

Acute promyelocytic leukemias (APLs) are characterized by the expression of the PML-RARA oncogene, which is a product of the 15;17 chromosomal translocation. Two lines of evidence suggest that this genetic alteration is involved in the initiation of promyelocytic leukemogenesis in vivo: i) the t(15;17) often represents the only cytogenetic abnormality in an otherwise-normal karyotype; ii) PML-RARA is able to initiate leukemogenesis in mouse models on expression in hematopoietic stem cells (HSCs) or progenitors, giving rise to a disease that recapitulates clinical and morphological features of human APLs.1 APL onset in mice, however, occurs after a long latency and with a low penetrance, suggesting that, like other cancers, APL is a multistep disease, whereby PML-RARA cooperates with secondary mutations for the full development of the leukemia phenotype. Notably, in the pre-leukemic phase of the disease, PML-RARA expression induces DNA damage in HSCs/progenitors and activates DNA repair, thus imparting a mutator phenotype that might contribute to disease progression.2, 3
Next-generation sequencing represents a powerful tool for the discovery of genetic alterations at high resolution. To identify gene mutations that might cooperate with PML-RARA in the leukemogenic process, we performed whole-exome sequencing of 5 leukemias that developed in PML-RARA transgenic mice (mouse APLs; mAPLs) and 11 patients' leukemias expressing PML-RARA (human APLs; hAPLs) (Supplementary Table 1). For the scope of this investigation, we only considered non-synonymous single-nucleotide variants (SNVs) and small insertions/deletions (indels) occurring in the tumor DNA with an allelic frequency ⩾25% (corresponding to a >50% frequency of cells carrying the mutation in the tumor sample, assuming the majority of mutations to be heterozygous). These experimental conditions allow identification of SNVs with a validation rate of 100%, as shown by an independent sequencing approach (Sanger sequencing; unpublished). We identified a total of 18 mutations in the 5 mAPLs (16 SNVs and 2 indels) and 73 mutations in the 11 hAPLs (59 SNVs and 14 indels) (Table 1 and Supplementary Table 2). We validated all the 16 mAPL SNVs by Sanger sequencing (the 2 indels were not amplifiable for technical reasons), 18/18 randomly selected human SNVs and 1/1 indel (Supplementary Table 2).
Table 1. Mutations identified by next-generation sequencing in APLs.
| Cases |
Mutations |
Study | |||
|---|---|---|---|---|---|
| nsSNVs | Indels | SJMs | Total | ||
| Mouse APLs | |||||
| mAPL#Mi1 | 0 | 0 | 0 | 0 | Present |
| mAPL#Mi2 | 1 | 0 | 0 | 1 | Present |
| mAPL#Mi3 | 9 | 1 | 0 | 10 | Present |
| mAPL#Mi4 | 6 | 1 | 0 | 7 | Present |
| mAPL#Mi5 | 0 | 0 | 0 | 0 | Present |
| mAPL | 3 | 0 | 0 | 3 | Wartman et al.4 |
| Total mutations (per Pt.) | 19 (3.16) | 2 (0.33) | 0 | 21 (3.50) | 6 cases total |
| Human APLs | |||||
| hAPL#Mi1 | 5 | 0 | 0 | 5 | Present |
| hAPL#Mi2 | 13 | 3 | 0 | 16 | Present |
| hAPL#Mi3 | 2 | 1 | 0 | 3 | Present |
| hAPL#Mi4 | 3 | 3 | 0 | 6 | Present |
| hAPL#Mi5 | 0 | 0 | 0 | 0 | Present |
