or WT NOD mice into irradiated young (~6 week-old) female NOD mice (n=6/group). Both CD73+CD4+ cells from TLR9-/-NOD and WT NOD donors significantly delayed diabetes development although CD73+CD4+ cells from TLR9-/-NOD mice induced further delay in diabetes onset (Fig 4E). In line with the delayed diabetes onset, CD73+CD4+ cells from TLR9-/-NOD mice also secreted higher amounts of TGF-β after anti-CD3 and anti-CD28 stimulation (p=0.023, Fig 4F). Furthermore, CD73+CD4+ cells from TLR9-/-NOD mice expressed higher levels of TGF-β mRNA and significantly lower mRNA expression levels of several transcription factors regulating inflammatory cytokines including the interferon regulatory factors, IRF1, IRF5, IRF7 and IRF8, compared to the same subset of T cells from WT NOD mice (Fig. 5A). These data suggest that CD73+CD4+ cells from TLR9-/-NOD mice exert stronger immunosuppressive function that is likely to be mediated by the higher levels of the anti-inflammatory cytokine TGF-β.
Figure 5.
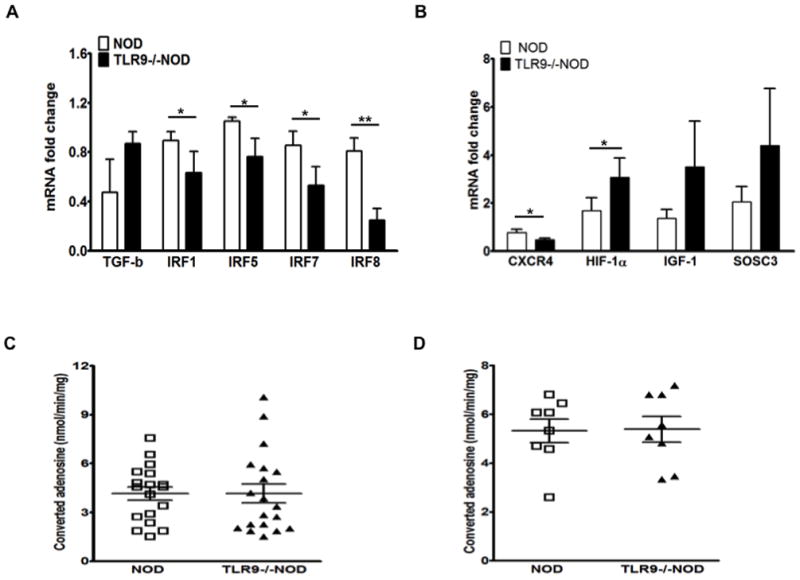
Gene expression of CD4 T cells in the presence or absence of TLR9.
(A) The levels of TGF-β, IRF1, IRF5, IRF7 and IRF8 mRNA expression were determined by qPCR. RNAs was prepared from sorted CD73+CD4+ or CD73- CD4+ cells from 7-8 week-old mice followed by reverse transcription. qPCR was performed to determine the mRNA levels of TGF-β, IRF1, IRF5, IRF7 and IRF8 which were normalized to the housekeeping gene GAPDH. The experiment was done 3 times and the mean ± SEM is presented. Student’s t test was used for statistical analysis, * p<0.05; ** p<0.01. (B) The levels of CXCR4, HIF-1α, IGF-1 and SOCS3 mRNA expression were determined by qPCR in purified splenic CD4+ T cells from 7-8 week-old NOD and TLR9-/-NOD mice (n=9/group, sex-matched males and females). GAPDH was used as the internal control for normalization. (C) Levels of calculated adenosine in serum and spleen cell lysates from 7-8 week-old mice (sex-matched, males and females). Adenosine deaminase (ADA) activity was measured and the concentration of adenosine was calculated as adenosine has a very short half-life. Comparison of serum adenosine levels between NOD and TLR9-/-NOD mice is shown on the left and comparison of adenosine levels in splenocyte lysates is shown on the right.
Mechanism by which CD73 is increased by TLR9 deficiency and effect on adenosine deaminase activity
Induction of CD73 is associated with several factors including hypoxia inducible factor 1α (HIF-1α) and insulin-like growth factor 1 (IGF-1) (37). IGF-1 can enhance HIF-1α expression, which in turn inhibits TLR9 and CXCR4 expression but elevates SOCS3 expression (38). To explore the mechanism by which CD73 expression is modulated, in particular potential HIF-1α-TLR9-CD73 cross talk, we performed qPCR to detect the gene expression in purified CD4+ T cells from NOD and TLR9-/-NOD mice. As shown in Fig. 5B, CD4+ cells from TLR9-/- mice had increased IGF-1 transcript, enhanced HIF-1α and SOCS3 expression, but reduced CXCR4 expression.
