

Abstract
Recent studies indicate that a subclass of APOBEC cytidine deaminases, which convert cytosine to uracil during RNA editing and retrovirus or retrotransposon restriction, may induce mutation clusters in human tumors. We show here that throughout cancer genomes APOBEC mutagenesis is pervasive and correlates with APOBEC mRNA levels. Mutation clusters in whole-genome and exome datasets conformed to stringent criteria indicative of an APOBEC mutation pattern. Applying these criteria to 954,247 mutations in 2,680 exomes of 14 cancer types, mostly from TCGA, revealed significant presence of the APOBEC mutation pattern in bladder, cervical, breast, head and neck and lung cancers, reaching 68% of all mutations in some samples. Within breast cancer, the HER2E subtype was clearly enriched with tumors displaying the APOBEC mutation pattern, suggesting this type of mutagenesis is functionally linked with cancer development. The APOBEC mutation pattern also extended to cancer-associated genes, implying that ubiquitous APOBEC mutagenesis is carcinogenic.
Genome instability triggers the development of many types of cancers1,2. Radiation and chemical damage are traditionally invoked as culprits in theories of carcinogenic mutagenesis3. However, normal enzymatic activities can also be a source of DNA damage and mutation. Cytidine deaminases, which convert cytosine bases (C) to uracil (U), likely contribute to DNA damage4. Activation-induced cytidine deaminase (AID), a key enzyme in adaptive immunity, not only initiates the hyper-mutation and class switch recombination of immunoglobulin genes, but also can mutate chromosomal DNA at a limited number of “secondary” targets, some of which have been implicated in carcinogenesis5. In addition to AID, the human genome encodes several homologous APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) cytidine deaminases that function in innate immunity as well as in RNA editing6. Prior human cell culture studies showed that a subclass of APOBECs with a tC mutational specificity (the mutated cytosine (C) is captalized) are capable of inducing mutations in chromosomal and mitochondrial DNA and therefore could play a role in carcinogenesis7–9 (“APOBEC” without the gene-specifying suffix is used hereafter to designate a subclass of cytidine deaminases with tC-specificity. Note: based on motif specificity, APOBEC3G and AID do not fall into this subclass). Supporting this, a mutation signature consistent with APOBEC editing was found in individual cancer-related genes10,11. Recently, clustered mutations (termed as kataegis in12) identified through next-generation sequencing suggested that APOBECs can induce base substitutions in tumor genomes12,13. Clustered mutations showed even higher preference to a more stringent tCw motif (where “W” corresponds to either adenine (A) or thymine (T)). Tightly linked strand-coordinated clustered mutations were often co-localized with rearrangement breakpoints, suggesting that this mutagenesis results from aberrant DNA double strand break (DSB) repair that produces single-stranded (ss) DNA: an ideal substrate for APOBEC enzymes6. The presence of base substitutions in the APOBEC motif was increased among mutations identified in whole-genome sequenced breast cancers12, as well as in multiple myeloma, prostate and head and neck cancers13. Interestingly, non-clustered substitutions in tCw were also enriched near rearrangement breakpoints across the genomes of several cancer types14. Analysis of breast cancer sequence and expression data suggested that specifically APOBEC3B may cause mutations in this cancer type9.
Despite the indication that APOBEC mutagenesis may play a role in cancer, it was unclear how strong of a mutagenic factor APOBEC enzymes are, whether APOBEC mutagenesis is a ubiquitous characteristic of many cancer types and cases, and whether it is associated with any specific tumor characteristics. Here, we have developed an analysis for evaluating the strength of an APOBEC mutation pattern in the individual samples from multiple whole-genome and exome mutation datasets such as TCGA. We found that the APOBEC mutation pattern is prominent and even prevailing in many samples within several types of cancer, as opposed to other cancer types where it is barely detectable, that it correlates with APOBEC mRNA levels and extends into a subset of genes considered by multiple criteria to be cancer “drivers.”
Results
A pattern for detecting APOBEC mutagenesis
Our approach to the statistical exploration of complex mutation spectra in multiple cancer samples involved the formulation of a single hypothesis surrounding a diagnostic mutation pattern that utilizes knowledge obtained in prior experiments as well as in data analyses and minimizes overlap with other known sequence-specific mutagenesis mechanisms. The first step in defining a measure for a pattern of APOBEC mutagenesis in a cancer sample was to find mutations that occur in the motif most likely to be an APOBEC-specific target. We chose the tCw motif as opposed to the less stringent tC, because of its demonstrated prevalence in mutagenesis caused by some APOBECs in model systems as well as in mutation clusters found in cancers (9,12,13 and refs. therein). The more stringent tCw motif also eliminates the potential overlap with a sequence-specific mutagenesis in highly mutable CpG sequences that would be occasionally preceded by a T. Secondly, we proposed that APOBEC-induced mutagenesis would involve primarily C to guanine (G) and/or to T substitutions with rare C to A changes. This substitution pattern is based on the tendency of trans-lesion synthesis to mis-incorporate C or A across from abasic sites (resulting in C→G and C→T mutations) that are generated frequently by uracil DNA-glycosylase15–17 activity towards the products of both spontaneous and APOBEC induced C-deamination, as well as the templated synthesis past a C-deamination-derived U (resulting in C→T changes). Thus, in the present analysis, we defined C→T and C→G substitutions in tCw as APOBEC-signature mutations. In order to identify samples that experienced APOBEC mutagenesis, we further defined an APOBEC mutagenesis pattern within a sample as a statistically significant enrichment of APOBEC-signature mutations when compared to that expected by random mutagenesis (See Methods). Enrichment for APOBEC signature mutations (tCw→tTw or →tGw and the complements wGa→wAa or →wCa) among all similar mutations of C or G (C→T or →G and G→A or →C) was calculated over the presence of the APOBEC mutation motif (tCw or wGa) in the +/− 20 nucleotide contexts surrounding mutated nucleotides. We utilized only the surrounding contexts in this calculation because APOBEC enzymes are thought to scan a limited area of ssDNA to deaminate C in a preferred motif18,19. This approach does not exclude any given area of the genome in general, but rather utilizes the areas within each sample, where mutagenesis has happened and then evaluates, if the mutagenesis in this sample is enriched with APOBEC signature mutations. To test the accuracy of our analysis, we compared our measure of the APOBEC mutation pattern (fold enrichment) to a previously reported measure of APOBEC mutagenesis obtained via a very different approach of mathematical decomposition and extraction of multiple mutation signatures from 21 breast cancer samples12. The results showed a very high level of correlation (Supplementary Fig. 1 and Supplementary Table 1) supporting the applicability of our method. Moreover, this analysis remained robust even when applied to samples containing small numbers of mutations. The fold enrichment of APOBEC mutagenesis among a subset of mutations representing exomes of the aforementioned 21 breast cancers (~2% of total mutations in the whole genome) correlated strongly with values obtained from the entire genome (Supplementary Fig. 2), suggesting this analysis may be effectively applied to mutations identified through exome-sequencing and thereby dramatically increase the number of cancer samples that are available for analysis.
