

Abstract
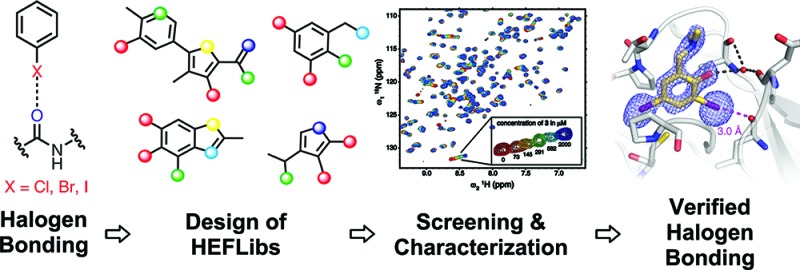
The destabilizing p53 cancer mutation Y220C creates a druggable surface crevice. We developed a strategy exploiting halogen bonding for lead discovery to stabilize the mutant with small molecules. We designed halogen-enriched fragment libraries (HEFLibs) as starting points to complement classical approaches. From screening of HEFLibs and subsequent structure-guided design, we developed substituted 2-(aminomethyl)-4-ethynyl-6-iodophenols as p53-Y220C stabilizers. Crystal structures of their complexes highlight two key features: (i) a central scaffold with a robust binding mode anchored by halogen bonding of an iodine with a main-chain carbonyl and (ii) an acetylene linker, enabling the targeting of an additional subsite in the crevice. The best binders showed induction of apoptosis in a human cancer cell line with homozygous Y220C mutation. Our structural and biophysical data suggest a more widespread applicability of HEFLibs in drug discovery.
Introduction
Halogen bonds have attracted increased interest in molecular design recently,1,2 offering alternatives to classical polar interactions such as hydrogen bonds. Halogen bonding is a noncovalent interaction of the type R–X···Y–R′, where X is chlorine, bromine or iodine acting as a Lewis acid and Y can be any kind of Lewis base. Because of a deficiency in electron density on the hind side of X along the R–X bond axis, chlorine, bromine and iodine possess a characteristic region of positive electrostatic potential, the σ-hole, which favors interaction with electron donors.3 Halogen bonds involving main-chain carbonyl oxygens were found to improve the binding of several ligands to their target protein significantly.4−7 The most frequently observed halogen bonds in protein–ligand structures are either halogen–carbonyl oxygen or halogen−π interactions.8 Quantum chemical calculations also suggest that affinity and selectivity of ligands can be increased by directed halogen–sulfur contacts with methionines.9 Despite this potential of halogen moieties to contribute favorably to ligand binding, the number of compounds containing heavy halides in standard libraries for fragment-based screening is very low.
Here, we have designed a halogen-enriched fragment library (HEFLib) and screened this library for molecules binding to the Y220C mutant of the p53 tumor suppressor. p53 is inactivated in virtually every cancer either through direct mutation or through perturbation of its associated pathways. Reactivation of p53 function in tumors has, therefore, become a prime target for therapeutic intervention.10−13 Most oncogenic p53 cancer mutations are located in the DNA-binding domain of the protein.14,15 About one-third of these mutations simply destabilize this only marginally stable domain, lowering its melting temperature so that it rapidly unfolds at body temperature.16,17 In theory, wild-type function of these mutants can be recovered by binding of molecules that shift the folding–unfolding equilibrium toward the folded state and also slow down the rate of unfolding. We have previously shown that the cancer hotspot mutant Y220C, which accounts for approximately 75 000 new cancer cases per year, is a particularly suitable test case for developing and validating such compounds.10 The mutation destabilizes the protein by creating a surface crevice at a site that is distant from the functional interfaces of the protein.18 From virtual screening, we discovered a carbazole-based small molecule, PhiKan083, that binds to this crevice with a KD of 150 μM and raises the melting temperature of the protein in vitro.19 Using halogen-enriched fragment libraries (HEFLibs), we were able to exploit halogen bonding for lead discovery and apply it to develop biologically active small molecules that stabilize the p53 mutant Y220C.
Results and Discussion
Design of a Halogen-Enriched Fragment Library and Lead Discovery
We used the previously solved structural features of the mutation-induced surface crevice in Y220C as starting points to design new leads. The central cavity has a number of water molecules that are displaced upon ligand binding (Figure 1).18,19 One water molecule forms a hydrogen bond with the main-chain oxygen of Leu145, which is embedded within a hydrophobic environment. The carbonyl is not saturated with hydrogen bonds in the complex of Y220C with the carbazole-based PhiKan083 compound (Figure 1b),19 whereas other small molecules interact with this oxygen via an amino group.20 Quantum chemical calculations at MP2/TZVPP level revealed that the carbonyl oxygen of Leu145 is poised for halogen-bond interactions with moieties binding to the central cavity, in addition to formation of hydrogen bonds (Figure 1c,d). We therefore designed a HEFLib to exploit this additional chemical space in our search for alternative scaffolds binding to the Y220C mutant.
Figure 1.

Mutation-induced cavity in p53 Y220C. (a) Molecular surface of the p53 mutant Y220C bound to the carbazole derivative PhiKan083 (PDB code 2VUK).19 The ligand is shown as a yellow stick model. The mutation-induced cavity can be subdivided into three parts: (i) a deep but narrow central cavity, colored in blue, which is occupied by the carbazole ring; (ii) an open rather shallow subsite 1, colored in red; and (iii) subsite 2, colored in green, which is flanked by several prolines and main-chain oxygens. The latter is not occupied by PhiKan083. (b) Ribbon diagram showing details of the binding mode of PhiKan083, in particular the role of Leu145 at the bottom of the central cavity.19 Also shown are structural water molecules in the ligand-free structure (PDB code 2J1X)18 that are displaced upon ligand binding. One of these water molecules sits at the bottom of the predominantly hydrophobic central cavity and forms a hydrogen bond with the main-chain oxygen of Leu145. (c and d) Potential for halogen-bond interactions with the carbonyl oxygen of Leu145. The iodine-interaction energy sphere is plotted onto the carbonyl oxygen of Leu145 in the Y220C-PhiKan083 structure, showing that this oxygen at the bottom of the central cavity is poised for interaction with iodine-containing ligands with maximum binding energy. Blue regions have the lowest energy; red regions have the highest energy (see Supporting Information for details).
