

Abstract
microRNAs (miRNAs) are non-coding small RNAs (sRNAs) capable of negatively regulating gene expression. Recently, microRNA-like small RNAs (milRNAs) were discovered in several filamentous fungi but not yet in Trichoderma reesei, an industrial filamentous fungus that can secrete abundant hydrolases. To explore the presence of milRNA in T. reesei and evaluate their expression under induction of cellulose, two T. reesei sRNA libraries of cellulose induction (IN) and non-induction (CON) were generated and sequenced using Solexa sequencing technology. A total of 726 and 631 sRNAs were obtained from the IN and CON samples, respectively. Global expression analysis showed an extensively differential expression of sRNAs in T. reesei under the two conditions. Thirteen predicted milRNAs were identified in T. reesei based on the short hairpin structure analysis. The milRNA profiles obtained in deep sequencing were further validated by RT-qPCR assay. Computational analysis predicted a number of potential targets relating to many processes including regulation of enzyme expression. The presence and differential expression of T. reesei milRNAs imply that milRNA might play a role in T. reesei growth and cellulase induction. This work lays foundation for further functional study of fungal milRNAs and their industrial application.
Introduction
microRNAs (miRNAs) are small non-coding RNAs of 18~25 nucleotides that negatively regulate gene expression by binding to the target mRNAs [1]. Mature miRNAs are processed from primary miRNA transcripts (pri-miRNA) by the endonuclease Drosha, producing a precursor hairpin structure of 60~70 nucleotides, termed pre-miRNAs. The pre-miRNAs are exported from nucleus to cytoplasm, where they are further cleaved by an endonuclease, Dicer, to yield mature miRNAs [2,3]. As a component of RNA-induced silencing complex (RISC), mature miRNAs guide the binding of RISC to mRNA targets, forcing mRNA degradation and/or translational inhibition [4].
Since the discovery of the first miRNA lin-4 in Caenorhabditis elegans [5], miRNAs have been identified in diverse organisms including animals, plants, and unicellular eukaryotes such as algae [6], Giardia lamblia [7], and Trichomonas vaginalis [8]. The presence of miRNAs in the unicellular organisms suggested that the miRNA pathway is an ancient mechanism of gene regulation [9]. Filamentous fungi are an important group of multicellular eukaryotes with over one billion years of evolution [10]. A variety of small RNAs (sRNAs) and its mediated RNA interference (RNAi) have been shown in filamentous fungi [11,12]. Recently, several groups reported that miRNA-like sRNAs (milRNAs) also exist in the filamentous fungi, including Neurospora crassa [13], Sclerotinia sclerotiorum [14] and Metarhizium anisopliae [15]. The success of siRNA-mediated gene silencing in T. reesei [16,17], an important industrial fungus capable of secreting a large amount of cellulolytic enzymes, suggests the RISC machinery and implies the presence of milRNAs in this fungus. However, neither miRNAs nor milRNAs have been reported in T. reesei, raising the question as to whether miRNAs exist in T. reesei.
Our purpose in this work is to explore the existence of milRNA in T. reesei and characterize their expression profile under cellulase induction. Two sRNA libraries of cellulose induction (IN) and non-induction (CON) were generated and sequenced using high-throughput sequencing technology.
Materials and Methods
T. reesei sample preparation
T. reesei strain QM9414 (ATCC 26921) were grown on potato dextrose agar (PDA) to obtain conidia. A total of 1×107conidia were inoculated into 25 ml of basal medium supplemented with 2% glucose at 28°C for 24 h with shaking at 250 rpm. The basal medium contains 0.4% KH2PO4, 0.28% (NH4)2SO4, 0.06% MgSO4, 0.08% CaCl2·2 H2O, 0.0005% FeSO4·7 H2O, 0.00016% MnSO4·H2O, 0.00017% ZnSO4·7 H2O, 0.00037% CoCl2·6H2O, 0.2% peptone and 0.1% Tween-80. For induction cultivation (IN), 2.5 ml mycelium supernatant was added into 50 ml of basal medium supplemented with 3% Avicel (Sigma PH101) at 28°C for 96 h. The same amount of mycelium supernatant was added into 50 ml of basal medium supplemented with 2% glucose for 72 h, which served as a non-induction control (CON).
Cellulase activity assays
The cellulase filter paper activity of T. reesei culture supernatant was measured with a method provided by Ghose [18].