An APOBEC mutagenesis pattern within mutation clusters
We evaluated the APOBEC mutation pattern in a large number of whole-genome and exome mutation datasets accumulated in The Cancer Genome Atlas (TCGA) as well as in several publications20,21. APOBECs are highly specific for ssDNA and are capable of simultaneously making multiple mutations, if a ssDNA region persists17,19. Such mutations are strand-coordinated as changes in cytosines occur within the same DNA strand. We and others have detected this APOBEC mutation pattern in C- and complementary G-strand-coordinated clusters from a limited number of whole-genome sequenced tumors12,13. These clusters often co-localized with rearrangement breakpoints12,13 (Fig. 1a and Supplementary Fig. 3), which agreed with mutagenesis occurring in ssDNA regions that are either prone to breakage and/or are formed during a DSB repair process. Clustered C or G mutations identified previously13 as well as in additional analysis of whole-genome sequenced colorectal adenocarcinomas22 presented here showed a strong APOBEC mutation pattern (i.e., highest enrichment with tCw and strong preference of C to T and C to G changes in this motif; Fig. 1a, Supplementary Fig. 3, and Supplementary Table 2).
Figure 1. APOBEC mutation pattern in clusters.

a. Whole-genome datasets. Analysis of all clusters identified among 23 multiple myeloma47, 2 head and neck squamous cell carcinoma21, 7 prostate carcinoma48 and 9 colorectal adenocarcinoma22 datasets. Co-localization of clusters with breakpoints was identified as described in13. The category “Close to breakpoints” includes clusters in which at least one mutation falls within 20 kb of a breakpoint. Fold-enrichment (shown above bars) of mutation motifs (mutated base is capitalized) was calculated for all three possible changes of C (or G) as described in On-line Methods. *** corresponds to Bonferroni-corrected q-values < 0.0001 as determined by a one-tailed Fisher’s Exact Test comparing the ratio of the number of C mutations at tCw and the number of C mutations not in the sequence tCw to the analogous ratio for all cytosines within a sample fraction of the genome. The bottom bar shows the numbers and fractions (above appropriate sections of the bar) of three different base substitutions of C (or G). b. Exome data sets. Analysis of clusters identified in 2,680 exomes of 14 different cancer types from TCGA as well as from other published sources20,21 (see details in the On-line Methods). Format and calculations are the same as in (a).
We next addressed whether an APOBEC mutation pattern is common among different cancer samples and types. We accumulated lists of cancer-specific mutations from the whole-exome sequencing of 2,680 tumors, mostly by the TCGA Research Network (Supplementary Table 3). While exome sequencing dramatically increases the number of samples available for analysis, its general specificity for protein coding regions results in only ~1% of total genomic DNA being assessed. To identify clusters from exome sequencing, we therefore estimated the total mutation load in a given tumor sample under the assumption that exome mutations constitute 1% of mutations in the entire genome and utilized this value to identify clusters using our previously described algorithm13. This method found 498 total clusters in the 2,680 sequenced exomes from 14 different cancer types. 218 C- or G-coordinated clusters were identified, occurring in every cancer type analyzed except acute myeloid leukemia (LAML) (Supplementary Fig. 4). Similar to results obtained by whole-genome analysis (compare Fig. 1a and 1b), these clusters showed a robust APOBEC mutation pattern, while other known mutagenic motifs involving C or G were depleted. Contrastingly, the APOBEC mutation pattern was barely detectable or undetectable in non-coordinated clustered C and G mutations and scattered mutations, respectively (Supplementary Fig. 5). The enrichment of APOBEC signature mutations in C- or G-coordinated clusters was more pronounced in clusters with >2 mutations (Supplementary Fig. 4), because clusters with only two mutations have a higher chance of occurring independently through non-APOBEC mechanisms.
The APOBEC mutagenesis pattern across 2,680 cancer exomes
The strength of the APOBEC mutation pattern in C- or G-coordinated clusters from our analysis of exome mutations (Fig. 1b) was comparable to that of clusters found in whole-genome mutation lists (Fig. 1a), suggesting that exome-wide mutation data may be sufficient to detect the APOBEC mutation pattern among all mutations in a sample’s exome. Indeed, the APOBEC mutation pattern was clearly present throughout many exomes indicating that APOBEC enzymes were likely a significant source of mutagenesis in these samples (Fig. 2a and Supplementary Table 4). Samples displaying this pattern occurred primarily within 6 cancer types while the other 8 types were deprived of this pattern even despite high general mutagenesis in many samples (p<0.0001; two-sided Chi-square comparison of the number of samples in each cancer type displaying fold enrichments of APOBEC signature mutations greater than the median fold enrichment among all samples; n=2,680). Bladder (BLCA), cervical (CESC), head and neck (HNSC), breast (BRCA) and lung cancers (LUAD and LUSC) were enriched in samples displaying a high level of APOBEC mutagenesis or greater odds-ratio as compared to the total range of APOBEC mutagenesis in exomes (Fig. 2 and Supplementary Fig. 6a and b). A motif-specific functional selection is unlikely to have caused the observed over-representation of the APOBEC mutation pattern, as corresponding calculations of fold enrichment among the silent and non-coding mutations in each sample produced similar results (Supplementary Fig. 7). Across all tumors analyzed, high fold APOBEC enrichment correlated strongly with a decreased Fisher’s q-value as well as an increase in the fraction of total mutations in a tumor that display the APOBEC signature (Supplementary Fig. 8). In individual tumors displaying a strong APOBEC pattern, the number of APOBEC-signature mutations was often large, making it the predominant source of mutations in the sample (Fig. 2b). Strikingly, some samples contained over a thousand APOBEC-signature mutations, constituting up to 68% of mutations in the exome.
