

Summary
High throughput sequencing of 16S ribosomal RNA gene amplicons is a cost-effective method for characterization of oral bacterial communities. However, before undertaking large-scale studies, it is necessary to understand the technique-associated limitations and intrinsic variability of the oral ecosystem. In this work we evaluated bias in species representation using an in vitro-assembled mock community of oral bacteria. We then characterized the bacterial communities in saliva and buccal mucosa of five healthy subjects to investigate the power of high throughput sequencing in revealing their diversity and biogeography patterns. Mock community analysis showed primer and DNA isolation biases and an overestimation of diversity that was reduced after eliminating singleton operational taxonomic units (OTUs). Sequencing of salivary and mucosal communities found a total of 455 OTUs (0.3% dissimilarity) with only 78 of these present in all subjects. We demonstrate that this variability was partly the result of incomplete richness coverage even at great sequencing depths, and so comparing communities by their structure was more effective than comparisons based solely on membership. With respect to oral biogeography, we found inter-subject variability in community structure was lower than site differences between salivary and mucosal communities within subjects. These differences were evident at very low sequencing depths and were mostly caused by the abundance of Streptococcus mitis and Gemella haemolysans in mucosa. In summary, we present an experimental and data analysis framework that will facilitate design and interpretation of pyrosequencing-based studies. Despite challenges associated with this technique, we demonstrate its power for evaluation of oral diversity and biogeography patterns.
Keywords: 454-pyrosequencing, biogeography, microbial communities, oral biodiversity
Introduction
Bacteria dominate the microbial communities that co-exist with humans. These assemblages of microorganisms are thought to play an important role in homeostasis, metabolic processes, nutrition and protection against deleterious infections (Mazmanian et al., 2008; Ismail et al., 2009; Kau et al., 2011). Indeed, disturbance of these communities and changes in their composition have been associated with the development of a variety of diseases (Eckburg & Relman, 2007; Chang et al., 2008; Turnbaugh & Gordon, 2009; Ravel et al., 2011). Most common oral diseases are also a consequence of changes in the structure of resident microbial communities, driven by an interplay between the microorganisms and the behavioral habits and immune system of the host (Marsh, 2003). Hence, an understanding of the composition and ecological events that drive changes in the structure, from health to disease, of oral microbial communities is an important step in the development of preventive strategies to promote oral health.
Highly parallel high throughput sequencing technologies, such as sequencing by synthesis in the 454 platform (454 Life Sciences/Roche Applied Sciences, Branford, CT), have opened a new era in microbial ecology. Obtaining sequences from amplicon libraries generated by universal amplification of portions of the 16S ribosomal RNA (rRNA) gene is now a cost-effective technique with thousands to hundreds of thousands of sequence reads generated in a single run. This approach allows an overview of the communities as a whole, overcoming the limited views that previously employed techniques offered. These advances are already generating open-ended studies of the variability in the oral microflora as it relates to oral diseases (Li et al., 2010; Belda-Ferre et al., 2011; Pushalkar et al., 2011). However, before large-scale studies are conducted, it is necessary to evaluate the oral microbiome composition during health because large inter-individual variability may limit the discovery of disease-associated biomarkers. Indeed, high throughput sequencing has already been used to characterize the bacterial microbiome of healthy subjects at different intra-oral niches (Zaura et al., 2009). This study sequenced V5–V6 variable regions of the 16S rRNA gene from intra-oral sites of three systemically and orally healthy individuals and found that subjects shared a great proportion of operational taxonomic units (OTUs), thereby supporting the concept of a core oral microbiome present during health. In contrast, other studies have reported that although a core microbiome exists, there is also great inter-subject variability in the microbial communities of humans (Eckburg et al., 2005; Diaz et al., 2006; Bik et al., 2010; Lazarevic et al., 2010). Despite the presence of common taxa at higher taxonomic ranks, it has been suggested that differences in the presence and abundance of lower rank taxa generate a unique microbiome signature for individuals (Diaz et al., 2006; Lazarevic et al., 2010). One important aspect in the detection of inter-individual variability is the coverage of species richness obtained after sampling. The lack of observation of a phylotype in a sample is not indicative of its absence if the richness in the sample is not fully covered. In this respect, the determination of the number of sequence reads needed to observe most phylotypes present in a sample becomes crucial for the proper design of studies aimed at defining the core microbiome and large clinical studies that investigate shifts in the microbial composition between health and disease or intend to discover disease-associated biomarkers.