RNA extraction and Solexa sequencing
The total RNAs of T. reesei IN and CON mycelium were extracted using the mirVana PARIS Kit (Ambion) according to the manufacturer’s instruction. sRNAs of T. reesei between 18~30 nucleotides were isolated using denatured polyacrylamide gel electrophoresis (PAGE). After ligated to 5’ and 3’ adapters, the sRNAs were reverse transcribed to cDNA using RT-PCR reaction. The PCR products were sequenced with an Illumina Genome Analyzer (BGI, Shenzhen, China).
Sequencing data analysis
The final clean reads were obtained by getting rid of the contaminant reads with 5' primer contaminants or polyA and those without 3' primer or insert tag. The sRNA sequences were mapped to the T. reesei genome [19] (http://genome.jgi-psf.org/Trire2/Trire2.home.html) using the Short Oligo Alignment Program (SOAP). We used the Rfam database (10.1) to eliminate the sRNAs originated from rRNA, tRNA, snRNA and snoRNA. The hairpin structures of the sRNAs were analyzed using the online tool Mfold [20] (http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form). Briefly, the sRNA sequence, as well as the 100 upstream nucleotides and 100 downstream nucleotides were folded using the Mfold software. The minimal free energy (MFE) of the hairpin structure was set as -15 kcal mol-1.
Globally differential expression of sRNAs
The global comparison of sRNA expression between the IN and CON samples was carried out. The procedure is as follows: (1) Normalize the expression of sRNAs in IN and CON samples to get the expression of transcript per million (TPM). Normalization formula: Normalized expression = actual miRNA count/total count of clean reads*1,000,000; (2) Calculate the log2 fold change (IN/CON); (3) evaluate the P-value using Fisher exact test; (4) graph a volcano plot that shows statistical significance versus fold change of expression on the y- and x-axes, respectively. P-values <0.01 were considered statistically significant. sRNAs were divided into three groups: (1) up-expressed sRNAs: log2 fold change≥1.0; (2) equally expressed sRNAs: -1.0<log2 fold change<1.0; (3) down-expressed sRNAs: log2 fold change ≤ -1.0. False discovery rates (FDR) were estimated, which was carried out by R statistical package version 3.0.1 (downloaded from http://www.r-project.org/). milRNAs with FDR< 0.05 for their expression in regression models were defined as differentially expressed miRNAs.
Analysis of milRNA expression using RT-qPCR method
We evaluated the expression of T. reesei milRNAs using the S-Poly(T) RT-qPCR method as described [21]. The 18S rRNA was used as a normalization control and the CON was served as the reference sample. The comparative Ct method (ΔΔCt) was exploited to calculate the relative expression levels of milRNAs. The probe and primers used were listed in Table S1. All analyses were performed in biological triplicate. Statistical analysis was performed with GraphPad Prism 5 using a two-tailed Student’s t-test. A P-value<0.05 was considered statistically significant.
milRNA target prediction and data analysis
The miRanda program was used to predict the potential targets of the T. reesei milRNAs. The parameters were set as follows: a gap opening penalty of 8; a gap extension penalty of 2; a score threshold of 90; an energy threshold of -23 kcal mol-1; and a scaling parameter of 2 [15]. We used the 1,000 bp sequences downstream of stop codon of all genes in T. reesei genome as database for the prediction analysis (http://genome.jgi-psf.org/Trire2/Trire2.home.html) due to the lacking of the 3’ UTR sequence database in T. reesei. The Gene Ontology (GO) annotations for predicted protein targets were available on JGI by search with protein ID (TreeseiV2_FrozenGeneCatalog20081022), and followed by functional classification using the WEGO software [22] (http://wego.genomics.org.cn/cgi-bin/wego/index.pl).
GEO accession numbers
The data obtained from sRNA deep sequencing studies were deposited in the Gene Expression Omnibus database at NCBI (http://www.ncbi.nlm.nih.gov/geo/). The accession number for GEO is GSE46679.
Results and Discussion
Overview of T. reesei sRNAs in Solexa sequencing
Accumulating evidence indicates the critical regulatory roles of miRNA in various biological processes in animals and plants. Recently, microRNA-like sRNAs (milRNAs) were discovered in fungi N. crassa [13], S. sclerotiorum [14] and M. anisopliae [15]. T. reesei is another important industrial fungus while miRNAs or milRNAs have not yet been discovered. To explore the existence of milRNAs in T. reesei, two separate T. reesei sRNA libraries were generated. First, T. reesei was cultivated in basal medium supplemented with 3% Avicel (IN) or 2% glucose (CON), respectively. The cellulase activities of the culture supernatant were measured and the results showed that the IN sample had a significant filter paper activity of approximately 1.10 FPU/ml, while the CON sample had no detectable activity (Figure S1). Next, the mycelium of both samples was subjected to RNA extraction. The quality of the RNA was measured with the Agilent 2100 Bioanalyzer (Table S2). Finally, two sRNA libraries of 18~30 nucleotides from IN and CON samples were prepared and sequenced using Solexa high-throughput sequencing technology.