As CD73 (ecto-5′-nucleotidase) catalyzes the terminal step in extracellular adenosine formation from AMP, we examined adenosine deaminase (ADA) activity in both sera and cell lysates of total splenocytes from NOD and TLR9-/NOD mice. The concentration of adenosine was calculated based on ADA activity. Despite the elevated CD73 expression in TLR9-/- NOD mice, we did not find any difference in adenosine levels in serum and lymphocyte lysates between WT and TLR9-/- NOD mice (Fig. 5C and 5D).
Up regulation of CD73 and reduction of diabetes development in TLR9-deficient NY8.3NOD mice
As CD8+ T cells also up-regulated CD73 expression at both mRNA and protein levels in the absence of TLR9 in NOD mice (Fig. 1), we studied CD73 expression and diabetes development in NY8.3 CD8 TCR transgenic NOD mice that develop highly accelerated diabetes. We generated TLR9-/-NY8.3 NOD mice by breeding NY8.3 NOD with TLR9-/-NOD mice and investigated the natural history of diabetes development in TLR9-/-NY8.3 NOD mice. There was no difference in the incidence of diabetes development between female TLR9-/-NY8.3 and NY8.3 mice; however, the onset of diabetes was significantly delayed in male TLR9-/-NY8.3 mice compared to wild type male NY8.3 mice (Fig. 6A). To investigate whether TLR9 deficiency in NY8.3 NOD mice affects CD73 expression, we examined CD73 expression in lymphocytes. Similar to the original TLR9-/-NOD mice, the frequency of CD73 expression was also significantly increased in NY8.3 CD8+ T cells and, to a lesser extent, increased in other immune cell subsets (B cells and CD4+ T cells) in male TLR9-/-NY8.3 mice compared to wild-type male NY8.3 mice (Fig. 6B). However, there was no significant difference in CD73 expression in all these immune cell subsets in female mice of these two strains (Fig. 6B). This gender difference in CD73 expression in the TLR9-/- NY8.3 mice was not observed in TLR9-/- NOD mice where diabetes development in both female and male TLR9-/- NOD mice was significantly reduced. In addition, male TLR9-/-NY8.3 NOD T cells produced significantly less pro-inflammatory IFN-γ and TNF-α compared to male NY8.3 NOD mice. This contrasted with moderate reduction in IFN-γ production by female CD8+ TLR9-/-NY8.3 NOD T cells where there was little difference in the secretion of pro-inflammatory cytokines from CD4+ and CD8+ T cells in female mice of the two strains (Fig. 6C and 6D).
Figure 6.
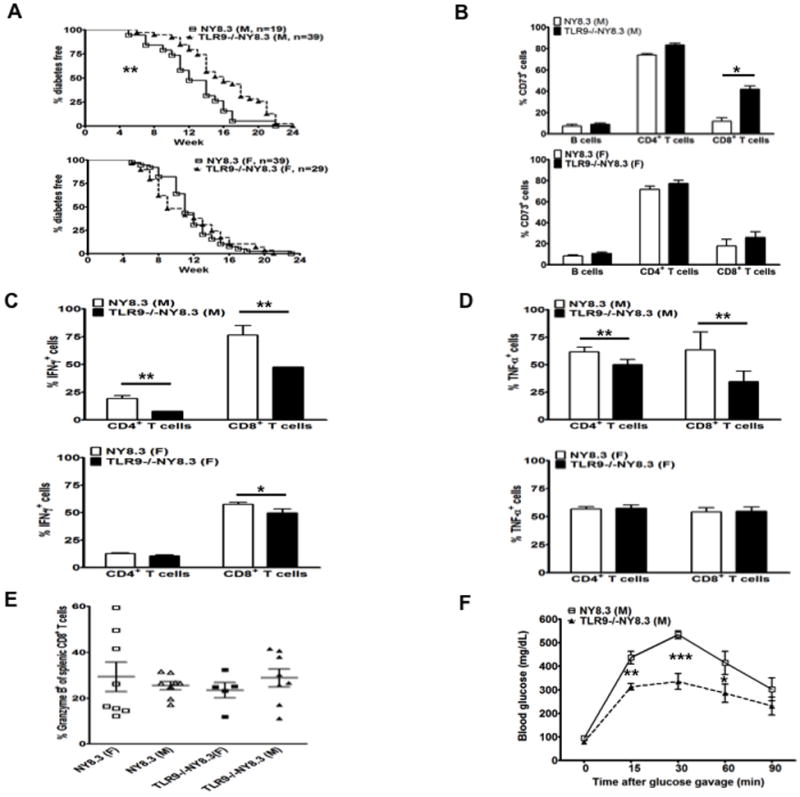
Male NY8.3 mice had delayed diabetes development and elevated CD73 expression in the absence of TLR9.