The biogeography of human microbial communities has also received considerable attention through studies that use 454-pyrosequencing (Costello et al., 2009). Resident microbial communities of humans assemble at body sites with dissimilar environmental conditions such as surface characteristics, humidity, oxygen tension, temperature and presence of body fluids. These communities differ in their membership and structure according to the body site sampled, an indication that specific environments select for certain types of microorganisms (Costello et al., 2009). This pattern may also be evident within a specific niche, with fine scale differences in community structure occurring over short distances. Indeed, the intra-niche biogeography of oral communities has been studied using both culturing and molecular approaches (Liljemark & Gibbons, 1971, 1972; Mager et al., 2003; Aas et al., 2005; Zaura et al., 2009). These investigations have revealed that the bacterial microflora differs markedly among intraoral surfaces. However, because most studies have pooled their data by site, it is not clear if inter-subject variability is greater than the variability among sites within the same subject.
Before undertaking large-scale studies to answer ecological or health-related questions, it is also imperative to understand the technical limitations and the intrinsic bias and variability inherent in 454-sequencing of 16S rRNA amplicon libraries. For example, it is known that targeting different regions of the 16S rRNA gene results in microbial communities with different structures (Sundquist et al., 2007; Kumar et al., 2011). Among the hypervariable regions of the 16S rRNA gene, those between the first 500 base pairs (bp) have been used for most molecular surveys of oral flora because they allow good taxonomic discrimination at the genus and even species level (Diaz et al., 2006; Sundquist et al., 2007). We and others have also demonstrated that DNA lysis procedures influence the composition of microbial communities assayed via molecular methods (Diaz et al., 2006; Morgan et al., 2010). Although such limitations may be insurmountable, their effects need to be assessed on a ‘mock’ community of oral organisms, an artificial community constructed in vitro with some of the same members encountered in vivo. This type of experiment constitutes an important first step in understanding the inherent biases in the technique chosen for a given study.
The great sampling depth possible with high throughput community sequencing may help to answer the question of how many phylotypes reside in the oral cavity of humans because it is possible to obtain data from rare phylotypes that are present at very low abundance. However, estimations of richness using high throughput sequencing are usually overinflated because of the inherent error in polymerase chain reactions (PCR) and sequencing, as well as by limitations in data analysis methodology (Reeder & Knight, 2009; Schloss et al., 2011). Ecological estimators commonly used in macroecology are applied to pyrosequencing datasets to predict the number of unseen phylotypes in a sample and to estimate the coverage obtained. However, the accuracy of these estimators is limited by the error-prone datasets provided as input. A mock community can help to ascertain error because the number of expected species is known a priori. With respect to the use of estimators to predict undetected species, it is also important to evaluate the behavior of the estimator at different sequencing depths to determine the minimum sequencing effort needed to reliably predict the total phylotypes present and the richness coverage obtained.