After removing the low quality and adaptor sequences, 7,519,676 and 8,185,677 clean reads were obtained, which represented 664,463 and 529,545 unique sRNA sequences for T. reesei IN and CON sample, respectively (Table 1, Figure 1). The comparative analysis showed that the common “unique sRNAs” between the two samples were 8.07% (104,801), which constituted 82.15% (12,902,389) of the total sRNAs (Figure 1). The large number of sRNA sequences corresponding to a minority of the reads in only IN or CON samples suggested that T. reesei has a high degree of sRNA sequence complexity.
Table 1. Statistical summary of sRNAs from T. reesei IN and CON samples.
| Reads | % | |
|---|---|---|
| IN | ||
| Total reads | 8,040,063 | |
| High quality | 7,970,582 | 100% |
| 3' adapter null | 6,855 | 0.09% |
| Insert null | 4,447 | 0.06% |
| 5' adapter contaminants | 22,916 | 0.29% |
| Smaller than 18 nt | 416,557 | 5.23% |
| PolyA | 131 | 0.00% |
| Clean reads | 7,519,676 | 94.34% |
| CON | ||
| Total reads | 8,640,163 | |
| High quality | 8,595,187 | 100% |
| 3' adapter null | 5,601 | 0.07% |
| Insert null | 4,239 | 0.05% |
| 5' adapter contaminants | 7,301 | 0.08% |
| Smaller than 18 nt | 392,333 | 4.56% |
| PolyA | 36 | 0.00% |
| Clean reads | 8,185,677 | 95.24% |
Figure 1. Common and specific sequences between T. reesei IN and CON samples.
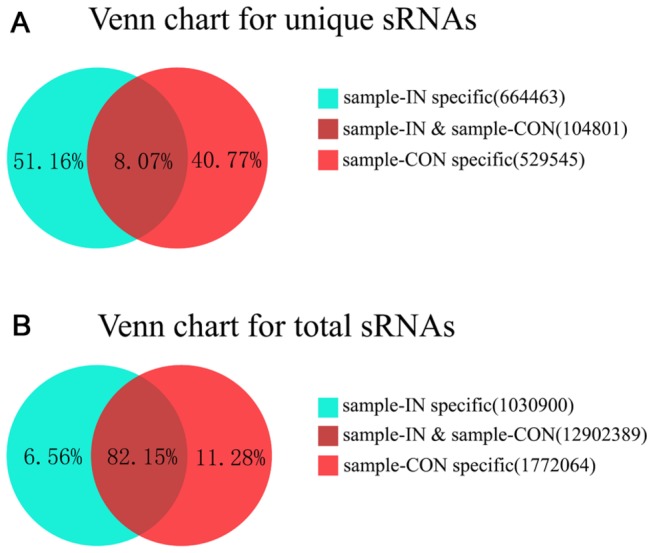
Venn charts show the summary of unique (A) and total (B) sRNAs sequence between T. reesei IN and CON samples. The number in brackets indicates the reads of the sequences.
All of the unique sequences were then compared with T. reesei genome, and the sequences that perfectly matched to the genomes were used for further analysis. The majority of sRNAs in IN and CON samples was 19~24 nucleotides in length (Figure S2). The length of sRNA peaked at 21 nucleotides in the IN library opposed to 19 nucleotides in the CON library, indicating a different predominance of sRNAs in length between the two samples. By aligning the sequences to the Rfam database, a total of 62,755 and 53,881 unique sRNAs from IN and CON samples, which originated from 50,338 and 46,267 rRNA, 10,072 and 6,157 tRNA, 1,981 and 1,340 snRNA, and 364 and 117 snoRNA, respectively, were removed (Figure S3). By doing a sequence alignment among the remaining sRNAs, many species of sRNAs were recognized as the truncated copies of the same sRNA molecule, with the sequence in the highest count representing the sequence of this sRNA. Finally, 726 and 631 sRNAs were obtained from IN and CON samples, respectively, which were subjected to analysis of global expression profile and RNA secondary structure.
