

Abstract
Signaling by vitamin A through its active metabolite retinoic acid (RA) is critical for the normal development and functions of the hematopoietic and immune systems. B cells, as both factories for antibody production and part of the immune regulatory system, are critical to a successful vaccination response. RA is a factor in the development and competence of mature B cells, in B cell proliferation, and in the regulation of transcription factors associated with B cell differentiation, class switch recombination, and the generation of antibody-secreting plasma cells. Emerging evidence suggests that RA can function alone and in combination with other immune system stimuli to augment the formation of germinal centers, leading to increased primary and secondary antibody responses. Taken together, RA could be a useful component in vaccine strategies and/or for immunotherapy.
I. Introduction
Vitamin A has long been implicated as an essential nutritional factor for normal immunity. In the 1920s, vitamin A was named “the anti-infective vitamin,” based on observations that vitamin A-deficient animals succumbed to infectious disease, while vitamin A-adequate animals recovered and survived. In humans, vitamin A deficiency is associated with increased mortality in children and pregnant women (Van et al., 2002; West, 2002). Providing vitamin A supplements to vitamin A-deficient children, ages 6–72 months, reduces all-cause mortality by 23%, measles-related mortality by 50%, and diarrheal disease mortality by 33% (WHO/UNICEF, 1998; and previous reviews). The anti-infective effect of vitamin A could be partially attributable to the prevention of VA deficiency. Enhanced immunity may also be involved, due to vitamin A or its active metabolite, retinoic acid (RA), which has gained attention due to its multiple effects on innate and adaptive immunity, including its ability to modulate cytokine production (Ma et al., 2005), promote the development of Th2 cells (Hoag et al., 2002), induce gut-homing T cells (Iwata et al., 2004) and T-regulatory cells (Benson et al., 2007), regulate Th-17 cells (Mucida et al., 2007), stimulate B-cell maturation (Chen and Ross, 2007; Wei et al., 2007), and increase primary and memory antibody responses (DeCicco and Ross, 2000; DeCicco et al., 2000; Ma et al., 2005).
In this chapter, we first discuss vitamin A and RA signaling, which is important in all organ systems, including the hematopoietic and immune systems. We then focus on B cells as a target of retinoid action. B cells are critical to a successful vaccination response, and antibodies are a central hallmark of adaptive immunity. All of the presently licensed vaccines “work” by eliciting antibodies, which recognize and bind to the pathogen and then activate the body’s immune system to destroy any of these micro-organisms that it later encounters. Hence, understanding the roles of vitamin A in the regulation of B cells and B-cell responses has important implications for vaccine development and effective immunization. B cells are not only important as “factories” for the production of antibodies, but are also one of the types of immune system cell classified as professional antigen-presenting cells (APC). Additionally, B cells may be more multi-functional than previously understood, with recent evident for populations of regulatory B cells (DiLillo et al., 2010). Here, we first discuss RA as a factor in the development and competence of mature B cells, then as a factor in B-cell proliferation, and in the regulation of transcription factors associated with B-cell differentiation, class switch recombination (CSR), and the generation of antibody-secreting plasma cells (PC). We conclude with emerging evidence that RA can function alone and in combination with other immune system stimuli to augment the formation of germinal centers (GC), leading to increased primary and secondary antibody responses. Taken together, the evidence supports the essentiality of vitamin A for normal functions, while suggesting that RA itself could be useful component in vaccine strategies and/or for immunotherapy.
II. The Vitamin A–Retinoic Acid Signaling System
A. Nutritional physiology and functions
Vitamin A is a fat-soluble micronutrient that is required in the diet of all chordates. Although retinol itself has no defined independent activity, it is essential as the precursor for the generation of retinal, a component of rhodopsin, and of all-trans-RA, the principal ligand for the nuclear retinoid receptors, RARα, β, and γ. These class 2 nuclear receptor proteins form a heterodimeric complex with retinoid X receptors (RXR) and together the RAR–RXR function as important transcription factors (Altucci and Gronemeyer, 2001; Bastien and Rochette-Egly, 2004; Wei, 2003). The target DNA sequences in retinoid-regulated target genes to which the RAR–RXR bind, termed RA response elements (RARE), often comprise a direct repeat of the hexanucleotide sequence A(G/A)GTCA with either two or five intervening nucleotides. Current evidence supports a model in which most RAR–RXR proteins are bound to target DNA sequences in an inactive, repressed state in the absence of ligand. The binding of ligand, for example, of physiologically produced all-trans-RA, or a suitable exogenous analog, to the RAR triggers a conformational change, specifically a large shift in the position of helix-12 of the RAR protein that, in turn, strengthens the binding of the RAR–RXR to DNA and to coactivator or core-pressor molecules that are part of a multiprotein complex that regulates the activity of DNA-dependent RNA polymerase (Altucci and Gronemeyer, 2001; Balmer and Blomhoff, 2002; Bastien and Rochette-Egly, 2004; Wei, 2003). In addition to all-trans-RA, numerous synthetic retinoid analogs possess agonistic activity, while other act as antagonistically. Several dozen genes are now considered bona fide direct targets of RA (Balmer and Blomhoff, 2002). However, several hundred other genes have been shown to respond in a physiological manner to RA, but direct or indirect mechanisms for these genes have not yet been established. Cell differentiation is often closely controlled by retinoid signaling through the RAR–RXR dimer, making retinoids of great interest in normal biology, as well as in the field of cancer prevention and differentiation therapy (Altucci and Gronemeyer, 2001; Fields et al., 2007; Vitoux et al., 2007). The RXR also form heterodimers with numerous other nuclear receptors including the vitamin D receptor, thyroid hormone receptor, peroxisome proliferator activator receptors (PPARs), lipid-activated receptors (LXR, FXR), and xenobiotic-activated receptors (PXR, CAR; Altucci and Gronemeyer, 2001). Hence, RXR signaling has both a retinoid-specific component mediated by RAR–RXR actions, and a very broad component due to the involvement of RXR with other nuclear receptors.
B. Clinical and experimental uses of RA
Besides being produced in vivo in a highly regulated manner (Napoli, 2000; Ross et al., 2001), RA is also used clinically, with applications in the treatment of skin disorders and certain cancers, including leukemias (Fields et al., 2007; Vitoux et al., 2007). It is important to keep in mind that physiologically produced and exogenously administered RA may act differently, due to differences in achieved concentrations or distribution profiles of pharmacological levels of RA (Muindi et al., 1994). Due to its lipophilic nature, RA is readily taken into cells by passive diffusion. In experimental studies, RA is often added directly to cells or organ cultures, sometimes at much higher concentrations than the 10–20 nM level present in normal plasma, or it may be administered to animals by a nonphysiological route, such as via injections (i.p., or s.c., resulting in a “bolus” effect), or in slow-release pellets implanted under the skin, which may deliver low or high doses depending on the retinoid loading and type of pellet. Even orally administered RA is not truly physiological since RA is not present at substantial levels in the diet (Ross, 2010). Despite these caveats, the use of RA administered by these routes may reveal potential mechanisms by which RA can act as a regulator of the immune system. However, it is also possible that the natural, local production of RA is integral to its actual physiological functions, in which case exogenous RA may exert actions that would not normally be observed in vivo. As important immunoregulatory roles for RA are further suggested, it will be important to integrate studies of the biological production and catabolism of RA into the larger picture, if a true understanding of the actions of vitamin A and RA in the immune system is to be achieved. Surprisingly, no systematic study has yet been conducted of VA concentrations in the organs of the immune system. However, based on analysis of casual samples, little retinyl ester is stored in the thymus, spleen, or lymph nodes. If this is correct, then immune system tissues depend on plasma retinol, and would be expected to be affected by low plasma levels of vitamin A, such as in situations of marginal or overt vitamin A deficiency. As discussed below, B cells are very sensitive to RA at physiological concentrations, with in vitro responses often produced by the addition of 10–20 nM of RA, similar to the physiological concentration of RA in plasma.