| hAPL#Mi6 | 5 | 1 | 0 | 6 | Present |
| hAPL#Mi7 | 12 | 2 | 0 | 14 | Present |
| hAPL#Mi8 | 1 | 1 | 0 | 2 | Present |
| hAPL#Mi9 | 4 | 1 | 0 | 5 | Present |
| hAPL#Mi10 | 7 | 1 | 0 | 8 | Present |
| hAPL#Mi11 | 7 | 1 | 0 | 8 | Present |
| hAPL#1 | 5 | 2 | 0 | 7 | Greif et al.5 |
| hAPL#2 | 3 | 0 | 0 | 3 | Greif et al.5 |
| hAPL#3 | 4 | 0 | 0 | 4 | Greif et al.5 |
| hAPL | 12 | 0 | 0 | 12 | Welch et al.6 |
| TCGA-AB-2803 | 12 | 1 | 0 | 13 | TCGA7 |
| TCGA-AB-2804 | 7 | 1 | 0 | 8 | TCGA7 |
| TCGA-AB-2823 | 0 | 1 | 0 | 1 | TCGA7 |
| TCGA-AB-2840 | 0 | 1 | 0 | 1 | TCGA7 |
| TCGA-AB-2841 | 4 | 0 | 0 | 4 | TCGA7 |
| TCGA-AB-2862 | 7 | 0 | 0 | 7 | TCGA7 |
| TCGA-AB-2872 | 9 | 1 | 0 | 10 | TCGA7 |
| TCGA-AB-2897 | 5 | 0 | 1 | 6 | TCGA7 |
| TCGA-AB-2905 | 15 | 2 | 0 | 17 | TCGA7 |
| TCGA-AB-2906 | 9 | 1 | 0 | 10 | TCGA7 |
| TCGA-AB-2980 | 3 | 1 | 0 | 4 | TCGA7 |
| TCGA-AB-2982 | 1 | 1 | 0 | 2 | TCGA7 |
| TCGA-AB-2991 | 10 | 0 | 0 | 10 | TCGA7 |
| TCGA-AB-2994 | 6 | 1 | 0 | 7 | TCGA7 |
| TCGA-AB-2997 | 9 | 1 | 0 | 10 | TCGA7 |
| TCGA-AB-2998 | 4 | 1 | 0 | 5 | TCGA7 |
| TCGA-AB-2999 | 9 | 0 | 0 | 9 | TCGA7 |
| TCGA-AB-3001 | 8 | 0 | 2 | 10 | TCGA7 |
| TCGA-AB-3007 | 5 | 1 | 1 | 7 | TCGA7 |
| TCGA-AB-3012 | 6 | 2 | 1 | 9 | TCGA7 |
| Total in hAPLs (per Pt.) | 212 (6.06) | 32 (0.91) | 5 (0.14) | 249 (7.11) | 35 cases |
| Total h+mAPLs (per Pt.) | 231 (5.63) | 34 (0.83) | 5 (0.12) | 270 (6.59) | 41 cases |
Abbreviations: APL, acute promyelocytic leukemia; h, human; indels, small insertion/deletions; m, mouse; nsSNV, non-synonymous single nucleotide variant; per Pt., per patient; SJM, splice junction mutation; TCGA, The Cancer Genome Atlas.
We next combined the results of our sequencing analyses with those previously published for APLs (1 mAPL and 24 hAPLs),4, 5, 6, 7 obtaining a data set of 41 APL samples (6 mAPLs and 35 hAPLs). Analysis of this data set showed a total of 270 mutations affecting 248 human genes (231 SNVs, 34 indels and 5 splice junction mutations—SJMs), with a low frequency of mutations per case (∼7.1 in hAPLs and ∼3.5 in mAPLs). In three cases (two mAPLs and one hAPL), we found no SNVs or indels with a frequency higher than 25%. Surprisingly, two hAPL samples showed several hundreds of low-frequency mutations, whose significance remains unclear (not shown).
Of the 248 mutated genes, 9 were found in ⩾2 patients (recurrent mutations), and 8 showed a significantly higher mutation rate (q<0.005; FLT3; WT1; KRAS; CALR; CSMD1; DDR2; REV3L and TCERG1L; Table 2 and Supplementary Table 3). FLT3 and KRAS have been already described as cooperators of PML-RARA in mouse models of APLs,8 whereas WT1 is infrequently mutated in hAPLs.9 Of the remaining five, the DDR2 (discoidin domain receptor 2) tyrosine kinase is mutated in a small subset of squamous cell lung cancer. Notably, DDR2 mutations are critical oncogenic events for these tumors and confer high sensitivity to the multi-targeted kinase inhibitor dasatinib.10 We found a total of 25 mutations affecting these eight genes (APL driver mutations), with FLT3 and WT1 being the most frequently involved (Table 2).