Figure 2. The presence of an APOBEC mutation pattern in exome datasets of different cancer types.
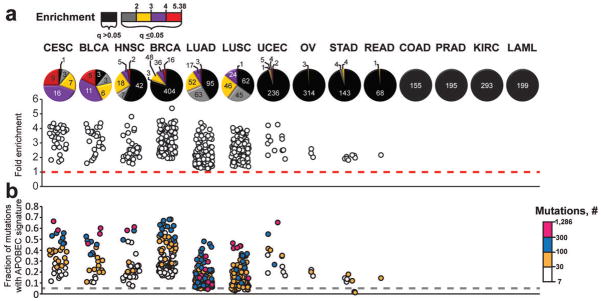
Cancer types are abbreviated as in TCGA: Cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), Bladder Urothelial Carcinoma (BLCA), Head and Neck squamous cell carcinoma (HNSC), Breast invasive carcinoma (BRCA), Lung adenocarcinoma (LUAD), Lung squamous cell carcinoma (LUSC), Uterine Corpus Endometrioid Carcinoma (UCEC), Ovarian serous cystadenocarcinoma (OV), Stomach adenocarcinoma (STAD), Rectum adenocarcinoma (READ), Colon adenocarcinoma (COAD), Prostate adenocarcinoma (PRAD), Kidney renal clear cell carcinoma (KIRC), and Acute Myeloid Leukemia (LAML). The fold enrichment (a) and mutation load (b) of the APOBEC mutation pattern was determined within each of 2,680 whole exome sequenced tumors, representing 14 cancer types. Samples were categorized by the statistical significance of the APOBEC mutation pattern (calculated by one-sided Fisher’s Exact Test comparing the ratio of the number of C→T or →G substitutions and complementary G→A or →C substitutions that occur in and out of the APOBEC target motif (tCw/wGa) to an analogous ratio for all cytosines or guanines that reside inside and outside of the tCw/wGa motif within a sample fraction of the genome; Benjamini-Hochberg corrected q-value < 0.05) and the magnitude of enrichment. The number of tumor samples in each category is presented on each pie graph in (a). Samples displaying q-values > 0.05 are represented in black. These samples are excluded from the scatter graphs in (a) and (b) that depict the range of enrichments and fractional mutation load, respectively. Color scales indicate the magnitude of enrichment (a) and the number of APOBEC signature mutations (b) for samples with q<0.05. Dashed lines indicate effects expected for random mutagenesis.
Importantly, in cancer types where an APOBEC mutation pattern was not noticeable within the exome data, the pattern was detectable in clusters of strand-coordinated C (or G) mutations from whole-genome data. Whole-genome data contains about 100 fold more mutations than exomes, which facilitates the detection of clusters. We previously reported such clusters to be enriched with the APOBEC mutation pattern among the mutations in whole-genome sequenced prostate carcinomas13 and show here the same pattern within 9 whole-genome colorectal cancer mutation datasets22 (Supplementary Fig. 3). In each of these data sets, many of the C- or G-coordinated clusters co-localized with chromosome rearrangement breakpoints, a phenomenon that supports the involvement of ssDNA (the exclusive substrate of APOBEC enzymes) in cluster formation23,24. Neither cancer type, however, showed a detectable presence of the APOBEC mutation pattern in exome data. Thus, the APOBEC mutagenesis pattern appears to be ubiquitous at a background level in all types of cancer, but is more prominent in particular types.
APOBEC mutagenesis correlates with an increase in APOBEC mRNA
Several cancer type-specific factors including the availability of ssDNA substrate and the expression level of APOBEC enzymes could contribute to the extent of APOBEC mutagenesis. Recently, a tumor-specific increase in the transcription of APOBEC3B, determined by qPCR, microarray, as well as RNA-seq in breast cancer samples, was shown to correlate with an increased number of C→T transitions9. C→T mutations are a relaxed measure of total deamination, which includes the APOBEC signature in tCw defined in our analysis as well as mutations stemming from other processes. We used RNA-seq expression data to address whether the expression of any of the eight APOBEC enzymes known to have biochemical deamination activity towards DNA correlate with the extent of the observed APOBEC mutagenesis. Consistent with the prior report in breast cancer, APOBEC3B expression was frequently increased in tumor samples over matched normal samples, however median APOBEC3H and APOBEC3A (Supplementary Figs. 9 and 10) expression were also increased more than 2 fold in tumors. Among the 483 breast cancers analyzed for both APOBEC mutagenesis and by RNA-seq, APOBEC3B as well as APOBEC3A expression correlated strongly with the total number of C→T mutations per exome (Supplementary Fig. 11a; Spearman r = 0.233; Bonferroni corrected q<0.001 and Spearman r = 0.1998; q<0.001, respectively). Importantly, when transcription levels were compared to the number of mutations conforming to our stringent definition of APOBEC mutagenesis (tCw→tTw or →tGw), the strength of the association increased for both enzymes (Fig. 3a; Spearman r = 0.3150; Bonferroni corrected q<0.001 and Spearman r = 0.3088; Bonferroni corrected q<0.001 for APOBEC3B and APOBEC3A, respectively). Extending this analysis to all 2048 tumors with available RNA-seq data across cancer types, expression of APOBEC3B again most strongly correlates with the number of tCw→tTw and →tGw mutations per exome (Fig. 3a and Supplementary Table 4; Spearman r = 0.2953, Bonferroni corrected q<0.001) with APOBEC1, 3A, 3F, and 3G also associating but to lesser extents (Supplementary Fig. 11b). Within individual cancer types, only APOBEC3A in breast cancer and APOBEC3B in breast cancer and lung adenocarcinomas displayed a positive correlation between expression and APOBEC mutagenesis (Supplementary Fig. 11b). However, in bladder and lung squamous cell cancers, the remaining 2 cancer types with available RNA-seq data and high APOBEC mutagenesis, the median APOBEC3B expression was elevated >3 fold in compared to the median of APOBEC3B expression among all samples (Bonferroni corrected Mann-Whitney q<0.001) (Fig. 3b). Thus, the APOBEC3B enzyme is likely the major candidate inducing the APOBEC mutation pattern across cancer types with the lesser correlations seen with APOBEC3A, 3F, and 3G possibly resulting from mis-attribution of some APOBEC3B RNA-seq reads from homologous mRNA regions.