The HEFLib was designed using criteria for fragment selection similar to the “rule of three” by Congreve et al.21 but employing an upper limit of the number of heavy atoms (up to 22) rather than the original molecular weight-based criterion (MW ≤ 300). As an additional cheminformatics selection rule, the molecules were required to contain heavy halides, such as bromine and, in particular, iodine. We chose 79 small, nonreactive, soluble halogenated aromatic compounds from standard vendors, tested them using a thermal shift assay (differential scanning fluorimetry, DSF) and performed secondary screenings with 1H/15N-HSQC NMR spectroscopy and isothermal titration calorimetry (ITC). The most promising lead obtained was 2,4-diiodo-6-((methyl(1-methylpiperidin-4-yl)amino)methyl)phenol 3, which bound to the mutation-induced cavity with a dissociation constant, KD, of 184 ± 23 μM as determined by NMR (Figure 2a).
Figure 2.
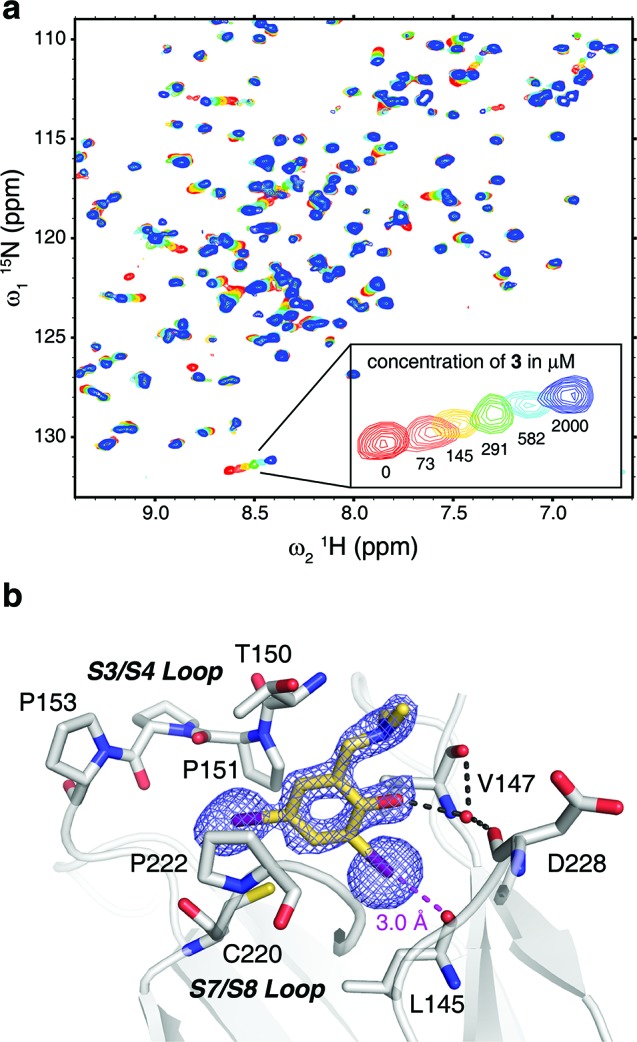
Binding mode of 3 in the mutation-induced pocket of the p53 mutant Y220C. (a) Overlay of 15N/1H HSQC spectra of T-p53C-Y220C without ligand (red) and increasing concentrations of 3 (73 μM, orange; 145 μM, yellow; 291 μM, green; 582 μM, cyan; 2000 μM, dark blue). (b) Crystal structure of the Y220C mutant bound to 3. The mutant is shown as a gray cartoon representation, with selected residues in the cavity shown as stick models. The ligand is shown as a yellow stick model, and polar interactions with the protein are highlighted with broken lines. An unbiased simulated-annealing omit (Fo – Fc) electron-density map for the ligand is shown at a contour level of 3.0 σ. The piperidine moiety of 3 protruding from the cavity was not resolved in the crystal structure and has been omitted from the model.
To verify whether 3 shows halogen bonding as an essential binding interaction and to guide our further ligand design, we solved the crystal structure of the Y220C mutant bound to 3 (1.7-Å resolution), revealing the detailed binding mode of the compound. The benzene moiety sits at the center of the mutation-induced cavity in Y220C, flanked by three prolines (Pro151, Pro222 and Pro223), Val147 and Thr150 (Figure 2b). The two iodine atoms are facing the bottom of the cavity. One iodine atom sits close to the sulfur atom of Cys220 (4.1-Å distance), whereas the other is further removed from the sulfur (5.4-Å distance) and, most importantly, forms an energetically favorable halogen bond with the main-chain oxygen of Leu145. The distance between the iodine and oxygen atoms is 3.0 Å, which is significantly shorter than the sum of the van der Waals’ radii of the two atoms, consistent with quantum chemical calculations on the nature of halogen bonds.22 The halogen-bond angle (C–I···O) is 172°. The phenol group interacts with a conserved structural water molecule, which in turn is stabilized by hydrogen bonds with main-chain atoms of Val147 and Asp228. In addition, the phenol hydroxyl forms an intramolecular hydrogen bond with the tertiary amine. There was no clear electron density to model the binding mode of the piperidine ring in 3, indicating a high flexibility of this moiety in the complex (Figure 2b).