Global expression analysis of T. reesei sRNAs
The differential expression of sRNAs between IN and CON samples were analyzed. The fold change of normalized expression of IN sample to normalized expression of CON sample was calculated for each sRNA. We set the fold change of 2.0 (log2 = ±1.0) as a cut-off and divided the sRNAs into three groups: (1) up-expressed sRNAs: log2 fold change≥1.0; (2) equally expressed sRNAs: -1.0<log2 fold change<1.0; (3) down-expressed sRNAs: log2 fold change ≤ -1.0. Among the sRNAs analyzed, there were 513 up-expressed sRNAs, 167 equally expressed sRNAs and 312 down-expressed sRNAs. The dramatic sRNA expression changes under different growth conditions indicate that sRNAs might play important roles in cellulose induced cellulase production (Figure 2).
Figure 2. Global expression analysis of T. reesei sRNAs.

A volcano plot shows the magnitude (fold change; x-axis) and significance (P-value; y-axis) of the unique sRNAs between IN and CON samples. The horizontal dashed line indicates the threshold of statistical significance (P-value= 0.01). The vertical dashed line shows the fold changes of 2.0 (log2 = ±1.0). sRNAs were divided into three groups based on the fold changes (IN/CON) : (1) up-expressed sRNAs: log2 fold change≥1.0; (2) equally expressed sRNAs: -1.0<log2 fold change<1.0; (3) down-expressed sRNAs: log2 fold change ≤ -1.0.
Identification of milRNAs in T. reesei
sRNAs were recognized as potential miRNAs when they were able to form hairpin structure with flanking nucleotide sequences in the genome [23]. To identify the milRNA in T. reesei, we used an online tool Mfold to analyze the hairpin structure of the sRNAs in both samples. Setting the criteria is of great importance in miRNA secondary structure prediction. Many criteria have ever been used for the miRNA prediction including the base pairs in a stem, the lengths of the whole hairpin and the hairpin loop, the minimal free energy (ΔG), etc [24]. However, the structures of pre-milRNAs in filamentous fungi might be different from those in animals and plants. For example, the length of pre-milRNAs was about 38 ~ 160 nucleotides in N. crassa which possesses a bigger range than that in plants [13]. In this work, we mainly focused on the length of the stem and the ΔG criteria during milRNA prediction. Additionally, GU is a wobble base pair with comparable thermodynamic stability to a Watson-Crick base pair. Thus, the GU base pair was permitted in the Mfold criteria as performed [25].
Thirteen candidate milRNAs were identified in T. reesei (Figure 3). The lengths of these T. reesei pre-milRNAs were similar to those in plants and animals [24] but much shorter than those reported in N. crassa [13]. The number of predicted milRNAs in T. reesei is much less than that in animals and plants, but is close to that in filamentous fungi. For example, four milRNAs were presented in N. crassa [13]. Two milRNAs and forty-two candidate milRNAs were reported in S. sclerotiorum [14], and fifteen in M. anisopliae [15]. Furthermore, no miRNA-miRNA* duplex was observed in T. reesei, which was yet found in a plant pathogenic fungus S. sclerotiorum [14]. Sequence search of T. reesei milRNAs on miRBase found no homologues in any other organisms including filamentous fungi, indicating a species-specific feature of T. reesei milRNAs.
Figure 3. Prediction of T. reesei pre-milRNAs hairpin structures.
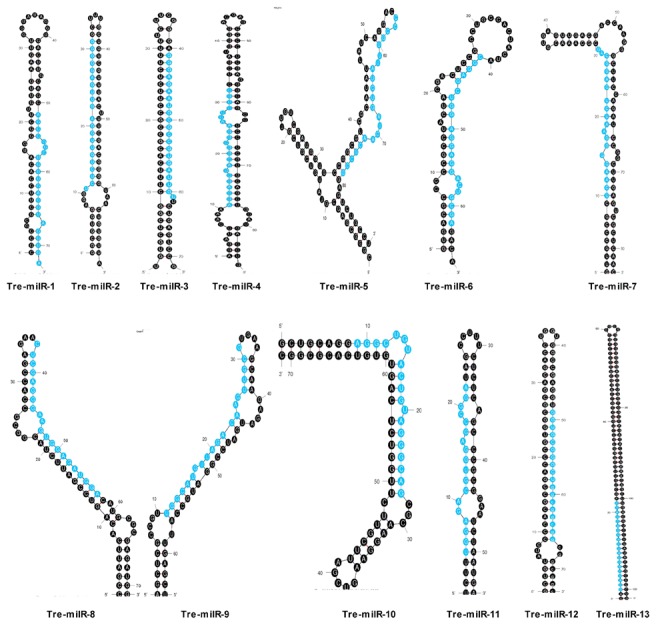
The secondary structures were analyzed with the Mfold software. The mature milRNA sequences were highlighted in blue.