Figure 3. APOBEC transcription level positively correlates with the number of APOBEC signature mutations.
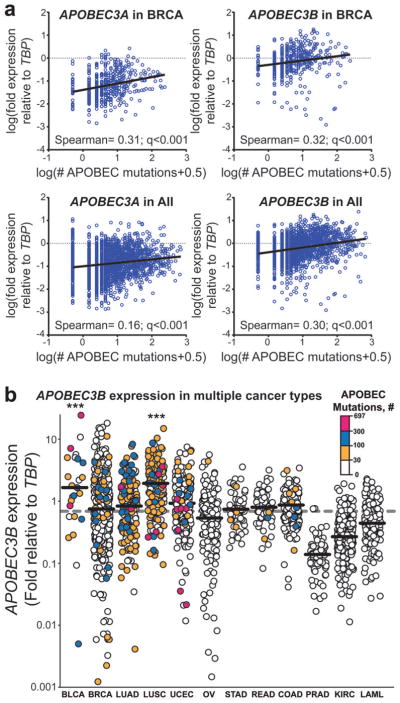
RNA-seq derived mRNA levels of each APOBEC family member with documented deaminase activity on DNA was standardized relative to TATA-binding protein (TBP). “APOBEC mutations” refers to number of tCw to tTw and tCw to tGw changes. (a) The expression (relative to TBP) of APOBEC3A and 3B was compared to the total number of APOBEC mutations in each exome (blue circles) among 483 breast cancers (“in BRCA”) and 2048 total tumor samples (“in All”) with available RNA-seq data by non-parametric Spearman correlation. Graphs display log transformed values with mutation values augmented by 0.5 to allow depiction of exomes with no observed APOBEC-signature mutations. Spearman coefficients and corresponding q-values (two-sided; corrected for multiple testing error by the Bonferroni method) are indicated. Black lines represent linear regressions. Correlation data for other APOBECs and individual cancer types are shown in Supplementary Figure 11. (b) APOBEC3B transcription relative to TBP in 2048 tumor samples separated by cancer type. Horizontal bars indicate the median expression level within a cancer type. Dashed grey line indicates the median APOBEC3B expression among all cancers analyzed. *** indicates that APOBEC3B expression in a cancer type is elevated (q<0.001 by pairwise two-sided Mann-Whitney comparison of a specific cancer type to the overall distribution and corrected for multiple analyses by the Bonferroni method). Color scales indicate the number of APOBEC-signature mutations in each individual exome. Individual cancer types are abbreviated as in Fig. 2.
The HER2E subtype of breast cancer is enriched with the APOBEC mutagenesis pattern
Several cancer types displayed high levels of the APOBEC mutation pattern as well as a wide variation among individual samples, which could reflect different biological pathways leading to carcinogenesis. The greatest range of variation was observed in breast cancer, which is often divided into subtypes based on differences in biomedical characteristics (see25 and therein). To determine whether the APOBEC mutagenesis pattern is associated with specific breast cancer subtypes, we subdivided the samples based on their PAM50 classification presented in25. The PAM50 algorithm utilizes mRNA levels of 50 differentially expressed genes to classify breast cancers into specific subtypes26. Four subtypes: luminal A (LumA), luminal B (LumB), basal-like, and HER2-enriched (HER2E) were significantly represented in our dataset. Each subtype contained samples with a prominent APOBEC mutation pattern and a correspondingly large number of APOBEC-signature mutations. However, such samples were unevenly distributed among the subtypes, occurring much more frequently in the HER2E class (Fig. 4 and Supplementary Fig. 12).
Figure 4. APOBEC mutation pattern in exome datasets of four breast cancer subtypes.

Cancer types are abbreviated as: luminal A (Lum A), Basal-like, luminal B (Lum B), and HER2-enriched (HER2E). The fold enrichment (a) and mutation load (b) of the APOBEC mutation pattern was determined within each of 507 whole-exome sequenced BRCA tumors. The number of samples above (blue) and below (red) the median for all 507 exomes (dashed red lines) was determined for each cancer subtype. The horizontal black bars indicate the median in each subtype. *** indicates that a cancer type is significantly enriched in samples containing a high presence of the APOBEC mutation pattern (q<0.001 by pairwise two-sided Chi-square comparison of a specific cancer type to the overall distribution and corrected for analysis of multiple subtypes by the Bonferroni method). Color scales indicate the magnitude of enrichment (a) and the number of APOBEC-signature mutations (b).
Unlike for the breast cancer as a whole (Fig. 3a), no correlation between the number of APOBEC-signature mutations and APOBEC mRNA levels was observed within the HER2E subtype (Supplementary Fig. 13a). This could result from consistently high APOBEC3B expression in HER2E samples (~3 fold greater than the median expression seen across all cancer types), which reduces the power of correlation analysis (Supplementary Fig. 13b). Interestingly, basal-like and luminal B cancers also have median APOBEC3B expression levels comparable to that of HER2E but display significantly less APOBEC mutagenesis, suggesting that additional factors are likely as important as expression.
The HER2E subtype is reportedly associated not only with amplification of the ERBB2 gene locus, but also with a high level of copy number variation (CNV) across the genome25. This feature as well as frequent co-localization of APOBEC signature mutations with chromosome rearrangements, suggested that a direct connection between the level of the APOBEC mutagenesis pattern and the number of segmental CNVs (i.e., CNVs originating from breakage) may exist. As shown in model studies27, increased APOBEC-induced deamination can lead to higher levels of breakage, which in turn could result in greater numbers of CNV. Alternatively, increased breakage could provide more ssDNA substrate for APOBEC deamination. However, comparison of the number of segmental CNV breakpoints with the fold enrichment of APOBEC-signature mutations in 449 breast cancer samples failed to identify any correlation (Supplementary Fig. 14). While the underlying reason for the enrichment of the APOBEC mutagenesis pattern in the HER2E subtype remains unclear, the association of this mutagenesis with a specific breast cancer subtype suggests that physiological aspects of this subtype are likely important.