Lead Optimization
With the binding mode of 3 confirmed by X-ray crystallography, we developed a lead optimization strategy as presented in Figure 3 to extend the ligand into subsites 1 and 2 of the Y220C binding pocket. Dissociation constants obtained by 1H/15N-HSQC NMR and ITC, as well as the increase in the melting temperature measured by differential scanning fluorimetry (DSF), are given in Table 1 for selected compounds at different stages of the lead optimization process. Compound 3 and its analogues are easily obtained by reductive amination of the commercially available building block 3,5-diiodosalicylaldehyde (1) with a range of different amines. The salicylic acid derivative of this building block 2 bound to the mutation-induced cavity with a KD of about 800 μM, which corresponds to a ligand efficiency23 of 0.34 kcal/mol per non-hydrogen atom. We generated computer models of all possible products using this building block and commercially available amines, and docked them into the Y220C structure to select suitable candidates for synthesis. We employed a rigid scaffold match constraint using the binding mode of 3 to fix the central scaffold, as halogen bonding is not generally implemented in current docking tools. Optimizing the amine moiety within subsite 1 (red areas in Figure 1a) by this structure-guided design increased compound affinities moderately. The two amines with highest binding affinity were 4 and 5, showing KDs measured by NMR of 104 and 87 μM, respectively. The crystal structure of the Y220C complex with 4 showed essentially the same binding mode of the central scaffold as observed for 3. In contrast to the crystal structure of 3, the piperidine ring was well-defined. It packs against residues 147–150, protruding from the central region of the pocket and extending into the shallow subsite 1 and out into solvent (Figure 4a and Supporting Information Figure S1).
Figure 3.
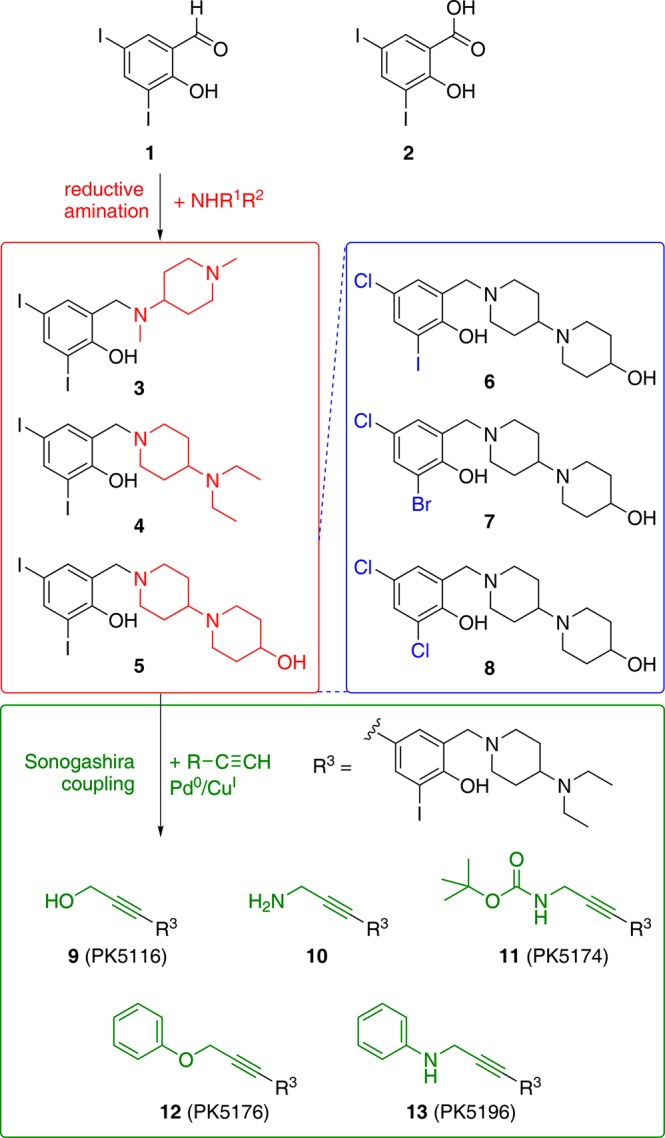
Overview of the synthetic strategy based on the initial hit 3 resulting from HEFLibs screening. Variations in the amine side chain (3–5) were created starting from building block 1 by reductive amination (red box). Variation in the halogen substitution pattern (6–8) for systematically studying the strength of the halogen–oxygen interactions was done by using different building blocks (blue box). The established scaffold was further extended into subsite 2 by Sonogashira coupling of 4 with different acetylenes yielding analogues 9–13 (green box).
Table 1. Biophysical characterization of designed p53-Y220C ligands.
| compound | DSF ΔTm [K] at 250 μMa |
NMR KD [μM] | ITC KD [μM] |
|---|---|---|---|
| 2 | n.d. | 819 ± 68 | n.d. |
| 3 | 0.55 | 184 ± 23 | 225 |
| 4 | 0.97 | 104 ± 23 | 105 |
| 5 | 1.10 | 87 ± 17 | 78 |
| 6 | 0.31 | 247 ± 44 | n.d. |
| 7 | 0.03 | 1040 ± 160 | n.d. |
| 8 | –0.05 | 4900 ± 2300 | n.d. |
| 9 (PhiKan5116) | 0.58 | 114 ± 8 | 107 |
| 10 | 0.64 | 1080 ± 360 | n.d. |
| 11 (PhiKan5174) | 3.21 | n.a.b | 15.5 |
| 12 (PhiKan5176) | 2.59 | n.a.b | 20.6 |
| 13 (PhiKan5196) | 3.61 | n.a.b | 9.7 |
ΔTm =Tm(ligand-bound protein) – Tm(free protein).
For binders with a KD in the low micromolar region, line broadening (intermediate exchange) was observed instead of chemical shift perturbation.
Figure 4.