The biogenesis of miRNAs is a multistep process, undergoing the pri-miRNA, pre-miRNA, and mature miRNA steps. Lee demonstrated that N. crassa generated milRNA by at least four different pathways involving components of Dicers, QDE-2, the exonuclease QIP, and an RNase III domain-containing protein, MRPL3 [13]. A homology search of the components in miRNA biogenesis showed that there were two Dicer-like and three Argonaute-like proteins in T. reesei genome (Table S3), providing evidences for the milRNA existence in this fungus. We thus termed the milRNAs in T. reesei as Tre-milRNAs concordant with the nomenclature used in N. crassa [13].
Differential expression of T. reesei milRNAs under different growth conditions
Another purpose of this study is to explore the role of milRNAs involved in the T. reesei cellulase production. We analyzed the Tre-milRNA expressions between IN and CON samples. Among the thirteen milRNAs, six were IN-specific, one was unique in CON, and six were present in both samples (Table 2). Of the six common milRNAs, Tre-milR-3, Tre-milR-5, Tre-milR-10 and Tre-milR-13 were up-regulated while Tre-milR-7 and Tre-milR-8 were down-regulated in IN sample when compared with that in CON sample. Among them, Tre-milR-5 and Tre-milR-10 were in high abundance in both samples, whereas other milRNAs expressed at a relatively low level. The different expression patterns of milRNAs suggested that milRNAs might be associated with the T. reesei metabolic processes, specifically with the cellulase induction.
Table 2. T. reesei milRNAs identified by Solexa sequencing.
| milRNA | Sequence (5’–3’) | Length (nt) | Total reads |
Location of milRNAs |
MFE (kcal mol-1) | P-value | FDR | |||
|---|---|---|---|---|---|---|---|---|---|---|
| IN | CON | Scaffold | Start | End | ||||||
| Tre-milR-1 | AGCCGGCTGTTGACGTAGGTGA | 22 | 5 | 0 | 16 | 434562 | 434583 | -22.6 | 7.36E-03 | 2.94E-02 |
| Tre-milR-2 | TCTCTGTTGGAGTTGAGGGGG | 21 | 9 | 0 | 4 | 95498 | 95478 | -23.3 | 1.27E-03 | 1.52E-03 |
| Tre-milR-3 | GGGAGAATGCGCCGTGATTGT | 21 | 100 | 53 | 20 | 508320 | 508340 | -31.3 | 3.47E-03 | 3.93E-03 |
| Tre-milR-4 | AGCAGCGACGGCGGAACTCTGC | 22 | 1,018 | 0 | 8 | 423216 | 423195 | -38.0 | 2.69E-27 | 1.33E-15 |
| Tre-milR-5 | CCCGTTTATCTGATCAACGCCG | 22 | 4,486 | 1,306 | 26 | 411357 | 411336 | -23.5 | 7.50E-07 | 1.55E-06 |
| Tre-milR-6 | CGGAGCTGGAGGAGGACTGCGA | 22 | 56 | 0 | 22 | 244532 | 244511 | -17.5 | 2.95E-20 | 1.40E-15 |
| Tre-milR-7 | TCAAGGGGAATCTGAGGCAG | 20 | 112 | 946 | 1 | 494569 | 494550 | -29.5 | 1.22E-06 | 2.10E-06 |
| Tre-milR-8 | CTCGAGGGAAGTGGAGATGGA | 21 | 22 | 55 | 6 | 551579 | 551599 | -23.5 | 6.21E-04 | 7.71E-04 |
| Tre-milR-9 | TGGCATGTTAGACAAGTTGCG | 21 | 6 | 0 | 7 | 628677 | 628695 | -18.6 | 4.22E-03 | 1.69E-02 |
| Tre-milR-10 | AGGCTGTACTGTAGGGCAG | 19 | 1,386 | 839 | 1 | 984928 | 984948 | -20.2 | 1.26E-03 | 1.49E-03 |
| Tre-milR-11 | TGGAGACGTGGAGCCGGA | 18 | 0 | 23 | 9 | 154864 | 154881 | -15.5 | 3.23E-07 | 5.86E-07 |
| Tre-milR-12 | GGTGCGGGCTGGCGGCGG | 18 | 54 | 0 | 13 | 231019 | 231036 | -36.2 | 4.67E-17 | 1.47E-15 |
| Tre-milR-13 | CCAGCAGGACTATGACGACG | 20 | 23 | 6 | 18 | 372972 | 372953 | -138.8 | 6.35E-04 | 8.13E-04 |
MFE: minimal free energy; FDR: false discovery rates.