(A) TLR9-/-NY8.3 mice were generated by breeding TLR9-/-NOD mice with NY8.3 NOD mice and the natural history of diabetes development was observed. The incidence of diabetes in TLR9-/-NY8.3 mice was compared to NY8.3 mice (males shown above, females shown below, **, p<0.01, survival curve). (B) CD73 expression was examined in TLR9-/-NY8.3 and wild type NY8.3 mice (males shown above, females shown below). Splenocytes from 3-month-old non-diabetic TLR9-/-NY8.3 and NY8.3 mice were stained with anti-CD73, anti-CD4, anti-CD8 and anti-B220 antibodies and analyzed by flow cytometry (n=4-5/group, *, p<0.05, Student t test). (C) IFN-γ production of CD4+ and CD8+ T cells. Splenocytes from WT NY8.3 or TLR9-/-NY8.3 NOD mice were used for ICC staining as described in Materials and Methods. The cells were also co-stained with anti-CD4 and anti-CD8. At least 4 mice were studied in each group. The results from male mice are shown above and females below. * p<0.05 and ** p<0.01, Student t test. (D) TNF-α production of CD4+ and CD8+ T cells was studied in the same way as for IFN-γ. The results from male mice are shown above and females below. At least 4 mice were studied in each group. **, p<0.01, Student t test. (E) Granzyme B expression in CD8+ T cells from NY8.3 and TLR9-/-NY8.3 NOD mice. Splenocytes from 2-3 month-old NY8.3NOD or TLR9-/-NY8.3 mice were stimulated with anti-CD3 (1:100 hybridoma supernatant) and anti-CD28 (1:300 hybridoma supernatant) overnight and followed by further stimulation with PMA/ionomycin in the presence of Golgi plug for an additional 4 hrs prior to surface (CD8 and TCRβ) and intracellular Granzyme B staining. The cells were analyzed by flow cytometry and the figure shows the percentage of Granzyme B expression in gated CD8+ T cells (TCRβ+). (F) Oral glucose tolerance test (OGTT). OGTT was performed in 3-month-old male non-diabetic NY8.3 and TLR9-/-NY8.3 mice after fasting overnight with free access to water. Each value represents mean ± SEM of at least 4 mice. Student’s t test was used for statistical analysis. *p<0.05; **p<0.01; ***p<0.001.
We next tested whether TLR9 deficiency affected NY8.3 CTL effector function, which may have contributed to the diabetes protected phenotype. Granzyme B expression was not different in CD8+ T cells between male and female TLR9-/-NY8.3 mice, nor between WT and TLR9-/- NY8.3 mice (Fig. 6E). Perforin expression was very low in male and female mice of both strains (data not shown). Our results suggest that diabetes protection in the absence of TLR9 is more likely to be associated with up-regulation of CD73 and reduction of inflammatory cytokines.
Improved islet beta cell function in TLR9-/-NY8.3 mice
To test islet β cell function, we performed OGTTs in non-diabetic male TLR9-/-NY8.3 and male NY8.3 mice. As expected, the TLR9-/-NY8.3 mice showed enhanced glucose tolerance compared to NY8.3 mice (Fig. 6F). Similar results were obtained with intraperitoneal glucose tolerance tests (IPGTT; data not shown). In contrast, there was no difference in the glucose tolerance test results between female NY8.3 mice and female TLR9-/-NY8.3 mice by OGTT and IPGTT (data not shown).
TLR9 blockade enhanced β-cell function in NOD mice
Our results presented above provide evidence that NOD or NY8.3 NOD mice had delayed and reduced diabetes development and expressed enhanced beta cell function in the absence of TLR9 through genetic targeting. To test whether blockade of TLR9 signaling using small molecules could also enhance beta cell function as described above, we treated NOD mice with chloroquine, a TLR9 inhibitor. We then tested β-cell function by OGTT. As shown in Figure 7A, inhibition of TLR9 by chloroquine significantly improved glucose tolerance. Interestingly, chloroquine treatment also induced CD73 expression on both CD4+ and CD8+ T cells although not to the same level as seen in TLR9 deficiency (Fig. 7B). Moreover, chloroquine treatment significantly suppressed insulitis development (Fig. 7C and 7D). Our results support a recent report that chloroquine can inhibit diabetes development (16).
Figure 7.