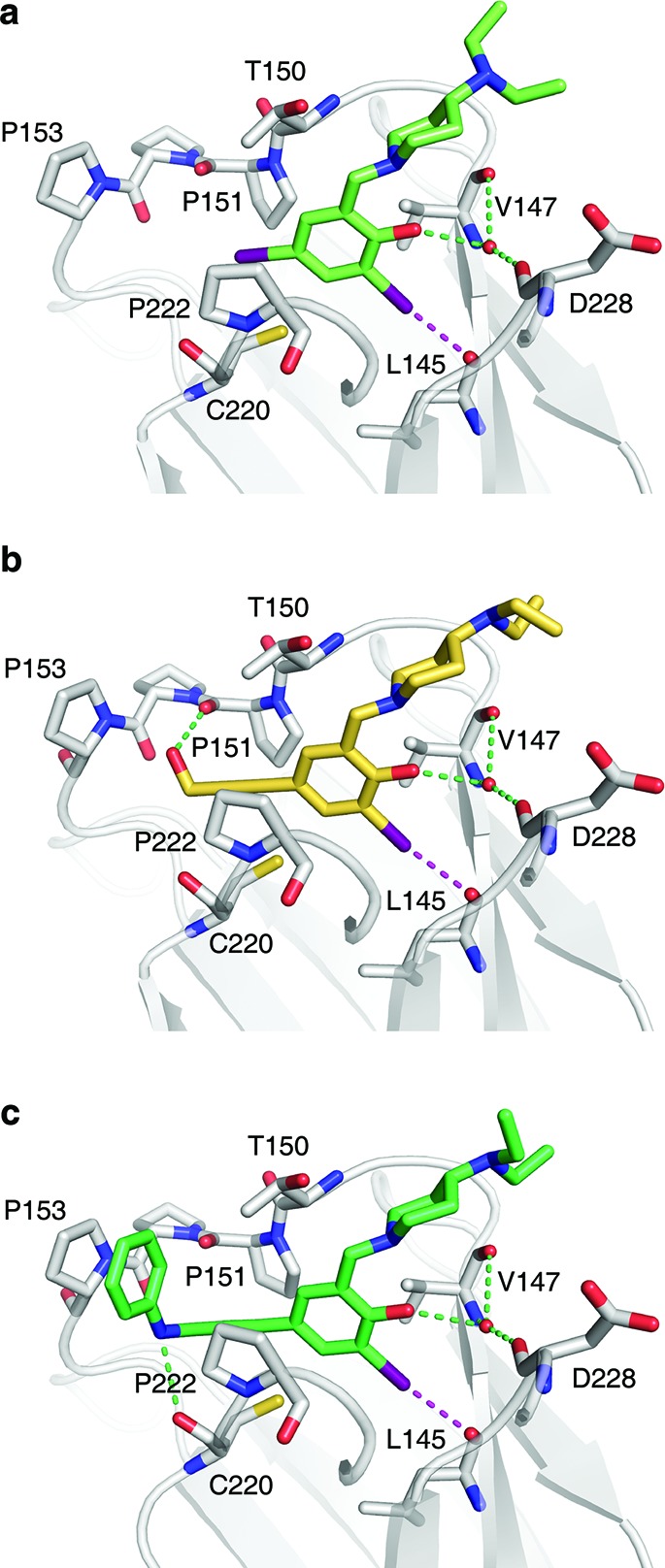
Crystal structures of Y220C–ligand complexes. Shown are the binding modes of 4 (a), PhiKan5116 (b), and PhiKan5196 (c). The protein is shown as a gray cartoon representation with selected residues highlighted as stick models. The halogen bond between the iodine and the carbonyl oxygen of Leu145 is indicated by a broken magenta line; additional polar interactions with the protein are shown as green broken lines.
To evaluate the contribution of halogen bonding to the observed ligand affinities, we replaced the iodine moieties in 5 with the lighter halogens bromine and chlorine, which are expected to form weaker halogen bonds based on theoretical calculations. Comparison of data for the three ligands 6, 7 and 8 with 5 (see Figure 3) demonstrates the importance of the iodine–oxygen halogen bond (Table 1); substituting the iodine moiety binding to Leu145 by bromine resulted in a 4-fold decrease in KD, while substitution with chlorine severely impaired binding of the compound to the Y220C crevice and resulted in a 20-fold loss in affinity. In addition to reduced halogen bonding, reduced space filling by the smaller chlorine compared to iodine may also affect binding affinity. Replacement of the other iodine in position 4 by chlorine resulted only in a 3-fold loss in affinity.
Next, we extended the ligand into subsite 2 (colored in green in Figure 1a), which required bridging the narrow gap between Cys220 and Pro151. A significant benefit of our HEFLibs strategy is that halogens not involved in essential interactions with the binding site can be utilized by a multitude of Pd-based cross-coupling reactions for developing and decorating the original scaffold. We evaluated various possibilities in silico and found an acetylene linker best suited to bridge this narrow gap between the central cavity and subsite 2. The acetylene linker was introduced by a reaction of the 4-iodo moiety on the central scaffold with terminal acetylenes in a Sonogashira coupling reaction.24,25 The first compound with acetylene linker to be tested was 9 (PhiKan5116), exhibiting very similar binding affinity to that of the parent di-iodo compound 4. Hence, loss of the bulky 4-iodo moiety in the central scaffold is compensated for by interactions formed by the new substituent in 9. Surprisingly, 10, which has a propargylamine moiety instead of the propargyl alcohol moiety in 9, bound only weakly to p53-Y220C, possibly due to heavy desolvation penalties for this highly polar compound (NMR KD ≈ 1.1 mM). In contrast, its t-BOC-protected precursor, 11 (PhiKan5174), exhibited very tight binding to the mutation-induced crevice (KD measured from ITC = 15.5 μM) and substantially stabilized the protein (Table 1, Figure 3). Substitution of the free terminal hydroxyl group in 9 by a phenoxy moiety resulting in compound 12 (PhiKan5176) led to a 10-fold increase in binding affinity predominantly due to a favorable CH-π stacking of the phenyl ring on Pro153. Finally, introducing an anilinic −NH– group instead of the phenol ether resulted in 13 (PhiKan5196), the most potent Y220C stabilizer identified to date. 13 bound to the mutation-induced cavity with a dissociation constant of 9.7 μM as determined by ITC and raised the melting point of the protein by about 3.6 K at 250 μM compound concentration (Figure 5).