We further validated the level of milRNAs that were present in both samples with RT-qPCR assay. As shown in Figure 4A, the expression of Tre-milR-3, Tre-milR-5, Tre-milR-10 and Tre-milR-13 in IN sample resulted in a 2.2-, 2.0-, 3.7- and 1.4-fold up-regulation, respectively, when compared with that in CON sample. Moreover, Tre-milR-7 and Tre-milR-8 were 5.2- and 2.4-fold down-regulated in IN relative to CON, respectively (Figure 4B). The results of the RT-qPCR assay showed a good coincidence with the data obtained by high-throughput sequencing.
Figure 4. Validation of differentially expressed milRNAs obtained in high-throughput sequencing with S-Poly(T) RT-qPCR method.

The expression levels of the selected milRNAs were assayed in both IN and CON. The 18S rRNA was used as a normalization control and the CON was served as the reference sample. All analyses were performed in biological triplicate. Statistical analysis was performed using a two-tailed Student’s t-test. Each bar represents mean ± SD. *P<0.05.
Target gene prediction of T. reesei milRNAs
We applied the miRanda software to predict the potential targets of T. reesei milRNAs. Since there was no T. reesei 3’ UTR database available, we chose the 1,000 bp sequences downstream of the stop codon of all genes in T. reesei genome as a hypothetical 3’ UTR database. The result showed that T. reesei milRNAs except Tre-milR-10 could bind to at least one target (Table S4). Surprisingly, Tre-milR-6 was able to match 101 target genes. Intriguingly, Tre-milR-4, an IN-specific milRNA, was predicted to target Cre1, a carbon catabolite repressor. The effects of Cre1 upon the negative regulation of cellulases and hemicellulases expression in T. reesei had been extensively reported [26-29]. Whether the significantly expression of Tre-milR-4 under inductive condition promotes the production of cellulases via targeting Cre1 needs further experimental study.
The WEGO software was used to perform a Gene Ontology (GO) analysis for functional classification of the predicted targets [22]. The result showed that the predicted targets were involved in different biological processes including substrate binding and catalysis, enzymatic transcriptional and translational regulation, transportation, growth, pigmentation, localization, response to stimulus and many other metabolic process (Figure S4).
Conclusion
In present work, thirteen predicted milRNAs were identified in T. reesei with high-throughput Solexa sequencing. The differential expression profile of milRNAs under inductive and non-inductive conditions suggests that milRNAs might play a role in T. reesei growth and cellulase production. This study will serve as a basis for further functional research of fungal milRNAs and their industrial application.
Supporting Information
T. reesei cellulase filter paper activity assay.
(DOCX)
Length distribution of sRNAs in T. reesei IN and CON samples.
(TIF)
Annotation of sRNAs of T. reesei IN (A) and CON (B). Pie chart showed the unique or total sequences matched to all categories of rRNA, tRNA, snRNA and snoRNA. The number in bracket showed the reads of the sequences.
(TIF)
GO classification of potential targets of T. reesei milRNAs. The results were summarized in three main categories as follows: cellular component, molecular function and biological process. In total, 196 genes have been assigned GO terms. In some cases, one gene has multiple terms.
(TIF)
Primers used in S-Poly(T) RT-qPCR assay.
(XLS)
Measurement of T. reesei total RNA.
(XLS)
Components involved in the sRNA biogenesis of T. reesei and two other filamentous fungi.
(XLS)
Predicted targets of T. reesei milRNAs.