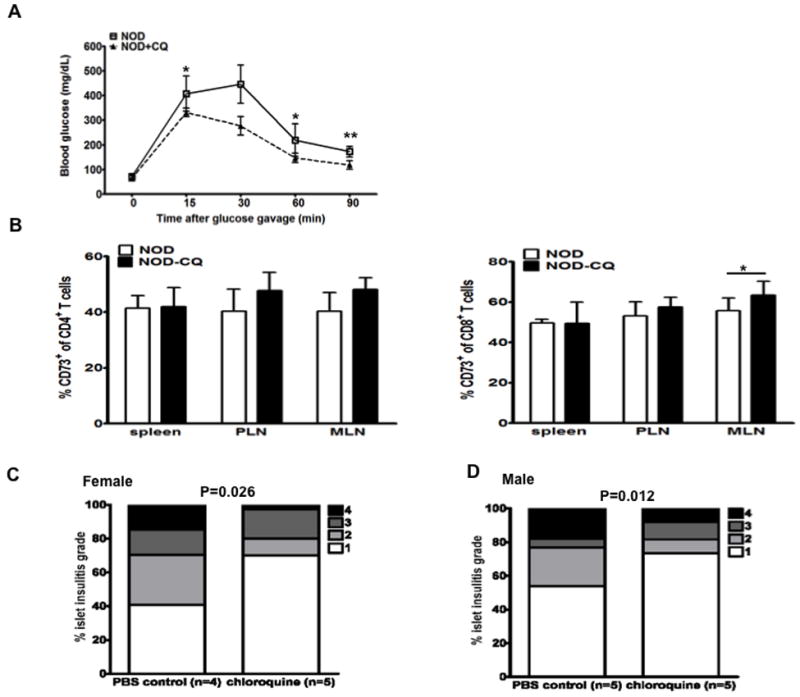
Blockade of TLR9 by chloroquine enhanced β-cell function and induced CD73 expression in T cells.
(A) Oral glucose tolerance test. Young NOD mice (4-week-old female, n=4) were treated with chloroquine (20 μg/g body weight) (CQ) for 4 weeks as described in Materials and Methods. OGTTs were performed to determine the effect of chloroquine on the β-cell function. *, p<0.05, **, p<0.01; Student’s t test for each time point. (B) Induced CD73 expression in CD4+ and CD8+ T cells in spleen, PLN and MLN from NOD mice (A) treated with PBS or CQ. Each value represents mean ± SEM and Student’s t test was used for statistical analysis. *p<0.05. (C, male; D, female) Severity of insulitis was ameliorated by chloroquine treatment. 1-month-old NOD mice were treated with chloroquine (20 μg/ml) or PBS for three months. Pancreata were fixed in formalin immediately ex vivo and embedded in paraffin. 8μm sections were cut and stained with H+E. Insulitis was examined under light microscopy (> 120 islets were examined for each group) and scored. 1: less than 25% infiltration; 2: 25-50% infiltration; 3: 50-75% infiltration; 4: greater than 75% infiltration. One-way ANOVA was used for statistical analysis.
Detection of CD73 expression in islet cells
To test whether islet β cells express CD73, we stained the cryo-sections of pancreas from 7-8 week-old WT and TLR9-/- NOD mice with anti-CD73 and anti-insulin, respectively. Whilst CD73 was expressed within islets, although not in insulin producing β cells (Fig 8A), the expression was not significantly different when WT or TLR9-/- NOD mice were compared (Fig 8A). To investigate which cells express CD73, we extracted infiltrating immune cells from islets of 5-month-old male mice and stained with mAbs to TCRβ, CD4, CD8, B220, CD19 and pDCA-1 (a marker for plasmacytoid dendritic cells, pDC). Interestingly, all the immune cells examined expressed CD73 and it is intriguing that over 80% of CD4+ T cells from islet infiltrates expressed CD73, more than the equivalent cells in the spleens and PLNs (Fig 8B and 8C).
Figure 8.
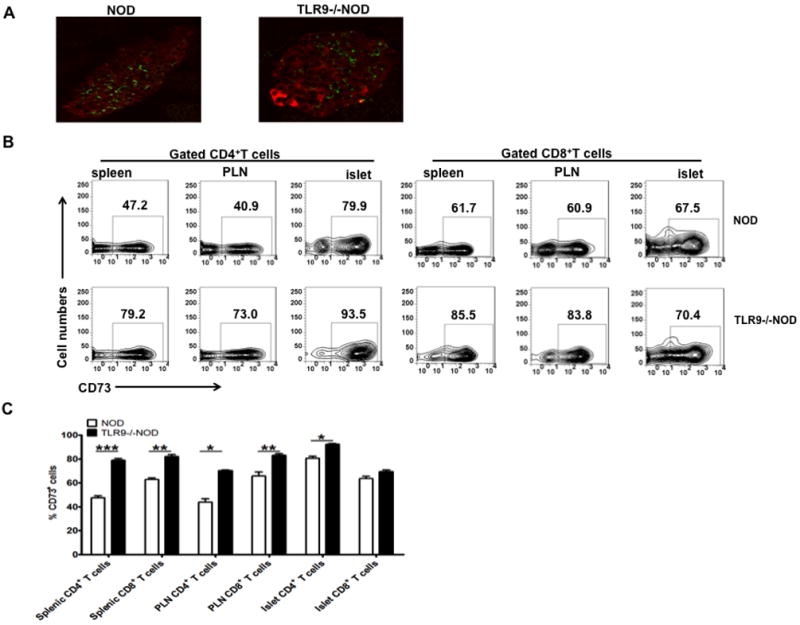
CD73 expression in pancreatic islets.