III. RA as a Factor in B-Cell Maturation, Activation, and Proliferation
A. Immunocompetence and initial activation
Retinoid signaling is important in all organ systems, including the hemato-poietic and immune systems (Ross, 1994). Vitamin A-deficient animals exhibit abnormalities of lymphocyte numbers in plasma and spleen, with reduced T cell and sometimes B-cell populations, and, generally, increases in myeloid cells and especially granulocytes (Kuwata et al., 2000; Zhao and Ross, 1995), whereas RA inhibits granulocyte–macrophage colony-stimulating factor production and granulocyte development (Smeland et al., 1994), and reverses the effects of vitamin A-deficient state in vivo (Zhao and Ross, 1995). Animal experiments conducted from several angles have demonstrated that RA signaling plays a critical role in B-lymphoid development. The B cell is the major cell type that mediates the humoral immune response. After lineage development in bone marrow, naive B cells enter the circulation and reside in the secondary lymphoid organs, such as lymph nodes, tonsils, and spleen, and become follicular and marginal zone B cells, depending on location, or they recirculate to the bone marrow to reside in sinuses, where they may receive signal from T cells and/or provide surveillance against the blood-borne antigens. Vitamin A and RA regulate the maturation and differentiation of B cells at multiple levels that, in combination, regulate and often potentiate antibody production overall.
Vitamin A deficiency has been shown to reduce the number of fetal B-cell progenitors, while the pan-RAR antagonist, LE540, inhibited both fetal and adult B lymphopoiesis, as studied in vitro (Chen et al., 2009). In another in vitro study that used RA at a physiological concentration, although RA inhibited the proliferation of normal B-cell progenitors of both mice and humans (Fahlman et al., 1995), it affected multiple stages of B lymphopoiesis and accelerated the generation of CD19+sIgM+ B cells (Chen et al., 2008). These results suggest that RA helps to provide a microenvironment that favors B-cell development and maintains a functional B-cell pool that is essential for the response to antigen (Fig. 5.1).
Figure 5.1.
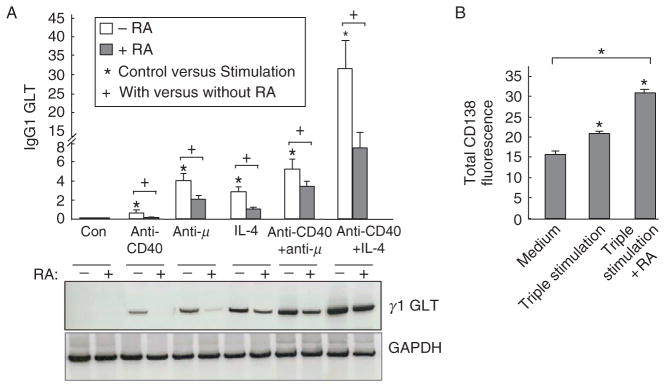
Retinoic acid decreased stimulation-induced IgG1 germline transcript expression. B cells were cultured in vitro with stimulation as indicated for different times. (A) RA inhibited CD40-ligation-induced γ1 GLT in B cells were variously stimulated with anti-CD40 (1 μg/ml), anti-μ (1 μg/ml), or IL-4 (2 ng/ml) with and without 20 nM RA for 48 h. A representative PCR gel image is showed along with the chart. Data shown were normalized to GAPDH mRNA. (B) RA increased CD138 expression on activated B cells. Flow-sorted CD138-negative B cells were cultured with medium alone or with triple stimulation (anti-μ, anti-CD40, and IL-4) in the presence and absence of 20 nM RA. After 5 days, cells were stained with anti-CD138-PE antibody. Mean ± SEM; ★P < 0.05. (Figure modified from Chen and Ross, 2007, with permission of Cellular Immunology.)
The majority of mature B cells enter the circulation and reside in the peripheral lymphoid organs. The ligation of the B-cell receptor (BCR) by cognate antigen initiates B-cell activation, which is positively or negatively modulated by the interaction of other signals generated through various receptors present on the surface of the B cell. Several important receptors include CD19, a coreceptor for the BCR (Ishiura et al., 2010); the Toll-like receptors, notably TLR4 which binds lipopolysaccharide (LPS), a well known direct mitogen for B cells; CD40, a receptor for the CD40 ligand expressed by activated T cells, which provides costimulation to B cells; B7 molecules (CD80 and CD86) which interact with CD28 on T cells; and additional molecules such as CD38 and cytokine receptors like the IL-4 receptor. Depending on the nature of stimuli or antigens and the strength of signaling, activated B cells will go through cell proliferation, CSR, and somatic hypermutation (SHM), and eventually become differentiated to antibody-secreting PCs to mount an effective immune response, or, if signaling is either too little or excessive, they will undergo apoptosis. Activation of the BCR triggers B-cell intrinsic signaling by the Src-family kinase, and through the Syk molecule, activating multiple signaling pathways, whereas BAFFR engagement activates the TNF receptor-associated factors. Both signaling pathways stimulate the classical and noncanonical NF-kB pathways, which are critical for B-cell survival, activation, and differentiation (Cancro, 2009; Stadanlick et al., 2008).
B. Proliferation
Cell proliferation is one of the earlier events of B-cell activation, which is necessary to expand the antigen-activated B-cell pool and ensure a sufficient level of immune response. B-cells proliferation can be triggered in vitro in multiple ways. The engagement of BCR serves as a primary stimulus but, in addition, several costimulatory molecules or accessory receptors, such as CD38, CD40, and CD19, can directly stimulate B-cell proliferation or reduce the threshold of B-cell activation by antigens (Barrington et al., 2009; Chen and Ross, 2005, 2007). The Toll-like receptor (TLR) agonists, such as LPS and CpG DNA, are multipotent mitogens that stimulate polyclonal B-cell proliferation via TLRs 4 and 9, respectively (Hoshino et al., 1999; Krieg et al., 1995). Recently, it has been shown that a group of glycolipid antigens can stimulate B-cell proliferation through the MHC class I-like molecule CD1d, present on certain B cells (Brigl and Brenner, 2010; Lang et al., 2008), as well as myeloid cells. The prototypical and most often studied antigen for CD1d is alpha-galactosylceramide, a lipid extracted from a marine sponge; however, endogenous glycolipid antigens of mammalian cells also activate CD1d (Zhou et al., 2004).
RA plays various roles to regulate B-cell activation and differentiation through its influences on these intrinsic signaling systems. Several lines of evidence have shown that the regulation of B-cell proliferation by RA depend on the nature of the stimulus encountered. At a physiological level (about 5–20 nM), RA inhibited the rate of proliferation of purified human peripheral blood B cells stimulated by anti-μ antibody (Blomhoff et al., 1992). In murine naïve B cells stimulated with anti-μ to initiate BCR signaling and with anti-CD38 for ligation of the CD38 molecule on the surface of B cells, proliferation was reduced in the population as a whole, but a group of larger sized, less cycling, and more differentiated B cells emerged over time, and these cells expressed more surface(s) Ig, indicative of enhanced progression toward becoming antibody-secreting PCs (Chen and Ross, 2005). In an in vitro model of T-cell dependent B-cell activation, RA reduced B-cell proliferation induced by ligation of the BCR and CD40, and by LPS (Chen and Ross, 2005, 2007). The reduction of B-cell proliferation by RA under conditions of various stimuli suggests the involvement of a common pathway resulting in the negative regulation of the cell cycle and growth, when B cells are stimulated by cross-linking of BCR-related receptors and TLR4. Naderi and Blomhoff (1999) showed that the reduction in B-cell proliferation in normal human peripheral B cells was preceded by cell cycle arrest, as evidenced by the altered expression of several cell cycle regulatory factors. A negative regulation of the NF-κB pathway may also contribute to the inhibitory effect of RA on cell proliferation, as NF-κB family members play major roles in controlling B-cell development and proliferation (Chen et al., 2002; Siebenlist et al., 2005). Studies of a B-lymphoid cell line in culture have also demonstrated that RA suppresses proliferation by blocking the ionized calcium channel, which mediates the early calcium response after BCR ligation (Bosma and Sidell, 1988).
In contrast to the inhibitory effect of RA on B-cell proliferation stimulated by BCR ligation and LPS as discussed above, RA increased the proliferation of memory B cells when B cells were stimulated with CpG DNA, which induces cell activation through TLR9 (Ertesvag et al., 2007). The increased rate of B-cell proliferation was accompanied by increased secretion of antibody. In a mechanistic study, Ertesvag et al. (2007) demonstrated that the enhanced proliferation and differentiation by RA corresponded to the activation of the p38 MAPK pathway that resulted in retinoblastoma protein phosphorylation and increased the level of cyclin D, factors that stimulate cell cycle progression. We also have also observed that RA increases the proliferation of purified murine spleen B cells stimulated by α-galactosylceramide, a ligand for the CD1d receptor, which was correlated with B-cell differentiation, evidenced by sIgG1 and CD138 expression (Q. Chen, unpublished data), while at the same time RA reduced the proliferation of identical B cells stimulated by LPS.