In this study, we provide experimental and data analysis frameworks to help researchers better understand the use of high throughput sequencing and inform the design of large clinical studies. We began by evaluating the bias of our DNA isolation, PCR amplification and sequencing protocols using a mock community of oral microorganisms. We also evaluated the error in OTU assignment using a recently developed data analysis pipeline (Schloss et al., 2011) and investigated the impact of removing singleton OTUs on decreasing this error. With this knowledge, we then characterized, using a deep-sequencing approach, the salivary and buccal mucosa communities of three healthy individuals and investigated the diversity at these sites. We then determined the sampling effort needed to cover most, or all, of the community richness and that to obtain an accurate estimate of the total richness in salivary and mucosal samples. Next, we sequenced the microbiome of two additional individuals and compared the β-diversity of salivary and buccal mucosa microbial communities at different sequencing efforts, followed by an OTU/phylotype-level analysis to explain the observed biodiversity patterns. The questions we wanted to answer were the following. What is the diversity present in oral bacterial communities of saliva and buccal mucosa? What is the sequencing effort necessary to cover most of the richness in oral microbial communities allowing membership-based community comparisons? Does 454 pyrosequencing reveal differences in biogeography similar to those reported in the literature using culture-based or other molecular approaches? What is the inter-individual variability in the oral microbial communities of healthy individuals and is this variability greater than the expected intra-individual biogeographical differences? And finally, how are these variability measures affected by sampling effort? By answering these questions, we demonstrate that community analysis by 454-pyrosequencing of 16S rRNA amplicon libraries is a technique that offers great advantages but also has its limitations. These studies represent a first step in the understanding of how to best capture comprehensive information on oral microbial communities using this powerful approach.
Methods
Preparation of mock communities of oral bacteria
Streptococcus oralis 34, Streptococcus mutans ATCC 10449, Lactobacillus casei LR1, Actinomyces oris T14v, Fusobacterium nucleatum ATCC 10953, Porphyromonas gingivalis ATCC 33277 and Veillonella sp. PK 1910 were grown at 37°C in appropriate media and environmental conditions until cultures reached late logarithmic phase. Streptococci, A. oris and L. casei were grown in brain–heart infusion (BHI) medium (Oxoid Ltd, Cambridge, UK) under aerobic static conditions. Anaerobes were grown under an atmosphere of 90% N2, 5% H2 and 5% CO2. F. nucleatum was grown in BHI supplemented with 0.5 g l−1 cysteine; P. gingivalis was grown in BHI supplemented with 0.5 g l−1 cysteine, 5 mg l−1 haemin and 1 mg l−1 vitamin K; and Veillonella sp. was grown in BHI supplemented with the same concentrations of cysteine and haemin and 0.6% (volume/volume) of lactic acid. Three types of mock communities were assembled containing these seven representative oral species. Mock 1 consisted of a mixture of genomic DNA from the seven organisms to obtain equal numbers of 16S rRNA gene copies per species. To accomplish this, we first identified the genome size (n) in bp for each organism and then calculated the mass of DNA (m) per genome using the formula m = (n) (1.096 × 10−21 g bp−1). We then normalized genome mass by the copy number of the 16S rRNA gene (ranging from three to five copies, depending on the organism) and calculated the grams of DNA containing the copy number of interest (1 × 105 16S rRNA molecules). Mock 2 was assembled by mixing the same number of cells from each species. Mock 3 was assembled to mimic unevenly distributed natural oral communities by mixing cells from the seven species in the following proportions: 30% cells of S. oralis; 15% cells each of F. nucleatum, Veillonella sp. and A. oris; and 8.3% cells each of S. mutans, L. casei and P. gingivalis. Cell numbers were determined by using a Petroff–Hausser counting chamber. Information on the number of 16S rRNA copies of the seven species was obtained from the Ribosomal RNA Operon Copy Number Database (RRNDB) (Klappenbach et al., 2001) and used to normalize DNA amounts added to mock 1 and to determine the expected number of sequence reads per taxon in mocks 2 and 3. Mock communities were assembled in duplicate and sequenced by combining triplicate amplicon libraries generated from each sample (see below).