(A) Frozen sections of pancreas from NOD and TLR9-/-NOD mice (n=3/group, 6-7 week-old, female) were stained with CD73 (green) and insulin (red) as described in Materials and Methods. The sections were examined and photographed using Olympus fluorescent microscope BX50 after staining. (B, C) CD73 expression in CD4+ and CD8+ T cells. Splenocytes, PLNs and isolated infiltrating immune cells from islets of 5-month-old male NOD mice were stained and examined by flow cytometry. (B) One representative FACS plot is shown. (C) Summary of the percentage of CD73 expressing cells in CD4+ and CD8+ T cells (n=5, *, p<0.05; **, p<0.01; ***, p<0.001).
Discussion
Our previous report showed that TLR9-/-NOD mice were significantly protected from development of autoimmune diabetes (15). In this study, we have demonstrated a novel mechanism of the protection that is mediated by CD73. In the absence of TLR9, a significant number of immune cells, especially CD4+ and CD8+ T cells, converted from CD73- to CD73+ in NOD mice. The conversion appeared to be unique to TLR9-/-NOD mice, as its expression in immune cells remained unchanged or was lower in other TLR, MyD88 and TRIF mutant NOD mice compared to WT NOD mice. Furthermore, the CD73 expression in an increased proportion of CD4+ and CD8+ T cells was limited to the NOD background, since CD73 expression was basically unchanged in diabetes-resistant TLR9-/-B6 mice compared to WT B6 mice. This increased expression of CD73 was associated with increased production TGF-β, together with decreased production of TNF-α and IFN-γ. Furthermore, CD73+CD4+T cells from the TLR9-/-NOD mice had an increased capacity to suppress the proliferation and inflammatory cytokine production of CD4+ antigen-specific BDC2.5 T cells in response to peptide. It was particularly interesting to note that when TLR9-/-NOD mice were crossed with the CD8+ TCR transgenic mouse NY8.3, a highly accelerated diabetes model, CD73 expression was significantly up-regulated in CD8+ T cells of male TLR9-/-NY8.3 mice, which was coincident with delay in autoimmune diabetes development in male TLR9-/-NY8.3NOD mice. Upregulation of CD73 was not observed in the female TLR9-/-NY8.3 mice that were not protected from diabetes. It is not clear at this stage what the reason is for this gender difference, a phenomenon not seen in TLR9-/-NOD mice.
CD73 is an ecto-5’-nucleotidase which converts 5’-adenosine monophosphate to adenosine that has immunoregulatory properties. The expression of CD73, together with CD39, which breaks down ADP to AMP, generates adenosine. Adenosine produced by these enzymes is one means by which T regulatory and other CD4+ T cells expressing CD73 can suppress effector T cells (28, 29, 39, 40). Thus, the production of adenosine might have been one explanation why CD73+CD4+ T cells, especially from TLR9-/-NOD mice, were immunosuppressive to diabetogenic T cells, reducing proliferation in vitro and preventing diabetes induction in vivo. However, to our surprise, TLR9 deficiency did not alter the level of adenosine production despite the fact that TLR9 deficiency specifically up-regulates CD73 expression. It is likely that other mechanisms including suppression of pro-inflammatory cytokine production by diabetogenic T cells may also be operative.
It was not clear at this point why CD73 was up-regulated in the absence of TLR9. We showed CD4+ cells from TLR9 deficient mice had increased IGF-1 transcripts, enhanced HIF-1α and SOCS3 expression, but reduced CXCR4 expression. This may suggest that TLR9 deficiency results in the induction of IGF-1, which induces HIF-1 expression and enhances CD73 expression while inhibiting CXCR4 expression and inducing SOCS3.
Our study suggests that TLR9 acts as a negative regulator for CD73 expression and immune regulatory function. Hall et al have recently reported that TLR9-/-B6 mice had increased CD4+Foxp3+ Treg cells, related to alterations in the balance of effector and regulatory cells induced by gut flora DNA (41). In our diabetes model systems, we did not observe an increase in Foxp3+ Treg cells, although we found a significant increase in CD73 expression on Foxp3+ Treg cells. It is interesting that CD39, the partner of CD73, remained unchanged in the presence or absence of TLR9 both in the NOD and B6 genetic backgrounds (data not shown). Further investigation is required to address how TLR9 signaling affects the expression of CD73 and its immunological and biological functions. Elucidation of these questions will bring new understanding of modulation of TLR9 signaling in innate immunity of T1D and possibly lead to novel immuno-therapies.