The APOBEC mutagenesis pattern includes cancer driver genes
An APOBEC mutagenesis pattern present in a sample or group of samples indicates that the level of this mutagenesis is significantly higher than expected if all base substitutions in C (or G) have occurred randomly. However, because of the sequence specificity of APOBEC deamination and its tight association with ssDNA, the fraction of the genome where carcinogenic mutations can occur may escape the bulk of APOBEC mutagenesis. We therefore examined the presence of APOBEC-signature mutations among mutations that are potential cancer “drivers.”. Three approaches were used to identify driver mutations. First, a stringent list of likely cancer driver mutations was assembled using the on-line software package CRAVAT28,29. Based on multiple parameters, including the occurrence of a mutation in the COSMIC database30, this software calculates a probability that a given missense mutation drives cancer. Mutations displaying FDR-corrected q-values of 0.05 or less were selected as likely drivers. We subsequently employed two additional “less stringent” criteria for identifying potentially carcinogenic mutations: (i) the presence of mutations in the COSMIC database (as indicated by CRAVAT) and (ii) mutations that affect a subset of genes from the Cancer Gene Census, i.e., genes in which missense or nonsense mutations are considered causative in cancer31. Both of these less stringent driver definitions extended the spectra of changes beyond missense mutations to include nonsense and synonymous mutations as potentially carcinogenic alterations31,32.
Using any of these three criteria, APOBEC signature mutations occurred at a higher frequency among carcinogenic mutations in the group of samples with high APOBEC presence as compared to samples in which the APOBEC mutation pattern was not detected (Fig. 5). This implies that APOBEC-signature mutations themselves can contribute to carcinogenesis in samples displaying a strong APOBEC mutation pattern. Further supporting this carcinogenic potential, many of APOBEC-signature mutations that are also CRAVAT driver mutations occur in genes that are highly mutated in the COSMIC database and are present in the Cancer Gene Census (Supplementary Table 5).
Figure 5. APOBEC signature mutations among potential cancer drivers.
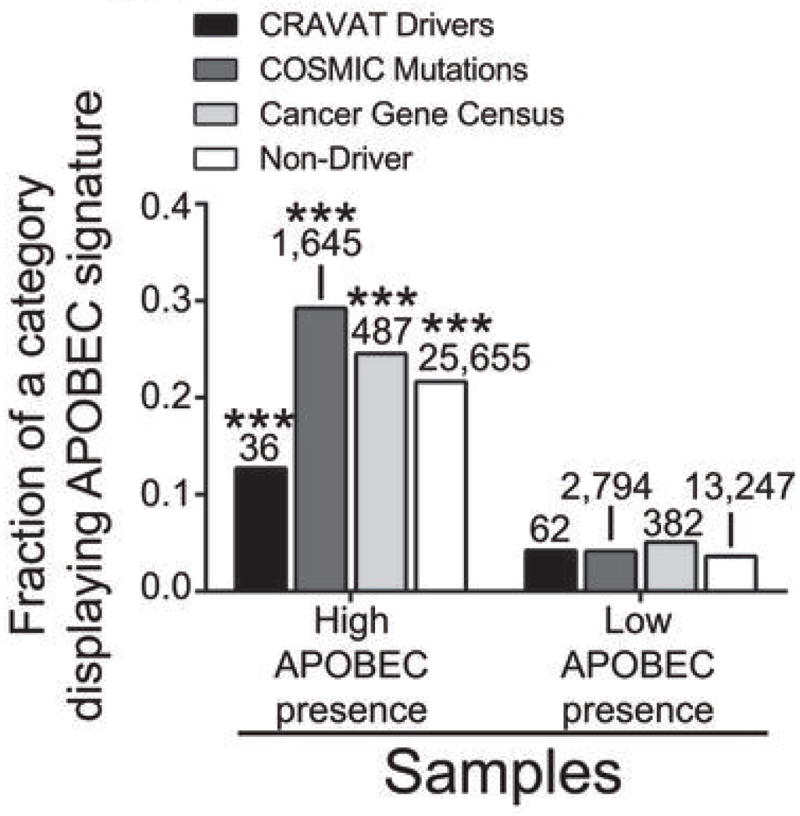
a. The fraction of potential cancer driving mutations that display an APOBEC signature was determined for samples with high (q-value for the enrichment of the APOBEC mutation pattern less than or equal to 0.05; see Fig. 2) and low (q-value >0.05) presence of an exome-wide APOBEC mutation pattern. Mutations were designated as potential cancer drivers by one of three criteria: 1) mutations displaying a Benjamini-Hochberg corrected q-value < 0.05 after CRAVAT analysis 2) mutations that are listed within the COSMIC database and 3) mutations that impact a subset of genes in the Cancer Gene Census whose alteration by missense or nonsense mutations can contribute to cancer. *** indicates p < 0.0001 for a two-sided Chi-square comparing the number of APOBEC and Non-APOBEC signature mutations among potential cancer drivers in samples with high and low presence of the APOBEC mutation pattern for a given criteria defining a driver. Corresponding analysis for non-driver mutations is provided for comparison. The specific mutated genes are presented in Supplementary Table 5.
Discussion
Determining the mutagenic factors that underlie the mix of mutations within tumors is important for a general understanding of carcinogenesis. However, this analysis is daunting as it often requires the testing of numerous poorly defined hypotheses. Here, we have developed a single detailed hypothesis, that APOBEC cytidine deaminases are a significant source of mutagenesis in human cancer genomes. This hypothesis is based on knowledge of the sequence- and single strand-specificity of APOBEC enzymes, their capacity to generate strand-coordinated mutation clusters in model systems and the impressive correlation between experimentally determined APOBEC mutagenesis patterns and the pattern of mutations in strand-coordinated clusters found in cancers. While formally, we cannot exclude that another mutagenic factor may closely mimic both the motif and mutagenic specificities of the APOBEC mutation pattern, there is yet no indication that such a factor exists. Furthermore, our observed correlation between the APOBEC mutagenesis pattern and APOBEC expression in cancer samples provides strong support for this hypothesis. Additional support could be sought in correlations with the germline genotype of patients as soon as such information would be available.