These contrasting results imply that RA affects B-cell proliferation differentially, in a manner that depends on the B-cell subpopulation as well as the stimulus. Whereas RA inhibits mature B-cell proliferation, which may facilitate their differentiation of the activated B cells toward PCs, RA promotes the expansion of a subset of B cells which undergo further differentiation (Chen and Ross, 2005), both processes leading to the promotion of antibody production. Additionally, whereas physiological levels of RA inhibited B-cell proliferation, RA at the same concentration also prevented spontaneous apoptosis of B lymphocytes (Lomo et al., 1998), further suggesting that although RA inhibits mature B-cell proliferation, it functions to maintain the functional B-cell pool, as required for an effective memory response. Further studies are needed to better define whether it is the stage of B-cell activation per se (naïve or memory) or the stimulus itself, or both, that determines whether RA promotes or inhibits B-cell cycling and proliferation.
IV. Transcription Factors, CSR, and B-Cell Differentiation Toward the PC Phenotype
A. Transcription factors promoting B-cell differentiation
Antigens, bacteria products, and T-cell signals can active B cells through engagement of BCR, TLR, and CD40 present on the B-cell surface, which variously trigger multiple pathways that involve activation/inactivation of many transcription factors. The concerted regulation of these factors is necessary to ensure sufficient and specific humoral immunity, while avoiding the generation of self-reactive antibodies. Four key transcription factors that coordinate the B-cell activation and differentiation are the paired box gene 5 (Pax5), B-cell lymphoma 6 (BCL-6), B lymphocyte-induced maturation protein-1 (BLIMP-1, gene PRDM1), and X-box binding protein 1 (XBP-1; Iwakoshi et al., 2003). As RA participates in the regulation of B-cell lymphopoiesis as well as B-cell activation, it differentially affects the activity of at least some of these B-cell transcription factors, according to the functional stages of the B cell. A schematic of B-cell development, and the effect of RA on various processes, is shown in Fig. 5.2.
Figure 5.2.
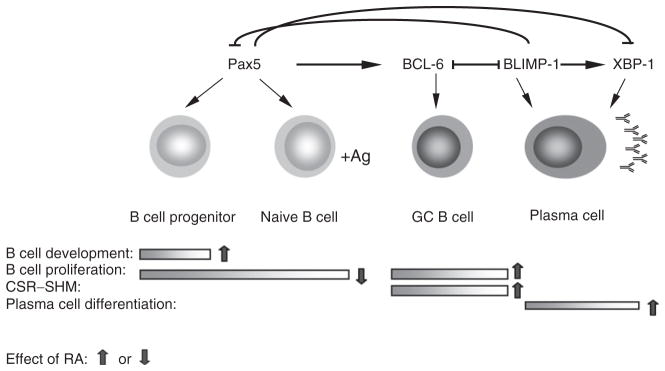
Schematic illustration of B-cell maturation/differentiation process under the control of transcriptional factors. Pax5 is indispensable for B-cell lineage development and proliferation, BCL-6 is essential for GC B-cell reaction that includes B-cell proliferation, CSR, and SHM, unselected cells go through apoptosis; BLIMP-1 is critical for plasma cell differentiation, it suppresses Pax5 expression, and together with XBP-1, ensues antibody production. RA regulates the process at multiple steps. As indicated by arrows, RA promotes B-cell lineage development, inhibits the mature B-cell proliferation, enhances CSR and SHM by increasing Aid gene expression, and augments the terminal differentiation of B cells towards plasma cell phenotype.
Pax5 is essential for B-cell lineage commitment, and is responsible for sustained B-cell lymphopoiesis and expansion of the B-cell pool (Northrup and Allman, 2008). Pax5, originally known as B-cell-lineage-specific activator protein (BASP), is required for B-cell lineage commitment and development as well as B-cell function through the GC stage (Horcher et al., 2001). Pax5 can activate many B-cell-related genes, including CD19, CD79A, B-cell linker (BLNK), and activation-induced cytidine deaminase (AID), an mRNA deaminase essential for B-cell identity, activation, and the GC reaction (Shapiro-Shelef and Calame, 2005). The major function of AID is to deaminate cytidine residues in the variable or switch regions of the Ig genes, thereby initiating SHM and CSR, respectively (de Yebenes and Ramiro, 2006). In murine B lymphopoiesis, RA markedly increased the Pax5 expression level early in the B-cell development phase (the progenitors), corresponding to promotion of the enrichment of the CD19+ sIgM+ B-cell population (Chen et al., 2008). In contrast, at the mature B-cell phase, the presence of RA decreased the Pax5 expression level, which favors the differentiation of sIgG1+ cells (Chen and Ross, 2005). Pax5 is known to repress genes related to antibody secretion, such as XBP1, IgH, IgL, and the J chain, thereby blocking the development of PCs (Calame et al., 2003). Inhibition of Pax5 downregulates IL-4/LPS-induced Ig class switching (Wakatsuki et al., 1994); in contrast, overexpression of Pax5 stimulates B-cell proliferation but suppresses Ig synthesis in both late B-cell lines and PC lines (Usui et al., 1997). Hence, Pax5 promotes the GC reaction but suppresses PC differentiation. In mouse B cells in vitro, RA significantly elevated the mRNA level of AID, suggesting that RA might enhance the isotype switching by inducing the expression of AID (Chen and Ross, 2005).
Oppositely, Blimp-1 is highly expressed in PCs and is known to control many genes that are important for PC differentiation. BLIMP-1 is a 98-kDa transcriptional repressor, expressed in all PCs and a subset of GC B cells, which plays an essential role in PC formation and Ig secretion. Introduction of Blimp-1 into B cells directly repressed genes involved in mature B-cell functions, including the genes for B-cell identity, BCR signaling components, and genes required for Ig class switching (Angelin-Duclos et al., 2000; Calame et al., 2003). Moreover, BLIMP-1 directly inhibited Pax5 and BCL-6, two major transcription factor genes essential for the GC formation. In contrast, BLIMP-1 induced several genes related to PC differentiation and Ig secretion, such as XBP-1 and J chain. Hence, BLIMP-1 functions to terminate the GC reaction, but it promotes plasmacytic cell differentiation by initiating and regulating a cascade of gene expression (Shaffer et al., 2002). BLIMP-1 also regulates XBP-1, which induces formation of the secretory apparatus necessary for the production of antibody, which is crucial for PC differentiation and Ig secretion (Hu et al., 2009). XBP-1 is a basic region leucine zipper protein and a member of the CREB/ATF family of transcription factors. Compared with other immature and mature B cells, the level of XBP-1 is much higher in PC lines. XBP-1 deficiency significantly impairs PC differentiation and severely reduced serum antibody levels, while oppositely, introduction of Xbp1 into B lineage cells initiates PC differentiation (Iwakoshi et al., 2003).
The accepted central concept is that along with B-cell differentiation, stimulation of B cells with antigen releases Blimp-1 from the suppression of BCL-6, increased expression of Blimp-1 suppressed the expression of Pax5 (Lin et al., 2002), leading to cessation of proliferation and, thence, to the terminal differentiation of B cells (Crotty et al., 2010; Tunyaplin et al., 2004). Bcl-6 is expressed predominantly in the GC B cells; however, it is undetectable in antibody-secreting PCs (Cattoretti et al., 1995). Bcl-6 is crucial in the induction of GC B-cell proliferation and the suppression of CSR/SHM; therefore Bcl-6 plays a central role in GC development and inhibition of PC differentiation (Fukuda et al., 1997; Shaffer et al., 2000). Also, for this reason, the elimination of Bcl-6 from PCs is necessary for the terminal differentiation of B cells. Although Bcl-6 mRNA in resting B cells and GC B cells are identical, BCL-6 protein was expressed about three to 34-fold higher in GC B cells than in resting B cells (Allman et al., 1996). The major function of BCL-6 is to inhibit the expression of Blimp-1, a transcription factor inducing PC differentiation, which allows the GC reaction to continue but prevents premature PC differentiation (Shapiro-Shelef and Calame, 2005). At this phase, increased expression of Xbp-1 ensures that the PCs secrete antibody (Hu et al., 2009). Besides the Pax-5/BCL-6/BLIMP-1/XBP-1 axis, another transcription factor, interferon (IFN) regulatory factor 4 (IRF-4) has emerged recently as a critical regulator for B-cell differentiation. A mechanistic study has demonstrated that IRF-4 directly upregulate Blimp-1 transcription to promote PC differentiation (Sciammas et al., 2006). An interesting model was drawn by Sciammas et al. (2006) in which a low level of IRF-4 could increase AID expression and promote CSR and SHM, whereas a high level of IRF-4 could increase Blimp-1, that promotes PC differentiation.