TLR9 appears to regulate islet beta cell function directly or indirectly in NOD mice. In the absence of TLR9-/-, NY8.3 NOD male mice showed significantly enhanced glucose tolerance compared to their WT counterparts, which suggests that β cell function was improved in these mice. Importantly, the incidence of diabetes in male TLR9-/-NY8.3 NOD mice was also significantly delayed. It is not clear at this point why TLR9 deficiency has this protective effect on male NY8.3NOD mice. However, blockade of TLR9 with chloroquine led to improved β cell function and significant reduction in insulitis in both male and female NOD mice. Our data showed that insulin-producing β cells do not express CD73; however, it is interesting that there are more CD73 expressing immune cells, in particular CD73+CD4 T cells, in pancreatic islets than in peripheral lymphoid organs and this was not obviously affected by the presence or absence of TLR9. The lack of difference in this CD73 expression could be related to the fact that CD73 expression is high on the immune cells in the islets of WT NOD mice. Regardless, our data suggest a very active immune-suppressive response in the target tissue. It is conceivable that CD73+ cells interact with islet beta cells directly or indirectly through production of anti-inflammatory cytokines, which result in the improvement of β cell function and protection of the mice from diabetes development. It is also possible that the increased expression of CD73 in immune cells does not contribute to the improved β cell function in TLR9-/- mice and that this is a separate, unlinked finding but contributes to the diabetes protected phenotype in the absence of TLR9.
There is a growing interest in targeting TLRs for the prevention and treatment of cancer, inflammation and autoimmune diseases. Several compounds are undergoing preclinical and clinical evaluation including TLR7 and TLR9 activators in boosting immune responses against infection and cancer, and inhibitors of TLR2, TLR4, TLR7 and TLR9 in the treatment of sepsis and inflammatory diseases (42-44). A recent study showed that systemic administration of TLR agonists could suppress both allergic and islet autoimmune responses and modify experimental asthma and spontaneous autoimmune diabetes in NOD mice (45). Our current study demonstrated a novel role of TLR9 in regulating CD73 expression, which mediates T1D suppression. In addition, our study and the study by Zhang et al (16) demonstrated that blockade of TLR9 with chloroquine may provide a novel therapeutic means to prevent T1D.
Conclusion
In summary, we report that TLR9 acts as a negative regulator of CD73 in NOD and NY8.3NOD mice and that by reducing TLR9, either in TLR9-/- mice or by treatment with a TLR9 inhibitor, CD73 was markedly up-regulated and the immunosuppressive function of CD73+ T cells was also significantly enhanced. Our findings suggest that CD73 plays an important role in diabetes protection and this study indicates that CD73, linked to TLR9, may be a novel target for therapeutic intervention for prevention of T1D in humans.
Supplementary Material
Acknowledgments
We are very grateful to Xiaojun Zhang (Yale University) for taking excellent care of the animals used in this study, to Jian Peng (Yale University) for genotyping of all the animals used in this study, to Monika Majewska-Szczepanik (Yale University) for helping with cytokine measurement.
This study was supported by NIH (RC1 DK087699 and RO1 DK088181 to LW) and JDRF (5-2010-664 and 4-2007-1059 to LW).
Abbreviations
- T1D
type 1 diabetes
- PLN
pancreatic lymph nodes
- WT
wild type
- AMP
adenosine monophosphate
- Treg
T regulatory
- ICC
intracellular cytokine
- OGTT
oral glucose tolerance test
- qPCR
quantitative real-time PCR
- ADA
adenosine deaminase
- IPGTT
intraperitoneal glucose test
Footnotes
Author Contributions
N.T. designed some of the experiments, performed research, analyzed data and wrote the manuscript; F.S.W. contributed to discussion, wrote the manuscript and reviewed/edited the manuscript; L.W. designed the study, analyzed some data, wrote the manuscript and reviewed/edited the manuscript.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Wong FS, Janeway CA., Jr The role of CD4 and CD8 T cells in type I diabetes in the NOD mouse. Res Immunol. 1997;148:327–332. doi: 10.1016/s0923-2494(97)87242-2. [DOI] [PubMed] [Google Scholar]