Our TCGA-based analysis indicates a widespread APOBEC mutagenesis pattern and suggests that this pattern is associated with biological mechanisms underlying carcinogenesis. With our approach, we establish a resource for identifying this pattern in the rapidly growing TCGA database as well as in other databases of genome- or exome-wide human mutations. In addition, the predominance of APOBEC-signature mutations across tumors of multiple cancer types sets the next round of questions to be resolved including the identification of the specific APOBEC proteins responsible for mutagenesis, the presence of this mutagenesis in other types and subtypes of cancers, identifying the stage(s) of cancer development that are most prone to APOBEC mutagenesis, and evaluation of the relative impact of this mutagenesis on genome changes that lead to cancer.
Multiple mechanisms could facilitate APOBEC mutagenesis. Environmental and physiological factors may trigger and/or support mutagenesis by (i) affecting the cellular abundance or activity of APOBEC proteins, (ii) altering access to nuclear DNA, and (iii) increasing the amount and/or persistence of ssDNA substrates for APOBEC cytidine deamination. Ours and previous analyses suggest that the level of APOBEC3B transcription impacts APOBEC mutagenesis. How increased 3B transcription levels are established remains unclear. Among the factors that could increase the amount of APOBEC(s) are the presence of viral and retrotransposable elements that these enzymes restrict6,33. Such factors can stimulate APOBEC expression through a complex network of innate immunity signaling including components like Toll-like receptors, interferons, interleukins and even the “usual suspect” in carcinogenesis, the p53 protein34–37. Infection with several viruses38 as well as retrotransposition39 are associated with carcinogenesis; however the mechanisms of this association are far from clear. A potential relationship between APOBEC mutagenesis and viral infection is appealing as cervical, bladder, and head and neck cancer, which are highly associated with HPV infection, display a strong enrichment in APOBEC mutagenesis.
Despite a positive correlation between APOBEC3B expression and APOBEC mutagenesis, the extent of the association is relatively small (Spearman r = 0.30). Thus other factors likely contribute more prominently to APOBEC mutagenesis. Factors which could increase the abundance and persistence of ssDNA include DNA damaging agents40,41 as well as defects in DNA transactions that impede break repair42,43 and replication integrity44,45. Our work in yeast demonstrated that proliferation in the presence of an alkylation agent leads to the formation of ssDNA at DSB sites and dysfunctional forks and subsequently to mutation clusters13. Importantly, a high level of APOBEC deamination may itself lead to DNA breakage27, which could generate a ssDNA substrate for APOBEC hyper-mutation. It is generally acknowledged that carcinogenesis requires the accumulation of multiple genetic changes46. As discussed in13, simultaneous mutations in scattered stretches of ssDNA formed at DSBs, replication forks and other cell contexts would be excellent substrates for APOBEC mutagenesis, which in turn may produce multiple changes without excessive genome-wide mutation and provide a means to accumulate multiple carcinogenic mutations in a single or a few generations.
URLs
TCGA data portal: https://tcga-data.nci.nih.gov/tcga/tcgaDownload.jsp
dbGaP: http://www.ncbi.nlm.nih.gov/gap
21 Breast cancer genomes: URL: ftp://ftp.sanger.ac.uk/pub/cancer/Nik-ZainalEtAl
9 Colorectal Adenocarcinomas: http://www.broadinstitute.org/~lawrence/crc/CRC9.genomic.v3.maf
CRAVAT: www.cravat.us
COSMIC database: http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/
Cancer Gene Census: http://cancer.sanger.ac.uk/cancergenome/projects/census/
On-Line Methods
Genome and exome datasets
Genome and exome datasets were obtained from publications20,21 or from TCGA data portal (https://tcga-data.nci.nih.gov/tcga/tcgaDownload.jsp; Controlled Data Access HTTP Directory). The catalogue of base substitutions identified by whole-genome sequencing in 21 breast cancers was downloaded from URL: ftp://ftp.sanger.ac.uk/pub/cancer/Nik-ZainalEtAl provided in12. Hyperlinks to TCGA datasets and references to published mutation lists are provided in Supplementary Table 3.
Cluster Analysis
Clusters and co-localization between clusters and rearrangement breakpoints in whole-genome data sets were identified as described in13. Analysis of mutation clustering in exomes was conducted similarly to whole-genome cluster finding. Briefly, we first filtered out mutations identical to dbSNPs. This generally constituted a small (0.9%-12.1%) percentage of all exome mutations for a given cancer type. However, lung squamous cell carcinoma (LUSC), kidney renal clear cell carcinoma (KIRC), prostate adenocarcinoma (PRAD) and stomach adenocarcinoma (STAD) samples contained somewhat higher numbers of mutations identical to dbSNPs (19.5%-25.1%). Importantly, each pre-filtered mutation was included in the total number of mutations in the genome, which could only increase the p-values of clusters (see below). We next identified groups of closely-spaced mutations (at most 10 nucleotides between neighbors), which we placed into a complex category. Complex mutations are likely to arise from a mutagenesis event triggered by trans-lesion synthesis across a single DNA lesion49,50. Each complex mutation was counted as a single mutation event. Then all groups of at least 2 mutations in which neighboring changes were separated by 10 kb or less were identified. The p-value for each group was calculated under the assumption that all mutations were distributed randomly across the genome. The total number of mutations in the genome was estimated as 100-fold greater than the number of exome mutations including those identical to dbSNPs.
Cluster p-value was defined as the probability of observing k−1 mutations in x−1 or fewer base pairs and was calculated using a negative binomial distribution as follows:
| (i) |
where x denotes the size of the mutation cluster (size is defined as the number of nucleotides in the region starting at the position of the first and ending at the last mutation of a cluster);
k - denotes the number of mutations observed in a cluster;
-
π - denotes the probability of finding a mutation at any random location in the genome calculated as:
(ii) where n denotes the total number of mutations in a genome and G denotes the total genome size (number of nucleotides).