Table 2. Genes with a significantly higher mutation rate in APLs and AMLs.
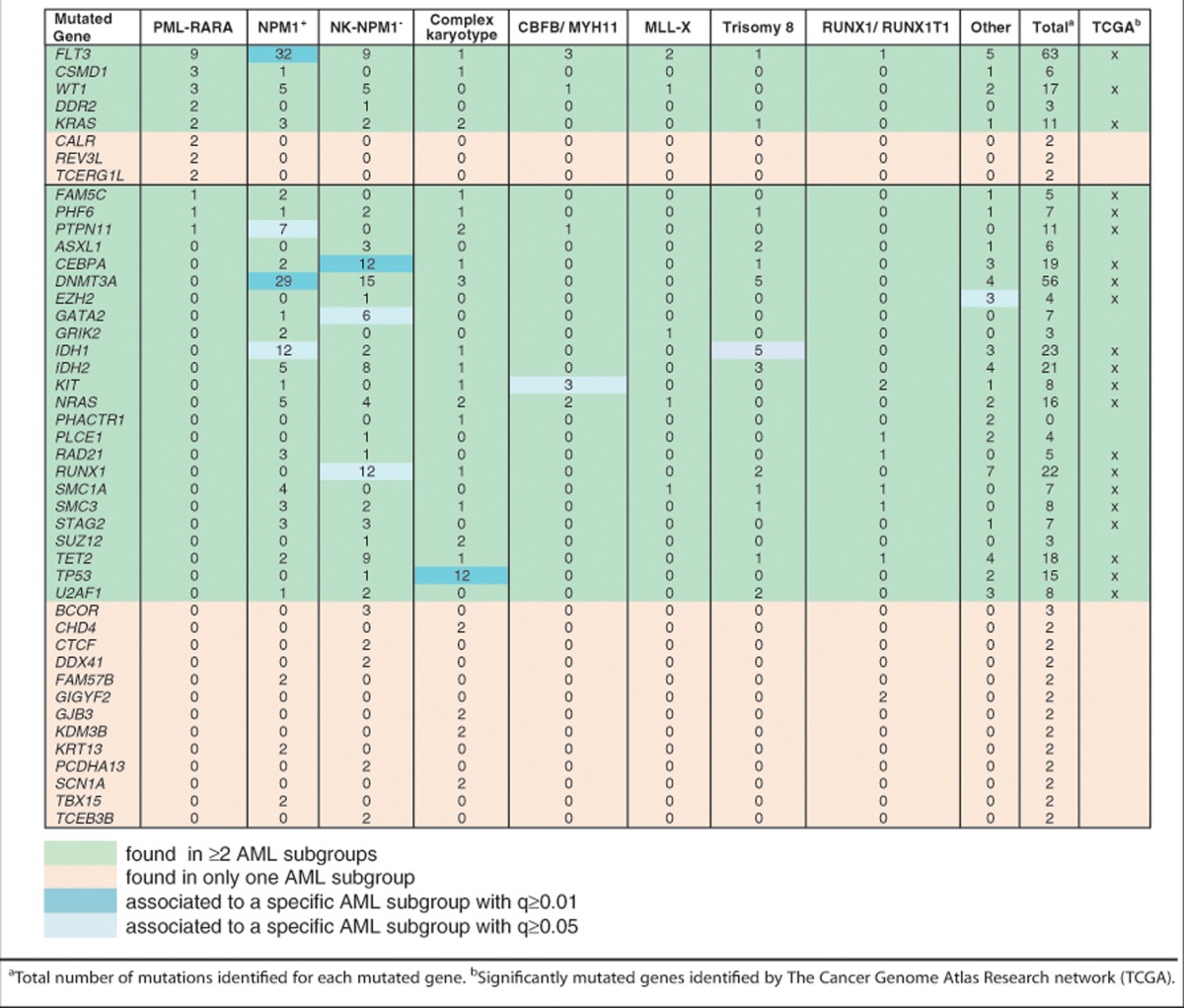
We next investigated the APL specificity of the identified mutations, as compared with other subgroups of acute myeloid leukemias (AMLs). First, we generated a data set of mutations in all the available AML samples (n=206; 196 previously published samples7, 11, 12, 13, 14 and 10 new samples from this study (Supplementary Tables 1 and 2)) and divided the AML samples in different genetic/cytogenetic subgroups: i) samples with mutations of nucleophosmin (NPM1+: 58 cases); ii) normal karyotype without NPM1 mutations (NK-NPM1−: 58 cases); iii) complex karyotype (n=22); iv) translocations or inversions affecting CBFB/MYH11 (n=11); v) t(8;21) RUNX1/RUNX1T1 (n=7); vi) trisomy 8 (n=8); vii) MLL-X translocations (n=10). Twenty-seven cases did not fall into any of these categories (‘Other'). As for PML-RARA, indirect evidence from mouse models suggests that mutated NPM115 and fusion proteins of CBFB, RUNX1 and MLL are initiating mutations for AMLs (reviewed in McCormack et al.8).