Figure 5.

Biophysical characterization of PhiKan5174 and PhiKan5196 by DSF and ITC. Concentration-dependent thermostabilization of the Y220C mutant by PhiKan5174 (a) and PhiKan5196 (b) measured by DSF. Binding of PhiKan5174 (c) and PhiKan5196 (d) to Y220C as characterized by ITC.
Binding Mode of Ligands with Acetylene Linker
To validate our ligand design results, we solved crystal structures of a series of Y220C–ligand complexes at 1.4–1.6 Å resolution (Figure 4 and Supporting Information Figure S2). Replacement of the iodine facing Cys220 with an acetylene linker in our third-generation compounds had virtually no effect on the binding mode of the central scaffold. In all ligand-bound protein structures, the key interactions of the central scaffold are conserved. The iodine–oxygen halogen bond distance is consistently between 3.0 and 3.1 Å (representing only ∼86–89% of the sum of the van der Waals radii), and the halogen-bond angles (C–I···O) are between 169° and 173°. This verifies our quantum chemical calculations regarding energetically favored iodine–oxygen halogen bonds (as shown in Figure 1c,d), which formed the basis for the design of HEFLibs. As anticipated, the acetylene group occupies the narrowest part of the cavity between residues Pro151, Cys220 and Pro222 that leads into the previously unoccupied subsite 2, thus widening the scope for additional interactions. The additional hydroxyl group in 9, for example, forms a hydrogen bond with the main-chain oxygen of Pro151 (Figure 4b). This oxygen is also hydrogen bonded with the amide of PhiKan5174 (Supporting Figure S2). Key structural features of the tightest binding ligands include the additional hydrophobic interactions linking Pro153 to Pro222. 12 and 13, for example, are stabilized by packing of a benzene ring against Pro153, allowing favorable CH−π stacking interaction, and hydrophobic interactions with Thr150 and Pro222 (Figure 4c and Supporting Information Figures S1 and S2). 11, however, combines a hydrogen bond with the carbonyl group of Pro151 and hydrophobic interactions of the tertiary butyl moiety with the aforementioned three residues (Supporting Information Figure S2). The higher affinity of 13 compared to 12 results from an additional, albeit weak, hydrogen bond formed between the benzamine moiety and the carbonyl oxygen of Cys220 (hydrogen-bond distance of 3.2 Å). Overall, the structural and biophysical data highlight the crucial role of the acetylene linker for designing high-affinity ligands occupying both subsites of the Y220C binding pocket.
Apoptotic Effects of PhiKan5174 and PhiKan5196 in Human Cancer Cell Lines
We tested the effects of 9, 11 and 13 on the human gastric cancer cell lines NUGC-3 (p53-Y220C+/+) and NUGC-4 (wild-type p53+/+) using a caspase 3/7 apoptosis assay at concentrations ranging from 6.25 to 100 μM (Figure 6). 9, the weakest binder of the three ligands tested, showed no effect on caspase activity in either NUGC-3 or NUGC-4 cells after 6 h at 37 °C. The more potent binders PhiKan5174 and PhiKan5196 (11 and 13), on the other hand, induced apoptosis in the Y220C-containing NUGC-3 cells in a dose-dependent manner. Onset of apoptosis with 13 was observed at a concentration of 50 μM, whereas 11 induced apoptosis only at 100 μM compound concentration, which is consistent with the 2-fold higher affinity of 13 toward the Y220C mutant measured in our in vitro studies. In contrast, no significant effect on apoptosis/caspase activity was observed in the NUGC-4 cell line with wild-type p53 under the same conditions, suggesting that the apoptotic effects of 11 and 13 in the NUGC-3 cell line are mutant-specific. The compounds showed cytotoxic effects at 50 and 100 μM in both cancer cell lines in a Y220C-independent manner (Supporting Information Figure S3). Future developments will be to increase the affinity to levels that ensure target selectivity. Structure-guided scaffold decoration, in particular to probe subsite 2 interactions (e.g., based on PhiKan5196, 13), should be ideally suited for this purpose.
Figure 6.

Effects of compounds in human cancer cell lines. Caspase-3/7 activities are shown at different concentrations of PhiKan5116 (a), PhiKan5174 (b), and PhiKan5196 (c) in NUGC-3 (p53-Y220C+/+) and NUGC-4 (p53-wt+/+) cell lines. While PhiKan5116 does not induce Caspase-3/7 activation, apoptosis is induced by the more potent binders PhiKan5174 and PhiKan5196 at high concentrations in Y220C mutant cells but not in wild-type cells.
Conclusions
We have designed a new class of biologically active small-molecule stabilizers of the p53 cancer mutant Y220C, resulting in a significant increase in affinity compared to the carbazole-based ligand PhiKan083 reported previously.19 This class of compounds has two key features: (i) a central scaffold anchored by halogen bonding and (ii) an acetylene linker which targets an additional subsite of the mutation-induced cavity. With its two iodine atoms, the lead fragment, 3, identified from screening of a halogen-enriched library is perfectly suited for the design of potent Y220C binders and provided a central scaffold with a very robust binding mode. While one iodine moiety facilitates ligand binding through energetically favorable halogen bonding, the other iodine can be exploited for carbon–carbon bond formation, such as the Sonogashira coupling performed here, to extend the ligand into subsite 2.
We propose a general strategy that uses halogen-enriched fragment libraries (HEFLibs) for lead discovery, in addition to halogen bonding having been successfully employed recently for scaffold decoration.1,2 Halogenated compounds are underrepresented in fragment libraries because of the significant atomic weight of bromine and iodine. However, HEFLibs share the advantages of regular fragments, and as molecular probes of small size they can explore binding sites for favorable halogen-bond interactions, to identify unique binding modes that are complementary to those obtained from classical fragment-based screening. We have demonstrated that highly directed iodine–oxygen contacts, in particular, are stable across a multistep structure-based design process. We therefore suggest a more widespread use of HEFLibs in molecular design and drug discovery. This strategy may not only be useful for tackling novel targets but also provide alternative leads for well-established pharmaceutically relevant target proteins and, thus, circumvent patent restrictions.