Human subject sampling
Subjects were enrolled via a protocol approved by the University of Connecticut Health Center Institutional Review Board. Criteria for inclusion of subjects included being 21 years of age or older and willing and able to provide informed consent. For the subset of subjects used in this analysis, no subject had been diagnosed with a systemic disease or was regularly taking any medication other than multivitamin supplements. All subjects had at least 25 teeth and were in good oral health, defined by the absence of mucosal disease, visible carious lesions or periodontal disease defined by a Community Periodontal Index of Treatment Needs ≥2 in any sextant of the mouth, with all teeth present evaluated at six sites (Ainamo et al., 1982). Additionally, no subject had taken systemic antibiotics within 2 months before sampling or used commercial probiotic supplements (≥108 organisms per day). Subjects were instructed not to perform any oral hygiene procedures for 4 h before sampling and to refrain from eating or drinking anything other than water for 1 h before sampling. Unstimulated saliva was collected by allowing saliva to flow freely for 5 min over a polypropylene tube. Saliva samples were immediately centrifuged at 6000 g and pellets were stored at −80°C until processed further. A mucosal swab sample was collected by passing a single catchall™ swab through the entire area of the right and left buccal mucosa for 10 s per side, avoiding contact with teeth. The swab was immediately swirled in a tube containing 500 μl of TE buffer (20 mm Tris–HCl pH 7.4, 2 mm EDTA) and pressed against the tube walls to transfer the material to the solution, which was stored at −80°C.
DNA isolation procedures
DNA was isolated by a protocol tested in preliminary experiments to efficiently disrupt difficult to lyse gram-positive oral organisms (data not shown). The protocol consisted of mixing the TE-resuspended sample (pure cultures, mock communities or human-derived) with lysozyme (final concentration of 20 mg ml−1) followed by incubation at 37°C for 30 min. This was followed by addition of buffer AL (Qiagen, Valencia, CA) and Proteinase K (final concentration 1.23 mg ml−1) and incubation at 56°C overnight. Samples were then incubated at 95°C for 5 min and DNA was isolated using a commercially available kit according to the instructions of the manufacturer (DNeasy Blood and Tissue kit; Qiagen). DNA was eluted in MD5 solution (MoBio Laboratories, Carlsbad, CA) and its concentration was measured using a NanoDrop instrument (ThermoScientific, Willmington, DE). A negative control containing only buffer was carried through extraction and quantification.
Preparation and sequencing of amplicon libraries
Amplicon libraries were prepared in triplicate from a ∼420-bp region of the 16S rRNA gene spanning V1 and V2 (the hypervariable regions that perform best when assigning species taxonomy to short sequence reads), using primers 8F 5′-agagtttgatcmtggctcag-3′ and 431R 5′-cyiactgctgcctcccgtag-3′ (Escherichia coli numeration) (Sundquist et al., 2007). These primers also included the 454 Life Sciences adapters A or B and in some cases a unique multiplex identifier sequence (MID). The PCR contained 10 ng purified DNA, 1 U platinum iTaq polymerase (Invitrogen, Carlsbad, CA), 1.5 mm MgCl2, 200 μm dNTPs, iTaq buffer (1×), 0.5 μm of each forward and reverse primer and molecular grade water to a final volume of 25 μl. Thermal cycler conditions were: initial step at 95°C for 3 min; 25 cycles of denaturation at 95°C for 30 s, annealing at 50°C for 30 s and extension at 72°C for 1 min; and a final extension step at 72°C for 9 min. A DNA isolation negative control and a PCR control without template were included. Following successful amplification (assayed by agarose gel electrophoresis), triplicate PCR were combined and PCR products were purified using the QIAquick PCR purification kit (Qiagen). Quantification and quality control of amplicon libraries was determined via Experion DNA 1K-chip analysis (BioRad Laboratories, Hercules, CA). Amplicon libraries were sequenced using 454 Titanium chemistry (454 Life Sciences) following emulsion PCR, bead recovery and enrichment. Sequences are available at the Short Reads Archive (accession number SRA048222).