(XLS)
Funding Statement
This study was supported in part by National Basic Research Program of China (973 Program, 2012CB124701); National Natural Science Foundation of China No. 81170047, 81370151 (to D.G.); Shenzhen overseas high-level talents innovation program No.YFZZ20111009 (to D.G.); Shenzhen Municipal Basic Research Program No.JC201006010725A (to D.G.); Shenzhen Municipal Basic Research Program No. JCYJ20130329120507746 (to K.K.); and Postdoctoral Science Foundation of China No. 2013M542203 (to K.K.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136: 215-233. doi:10.1016/j.cell.2009.01.002. PubMed: 19167326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. He L, Hannon GJ (2004) MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5: 522-531. doi:10.1038/nrg1379. PubMed: 15211354. [DOI] [PubMed] [Google Scholar]
- 3. Roush S, Slack FJ (2008) The let-7 family of microRNAs. Trends Cell Biol 18: 505-516. doi:10.1016/j.tcb.2008.07.007. PubMed: 18774294. [DOI] [PubMed] [Google Scholar]
- 4. Bader AG, Brown D, Winkler M (2010) The promise of microRNA replacement therapy. Cancer Res 70: 7027-7030. doi:10.1158/0008-5472.CAN-10-2010. PubMed: 20807816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843-854. doi:10.1016/0092-8674(93)90529-Y. PubMed: 8252621. [DOI] [PubMed] [Google Scholar]
- 6. Molnár A, Schwach F, Studholme DJ, Thuenemann EC, Baulcombe DC (2007) miRNAs control gene expression in the single-cell alga Chlamydomonas reinhardtii. Nature 447: 1126-1129. doi:10.1038/nature05903. PubMed: 17538623. [DOI] [PubMed] [Google Scholar]
- 7. Zhang YQ, Chen DL, Tian HF, Zhang BH, Wen JF (2009) Genome-wide computational identification of microRNAs and their targets in the deep-branching eukaryote Giardia lamblia. Comput Biol Chem 33: 391-396. doi:10.1016/j.compbiolchem.2009.07.013. PubMed: 19716768. [DOI] [PubMed] [Google Scholar]
- 8. Lin WC, Li SC, Shin JW, Hu SN, Yu XM et al. (2009) Identification of microRNA in the protist Trichomonas vaginalis. Genomics 93: 487-493. doi:10.1016/j.ygeno.2009.01.004. PubMed: 19442639. [DOI] [PubMed] [Google Scholar]
- 9. Zhao T, Li G, Mi S, Li S, Hannon GJ et al. (2007) A complex system of small RNAs in the unicellular green alga Chlamydomonas reinhardtii. Genes Dev 21: 1190-1203. doi:10.1101/gad.1543507. PubMed: 17470535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li L, Wright SJ, Krystofova S, Park G, Borkovich KA (2007) Heterotrimeric G protein signaling in filamentous fungi. Annu Rev Microbiol 61: 423-452. doi:10.1146/annurev.micro.61.080706.093432. PubMed: 17506673. [DOI] [PubMed] [Google Scholar]
- 11. Dang Y, Yang Q, Xue Z, Liu Y (2011) RNA interference in fungi: pathways, functions, and applications. Eukaryot Cell 10: 1148-1155. doi:10.1128/EC.05109-11. PubMed: 21724934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fulci V, Macino G (2007) Quelling: post-transcriptional gene silencing guided by small RNAs in Neurospora crassa. Curr Opin Microbiol 10: 199-203. doi:10.1016/j.mib.2007.03.016. PubMed: 17395524. [DOI] [PubMed] [Google Scholar]
- 13. Lee HC, Li L, Gu W, Xue Z, Crosthwaite SK et al. (2010) Diverse pathways generate microRNA-like RNAs and Dicer-independent small interfering RNAs in fungi. Mol Cell 38: 803-814. doi:10.1016/j.molcel.2010.04.005. PubMed: 20417140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou J, Fu Y, Xie J, Li B, Jiang D et al. (2012) Identification of microRNA-like RNAs in a plant pathogenic fungus Sclerotinia sclerotiorum by high-throughput sequencing. Mol Genet Genomics 287: 275-282. doi:10.1007/s00438-012-0678-8. PubMed: 22314800. [DOI] [PubMed] [Google Scholar]
- 15. Zhou Q, Wang Z, Zhang J, Meng H, Huang B (2012) Genome-wide identification and profiling of microRNA-like RNAs from Metarhizium anisopliae during development. Fungal Biol 116: 1156-1162. doi:10.1016/j.funbio.2012.09.001. PubMed: 23153806. [DOI] [PubMed] [Google Scholar]
- 16. Qin LN, Cai FR, Dong XR, Huang ZB, Tao Y et al. (2012) Improved production of heterologous lipase in Trichoderma reesei by RNAi mediated gene silencing of an endogenic highly expressed gene. Bioresour Technol 109: 116-122. doi:10.1016/j.biortech.2012.01.013. PubMed: 22305540. [DOI] [PubMed] [Google Scholar]