- 2.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 3.D’Alise AM, Auyeung V, Feuerer M, Nishio J, Fontenot J, Benoist C, Mathis D. The defect in T-cell regulation in NOD mice is an effect on the T-cell effectors. Proc Natl Acad Sci U S A. 2008;105:19857–19862. doi: 10.1073/pnas.0810713105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papadimitraki ED, Bertsias GK, Boumpas DT. Toll like receptors and autoimmunity: a critical appraisal. J Autoimmun. 2007;29:310–318. doi: 10.1016/j.jaut.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Allam R, Anders HJ. The role of innate immunity in autoimmune tissue injury. Curr Opin Rheumatol. 2008;20:538–544. doi: 10.1097/BOR.0b013e3283025ed4. [DOI] [PubMed] [Google Scholar]
- 6.Lampropoulou V, Hoehlig K, Roch T, Neves P, Calderon Gomez E, Sweenie CH, Hao Y, Freitas AA, Steinhoff U, Anderton SM, Fillatreau S. TLR-activated B cells suppress T cell-mediated autoimmunity. J Immunol. 2008;180:4763–4773. doi: 10.4049/jimmunol.180.7.4763. [DOI] [PubMed] [Google Scholar]
- 7.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, Chervonsky AV. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brentano F, Kyburz D, Gay S. Toll-like receptors and rheumatoid arthritis. Methods Mol Biol. 2009;517:329–343. doi: 10.1007/978-1-59745-541-1_20. [DOI] [PubMed] [Google Scholar]
- 9.Lien E, Zipris D. The role of Toll-like receptor pathways in the mechanism of type 1 diabetes. Curr Mol Med. 2009;9:52–68. doi: 10.2174/156652409787314453. [DOI] [PubMed] [Google Scholar]
- 10.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 11.Takeshita F, Leifer CA, Gursel I, Ishii KJ, Takeshita S, Gursel M, Klinman DM. Cutting edge: Role of Toll-like receptor 9 in CpG DNA-induced activation of human cells. J Immunol. 2001;167:3555–3558. doi: 10.4049/jimmunol.167.7.3555. [DOI] [PubMed] [Google Scholar]
- 12.Kaisho T, Akira S. Toll-like receptors and their signaling mechanism in innate immunity. Acta Odontol Scand. 2001;59:124–130. doi: 10.1080/000163501750266701. [DOI] [PubMed] [Google Scholar]
- 13.Kaisho T, Akira S. Dendritic-cell function in Toll-like receptor- and MyD88-knockout mice. Trends Immunol. 2001;22:78–83. doi: 10.1016/s1471-4906(00)01811-1. [DOI] [PubMed] [Google Scholar]
- 14.Chuang TH, Lee J, Kline L, Mathison JC, Ulevitch RJ. Toll-like receptor 9 mediates CpG-DNA signaling. J Leukoc Biol. 2002;71:538–544. [PubMed] [Google Scholar]
- 15.Wong FS, Hu C, Zhang L, Du W, Alexopoulou L, Flavell RA, Wen L. The role of Toll-like receptors 3 and 9 in the development of autoimmune diabetes in NOD mice. Ann N Y Acad Sci. 2008;1150:146–148. doi: 10.1196/annals.1447.039. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Lee AS, Shameli A, Geng X, Finegood D, Santamaria P, Dutz JP. TLR9 blockade inhibits activation of diabetogenic CD8+ T cells and delays autoimmune diabetes. J Immunol. 2010;184:5645–5653. doi: 10.4049/jimmunol.0901814. [DOI] [PubMed] [Google Scholar]
- 17.Wong FS, Wen L. IFN-alpha can both protect against and promote the development of type 1 diabetes. Ann N Y Acad Sci. 2008;1150:187–189. doi: 10.1196/annals.1447.031. [DOI] [PubMed] [Google Scholar]
- 18.Lennon PF, Taylor CT, Stahl GL, Colgan SP. Neutrophil-derived 5’-adenosine monophosphate promotes endothelial barrier function via CD73-mediated conversion to adenosine and endothelial A2B receptor activation. J Exp Med. 1998;188:1433–1443. doi: 10.1084/jem.188.8.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Resta R, Yamashita Y, Thompson LF. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol Rev. 1998;161:95–109. doi: 10.1111/j.1600-065x.1998.tb01574.x. [DOI] [PubMed] [Google Scholar]
- 20.Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim Biophys Acta. 2008;1783:673–694. doi: 10.1016/j.bbamcr.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 21.Linden J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu Rev Pharmacol Toxicol. 2001;41:775–787. doi: 10.1146/annurev.pharmtox.41.1.775. [DOI] [PubMed] [Google Scholar]
- 22.Thiel M, Caldwell CC, Sitkovsky MV. The critical role of adenosine A2A receptors in downregulation of inflammation and immunity in the pathogenesis of infectious diseases. Microbes Infect. 2003;5:515–526. doi: 10.1016/s1286-4579(03)00068-6. [DOI] [PubMed] [Google Scholar]