Data analysis
Sequences were preprocessed following the protocols described by Schloss et al. (2011), using mothur (Schloss et al., 2009). First, primers and barcodes were trimmed followed by removal of sequences shorter than 200 bp, or with homopolymers greater than eight nucleotides or with ambiguous base calls. Sequences were then filtered according to quality scores using the sliding window approach, which trims sequences when the average quality score over a 50-bp sliding window drops below 35. Unique sequences were aligned using the SILVA database as a reference (Schloss, 2010) and trimmed so that sequences only included a comparable anchor region. Sequences were further denoized by a modification of the single linkage algorithm (Huse et al., 2010; Schloss et al., 2011) to find sequences with up to 2 bp difference from a more abundant sequence and then merge their counts. This step reduces variability but also diminishes errors caused by pyrosequencing. Chimeric sequences were then removed by applying the UChime algorithm (Edgar et al., 2011), as implemented in mothur.
To group similar sequences into clusters that may represent biological species (OTUs), a distance matrix was generated by calculating uncorrected pair-wise distances using default settings in mothur penalizing consecutive gaps as one gap. Sequences were then clustered into OTUs using the average neighbor algorithm (Schloss & Westcott, 2011) and a 3% dissimilarity cutoff. Sequences were individually classified using the Ribosomal Database Project (RDP) classifier (Wang et al., 2007), which uses a Bayesian approach and also runs a bootstrapping algorithm. The threshold for bootstrapping assignment to a specific taxonomy was set at 80%. Template taxonomies used were the large RDP reference dataset and the Human Oral Microbiome Database (HOMD), a curated dataset for oral taxa (Dewhirst et al., 2010). The OTUs were assigned a taxonomic classification based on the consensus taxonomic assignment for the majority of sequences within that OTU. If a consensus taxonomic assignment was not possible at the species level, then the nearest taxonomical level where a consensus was obtained was reported. Classified sequences were also used to group sequences into phylotypes (from genus to phylum level) based on taxonomic identity. In some cases OTUs with only one sequence across all datasets (singletons) were eliminated.
The α-diversity was calculated by the reciprocal of the Simpson Index (Simpson, 1949; Marrugan & McGill, 2011), the non-parametric Shannon Index (Chao & Shen, 2003) and the Shannon evenness index [EShannon = DShannon/ln(S)], as described in Marrugan & McGill (2011) and implemented in mothur. We observed that these estimators were not sensitive to sequencing effort and thus comparison of samples with different sampling depths was possible. Rarefaction curves were constructed using output from mothur. Coverage of richness at a given sampling effort was determined via the Good–Turing estimator (Good, 1953). Total richness was also estimated via catchall (Bunge, 2011), as implemented in mothur. The number of sequence reads needed to observe all the OTUs estimated by catchall to exist in a given sample was calculated by assuming a logarithmic dependency of the number of OTUs y on the number of sequence reads x(y = a ln (x) + b). Parameters a and b of this model were estimated by least squares best fit of observed data points. This dependency function gave the best fit to our data among other functions with the same number (two) of parameters.
β-diversity was measured by the incidence-based Jaccard Index for comparisons of communities based on membership and the θYC distance (Yue & Clayton, 2005) for comparisons of communities based on structure. A phylogenetic tree was constructed with clearcut (Evans et al., 2006) as implemented in mothur, using the neighbor-joining algorithm. Communities were then compared based on phylogenetic distances using the unifrac weighted and unweighted metrics (Lozupone & Knight, 2005). Principal Coordinate Analysis was performed in mothur and graphs were visualized using the rgl application within the R package. Relative abundances of OTUs or phylotypes were compared among saliva and mucosal sites and tested for statistical significance using metastats and lefse (White et al., 2009; Segata et al., 2011).
Results
Elimination of singleton OTUs decreases the number of erroneous OTUs in pyrosequencing datasets
The 454-pyrosequencing of amplicon libraries produces significant errors with sequence datasets yielding more OTUs than those existing in reality (Kunin et al., 2010; Schloss et al., 2011). As a consequence, application of a strict pipeline for dataset curation is a crucial component of data analysis. In this study, we evaluated the error in amplicon pyrosequencing methods using laboratory-created mock communities of oral microorganisms with defined compositions as a training set. Table 1 lists the libraries from mock communities sequenced in this study. Although sequence curation eliminated ∼35% of low-quality/chimeric sequences, some of these curated datasets still generated more OTUs than the seven expected OTUs contained in mock communities (from 0 to +12 extra OTUs). We have frequently observed that singleton OTUs, defined as OTUs containing only one sequence across datasets, can be manually identified as chimeric sequences. To correct for this, we have added a step to our analysis pipeline to eliminate singleton OTUs. As Table 1 shows, this step decreases the erroneous OTUs appearing in sequenced libraries.