AML mutations were separately analyzed in each of the AML subgroups. We found a total of 1360 mutated genes (∼9 mutations per case), of which 153 were recurrently mutated (⩾2 patients in the same subgroup) and 40 showed a significantly higher mutation rate (q<0.005; Table 2 and Supplementary Table 3). Notably, included in the 40 genes were 21 of the 22 significantly mutated genes identified in a recent analysis of 200 AMLs (also part of our data set7), and additional mutated genes critical for AMLs (BCOR, ASXL1, GATA2, SUZ12 and DDX41) or for selected epithelial cancers (CTCF, PLCE1 and CHD4). The most frequently mutated genes were also significantly associated with specific AML subgroups: FLT3, IDH1, DNMT3A and PTPN11 with NPM1+ AMLs; RUNX1, CEBPA and GATA2 with NK-NPM1− AMLs; TP53 with AMLs with complex karyotypes; KIT with CBFB/MYH11 AMLs; IDH1 with AMLs with trisomy 8 (Table 2). Moreover, mutations in SF3B1, PTPN11, DNAH9, are present in both human and mouse leukemias.
We then analyzed the distribution of the significantly mutated APL and AML genes (n=44) across all samples (n=239). Twenty-eight genes (∼64%) were mutated in more than one cytogenetic subgroup, covering 383 of the 416 mutations identified in all samples (∼92%). In the remaining 16 genes (∼36%), mutations were instead associated with a specific subgroup, corresponding to just 33 of the identified mutations (∼8%) and suggesting that subgroup specificity might be due to their low frequency. Indeed, we found a significant correlation between frequency of mutations per gene and numbers of subgroups where it is mutated (Spearman's coefficient value of 0.93). As regards APLs, 5 of the 8 significantly mutated genes were also found in other AML cytogenetic subgroups (FLT3; WT1; KRAS; CSMD1 and DDR2), covering a total of 100 mutations, while 3 (CALR; REV3L and TCERG1L) were only found in APLs, covering 6 mutations. Three genes that were found significantly mutated in other AML subgroups were also mutated in APLs (PHF6, FAM5C and PTPN11).
Together, these results imply that different myeloid leukemias, including APLs, share the same subset of cooperating mutations, and are consistent with a scenario whereby specific initiating mutations interact with a common pool of highly heterogeneous, yet phenotypically equivalent, cooperating mutations. Indirect evidence, however, suggests that the pool of cooperating mutations in AMLs is not yet entirely defined, and that sequencing of additional AMLs is needed. In fact, due to the limited size of some samples (for example, in selected AML subgroups such as those with rearrangements of CBFB, RUNX1 or MLL), it is likely that our statistical analyses do not allow the identification of all the driver mutations in AML. Among all the mutated genes (n=1559), ∼3% (n=45) were mutated at statistically significant frequency (driver mutations). Among the others (passenger mutations), however, we identified mutations that have been causally implicated in the pathogenesis of AMLs (for example, ETV6, JAK2, NOTCH1, NUMA1, PRDM16, CBL, CBFB, CHIC2, ELF4, NSD1 and PDGFRB) or other cancers (for example, PTEN, MYC, ARID1A, SF3B1, EGFR, NF1, THRAP3, MED12, KDR, IKZF1, DAXX and SETD2).