Experimental Section
Molecular Modeling
All docking experiments were performed using the GOLD v3.226 suite of programs together with the scoring functions Goldscore27 and Chemscore28 at default parameters, as described previously.29 The search efficiency for the genetic algorithm was increased to 200% in automatic mode. Binding site residues were defined automatically by providing the co-crystal structures of PhiKan083 (PDB entry 2VUK) or 3 as a template and using flood fill (“detect cavity” option enabled) with an active site radius of 12 Å. For docking of halogen-bond forming compounds derived from 3, a scaffold match constraint was used, with the central scaffold of 3 as a template. Further details of computational methods, and a description of the quantum chemical calculations used to plot the iodine–oxygen interaction energy sphere (Figure 1c,d) are given in the Supporting Information.
Protein Expression and Purification
The stabilized DNA-binding domain of the p53 mutant Y220C, T-p53-Y220C, was expressed and purified as described previously.19 For the expression of 15N-labeled protein for NMR experiments, M9 minimal medium with 15NH4Cl (1 g/L) as the sole nitrogen source was used.
Chemical Compounds
Compound 2 was purchased from Sigma-Aldrich and >99% pure. The screening compound 3 was purchased from Chembridge (San Diego, CA) with >95% guaranteed purity. The remaining compounds were synthesized within the framework of custom synthesis contracts. 4 was synthesized by Enamine (Kiev, Ukraine). Compounds 5–8 were synthesized by Chembridge (San Diego, CA), and compounds 9–13 were synthesized by Roowin (Romainville, France). For all compounds, compound identity and >95% purity were guaranteed by the supplier.
Differential Scanning Fluorimetry (DSF)
The effect of compounds on the melting temperature of T-p53C-Y220C was monitored using SYPRO Orange (Invitrogen) as the fluorescent probe, which quantitatively binds to the hydrophobic protein patches exposed upon thermal denaturation. Real-time melt analysis was performed using a Corbett Rotor-Gene 6000 real-time qPCR thermocycler. Excitation and emission filters were set to 460 and 510 nm, respectively. Heating from 28 to 60 °C, a constant heating rate of 270 K/h was applied. The protein (final concentration of 10 μM) was briefly mixed with SYPRO orange (10×) in buffer (25 mM KPi pH 7.2, 150 mM NaCl, 1 mM TCEP), and compound (5 mM) dissolved in DMSO was added to give a final compound concentration of 250 μM in 5% (v/v) DMSO. The melting temperature (Tm) of the protein (10 μM) in presence of compounds was determined from the inflection point of the melting curve. Melting temperatures were compared with control samples without compound (yielding ΔTm DSF). All samples were measured in triplicate.
NMR Spectroscopy
1H/15N-HSQC spectra of uniformly 15N-labeled T-p53-Y220C (75 μM) with and without compounds were acquired at 20 °C on a Bruker Avance-800 spectrometer using a 5-mm inverse cryogenic probe. Samples were prepared by adding dilutions of compound from stock solutions in DMSO-d6 to a final concentration of 5% (v/v) DMSO-d6 in buffer. All HSQC spectra were acquired with 8 transients per t1 data point, 1024 data points in t2, and 64 complex data points in t1, with spectral widths of 11.0 kHz for 1H and 2.7 kHz for 15N, and a recycle delay of 800 ms. After zero filling, forward complex linear prediction in f1 and Fourier transformation, the digital resolution was 0.01 ppm/point for 1H and 0.13 ppm/point for 15N. Chemical shifts were considered significant if the average weighted 1H/15N chemical shift difference Δδ(1H/15N) = [(Δδ(1H))2 + (Δδ(15N)/5)2]1/2 was greater than 0.04 ppm. To determine dissociation constants, at least five 15N/1H HSQC spectra at different compound concentrations were measured. Spectra analysis was performed using Sparky 3.11430 and Bruker Topspin 2.0 software. To derive KD values, a quadratic saturation binding equation was fitted to the concentration-dependent chemical shift changes of the relevant shifting peaks:
Isothermal Titration Calorimetry (ITC)
ITC experiments were conducted using a MicroCal (Amherst) iTC200 calorimeter. Protein samples used in the cell unit were prepared to a final concentration of 50–200 μM in 25 mM KPi, pH 7.2, 150 mM NaCl, 1 mM TCEP in 5% (v/v) DMSO. Compounds for use in the syringe unit were dissolved in the same buffer at 5% (v/v) DMSO. Measurements were performed at 20 °C using injection steps of 2 μL at a rate of 0.5 μL/s (initial injection: 0.5 μL) and 120 s spacing. Data analysis was performed using MicroCal Origin software.
X-ray Crystallography
Crystals of T-p53C-Y220C were grown by the sitting drop vapor diffusion method as described previously.18 They were soaked in solutions of compound (30 mM or saturated solution) in cryo buffer (19% polyethylene glycol 4000, 20% glycerol, 10 mM sodium phosphate, pH 7.2, 100 mM HEPES, pH 7.2, 150 mM KCl, 10 mM DTT) for 1–3 h and flash frozen in liquid nitrogen. X-ray data sets were collected at 100 K at the Diamond Light Source, Oxford (beamlines I02, I03 and I04). The data sets were processed with either MOSFLM31 or XDS,32 and SCALA.33 The structures were solved by rigid body refinement with PHENIX34 using the structure of the ligand-free mutant (PDB ID 2J1X)18 as starting model. Models were built and refined using Coot35 and PHENIX. Data collection and refinement statistics are shown in Table 2. The atomic coordinates and structure factors of the Y220C–ligand complexes have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4AGL, 4AGM, 4AGN, 4AGO, 4AGP, and 4AGQ). Structural figures were prepared using PyMOL (www.pymol.org).