Table 1. Mock communities sequenced.
| Library name | Composition | Reads obtained | Reads used for analysis | Number of extra OTUs | Number of extra OTUs without singletons |
|---|---|---|---|---|---|
| Mock 1a | Equal number of 16S rRNA molecules for seven species | 6471 | 4004 | 0 | 0 |
| Mock 1b | Equal number of 16S rRNA molecules for seven species | 6833 | 4239 | 1 | 1 |
| Mock 2a | Equal number of cells for seven species | 6131 | 3778 | 0 | 0 |
| Mock 2b | Equal number of cells for seven species | 6186 | 4222 | 12 | 4 |
| Mock 3a | Unequal number of cells for seven species | 5607 | 3778 | 3 | 1 |
| Mock 3b | Unequal number of cells for seven species | 5132 | 3268 | 0 | 0 |
OTU, operational taxonomic unit.
Evaluation of bias in species representation using mock communities
Using data analysed as described above, we evaluated the accuracy of 454-pyrosequencing in estimating the relative abundance of species in a community. We used three types of mock communities containing equal numbers of 16S rRNA molecules (mock 1), equal numbers of cells (mock 2) or unequal numbers of cells (mock 3) for seven different oral microorganisms (Table 1). Mock 1 is comprised of genomic DNA and is expected to yield an equal number of reads for each species if PCR and sequencing bias are not present. Mocks 2 and 3 could be affected by both PCR/sequencing bias and by differences in cell lysis procedures. As shown in Fig. 1, mock 1 yielded a greater than expected number of reads for F. nucleatum and lower than expected read numbers for A. oris and L. casei. Starting with known numbers of cells, as in mocks 2 and 3, showed both F. nucleatum and S. oralis as over-represented, a finding that suggests that S. oralis was more easily lysed than other organisms. In addition to being under-represented in mock 1, A. oris and L. casei were also under-represented in mocks 2 and 3, as predicted from PCR bias. Both S. mutans and P. gingivalis appeared in lower abundance than expected only in mocks 2 and 3, a finding that suggests that these organisms are less efficiently lysed. These results demonstrated that although 454-pyrosequencing of amplicon libraries is a powerful technique simultaneously detecting all members in a microbial community, species abundance is subject to empirical bias introduced through methods for DNA isolation and amplification.
Figure 1.

Accuracy of 16S ribosomal RNA (rRNA) amplification followed by 454-pyrosequencing in estimating species abundance. Graph depicts expected and obtained sequence reads for each species in three different types of mock communities. Mock 1 is a community formed by mixing equal amounts of 16S rRNA molecules for seven organisms. Mock 2 is formed by mixing equal numbers of bacterial cells from each species. Mock 3 is formed by mixing unequal number of bacterial cells to obtain a community where some species are more abundant than others. Expected numbers of sequence reads for mocks 2 and 3 were normalized according to the number of 16S rRNA copies in the genome of each organism. Number of total reads per sample was normalized to 3268 reads to allow comparisons. Duplicate libraries are indicated by the letters a and b.