Groups of mutations were identified as clusters if the calculated p-value was no greater than 10−4. A recursive algorithm was used so that all clusters that met the p-value criteria were identified, even if they were part of a larger group that fit the spacing criterion but did not meet the probability cutoff. Individual mutations and clusters with p-values no greater than 10−4 were classified as follows: Clusters in which all mutations resulted from a change of the same kind of nucleotide were defined as strand-coordinated, while clusters containing mutations of at least two different kinds of bases were called non-coordinated. Mutations that did not belong to a cluster were classified as scattered, while the other category was named clustered.
Detecting an APOBEC mutation pattern
Enrichment
The numeric value of enrichment, E, characterizing the strength of mutagenesis at the tCw motif in mutation clusters was calculated as:
MUTATIONStCw - the number of mutated cytosines (and guanines) falling into tCw (or wGa motif) (W stands for either A or T);
MUTATIONSC or G - the total number of mutated cytosines (and guanines)
CONTEXTtcw - the total number of tcw (or wga) motifs within the area +/−20 nucleotides around mutated cytosines (and guanines)
CONTEXTc or g - the total number of cytosines (or guanines) within the area +/−20 nucleotides around mutated cytosines (and guanines)
For determining the presence of the APOBEC mutagenesis pattern, Enrichment was calculated as above, except only specific base substitutions (tCw→tTw or →tGw, wGa→wAa or →wCa, C→T or →G, and G→A or →C) were included.
Fisher’s Exact Test
Statistical evaluation of the over-representation of APOBEC signature mutations in each sample was performed using a one-sided Fisher’s Exact Test comparing the ratio of the number of C→T or →G substitutions and G→A or →C substitutions that occur in and out of the APOBEC target motif (tCw/wGa) to an analogous ratio for all cytosines and guanines that reside inside and outside of the tCw/wGa motif within a sample fraction of the genome. P-values calculated for multiple samples or multiple comparisons were corrected using the Benjamini-Hochberg method51. Only corrected q-values < 0.05 were considered significant.
Determining the number of breakpoints associated with segmental copy number variations (CNV)
The number of breakpoints associated with segmental CNVs was determined based on TCGA SNP6.0 analysis of 449 breast cancer (BRCA) samples. Breakpoints were identified as pairs of adjacent segments on the same chromosome with a difference in copy-ratio > 0.1. Any segments with fewer than 5 probes were removed from analysis, as being likely due to technical noise.
Defining Cancer Driver Mutations
The on-line software package, CRAVAT (28,29, www.cravat.us), was used to identify potential cancer driving mutations among missense mutations. For acute myeloid leukemia (LAML), breast (BRCA), colorectal (COAD), ovarian (OV), rectal (READ), stomach (STAD), and uterine endometrial (UCEC) cancers, the matched tissue-specific passenger mutation profile provided within CRAVAT package was utilized. For all other cancer types, for which a tissue-specific profile was unavailable, a generic profile was used. CRAVAT outputs include a CHASM score, p-value indicating the likelihood of a mutation being a driver, and a Benjamini-Hochberg (FDR) q-value to correct for multiple hypotheses testing. In our analysis, potential cancer drivers were identified as those mutations with a Benjamini-Hochberg q-value no greater than 0.05. In addition to CRAVAT analysis, two other metrics to identify driver mutations were considered: mutations that occur in the COSMIC database and mutations that alter genes listed in the Cancer Gene Census31, a curated list of genes whose alteration has been shown to be causative in at least some cancers. For the latter metric, only genes in the Cancer Gene Census where missense and nonsense muations are known to be involved in carcinogenesis were used to identify potential drivers.
Analysis of controlled access data
The complete list of analyzed mutations used for making all figures and conclusions in this paper will be submitted as a TCGA sub-study and be available through controlled access to dbGaP (http://www.ncbi.nlm.nih.gov/gap) study phs000178.v7.p6. The file will be in the TCGA MAF format. In addition to the information from the original TCGA MAFs (Supplementary Table 3), the file will contain results of mutation cluster analysis, sequence context of mutations, and CRAVAT analysis. Before the acceptance of the sub-study by TCGA, the file will be available to investigators after they acquire access to controlled TCGA data levels in coordination with DAG.
Supplementary Material
Acknowledgments
We would like to thank Drs. Jack Taylor, Paul Wade, and Dmitri Zaykin for helpful discussions and critical reading of the manuscript. The results published here are in part based upon data generated by The Cancer Genome Atlas project established by the NCI and NHGRI (dbGaP Study Accession: phs000178.v7.p6). The work was supported in part by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (project ES065073 to M.A.R.; Contract GS-23F-9806H and Order: HHSN273201000086U to R.R.S.) and by National Human Genome Research Institute grant U54HG003067 to G.G.