Table 2. X-ray Data Collection and Refinement Statistics of p53-Y220C:Ligand Complexes.
| compound | 3 | 4 | 9 | 11 | 12 | 13 |
|---|---|---|---|---|---|---|
| Data Collection | ||||||
| Space Group | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 |
| a (Å) | 65.33 | 64.98 | 65.04 | 65.05 | 65.08 | 65.04 |
| b (Å) | 71.04 | 71.17 | 71.26 | 71.13 | 71.17 | 71.24 |
| c (Å) | 105.65 | 104.96 | 104.88 | 105.15 | 104.98 | 104.94 |
| Molecules/AU | 2 | 2 | 2 | 2 | 2 | 2 |
| Resolution (Å)a | 42.4–1.70 | 35.4–1.52 | 35.4–1.60 | 35.5–1.45 | 35.4–1.50 | 29.6–1.42 |
| (1.79–1.70) | (1.60–1.52) | (1.69–1.60) | (1.53–1.45) | (1.58–1.50) | (1.50–1.42) | |
| Unique reflections | 54,754 | 73,697 | 64,327 | 84,513 | 79,145 | 91,185 |
| Completeness (%)a | 99.8 (99.6) | 97.8 (94.2) | 99.0 (97.0) | 97.3 (93.2) | 99.9 (99.9) | 98.7 (94.7) |
| Multiplicitya | 5.4 (5.4) | 5.7 (5.5) | 5.4 (5.3) | 5.5 (5.7) | 5.7 (5.5) | 5.2 (4.7) |
| Rmerge(%)a,b | 10.0 (31.1) | 7.7 (39.4) | 7.8 (31.4) | 7.2 (31.4) | 6.7 (22.9) | 5.0 (38.1) |
| <I/σI>a | 9.9 (4.3) | 13.8 (4.1) | 12.9 (4.4) | 14.4 (5.0) | 16.3 (6.7) | 17.0 (3.8) |
| Wilson B value (Å2) | 13.6 | 12.8 | 13.4 | 10.8 | 11.5 | 14.5 |
| Refinement | ||||||
| No. of protein atomsc | 3124 | 3150 | 3160 | 3126 | 3137 | 3127 |
| No. of water atoms | 638 | 607 | 609 | 557 | 577 | 558 |
| No. of zinc atoms | 2 | 2 | 2 | 2 | 2 | 2 |
| No. of ligand atoms | 26 | 42 | 48 | 62 | 60 | 60 |
| Overall B value (Å2) | 17.9 | 16.2 | 16.8 | 13.6 | 14.2 | 16.8 |
| Rcryst, (%)d | 16.3 | 17.3 | 17.6 | 19.4 | 16.8 | 18.0 |
| Rfree, (%)d | 19.3 | 19.7 | 19.3 | 21.6 | 18.7 | 19.5 |
| rmsd bonds (Å) | 0.007 | 0.006 | 0.005 | 0.006 | 0.006 | 0.006 |
| rmsd angles (°) | 1.0 | 1.1 | 1.0 | 1.1 | 1.1 | 1.1 |
| PDB code | 4AGL | 4AGM | 4AGN | 4AGO | 4AGP | 4AGQ |
Values in parentheses are for the highest-resolution shell.
Rmerge = ∑(Ih,i – <Ih>)/∑Ih,i.
Number includes alternative conformations.
Rcryst and Rfree = ∑||Fobs| – |Fcalc||/∑|Fobs| where Rfree was calculated over 5% of the amplitudes chosen at random and not used in the refinement.
Cancer Cell Assays
Human gastric cancer cell lines NUGC-3 (p53-Y220C+/+, registration no. JCRB0822) and NUGC-4 (wild-type p53+/+, registration no. JCRB0834) were purchased from Japan Health Science Research Resources Bank. They were maintained in RPMI1640 medium with 10% fetal calf serum and 1% antibiotic stock mix (10 000 U/mL penicillin, 10 000 U/mL streptomycin). Both cell lines were incubated in a humidified incubator at 37 °C with 5% CO2. Cell viability, cytotoxicity and apoptosis were measured using the ApoTox-Glo Triplex assay kit (Promega) following the manufacturer’s instructions. Briefly, cells were seeded at 1 × 104 cells/well in Packard ViewPlate blank clear-bottom 96-well microtiter plates. After 18 h, cells were incubated with compounds at different concentrations for 6 h at 37 °C and 5% CO2. After treatment, 20 μL of a mixture containing the cell-permeable protease substrate GF-AFC (marker for viability) and the cell-impermeable protease substrate bis-AAF-R11 (marker for cytotoxicity) was added to each well. The content was mixed by orbital shaking for 1 min, and cells were incubated for another 1 h at 37 °C. The fluorescence signals to determine cell viability and cytotoxicity were recorded on a Pherastar plate reader using a 400/500 nm and a 480/520 nm optic module, respectively. Luminescence was recorded using an Orion microplate Luminometer (Berthold Detection Systems, Germany) after 30 min incubation with Caspase-Glo 3/7 reagent (100 μL/well) at room temperature. Experiments were performed in triplicate.
Acknowledgments
We thank the staff at the Diamond Light Source for technical assistance during data collection. This work was funded by the ERC Advanced Grant “Tumour suppressor p53: structure, stability and novel anti-cancer drug development” and the federal state of Baden-Wuerttemberg, Germany.