Deep-sequencing of salivary and mucosal bacterial communities to determine α-diversity
We next investigated whether 454-pyrosequencing of amplicon libraries can be used to estimate the number of taxa in the oral cavity of individuals. This knowledge is not only required to understand differences among sites, subjects and disease states, but has important experimental implications because most studies using 454 amplicon sequencing are conducted via a multiplexing approach, where multiple samples are sequenced in parallel at a decreased sequencing depth. Hence, it is necessary to be aware of the number of undetected species when interpreting results, especially if communities are compared based solely on membership. From ecological patterns followed by most microbial communities, it is expected that rare species will remain unseen even at a great sequencing depth, because species abundance distribution curves usually show a long tail with most species being rare (Marrugan & McGill, 2011). In ecological terms, the number of species in a given community is known as richness. To investigate richness at two oral sites, we conducted a moderately deep-sequencing survey (at least 30,000 sequences per sample) of the microbial communities in saliva and buccal mucosa of three subjects. We then tested the performance of two estimators of richness and coverage as a function of increasing sequencing efforts. The number of observed OTUs and measures of diversity at maximum sequencing effort are shown in Table 2, and the rarefaction curves for these samples are depicted in Fig. 2.
Table 2. Subject-derived amplicon libraries.
| Subject (library name) | Site | Reads obtained | Reads used for analysis | Good–Turing's coverage (%) |
|---|---|---|---|---|
| Deep sequencing | ||||
| 1 (1S) | Saliva | 57,592 | 34,936 | 99.9 |
| 2 (2S) | Saliva | 68,786 | 39,785 | 99.9 |
| 3 (3Sa) | Saliva | 45,792 | 24,835 | 99.8 |
| 3 (3Sb)1 | Saliva | 55,401 | 36,456 | 99.9 |
| 1 (1M) | Buccal mucosa | 31,179 | 16,917 | 99.8 |
| 2 (2M) | Buccal mucosa | 135,216 | 51,911 | 99.9 |
| 3 (3Ma) | Buccal mucosa | 46,654 | 24,361 | 99.9 |
| 3 (3Mb)1 | Buccal mucosa | 69,422 | 51,107 | 99.9 |
| Multiplex sequencing | ||||
| 4 (4S) | Saliva | 8595 | 5545 | 99.6 |
| 5 (5S) | Saliva | 9741 | 6166 | 99.6 |
| 4 (4M) | Buccal mucosa | 6950 | 4631 | 99.5 |
| 5 (5M) | Buccal mucosa | 6043 | 3866 | 99.0 |
Technical replicates obtained by a new amplification and sequencing of DNA previously isolated from subject 3.
Figure 2.
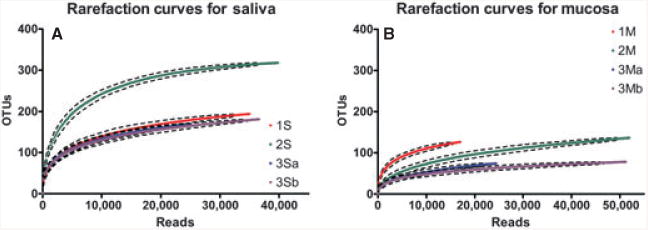
Rarefaction curves for deep-sequenced saliva (S) and buccal mucosa (M) communities from three subjects (1–3).
The Good–Turing estimator, which calculates the percentage of observed OTUs with two or more sequences, is most commonly used in microbial ecology studies to predict coverage (Eckburg et al., 2005; Lemos et al., 2011). According to the Good–Turing estimator, richness in all samples was covered to a minimum of 99% (Table 2). Using this estimator, we calculated the minimal number of sequence reads needed to achieve an acceptable level of coverage of 98% (Table 3). Assuming that the number of OTUs at the maximum sequencing effort closely approximates total richness (sampling universe), we then determined the percentage of OTUs (based on total number of OTUs observed), that were actually detected at the sequencing effort predicted to yield 98% coverage. As Table 3 illustrates, if sampling was to terminate at a level of 98% Good–Turing coverage, a great proportion of richness would not be captured as the result of insufficient sampling. These results demonstrate that despite its broad application, Good–Turing alone is not a sufficient measure of richness coverage in microbial community sampling. Moreover, because Good–Turing is based on the number of singletons, its use as an estimator of coverage appears particularly inadequate for less evenly distributed communities, such as those from buccal mucosa (Table 3).