Our studies in a model of murine splenic B-cell differentiation have shown that addition of a physiological concentration of RA induces B-cell differentiation after ligation of the BCR, CD38, or CD40. A decreased rate of cell proliferation, located in a population of larger activated B cells, was accompanied by a reduction in Pax5 and an increase in Aid and Blimp-1 expression levels, which led with time in culture to the development of the PC phenotype, with a higher level of sIgG1 expression and CD138 (syn-decan-1), known as a hallmark of antibody-secreting cells (Chen and Ross, 2005, 2007). Syndecan-1, also called CD138, is a heparin sulfate-rich proteoglycan present on the plasma membrane. Although it is expressed on several types of cells, the expression of syndecan-1 on B cells is commonly used to identify PCs, as thus serves as a marker of terminally differentiated plasmacytic cells (Sanderson et al., 1989). During normal B-cell differentiation, syndecan-1 is temporarily expressed on pre-B cells, lost on circulating B cells, and then reexpressed on PCs. The onset of syndecan-1 expression on PCs in mice correlates closely with immunoglob-ulin secretion. In murine B cells in the presence of a physiological concentration of RA, activated spleen B cells expressed a higher level of CD138 that correlated with the increased level of sIgG1 expression, and the cessation of B-cell proliferation, indicating a more differentiated B-cell phenotype.
Although the detailed mechanisms are not yet clear, the involvement of RA in the regulation of multiple signaling pathways, such as NF-κB, MAPK, and cell cycle regulation, may help to explain its regulatory role. It is worthwhile to note that activation of NF-κB is essential for B-cell proliferation through transactivation of cell growth-related genes, but on the other hand, it also activates prmd-1/Blimp-1 gene expression, and is important in PC differentiation (Morgan et al., 2009), suggesting that the spatiotemporal-specific expression of the transcription factors is especially critical.
So far, little is known regarding the regulation by RA of the expression of Xbp-1 and IRF-4. It will be interesting in future studies to further identify the role of RA in this important autoregulatory loop that coordinates the process of PC differentiation.
B. Class switch recombination
After activation by antigen, mature B lymphocytes in the peripheral organs go through CSR and SHM, processes that diversify the immunoglobulin (Ig) genes and increase the affinity of antibody, respectively. Both Ig CSR and SHM are tightly controlled events that are stimulus specific as well as activation stage specific. CSR is a deletional DNA recombination that occurs between two switch (S) regions located upstream of each heavy-chain constant region (CH). CSR results in replacement of the Cμ gene by one of the downstream CH genes (Cγ, Cα, or Cε), which consequently leads to the production of IgG, IgA, and IgE (Zhang, 2003). AID, which is expressed at high level mainly in activated mature B cells undergoing CSR and SHM, plays an essential role in both of these processes (de Yebenes and Ramiro, 2006). The expression of AID in B cells can be induced by stimulation with bacterial products, like LPS, cytokines such as IL-4, transforming growth factor-β (TGF-β) and IFN-γ, and the ligand for the costimulatory molecule CD40; all of these stimuli more or less participate in B-cell CSR and SHM to certain levels (Xu et al., 2007). By examining at the promoter of Aid gene, Tran et al. (2010) reported a region that is responsible for cytokine or B-cell-specific activator mediated transcription activation of the Aid gene. It is also a region that binds to the transcription factors Pax-5 and E47 to maintain a low level of Aid expression in the steady state. Interestingly, RA increased Aid gene expression in BCR-stimulated B cells, suggesting its positive role in regulation of CSR (Chen and Ross, 2005). Other studies have shown that RA can synergize with TGF-β1 to promote IgA CSR, a process relevant to mucosal immunity (Watanabe et al., 2010). RA also increased the CD40 and IL-4-induced IgG CSR, but inhibited the CD40 and IL-4-induced IgE CSR (Chen and Ross, 2007; Scheffel et al., 2005), indicating that RA can affect the balance of Ig classes produced by antigen-stimulated B cells.
During CSR, the germline transcript (GLT) is first synthesized and then processed to a mature Ig transcript that leads to class-switched Ig production. We have observed that upon stimulation of normal murine naïve B cells, such as by ligation of the BCR, CD38, or CD40 ligation, the stimulated B cells express a higher level of Aid mRNA as well as increased γ1 GLT, which can be detected within 24 h after stimulation. The addition of a physiological concentration of RA (20 nM) increased the Aid mRNA level. However, conversely, RA dramatically decreased the level of γ1GLT and, similarly, the level of Pax5 transcript (Chen and Ross, 2005). After day 3 of stimulation, the surface IgG1 level was increased in the presence of RA regardless of the suppression of γ1 GLT level. This result suggests that, although GLT formation is essential for B cells to undergo class switching, there is not necessarily a direct relationship between the level of γ1 GLT formation and the outcome in terms of IgG1-expressing B cells. Overall, we propose that RA promotes CSR by upregulation of Aid and downregula-tion of Pax5 gene expression, while the GLT is a temporary product that provides a signal for the initiation of CSR, but is not a quantitatively regulated in a manner that predicts B-cell Ig production. Under the same conditions, RA promotes an increase in syndecan-1 expression, which reflects maturation toward the PC phenotype.
V. RA as a Factor in Germinal Center Formation
To understand the mechanisms regarding RA’s effect on the antibody response to T-cell dependent antigens, examination of GC formation is essential. Within the GC, several essential molecular processes take place, such as expansion of B cells, CSR, and SHM, and then the differentiation of memory B cells and PCs, which all are necessary for the evolution of prolonged humoral immunity (Klein and Dalla-Favera, 2008). Upon activation by T-cell dependent antigens (e.g., proteins), some B cells directly go through isotype switching and differentiate into low-affinity PCs. However, many activated B cells migrate into primary follicles, and then rapidly expand to form secondary follicles. About 1 week after antigen priming, the secondary follicle polarizes into the dark zone and the light zone, forming a dynamic structure called the GC, which is primarily comprised of antigen-specific B cells and T-helper cells, follicular dendritic cells (FDC), a type of stromal cell, as well as macrophages (Allen and Cyster, 2008). Histologically, the GC exhibits polarization into two zonal regions termed the dark zone and the light zone. In the dark zone, newly stimulated and still relatively small B cells proliferate rapidly and undergo somatic mutation by the CSR reaction discussed above. The progeny of these B cells, termed centrocytes, migrate into the light zone, where FDC together with antigen-activated Th cells, provide essential signals for B cell survival, CSR, affinity maturation (SHM), and differentiation into long-lived PCs or memory B cells (Benson et al., 2007; Chappell and Jacob, 2007; McHeyzer-Williams et al., 2006; Stavnezer et al., 2008). Hence, the formation of the GC structure and the cellular and molecular processes that occur within the GC are essential for the generation of B cells expressing antibodies of the IgG, IgA, or IgE classes, with high-affinity antigen-combining sites, as well as for the production of memory B cells (McHeyzer-Williams et al., 2006).
The GC reaction and the differentiation of activated B cells into PCs and memory B cells are regulated by the transcription factors described above, that form an autoregulatory loop controlled by Pax5/Bcl-6/Blimp-1. Downregulation of Pax5 and the sequential expression of Bcl-6 and Blimp-1 are required for the induction of PC development. IRF-4 and XBP-1 are also essential to function together with BLIMP-1 in promoting centrocytes to differentiate to PCs (Kallies and Nutt, 2007; Saito et al., 2007). Regarding the role of RA, evidence suggests that RA affects several cell processes that may be expected to facilitate the GC reaction. RA increases CD40 expression on DC that, in turn, enhances the activation of B cells (Park et al., 2004). RA also increases the expression of homing molecules such as the integrin family proteins that promote B-cell migration to the GC (Mora et al., 2006). Moreover, RA affects the FDC to increase the efficiency of antigen presentation and antibody production (Suzuki et al., 2010). Furthermore, as discussed above, RA also regulates the expression of transcription factors that favor the differentiation of PCs and memory B cells (Chen and Ross, 2005). All these processes together may help to explain the enhanced antibody production by RA observed in animal studies in a T-cell dependent antigen immunization model (described below; Ma et al., 2005). In Peyer’s patches, the activation of RAR and TLR signaling by the presence of RA with bacteria products activates FDC within GC, and leads to enhanced expression of the chemokine CXCL13 and the survival factor BAFF/April, which then facilitate the secretion of the TGF-β1, the major cytokine promoting IgA class switching in Peyer’s patches. These factors together increase the numbers of GC B cells and promote the generation of IgA (+) B cells within GCs (Suzuki et al., 2010).