- 17. Brody H, Maiyuran S (2009) RNAi-mediated gene silencing of highly expressed genes in the industrial fungi Trichoderma reesei and Aspergillus niger. Ind Biotechnol 5: 53-60. doi:10.1089/ind.2009.5.53. [Google Scholar]
- 18. Ghose T (1987) Measurement of cellulase activities. Pure Appl Chem 59: 257-268. doi:10.1351/pac198759020257. [Google Scholar]
- 19. Martinez D, Berka RM, Henrissat B, Saloheimo M, Arvas M et al. (2008) Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat Biotechnol 26: 553-560. doi:10.1038/nbt1403. PubMed: 18454138. [DOI] [PubMed] [Google Scholar]
- 20. Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406-3415. doi:10.1093/nar/gkg595. PubMed: 12824337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kang K, Zhang X, Liu H, Wang Z, Zhong J et al. (2012) A Novel Real-Time PCR Assay of microRNAs Using S-Poly(T), a Specific Oligo(dT) Reverse Transcription Primer with Excellent Sensitivity and Specificity. PLOS ONE 7: e48536-48545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ye J, Fang L, Zheng H, Zhang Y, Chen J et al. (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34: W293-W297. doi:10.1093/nar/gkl031. PubMed: 16845012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee HJ, Hong SH (2012) Analysis of microRNA-size, small RNAs in Streptococcus mutans by deep sequencing. FEMS Microbiol Lett 326: 131-136. doi:10.1111/j.1574-6968.2011.02441.x. PubMed: 22092283. [DOI] [PubMed] [Google Scholar]
- 24. Fu Y, Shi Z, Wu M, Zhang J, Jia L et al. (2011) Identification and differential expression of microRNAs during metamorphosis of the Japanese flounder (Paralichthys olivaceus). PLOS ONE 6: e22957. doi:10.1371/journal.pone.0022957. PubMed: 21818405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hancock MH, Tirabassi RS, Nelson JA (2012) Rhesus cytomegalovirus encodes seventeen microRNAs that are differentially expressed in vitro and in vivo. Virology 425: 133-142. doi:10.1016/j.virol.2012.01.009. PubMed: 22305624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cziferszky A, Mach RL, Kubicek CP (2002) Phosphorylation positively regulates DNA binding of the carbon catabolite repressor Cre1 of Hypocrea jecorina (Trichoderma reesei). J Biol Chem 277: 14688-14694. doi:10.1074/jbc.M200744200. PubMed: 11850429. [DOI] [PubMed] [Google Scholar]
- 27. Portnoy T, Margeot A, Linke R, Atanasova L, Fekete E et al. (2011) The CRE1 carbon catabolite repressor of the fungus Trichoderma reesei: a master regulator of carbon assimilation. BMC Genomics 12: 269. doi:10.1186/1471-2164-12-269. PubMed: 21619626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ilmén M, Thrane C, Penttilä M (1996) The glucose repressor gene cre1 of Trichoderma: isolation and expression of a full-length and a truncated mutant form. Mol Gen Genet 251: 451-460. doi:10.1007/s004380050189. PubMed: 8709949. [DOI] [PubMed] [Google Scholar]
- 29. Kubicek CP, Mikus M, Schuster A, Schmoll M, Seiboth B (2009) Metabolic engineering strategies for the improvement of cellulase production by Hypocrea jecorina. Biotechnol Biofuels 2: 19. doi:10.1186/1754-6834-2-19. PubMed: 19723296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
T. reesei cellulase filter paper activity assay.
(DOCX)
Length distribution of sRNAs in T. reesei IN and CON samples.
(TIF)
Annotation of sRNAs of T. reesei IN (A) and CON (B). Pie chart showed the unique or total sequences matched to all categories of rRNA, tRNA, snRNA and snoRNA. The number in bracket showed the reads of the sequences.
(TIF)
GO classification of potential targets of T. reesei milRNAs. The results were summarized in three main categories as follows: cellular component, molecular function and biological process. In total, 196 genes have been assigned GO terms. In some cases, one gene has multiple terms.
(TIF)
Primers used in S-Poly(T) RT-qPCR assay.
(XLS)
Measurement of T. reesei total RNA.
(XLS)
Components involved in the sRNA biogenesis of T. reesei and two other filamentous fungi.
(XLS)
Predicted targets of T. reesei milRNAs.
(XLS)