Acknowledgments
LR was supported by a Reintegration AIRC/Marie Curie International Fellowship in Cancer Research. This work was supported by grants from Fondazione Cariplo to LR; Associazione Italiana Ricerca sul Cancro (AIRC) and Italian Ministry of Health to PGP; European Commission (FP7) to GM. We thank Paola Dalton for critical review of the manuscript.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Blood Cancer Journal website (http://www.nature.com/bcj)
Supplementary Material
References
- Grisolano JL, Wesselschmidt RL, Pelicci PG, Ley TJ. Altered myeloid development and acute leukemia in transgenic mice expressing PML-RAR alpha under control of cathepsin G regulatory sequences. Blood. 1997;89:376–387. [PubMed] [Google Scholar]
- Viale A, De Franco F, Orleth A, Cambiaghi V, Giuliani V, Bossi D, et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature. 2009;457:51–56. doi: 10.1038/nature07618. [DOI] [PubMed] [Google Scholar]
- Insinga A, Cicalese A, Faretta M, Gallo B, Albano L, Ronzoni S, et al. DNA damage in stem cells activates p21, inhibits p53, and induces symmetric self-renewing divisions. Proc Natl Acad Sci USA. 2013;110:3931–3936. doi: 10.1073/pnas.1213394110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartman LD, Larson DE, Xiang Z, Ding L, Chen K, Lin L, et al. Sequencing a mouse acute promyelocytic leukemia genome reveals genetic events relevant for disease progression. J Clin Invest. 2011;121:1445–1455. doi: 10.1172/JCI45284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greif PA, Yaghmaie M, Konstandin NP, Ksienzyk B, Alimoghaddam K, Ghavamzadeh A, et al. Somatic mutations in acute promyelocytic leukemia (APL) identified by exome sequencing. Leukemia. 2011;25:1519–1522. doi: 10.1038/leu.2011.114. [DOI] [PubMed] [Google Scholar]
- Welch JS, Westervelt P, Ding L, Larson DE, Klco JM, Kulkarni S, et al. Use of whole-genome sequencing to diagnose a cryptic fusion oncogene. JAMA. 2011;305:1577–1584. doi: 10.1001/jama.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack E, Bruserud O, Gjertsen BT. Review: genetic models of acute myeloid leukaemia. Oncogene. 2008;27:3765–3779. doi: 10.1038/onc.2008.16. [DOI] [PubMed] [Google Scholar]
- Gaur GC, Ramadan SM, Cicconi L, Noguera NI, Luna I, Such E, et al. Analysis of mutational status, SNP rs16754, and expression levels of Wilms tumor 1 (WT1) gene in acute promyelocytic leukemia. Ann Hematol. 2012;91:1855–1860. doi: 10.1007/s00277-012-1546-7. [DOI] [PubMed] [Google Scholar]
- Hammerman PS, Sos ML, Ramos AH, Xu C, Dutt A, Zhou W, et al. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov. 2011;1:78–89. doi: 10.1158/2159-8274.CD-11-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greif PA, Dufour A, Konstandin NP, Ksienzyk B, Zellmeier E, Tizazu B, et al. GATA2 zinc finger 1 mutations associated with biallelic CEBPA mutations define a unique genetic entity of acute myeloid leukemia. Blood. 2012;120:395–403. doi: 10.1182/blood-2012-01-403220. [DOI] [PubMed] [Google Scholar]
- Greif PA, Eck SH, Konstandin NP, Benet-Pages A, Ksienzyk B, Dufour A, et al. Identification of recurring tumor-specific somatic mutations in acute myeloid leukemia by transcriptome sequencing. Leukemia. 2011;25:821–827. doi: 10.1038/leu.2011.19. [DOI] [PubMed] [Google Scholar]
- Grossmann V, Tiacci E, Holmes AB, Kohlmann A, Martelli MP, Kern W, et al. Whole-exome sequencing identifies mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118:6153–6163. doi: 10.1182/blood-2011-07-365320. [DOI] [PubMed] [Google Scholar]
- Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43:309–315. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- Vassiliou GS, Cooper JL, Rad R, Li J, Rice S, Uren A, et al. Mutant nucleophosmin and cooperating pathways drive leukemia initiation and progression in mice. Nat Genet. 2011;43:470–475. doi: 10.1038/ng.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.