Previous studies demonstrated that the combination of RA and polyino-sinic acid:polycytidylic acid (PIC), a strong inducer of type I IFN and IFN-γ and other cytokines, significantly enhanced T-cell dependent antibody production in vitamin A-deficient (DeCicco et al., 2000) and vitamin A-adequate rats (DeCicco et al., 2001), and in both adult and neonatal mice immunized with tetanus toxoid (Ma and Ross, 2005; Ma et al., 2005; see Ross et al., 2009 for further review). On one hand, PIC was shown to potentiate the primary antibody response, but have little impact on the memory response (DeCicco et al., 2000). On the other hand, RA enhanced both the primary and secondary responses, but the combination of RA + PIC produced a powerful increase in both the primary and secondary antibody responses (DeCicco et al., 2000, 2001; Ma and Ross, 2005; Ma et al., 2005), although mice were only treated with RA and PIC at the time of priming. From these studies, we proposed that RA both augments and “imprints” the immune response (as shown by Iwata et al., 2004 for gut-homing T cells), whereas PIC through the rapid but transient production of IFNs and other cytokines, affects the initial reactions of APCs and activated T and B cells, but cannot by itself promote the differentiation of memory cells. With these results pointing to the importance of RA and PIC at the time or priming, it was of interest to determine how RA, PIC, and the combination, which produced the strongest impact on antibody production, might affect the GC reaction.
A. Costimulation with RA and PIC enhance antigen-induced GC formation
GC B cells can be identified using two surface markers: B220 and peanut agglutinin (PNA). PNA is a plant lectin that specifically binds to lymphocyte glycoprotein on terminal galactosyl residues (Reichert et al., 1983). PNA was first reported as a surface marker for immature (cortical) thymo-cytes, which could bind over 90% of thymus cells (Lahvis and Cerny, 1997; Rose et al., 1980). Later studies showed that PNA also selectively bound to GC cells in peripheral lymphoid organs. Compared with other B cells and T cells, GC B cells bind about 10–30 times more of PNA. Based on these results, PNA is used as a major marker of GC B cells, and GC B cells are defined as B220+PNAHi cells.
Based on the observations described above that RA and PIC administered at the time of antigen priming promoted a robust primary and secondary antibody response, we hypothesized that RA and PIC alone, and especially in combination, could act as a promising vaccine adjuvant, which might stimulate GC formation. Thus, studies were conducted in normal adult mice that were immunized with tetanus toxoid as a prototypical and clinically relevant T-cell dependent antigen, and treated at the time of priming with RA, PIC, or both in combination (RA + PIC). Immunization with tetanus toxoid alone induced a weak but detectable GC reaction, visualized as relatively small PNA-positive GC, with fewer than 20% of the B-cell follicles containing a visible PNA-positive GC (Fig. 5.3A). However, RA, PIC, and RA+PIC increased the number of GC and elevated the GC-to-B-cell follicle ratio about two- to threefold (Fig. 5.3B), while, in addition, the average size of the GC was increased. The enhanced GC response depended on the antigen challenge, because RA and PIC did not induce the GC formation in naïve mice. These treatments increased the plasma titers of antitetanus IgG1, as expected, and by linear regression analysis the plasma antitetanus IgG1 titers were well correlated with both the fraction of B-cell follicles with a GC (R2 = 0.69; p < 0.01; Fig. 5.3C) and the size of GC (R2 = 0.51, p < 0.05; see Ma and Ross, 2009). Therefore, RA and PIC alone and especially combined promoted the tetanus toxoid-induced GC response, which may have directly contributed to the enhanced antitetanus IgG response measured in plasma.
Figure 5.3.

RA and PIC promote TT-induced GC formation. Mice were immunized with tetanus toxoid (TT) and treated with RA orally for 5 days and with PIC at the time of antigen priming. (A) On day 10, fixed spleen sections were prepared and 7-μm sections were stained with biotinylated peanut agglutinin (PNA) followed by incubation with Alexa-568-Streptavidin (red) to detect GC, and with FITC-anti-mouse IgD to identify B-cell follicles (green). Results for nonimmunized mice showed no substantial GC formation. (B) The proportion of B-cell follicles containing a GC, determined by microscopic imaging and counting (Ma and Ross, 2009). The number and relative size of GC were determined by imaging a minimum of 20 GC in each spleen. Bars show mean ± SE, n = 4 mice/group. Different letters above bars within panels indicate significant differences (P < 0.05, a < b). Results of two-way ANOVA are also shown in each panel. (C) Linear regression analysis of the plasma anti-TT IgG response versus the proportion of follicles with a GC, determined for the same animals. (D) GC staining of the same spleen samples with biotinylated PNA (red stain), and monoclonal antibody against FDC (FDC-M1, green stain). (Figure modified from (Ma and Ross, 2009), with permission of Clinical and Vaccine Immunology.)
B. FDC network formation
Because FDCs, as stromal cells in the GC, play a critical role in the GC response (Cyster et al., 2000), and are particularly important for the positive selection of high-affinity B cells (Park and Choi, 2005), and RA and/or PIC significantly increased antigen-induced GC formation and antitetanus IgG production, we tested their effects on the formation of FDC networks, visualized by staining with anti-mouse FDC-M1, a marker of FDC. The FDC networks were located on the one side of GC (see Fig. 5.3D arrows; RA + PIC treatment), consistent with reports that FDC networks mostly occupy the light zone of the polarized GC (Cozine et al., 2005; MacLennan et al., 1992; Steiniger and Barth, 2000). Although RA and or PIC did not affect FDC number, they enlarged the average area of FDC networks about 30–40%, whereas in the immunized control group, the FDC networks were small and dim (see Fig. 5.3D). After administration of PIC and RA + PIC, there was a robust enhancement in the formation of FDC networks in GC, as shown by more intense staining and an increase of about 3 times in the average size of the FDC network; these results also correlated well by linear regression with the titers of plasma anti-TT IgG. Therefore, the expansion of FDC networks in GC, mainly influenced by PIC but supported by RA, could directly contribute to the increase in antigen-specific antibody response by modulating GC microarchitecture and promoting the GC response, which is known to enhance the formation of long-lived antibody-secreting cells (Ma and Ross, 2009).
In addition to enhancing the antigen-triggered GC response, RA and/or PIC increased the number of IgG1+ PCs in the periarteriolar lymphoid sheath (PALS) region, a T-cell zone in spleen where naïve B cells are exposed to antigen and become activated. Upon activation, some T-cell dependent antigen-specific B cells remain in the PALS region and differentiate into short-lived PCs (Angelin-Duclos et al., 2000; Jacob et al., 1991). Compared with the long-lived PC, these short-lived PC produce IgM and IgG antibodies with lower affinity and a shorter half-life. Hence, the PCs generated in the PALS region during the primary response contribute to an early adaptive immune response against antigens and pathogens. Thus it was of interest that RA, PIC, and RA plus PIC significantly increased the generation of PCs in the PALS region, suggesting that the treatments also enhanced the early and short-lived antibody production (Ma and Ross, 2009).
C. Future directions
Additional research is needed on the mechanisms by which RA and costi-muli to enhance the recruitment of B and T cells to the GC, and/or their expansion through proliferation once recruited. RA plays an essential role in directing T cells to the gut (Iwata et al., 2004), and in promote B-cell migration to the GC (Mora et al., 2006). However, the molecular mechanisms by which RA promotes B- and T-cell homing to lymphoid follicles is presently unknown. Therefore, it will be interesting to determine if RA and stimuli such as PIC regulate GC B- and T-cell recruitment, and which genes and pathways are involved. In addition, the effect of RA and PIC on cell survival and cell death within GCs remains to be determined. Although there is little information on the regulation by RA of the expression of Bcl-6, which is the master regulator of GC reaction, the involvement of a common corepressor has been reported, which also interacted with RAR–RXR transcriptional activity (Dhordain et al., 1997; Yamamoto et al., 2010). While some research has addressed isotype switching through the CSR process, as discussed earlier, essentially nothing is known regarding whether RA affects affinity maturation through the SHM process, and therefore has any effect on the quality of the antibodies produced, through affinity maturation. Given indications as discussed above from studies of isolated B cells that RA can regulate AID (Chen and Ross, 2005, 2007), and that AID also mediates SHM, it would seem reasonable to expect that RA also affects SHM, but direct studies are still necessary. Further studies of AID in the context of GC formation in vivo will also be important for better defining the roles of vitamin A and RA in promoting humoral immunity.