Footnotes
Author Contributions
S.A.R., G.G., and D.A.G designed the study. S.A.R., M.S.L., L.J. K., S.A.G., D.F., P.S., A.K., G.V.K., S.L.C., G.S., S.H., R.R.S., M.A.R., G.G., and D.A.G contributed to data analysis. S.A.R. and D.A.G. wrote the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Loeb LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991;51:3075–9. [PubMed] [Google Scholar]
- 3.Luch A. Nature and nurture - lessons from chemical carcinogenesis. Nat Rev Cancer. 2005;5:113–25. doi: 10.1038/nrc1546. [DOI] [PubMed] [Google Scholar]
- 4.Conticello SG. Creative deaminases, self-inflicted damage, and genome evolution. Ann N Y Acad Sci. 2012;1267:79–85. doi: 10.1111/j.1749-6632.2012.06614.x. [DOI] [PubMed] [Google Scholar]
- 5.Pavri R, Nussenzweig MC. AID targeting in antibody diversity. Advances in immunology. 2011;110:1–26. doi: 10.1016/B978-0-12-387663-8.00005-3. [DOI] [PubMed] [Google Scholar]
- 6.Smith HC, Bennett RP, Kizilyer A, McDougall WM, Prohaska KM. Functions and regulation of the APOBEC family of proteins. Semin Cell Dev Biol. 2012;23:258–68. doi: 10.1016/j.semcdb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suspene R, et al. Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4858–63. doi: 10.1073/pnas.1009687108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shinohara M, et al. APOBEC3B can impair genomic stability by inducing base substitutions in genomic DNA in human cells. Sci Rep. 2012;2:806. doi: 10.1038/srep00806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burns MB, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494:366–70. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stephens P, et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nature genetics. 2005;37:590–2. doi: 10.1038/ng1571. [DOI] [PubMed] [Google Scholar]
- 11.Beale RC, et al. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. Journal of molecular biology. 2004;337:585–96. doi: 10.1016/j.jmb.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 12.Nik-Zainal S, et al. Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roberts SA, et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Molecular cell. 2012;46:424–35. doi: 10.1016/j.molcel.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drier Y, et al. Somatic rearrangements across cancer reveal classes of samples with distinct patterns of DNA breakage and rearrangement-induced hypermutability. Genome Res. 2012 doi: 10.1101/gr.141382.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibbs PE, Lawrence CW. Novel mutagenic properties of abasic sites in Saccharomyces cerevisiae. Journal of molecular biology. 1995;251:229–36. doi: 10.1006/jmbi.1995.0430. [DOI] [PubMed] [Google Scholar]
- 16.Simonelli V, Narciso L, Dogliotti E, Fortini P. Base excision repair intermediates are mutagenic in mammalian cells. Nucleic acids research. 2005;33:4404–11. doi: 10.1093/nar/gki749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan K, et al. Base Damage within Single-Strand DNA Underlies In Vivo Hypermutability Induced by a Ubiquitous Environmental Agent. PLoS genetics. 2012;8:e1003149. doi: 10.1371/journal.pgen.1003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Senavirathne G, et al. Single-stranded DNA scanning and deamination by APOBEC3G cytidine deaminase at single molecule resolution. J Biol Chem. 2012;287:15826–35. doi: 10.1074/jbc.M112.342790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chelico L, Pham P, Goodman MF. Mechanisms of APOBEC3G-catalyzed processive deamination of deoxycytidine on single-stranded DNA. Nature structural & molecular biology. 2009;16:454–5. doi: 10.1038/nsmb0509-454. author reply 455–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barbieri CE, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nature genetics. 2012;44:685–9. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stransky N, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bass AJ, et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nature genetics. 2011;43:964–8. doi: 10.1038/ng.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shammas MA, et al. Dysfunctional homologous recombination mediates genomic instability and progression in myeloma. Blood. 2009;113:2290–7. doi: 10.1182/blood-2007-05-089193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu P, Carvalho CM, Hastings PJ, Lupski JR. Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev. 2012;22:211–20. doi: 10.1016/j.gde.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.TCGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parker JS, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27:1160–7. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landry S, Narvaiza I, Linfesty DC, Weitzman MD. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO reports. 2011;12:444–450. doi: 10.1038/embor.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carter H, et al. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res. 2009;69:6660–7. doi: 10.1158/0008-5472.CAN-09-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Douville C, et al. CRAVAT: Cancer-Related Analysis of VAriants Toolkit. Bioinformatics. 2013 doi: 10.1093/bioinformatics/btt017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forbes SA, et al. The Catalogue of Somatic Mutations in Cancer (COSMIC) Curr Protoc Hum Genet. 2008;Chapter 10(Unit 10):11. doi: 10.1002/0471142905.hg1011s57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Futreal PA, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–83. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lampson BL, et al. Rare Codons Regulate KRas Oncogenesis. Current biology: CB. 2012 doi: 10.1016/j.cub.2012.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schumacher AJ, Nissley DV, Harris RS. APOBEC3G hypermutates genomic DNA and inhibits Ty1 retrotransposition in yeast. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:9854–9. doi: 10.1073/pnas.0501694102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Einav U, et al. Gene expression analysis reveals a strong signature of an interferon-induced pathway in childhood lymphoblastic leukemia as well as in breast and ovarian cancer. Oncogene. 2005;24:6367–75. doi: 10.1038/sj.onc.1208797. [DOI] [PubMed] [Google Scholar]
- 35.Refsland EW, et al. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic acids research. 2010;38:4274–84. doi: 10.1093/nar/gkq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Menendez D, Shatz M, Resnick MA. Interactions between the tumor suppressor p53 and immune responses. Current Opinion in Oncology. 2013;25:85–92. doi: 10.1097/CCO.0b013e32835b6386. [DOI] [PubMed] [Google Scholar]
- 37.Zhou L, et al. Activation of toll-like receptor-3 induces interferon-lambda expression in human neuronal cells. Neuroscience. 2009;159:629–37. doi: 10.1016/j.neuroscience.2008.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biological agents. Volume 100 B. A review of human carcinogens. IARC Monogr Eval Carcinog Risks Hum. 2012;100:1–441. [PMC free article] [PubMed] [Google Scholar]
- 39.Lee E, et al. Landscape of somatic retrotransposition in human cancers. Science. 2012;337:967–71. doi: 10.1126/science.1222077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lopes M, Foiani M, Sogo JM. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Molecular cell. 2006;21:15–27. doi: 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 41.Pages V, Fuchs RP. Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science. 2003;300:1300–3. doi: 10.1126/science.1083964. [DOI] [PubMed] [Google Scholar]
- 42.Bouwman P, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nature structural & molecular biology. 2010;17:688–95. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bunting SF, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–54. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bando M, et al. Csm3, Tof1, and Mrc1 form a heterotrimeric mediator complex that associates with DNA replication forks. The Journal of biological chemistry. 2009;284:34355–65. doi: 10.1074/jbc.M109.065730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Katou Y, et al. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature. 2003;424:1078–83. doi: 10.1038/nature01900. [DOI] [PubMed] [Google Scholar]
- 46.Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet. 2012;13:795–806. doi: 10.1038/nrg3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chapman MA, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–72. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berger MF, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–20. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harfe BD, Jinks-Robertson S. DNA polymerase zeta introduces multiple mutations when bypassing spontaneous DNA damage in Saccharomyces cerevisiae. Molecular cell. 2000;6:1491–9. doi: 10.1016/s1097-2765(00)00145-3. [DOI] [PubMed] [Google Scholar]
- 50.Sakamoto AN, et al. Mutator alleles of yeast DNA polymerase zeta. DNA Repair (Amst) 2007;6:1829–38. doi: 10.1016/j.dnarep.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Methodological. 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.