Supporting Information Available
Supplementary computational procedures, Figures S1–S3, spectroscopic data of compounds 4–13. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Lu Y.; Shi T.; Wang Y.; Yang H.; Yan X.; Luo X.; Jiang H.; Zhu W. J. Med. Chem. 2009, 52, 2854. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Liu Z.; Chen T.; Wang Z.; Tian G.; Shi J.; Wang X.; Lu Y.; Yan X.; Wang G.; Jiang H.; Chen K.; Wang S.; Xu Y.; Shen J.; Zhu W. J. Med. Chem. 2011, 54, 5607. [DOI] [PubMed] [Google Scholar]
- Clark T.; Hennemann M.; Murray J.; Politzer P. J. Mol. Model. 2007, 13, 291. [DOI] [PubMed] [Google Scholar]
- Grasberger B. L.; Lu T.; Schubert C.; Parks D. J.; Carver T. E.; Koblish H. K.; Cummings M. D.; LaFrance L. V.; Milkiewicz K. L.; Calvo R. R.; Maguire D.; Lattanze J.; Franks C. F.; Zhao S.; Ramachandren K.; Bylebyl G. R.; Zhang M.; Manthey C. L.; Petrella E. C.; Pantoliano M. W.; Deckman I. C.; Spurlino J. C.; Maroney A. C.; Tomczuk B. E.; Molloy C. J.; Bone R. F. J. Med. Chem. 2005, 48, 909. [DOI] [PubMed] [Google Scholar]
- Parks D. J.; Lafrance L. V.; Calvo R. R.; Milkiewicz K. L.; Gupta V.; Lattanze J.; Ramachandren K.; Carver T. E.; Petrella E. C.; Cummings M. D.; Maguire D.; Grasberger B. L.; Lu T. Bioorg. Med. Chem. Lett. 2005, 15, 765. [DOI] [PubMed] [Google Scholar]
- Fedorov O.; Huber K.; Eisenreich A.; Filippakopoulos P.; King O.; Bullock A. N.; Szklarczyk D.; Jensen L. J.; Fabbro D.; Trappe J.; Rauch U.; Bracher F.; Knapp S. Chem. Biol. 2011, 18, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardegger L. A.; Kuhn B.; Spinnler B.; Anselm L.; Ecabert R.; Stihle M.; Gsell B.; Thoma R.; Diez J.; Benz J.; Plancher J.-M.; Hartmann G.; Isshiki Y.; Morikami K.; Shimma N.; Haap W.; Banner D. W.; Diederich F. ChemMedChem 2011, 6, 2048. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Wang Y.; Zhu W. Phys. Chem. Chem. Phys. 2010, 12, 4543. [DOI] [PubMed] [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Lange A.; Zahn S.; Kirchner B.; Boeckler F. M. J. Chem. Theory Comput. 2011, 7, 2307. [DOI] [PubMed] [Google Scholar]
- Joerger A. C.; Fersht A. R. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown C. J.; Lain S.; Verma C. S.; Fersht A. R.; Lane D. P. Nat. Rev. Cancer 2009, 9, 862. [DOI] [PubMed] [Google Scholar]
- Wiman K. G. Oncogene 2010, 29, 4245. [DOI] [PubMed] [Google Scholar]
- Popowicz G. M.; Domling A.; Holak T. A. Angew. Chem., Int. Ed. 2011, 50, 2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitjean A.; Mathe E.; Kato S.; Ishioka C.; Tavtigian S. V.; Hainaut P.; Olivier M. Hum. Mutat. 2007, 28, 622. [DOI] [PubMed] [Google Scholar]
- Joerger A. C.; Fersht A. R. Oncogene 2007, 26, 2226. [DOI] [PubMed] [Google Scholar]
- Bullock A. N.; Henckel J.; Fersht A. R. Oncogene 2000, 19, 1245. [DOI] [PubMed] [Google Scholar]
- Friedler A.; Veprintsev D. B.; Hansson L. O.; Fersht A. R. J. Biol. Chem. 2003, 278, 24108. [DOI] [PubMed] [Google Scholar]
- Joerger A. C.; Ang H. C.; Fersht A. R. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 15056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckler F. M.; Joerger A. C.; Jaggi G.; Rutherford T. J.; Veprintsev D. B.; Fersht A. R. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basse N.; Kaar J. L.; Settanni G.; Joerger A. C.; Rutherford T. J.; Fersht A. R. Chem. Biol. 2010, 17, 46. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Carr R.; Murray C.; Jhoti H. Drug Discovery Today 2003, 8, 876. [DOI] [PubMed] [Google Scholar]
- Hardegger L. A.; Kuhn B.; Spinnler B.; Anselm L.; Ecabert R.; Stihle M.; Gsell B.; Thoma R.; Diez J.; Benz J.; Plancher J.-M.; Hartmann G.; Banner D. W.; Haap W.; Diederich F. Angew. Chem., Int. Ed. 2011, 50, 314. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Drug Discovery Today 2004, 9, 430. [DOI] [PubMed] [Google Scholar]
- Sonogashira K.; Tohda Y.; Hagihara N. Tetrahedron Lett. 1975, 16, 4467. [Google Scholar]
- Doucet H.; Hierso J.-C. Angew. Chem., Int. Ed. 2007, 46, 834. [DOI] [PubMed] [Google Scholar]
- GOLD, version 3.2; The Cambridge Crystallographic Data Centre (CCDC): Cambridge, U.K, 2005.
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. J. Mol. Biol. 1997, 267, 727. [DOI] [PubMed] [Google Scholar]
- Eldridge M. D.; Murray C. W.; Auton T. R.; Paolini G. V.; Mee R. P. J. Comput.-Aided Mol. Des. 1997, 11, 425. [DOI] [PubMed] [Google Scholar]
- Vogel S. M.; Bauer M. R.; Boeckler F. M. J. Chem. Inf. Model. 2011, 51, 2650. [DOI] [PubMed] [Google Scholar]
- Goddard T. D.; Kneller D. G.. Sparky 3; University of California: San Francisco, CA, 2008.
- Leslie A. G. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999, 55, 1696. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 72. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Grosse-Kunstleve R. W.; Hung L. W.; Ioerger T. R.; McCoy A. J.; Moriarty N. W.; Read R. J.; Sacchettini J. C.; Sauter N. K.; Terwilliger T. C. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2002, 58, 1948. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.