Previous studies have shown that RA and PIC cooperatively enhance the secondary antibody response, as well as the primary antibody response in both VA-deficient and VA-sufficient animals (DeCicco et al., 2000, 2001; Ma and Ross, 2005; Ma et al., 2005; Ross et al., 2009). Since GC formation is also important for B memory cell differentiation, further studies are needed to understand how RA and immunomodulatory agents like PIC, even when administered only at the time of priming as in the studies discussed above, can regulate the formation of B-cell memory and generation of long-lived cells that, upon reactivation, mediate the recall response to antigen or pathogen at a later time. Such questions are central to understanding how vitamin A and RA may be used to improve vaccination strategies in the future.
Abbreviations
- APC
antigen-presenting cell(s)
- BCR
B-cell receptor
- CSR
class switch recombination
- DC
dendritic cell(s)
- GC
germinal center(s)
- Ig
immunoglobulin
- LPS
lipopolysaccharide
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- PC
plasma cell(s)
- PNA
peanut agglutinin
- RA
retinoic acid
- RAR
retinoic acid receptor(s)
- RXR
retinoid X receptor(s)
- SHM
somatic hypermutation
- TLR
Toll-like receptor
- TNFα
tumor necrosis factor alpha
References
- Allen CD, Cyster JG. Follicular dendritic cell networks of primary follicles and germinal centers: Phenotype and function. Semin Immunol. 2008;20:14–25. doi: 10.1016/j.smim.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allman D, Jain A, Dent A, Maile RR, Selvaggi T, Kehry MR, Staudt LM. BCL-6 expression during B-cell activation. Blood. 1996;87:5257–5268. [PubMed] [Google Scholar]
- Altucci L, Gronemeyer H. Nuclear receptors in cell life and death. Trends Endocrinol Metab. 2001;12:460–468. doi: 10.1016/s1043-2760(01)00502-1. [DOI] [PubMed] [Google Scholar]
- Angelin-Duclos C, Cattoretti G, Lin KI, Calame K. Commitment of B lymphocytes to a plasma cell fate is associated with Blimp-1 expression in vivo. J Immunol. 2000;165:5462–5471. doi: 10.4049/jimmunol.165.10.5462. [DOI] [PubMed] [Google Scholar]
- Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res. 2002;43:1773–1808. doi: 10.1194/jlr.r100015-jlr200. [DOI] [PubMed] [Google Scholar]
- Barrington RA, Schneider TJ, Pitcher LA, Mempel TR, Ma M, Barteneva NS, Carroll MC. Uncoupling CD21 and CD19 of the B-cell coreceptor. Proc Natl Acad Sci USA. 2009;106:14490–14495. doi: 10.1073/pnas.0903477106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastien J, Rochette-Egly C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene. 2004;328:1–16. doi: 10.1016/j.gene.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007;204:1765–1774. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomhoff HK, Smeland EB, Erikstein B, Rasmussen AM, Skrede B, Skjonsberg C, Blomhoff R. Vitamin A is a key regulator for cell growth, cytokine production, and differentiation in normal B cells. J Biol Chem. 1992;267:23988–23992. [PubMed] [Google Scholar]
- Bosma M, Sidell N. Retinoic acid inhibits Ca2+ currents and cell proliferation in a B-lymphocyte cell line. J Cell Physiol. 1988;135:317–323. doi: 10.1002/jcp.1041350220. [DOI] [PubMed] [Google Scholar]
- Brigl M, Brenner MB. How invariant natural killer T cells respond to infection by recognizing microbial or endogenous lipid antigens. Semin Immunol. 2010;22:79–86. doi: 10.1016/j.smim.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Calame KL, Lin KI, Tunyaplin C. Regulatory mechanisms that determine the development and function of plasma cells. Annu Rev Immunol. 2003;21:205–230. doi: 10.1146/annurev.immunol.21.120601.141138. [DOI] [PubMed] [Google Scholar]
- Cancro MP. Signalling crosstalk in B cells: Managing worth and need. Nat Rev Immunol. 2009;9:657–661. doi: 10.1038/nri2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattoretti G, Chang CC, Cechova K, Zhang J, Ye BH, Falini B, Louie DC, Offit K, Chaganti RS, Dalla-Favera R. BCL-6 protein is expressed in germinal-center B cells. Blood. 1995;86:45–53. [PubMed] [Google Scholar]
- Chappell CP, Jacob J. Germinal-center-derived B-cell memory. Adv Exp Med Biol. 2007;590:139–148. doi: 10.1007/978-0-387-34814-8_10. [DOI] [PubMed] [Google Scholar]
- Chen Q, Ross AC. Vitamin A and immune function: Retinoic acid modulates population dynamics in antigen receptor and CD38-stimulated splenic B cells. Proc Natl Acad Sci USA. 2005;102:14142–14149. doi: 10.1073/pnas.0505018102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Ross AC. Retinoic acid promotes mouse splenic B cell surface IgG expression and maturation stimulated by CD40 and IL-4. Cell Immunol. 2007;249:37–45. doi: 10.1016/j.cellimm.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Ma Y, Ross AC. Opposing cytokine-specific effects of all trans-retinoic acid on the activation and expression of signal transducer and activator of transcription (STAT)-1 in THP-1 cells. Immunology. 2002;107:199–208. doi: 10.1046/j.1365-2567.2002.01485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Esplin BL, Garrett KP, Welner RS, Webb CF, Kincade PW. Retinoids accelerate B lineage lymphoid differentiation. J Immunol. 2008;180:138–145. doi: 10.4049/jimmunol.180.1.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Welner RS, Kincade PW. A possible contribution of retinoids to regulation of fetal B lymphopoiesis. Eur J Immunol. 2009;39:2515–2524. doi: 10.1002/eji.200939374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozine CL, Wolniak KL, Waldschmidt TJ. The primary germinal center response in mice. Curr Opin Immunol. 2005;17:298–302. doi: 10.1016/j.coi.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Crotty S, Johnston RJ, Schoenberger SP. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol. 2010;11:114–120. doi: 10.1038/ni.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyster JG, Ansel KM, Reif K, Ekland EH, Hyman PL, Tang HL, Luther SA, Ngo VN. Follicular stromal cells and lymphocyte homing to follicles. Immunol Rev. 2000;176:181–193. doi: 10.1034/j.1600-065x.2000.00618.x. [DOI] [PubMed] [Google Scholar]
- de Yebenes VG, Ramiro AR. Activation-induced deaminase: Light and dark sides. Trends Mol Med. 2006;12:432–439. doi: 10.1016/j.molmed.2006.07.001. [DOI] [PubMed] [Google Scholar]
- DeCicco KL, Ross AC. All-trans-retinoic acid and polyriboinosinoic: polyribocytidylic acid cooperate to elevate anti-tetanus immunoglobulin G and immunoglobulin M responses in vitamin A-deficient Lewis rats and Balb/c mice. Proc Nutr Soc. 2000;59:519–529. doi: 10.1017/s0029665100000756. [DOI] [PubMed] [Google Scholar]
- DeCicco KL, Zolfaghari R, Li N, Ross AC. Retinoic acid and poly-riboinosinic acid act synergistically to enhance the antibody response to tetanus toxoid during vitamin A deficiency: Possible involvement of interleukin-2 receptor-beta, signal transducer and activator of transcription-1, and interferon regulatory factor-1. J Infect Dis. 2000;182(Suppl 1):S29–S36. doi: 10.1086/315908. [DOI] [PubMed] [Google Scholar]
- DeCicco KL, Youngdahl JD, Ross AC. All-trans-retinoic acid and polyriboinosinic:polyribocytidylic acid in combination potentiate specific antibody production and cell-mediated immunity. Immunology. 2001;104:341–348. doi: 10.1046/j.1365-2567.2001.01317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhordain P, Albagli O, Lin RJ, Ansieau S, Quief S, Leutz A, Kerckaert JP, Evans RM, Leprince D. Corepressor SMRT binds the BTB/POZ repressing domain of the LAZ3/BCL6 oncoprotein. Proc Natl Acad Sci USA. 1997;94:10762–10767. doi: 10.1073/pnas.94.20.10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiLillo DJ, Matsushita T, Tedder TF. B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Ann N Y Acad Sci. 2010;1183:38–57. doi: 10.1111/j.1749-6632.2009.05137.x. [DOI] [PubMed] [Google Scholar]