- 23.Hasegawa T, Bouis D, Liao H, Visovatti SH, Pinsky DJ. Ecto-5’ nucleotidase (CD73)-mediated adenosine generation and signaling in murine cardiac allograft vasculopathy. Circ Res. 2008;103:1410–1421. doi: 10.1161/CIRCRESAHA.108.180059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng Z, Fernandez P, Wilder T, Yee H, Chiriboga L, Chan ES, Cronstein BN. Ecto-5’-nucleotidase (CD73) -mediated extracellular adenosine production plays a critical role in hepatic fibrosis. FASEB J. 2008;22:2263–2272. doi: 10.1096/fj.07-100685. [DOI] [PubMed] [Google Scholar]
- 25.Reutershan J, Vollmer I, Stark S, Wagner R, Ngamsri KC, Eltzschig HK. Adenosine and inflammation: CD39 and CD73 are critical mediators in LPS-induced PMN trafficking into the lungs. FASEB J. 2009;23:473–482. doi: 10.1096/fj.08-119701. [DOI] [PubMed] [Google Scholar]
- 26.Takedachi M, Qu D, Ebisuno Y, Oohara H, Joachims ML, McGee ST, Maeda E, McEver RP, Tanaka T, Miyasaka M, Murakami S, Krahn T, Blackburn MR, Thompson LF. CD73-generated adenosine restricts lymphocyte migration into draining lymph nodes. J Immunol. 2008;180:6288–6296. doi: 10.4049/jimmunol.180.9.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Airas L, Jalkanen S. CD73 mediates adhesion of B cells to follicular dendritic cells. Blood. 1996;88:1755–1764. [PubMed] [Google Scholar]
- 28.Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, Lang S, Jackson EK, Gorelik E, Whiteside TL. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. J Biol Chem. 2010;285:7176–7186. doi: 10.1074/jbc.M109.047423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romio M, Reinbeck B, Bongardt S, Huls S, Burghoff S, Schrader J. Extracellular purine metabolism and signaling of CD73-derived adenosine in murine Treg and Teff cells. Am J Physiol Cell Physiol. 2011;301:C530–539. doi: 10.1152/ajpcell.00385.2010. [DOI] [PubMed] [Google Scholar]
- 30.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 31.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 32.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 33.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 34.Wooten RM, Ma Y, Yoder RA, Brown JP, Weis JH, Zachary JF, Kirschning CJ, Weis JJ. Toll-like receptor 2 is required for innate, but not acquired, host defense to Borrelia burgdorferi. J Immunol. 2002;168:348–355. doi: 10.4049/jimmunol.168.1.348. [DOI] [PubMed] [Google Scholar]
- 35.Verdaguer J, Schmidt D, Amrani A, Anderson B, Averill N, Santamaria P. Spontaneous autoimmune diabetes in monoclonal T cell nonobese diabetic mice. J Exp Med. 1997;186:1663–1676. doi: 10.1084/jem.186.10.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mishra OP, Gupta BL, Ali Z, Nath G, Chandra L. Adenosine deaminase activity in typhoid fever. Indian Pediatr. 1994;31:1379–1384. [PubMed] [Google Scholar]
- 37.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sinha S, Koul N, Dixit D, Sharma V, Sen E. IGF-1 induced HIF-1alpha-TLR9 cross talk regulates inflammatory responses in glioma. Cell Signal. 2011;23:1869–1875. doi: 10.1016/j.cellsig.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 39.Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, Enjyoji K, Linden J, Oukka M, Kuchroo VK, Strom TB, Robson SC. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5’-adenosine monophosphate to adenosine. J Immunol. 2006;177:6780–6786. doi: 10.4049/jimmunol.177.10.6780. [DOI] [PubMed] [Google Scholar]
- 41.Hall JA, Bouladoux N, Sun CM, Wohlfert EA, Blank RB, Zhu Q, Grigg ME, Berzofsky JA, Belkaid Y. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity. 2008;29:637–649. doi: 10.1016/j.immuni.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dunne A, Marshall NA, Mills KH. TLR based therapeutics. Curr Opin Pharmacol. 2011;11:404–411. doi: 10.1016/j.coph.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 43.Gomariz RP, Gutierrez-Canas I, Arranz A, Carrion M, Juarranz Y, Leceta J, Martinez C. Peptides targeting Toll-like receptor signalling pathways for novel immune therapeutics. Curr Pharm Des. 2010;16:1063–1080. doi: 10.2174/138161210790963841. [DOI] [PubMed] [Google Scholar]
- 44.Wiersinga WJ. Current insights in sepsis: from pathogenesis to new treatment targets. Curr Opin Crit Care. 2011;17:480–486. doi: 10.1097/MCC.0b013e32834a4aeb. [DOI] [PubMed] [Google Scholar]
- 45.Aumeunier A, Grela F, Ramadan A, Pham Van L, Bardel E, Gomez Alcala A, Jeannin P, Akira S, Bach JF, Thieblemont N. Systemic Toll-like receptor stimulation suppresses experimental allergic asthma and autoimmune diabetes in NOD mice. PLoS One. 2010;5:e11484. doi: 10.1371/journal.pone.0011484. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.