- Ertesvag A, Aasheim HC, Naderi S, Blomhoff HK. Vitamin A potentiates CpG-mediated memory B-cell proliferation and differentiation: Involvement of early activation of p38MAPK. Blood. 2007;109:3865–3872. doi: 10.1182/blood-2006-09-046748. [DOI] [PubMed] [Google Scholar]
- Fahlman C, Jacobsen SE, Smeland EB, Lomo J, Naess CE, Funderud S, Blomhoff HK. All-trans- and 9-cis-retinoic acid inhibit growth of normal human and murine B cell precursors. J Immunol. 1995;155:58–65. [PubMed] [Google Scholar]
- Fields AL, Soprano DR, Soprano KJ. Retinoids in biological control and cancer. J Cell Biochem. 2007;102:886–898. doi: 10.1002/jcb.21530. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Yoshida T, Okada S, Hatano M, Miki T, Ishibashi K, Okabe S, Koseki H, Hirosawa S, Taniguchi M, Miyasaka N, Tokuhisa T. Disruption of the Bcl6 gene results in an impaired germinal center formation. J Exp Med. 1997;186:439–448. doi: 10.1084/jem.186.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoag KA, Nashold FE, Goverman J, Hayes CE. Retinoic acid enhances the T helper 2 cell development that is essential for robust antibody responses through its action on antigen-presenting cells. J Nutr. 2002;132:3736–3739. doi: 10.1093/jn/132.12.3736. [DOI] [PubMed] [Google Scholar]
- Horcher M, Souabni A, Busslinger M. Pax5/BSAP maintains the identity of B cells in late B lymphopoiesis. Immunity. 2001;14:779–790. doi: 10.1016/s1074-7613(01)00153-4. [DOI] [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hypor-esponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- Hu CC, Dougan SK, McGehee AM, Love JC, Ploegh HL. XBP-1 regulates signal transduction, transcription factors and bone marrow colonization in B cells. EMBO J. 2009;28:1624–1636. doi: 10.1038/emboj.2009.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiura N, Nakashima H, Watanabe R, Kuwano Y, Adachi T, Takahashi Y, Tsubata T, Okochi H, Tamaki K, Tedder TF, Fujimoto M. Differential phosphorylation of functional tyrosines in CD19 modulates B-lymphocyte activation. Eur J Immunol. 2010;40:1192–1204. doi: 10.1002/eji.200939848. [DOI] [PubMed] [Google Scholar]
- Iwakoshi NN, Lee AH, Glimcher LH. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol Rev. 2003;194:29–38. doi: 10.1034/j.1600-065x.2003.00057.x. [DOI] [PubMed] [Google Scholar]
- Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21:527–538. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Jacob J, Kassir R, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I The architecture and dynamics of responding cell populations. J Exp Med. 1991;173:1165–1175. doi: 10.1084/jem.173.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallies A, Nutt SL. Terminal differentiation of lymphocytes depends on Blimp-1. Curr Opin Immunol. 2007;19:156–162. doi: 10.1016/j.coi.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Klein U, Dalla-Favera R. Germinal centres: Role in B-cell physiology and malignancy. Nat Rev Immunol. 2008;8:22–33. doi: 10.1038/nri2217. [DOI] [PubMed] [Google Scholar]
- Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- Kuwata T, Wang IM, Tamura T, Ponnamperuma RM, Levine R, Holmes KL, Morse HC, III, DeLuca LM, Ozato K. Vitamin A deficiency in mice causes a systemic expansion of myeloid cells. Blood. 2000;95:3349–3356. [PubMed] [Google Scholar]
- Lahvis GP, Cerny J. Induction of germinal center B cell markers in vitro by activated CD4+ T lymphocytes: The role of CD40 ligand, soluble factors, and B cell antigen receptor cross-linking. J Immunol. 1997;159:1783–1793. [PubMed] [Google Scholar]
- Lang GA, Devera TS, Lang ML. Requirement for CD1d expression by B cells to stimulate NKT cell-enhanced antibody production. Blood. 2008;111:2158–2162. doi: 10.1182/blood-2007-10-117309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KI, Angelin-Duclos C, Kuo TC, Calame K. Blimp-1-dependent repression of Pax-5 is required for differentiation of B cells to immunoglobulin M-secreting plasma cells. Mol Cell Biol. 2002;22:4771–4780. doi: 10.1128/MCB.22.13.4771-4780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomo J, Smeland EB, Ulven S, Natarajan V, Blomhoff R, Gandhi U, Dawson MI, Blomhoff HK. RAR-, not RXR, ligands inhibit cell activation and prevent apoptosis in B-lymphocytes. J Cell Physiol. 1998;175:68–77. doi: 10.1002/(SICI)1097-4652(199804)175:1<68::AID-JCP8>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Ma Y, Ross AC. The anti-tetanus immune response of neonatal mice is augmented by retinoic acid combined with polyriboinosinic:polyribocytidylic acid. Proc Natl Acad Sci USA. 2005;102:13556–13561. doi: 10.1073/pnas.0506438102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Ross AC. Toll-like receptor 3 ligand and retinoic acid enhance germinal center formation and increase the tetanus toxoid vaccine response. Clin Vaccine Immunol. 2009;16:1476–1484. doi: 10.1128/CVI.00282-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Chen Q, Ross AC. Retinoic acid and polyriboinosinic:polyribocy-tidylic acid stimulate robust anti-tetanus antibody production while differentially regulating type 1/type 2 cytokines and lymphocyte populations. J Immunol. 2005;174:7961–7969. doi: 10.4049/jimmunol.174.12.7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLennan IC, Liu YJ, Johnson GD. Maturation and dispersal of B-cell clones during T cell-dependent antibody responses. Immunol Rev. 1992;126:143–161. doi: 10.1111/j.1600-065x.1992.tb00635.x. [DOI] [PubMed] [Google Scholar]
- McHeyzer-Williams LJ, Malherbe LP, McHeyzer-Williams MG. Helper T cell-regulated B cell immunity. Curr Top Microbiol Immunol. 2006;311:59–83. doi: 10.1007/3-540-32636-7_3. [DOI] [PubMed] [Google Scholar]
- Mora JR, Iwata M, Eksteen B, Song SY, Junt T, Senman B, Otipoby KL, Yokota A, Takeuchi H, Ricciardi-Castagnoli P, Rajewsky K, Adams DH, et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science. 2006;314:1157–1160. doi: 10.1126/science.1132742. [DOI] [PubMed] [Google Scholar]
- Morgan MA, Magnusdottir E, Kuo TC, Tunyaplin C, Harper J, Arnold SJ, Calame K, Robertson EJ, Bikoff EK. Blimp-1/Prdm1 alternative promoter usage during mouse development and plasma cell differentiation. Mol Cell Biol. 2009;29:5813–5827. doi: 10.1128/MCB.00670-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reciprocal Th-17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;17:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- Muindi JRF, Young CW, Warrell RP., Jr Clinical pharmacology of all-trans retinoic acid. Leukemia. 1994;8:1807–1812. [PubMed] [Google Scholar]
- Naderi S, Blomhoff HK. Retinoic acid prevents phosphorylation of pRB in normal human B lymphocytes: Regulation of cyclin E, cyclin A, and p21(Cip1) Blood. 1999;94:1348–1358. [PubMed] [Google Scholar]
- Napoli JL. Enzymology and biogenesis of retinoic acid. In: Livrea MA, editor. Vitamin A and Retinoids: An Update of Biological Aspects and Clinical Applications. Birkhèuser Verlag; Basel: 2000. pp. 17–27. [Google Scholar]
- Northrup DL, Allman D. Transcriptional regulation of early B cell development. Immunol Res. 2008;42:106–117. doi: 10.1007/s12026-008-8043-z. [DOI] [PubMed] [Google Scholar]
- Park CS, Choi YS. How do follicular dendritic cells interact intimately with B cells in the germinal centre? Immunology. 2005;114:2–10. doi: 10.1111/j.1365-2567.2004.02075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HY, Park JY, Kim JW, Lee MJ, Jang MJ, Lee SY, Baek DW, Park YM, Lee SW, Yoon S, Bae YS, Kwak JY. Differential expression of dendritic cell markers by all-trans retinoic acid on human acute promye-locytic leukemic cell line. Int Immunopharmacol. 2004;4:1587–1601. doi: 10.1016/j.intimp.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Reichert RA, Gallatin WM, Weissman IL, Butcher EC. Germinal center B cells lack homing receptors necessary for normal lymphocyte recirculation. J Exp Med. 1983;157:813–827. doi: 10.1084/jem.157.3.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose ML, Birbeck MS, Wallis VJ, Forrester JA, Davies AJ. Peanut lectin binding properties of germinal centres of mouse lymphoid tissue. Nature. 1980;284:364–366. doi: 10.1038/284364a0. [DOI] [PubMed] [Google Scholar]
- Ross AC. The Retinoids: Biology, Chemistry, and Medicine. 2. Raven Press; New York: 1994. Retinoids and the immune system; pp. 521–543. [Google Scholar]
- Ross AC. Diet in vitamin A research. In: Sun H, Travis GH, editors. Retinoids, Methods and Protocols. Humana Press, Springer; New York: 2010. pp. 295–313. [Google Scholar]
- Ross AC, Zolfaghari R, Weisz J. Vitamin A: Recent advances in the biotrans-formation, transport, and metabolism of retinoids. Curr Opin Gastroenterol. 2001;17:184–192. doi: 10.1097/00001574-200103000-00015. [DOI] [PubMed] [Google Scholar]
- Ross AC, Chen Q, Ma Y. Augmentation of antibody responses by retinoic acid and costimulatory molecules. Semin Immunol. 2009;21:42–50. doi: 10.1016/j.smim.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Gao J, Basso K, Kitagawa Y, Smith PM, Bhagat G, Pernis A, Pasqualucci L, Dalla-Favera R. A signaling pathway mediating down-regulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell. 2007;12:280–292. doi: 10.1016/j.ccr.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Sanderson RD, Lalor P, Bernfield M. B lymphocytes express and lose syndecan at specific stages of differentiation. Cell Regul. 1989;1:27–35. doi: 10.1091/mbc.1.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffel F, Heine G, Henz BM, Worm M. Retinoic acid inhibits CD40 plus IL-4 mediated IgE production through alterations of sCD23, sCD54 and IL-6 production. Inflamm Res. 2005;54:113–118. doi: 10.1007/s00011-004-1331-8. [DOI] [PubMed] [Google Scholar]
- Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity. 2006;25:225–236. doi: 10.1016/j.immuni.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Yu X, He Y, Boldrick J, Chan EP, Staudt LM. BCL-6 represses genes that function in lymphocyte differentiation, inflammation, and cell cycle control. Immunity. 2000;13:199–212. doi: 10.1016/s1074-7613(00)00020-0. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, Giltnane JM, Yang L, Zhao H, Calame K, Staudt LM. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002;17:51–62. doi: 10.1016/s1074-7613(02)00335-7. [DOI] [PubMed] [Google Scholar]
- Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol. 2005;5:230–242. doi: 10.1038/nri1572. [DOI] [PubMed] [Google Scholar]
- Siebenlist U, Brown K, Claudio E. Control of lymphocyte development by nuclear factor-kappaB. Nat Rev Immunol. 2005;5:435–445. doi: 10.1038/nri1629. [DOI] [PubMed] [Google Scholar]
- Smeland EB, Rusten L, Jacobsen SEW, Skrede B, Blomhoff R, Wang MY, Funderud S, Kvalheim G, Blomhoff HK. All-trans retinoic acid inhibits granulocyte colony-stimulating factor-induced proliferation of CD34 human hemato-poietic progenitor cells. Blood. 1994;9:2940–2945. [PubMed] [Google Scholar]
- Stadanlick JE, Kaileh M, Karnell FG, Scholz JL, Miller JP, Quinn WJ, III, Brezski RJ, Treml LS, Jordan KA, Monroe JG, Sen R, Cancro MP. Tonic B cell antigen receptor signals supply an NF-kappaB substrate for prosur-vival BLyS signaling. Nat Immunol. 2008;9:1379–1387. doi: 10.1038/ni.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiniger B, Barth P. Microanatomy and function of the spleen. Adv Anat Embryol Cell Biol. 2000;151(III–IX):1–101. doi: 10.1007/978-3-642-57088-9. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Maruya M, Kawamoto S, Sitnik K, Kitamura H, Agace WW, Fagarasan S. The sensing of environmental stimuli by follicular dendritic cells promotes immunoglobulin A generation in the gut. Immunity. 2010;33:71–83. doi: 10.1016/j.immuni.2010.07.003. [DOI] [PubMed] [Google Scholar]
- Tran TH, Nakata M, Suzuki K, Begum NA, Shinkura R, Fagarasan S, Honjo T, Nagaoka H. B cell-specific and stimulation-responsive enhancers derepress Aicda by overcoming the effects of silencers. Nat Immunol. 2010;11:148–154. doi: 10.1038/ni.1829. [DOI] [PubMed] [Google Scholar]
- Tunyaplin C, Shaffer AL, Angelin-Duclos CD, Yu X, Staudt LM, Calame KL. Direct repression of prdm1 by Bcl-6 inhibits plasmacytic differentiation. J Immunol. 2004;173:1158–1165. doi: 10.4049/jimmunol.173.2.1158. [DOI] [PubMed] [Google Scholar]
- Usui T, Wakatsuki Y, Matsunaga Y, Kaneko S, Koseki H, Kita T. Overexpression of B cell-specific activator protein (BSAP/Pax-5) in a late B cell is sufficient to suppress differentiation to an Ig high producer cell with plasma cell phenotype. J Immunol. 1997;158:3197–3204. [PubMed] [Google Scholar]
- Van DE, Kulier R, Gülmezoglu AM, Villar J. Vitamin A supplementation during pregnancy. Cochrane Database Syst Rev. 2002;4:CD001996. doi: 10.1002/14651858.CD001996. [DOI] [PubMed] [Google Scholar]
- Vitoux D, Nasr R, de The H. Acute prmyelocytic leukemia: New issues on pathogenesis and treatment response. Int J Biochem Cell Biol. 2007;39:1063–1070. doi: 10.1016/j.biocel.2007.01.028. [DOI] [PubMed] [Google Scholar]
- Wakatsuki Y, Neurath MF, Max EE, Strober W. The B cell-specific transcription factor BSAP regulates B cell proliferation. J Exp Med. 1994;179:1099–1108. doi: 10.1084/jem.179.4.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Sugai M, Nambu Y, Osato M, Hayashi T, Kawaguchi M, Komori T, Ito Y, Shimizu A. Requirement for Runx proteins in IgA class switching acting downstream of TGF-beta 1 and retinoic acid signaling. J Immunol. 2010;184:2785–2792. doi: 10.4049/jimmunol.0901823. [DOI] [PubMed] [Google Scholar]
- Wei LN. Retinoid receptors and their coregulators. Annu Rev Pharmacol Toxicol. 2003;43:47–72. doi: 10.1146/annurev.pharmtox.43.100901.140301. [DOI] [PubMed] [Google Scholar]
- Wei D, Yang Y, Wang W. The expression of retinoic acid receptors in lymph nodes of young children and the effect of all-trans-retinoic acid on the B cells from lymph nodes. J Clin Immunol. 2007;27:88–94. doi: 10.1007/s10875-006-9059-6. [DOI] [PubMed] [Google Scholar]
- West KP., Jr Extent of vitamin A deficiency among preschool children and women of reproductive age. J Nutr. 2002;132:2857S–2866S. doi: 10.1093/jn/132.9.2857S. [DOI] [PubMed] [Google Scholar]
- WHO/UNICEF. Integration of vitamin A supplementation with immunization: Policy and programme implication. Report of a meeting; 12–13 January 1998. New York: UNICEF; 1998. pp. 1–7. [Google Scholar]
- Xu Z, Pone EJ, Al-Qahtani A, Park SR, Zan H, Casali P. Regulation of aicda expression and AID activity: Relevance to somatic hypermutation and class switch DNA recombination. Crit Rev Immunol. 2007;27:367–397. doi: 10.1615/critrevimmunol.v27.i4.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Tsuzuki S, Tsuzuki M, Handa K, Inaguma Y, Emi N. BCOR as a novel fusion partner of retinoic acid receptor alpha in a t(X;17)(p11;q12) variant of acute promyelocytic leukemia. Blood. 2010;116:4274–4283. doi: 10.1182/blood-2010-01-264432. [DOI] [PubMed] [Google Scholar]
- Zhang K. Accessibility control and machinery of immunoglobulin class switch recombination. J Leukoc Biol. 2003;73:323–332. doi: 10.1189/jlb.0702339. [DOI] [PubMed] [Google Scholar]
- Zhao ZR, Ross AC. Retinoic acid repletion restores the number of leukocytes and their subsets and stimulates natural cytotoxicity in vitamin A-deficient rats. J Nutr. 1995;125:2064–2073. doi: 10.1093/jn/125.8.2064. [DOI] [PubMed] [Google Scholar]
- Zhou D, Mattner J, Cantu C, III, Schrantz N, Yin N, Gao Y, Sagiv Y, Hudspeth K, Wu YP, Yamashita T, Teneberg S, Wang D, et al. Lysosomal glycosphingolipid recognition by NKT cells. Science. 2004;306:1786–1789. doi: 10.1126/science.1103440. [DOI] [PubMed] [Google Scholar]