Table 3. Alpha diversity estimates for deep-sequenced microbial communities.
| Sample | Sobs1 | Reads needed for 98% Good–Turing's coverage | Sobs (%) at sequencing effort in previous column2 | Scatchall (lci–uci) | Richness coverage (%) at maximum sequencing effort according to catchall3 | Number of sequences needed for 98% catchall richness coverage | DInvSimpson | DNpShannon | EShannon |
|---|---|---|---|---|---|---|---|---|---|
| 1S | 194 | 416 | 22.2 | 282 (242–355) | 69 (55–80) | 7.5E04 (5.0E04–1.4E05) | 6.8 | 2.6 | 0.5 |
| 2S | 318 | 3342 | 54.4 | 369 (351–397) | 86 (80–91) | 4.3E04 (3.6E04–5.3E04) | 19.9 | 3.6 | 0.6 |
| 3Sa | 181 | 1048 | 34.3 | 228 (206–264) | 79 (69–88) | 5.2E04 (3.7E04–8.2E04) | 10.5 | 3.0 | 0.6 |
| 3Sb | 169 | 998 | 35.0 | 248 (223–288) | 68 (59–76) | 7.1E04 (5.1E04–1.2E05) | 8.9 | 2.9 | 0.6 |
| 1M | 126 | 939 | 40.5 | 197 (162–264) | 64 (48–78) | 5.8E04 (3.1E04–1.4E05) | 4.1 | 2.2 | 0.5 |
| 2M | 136 | 416 | 13.2 | 377 (360–395) | 36 (34–38) | 5.6E05 (5.1E05–6.3E05) | 1.6 | 1.0 | 0.2 |
| 3Ma | 78 | 150 | 12.8 | 161 (111–280) | 49 (28–70) | 1.6E05 (5.8E04–6.9E05) | 3.2 | 1.6 | 0.4 |
| 3Mb | 74 | 50 | 8.1 | 141 (104–228) | 53 (32–71) | 2.0E05 (8.3E04–7.9E05) | 2.9 | 1.5 | 0.4 |
Sobs are operational taxonomic units (OTUs) observed at maximum sequencing effort and defined at 3% dissimilarity.
This column represents the percentage of observed OTUs, based on total observed OTUs, if sampling efforts were stopped at 98% Good–Turing's coverage.
Coverage of richness, calculated as the percentage Sobs from those predicted by catchall.
We also measured richness coverage using the parametric estimator of diversity catchall, which estimates species based on finite mixture models (Bunge, 2011). As seen in Table 3, our sequencing effort covered only a percentage (36–86%) of the number of OTUs predicted by catchall to exist in our samples. We then calculated the number of sequences needed to cover at least 98% of the catchall-predicted number of OTUs. As Table 3 shows, covering 98% of the predicted richness requires 10–100 times more sequences than those required for 98% Good–Turing's coverage. Furthermore, in contrast to Good–Turing's estimator, catchall demonstrated that the less rich but more uneven mucosal communities would require greater sequencing effort than salivary communities.
We next evaluated the minimum number of sequences required to reliably use catchall as an estimator of total richness and asked whether catchall is affected by a possible increase in the number of erroneous OTUs as sequencing effort increases. As seen in Fig. 3, the number of OTUs estimated by catchall increased initially with sequencing effort, but reached relative stability around 3000–5000 sequence reads, defining the minimum sampling effort needed to predict the richness in an oral sample. Furthermore, increasing sampling effort, which may also increase error, did not affect the estimator.
Figure 3.

Stability of the richness estimator catchall at different sampling efforts. Graphs depict catchall-estimated OTUs present in salivary (S) and mucosal (M) communities of three individuals (1–3) as a function of sampling effort demonstrating that the estimator reaches stability relatively early.
Taken together, these results demonstrate that a great sequencing effort is needed to display all the richness contained in an individual oral sample. However, using an accurate estimator of richness, such as catchall, allows prediction of the number of unseen phylotypes in a given sample, provided enough sequences are obtained for the estimator to be accurate.
Comparing inter-subject and inter-site variability in salivary and buccal mucosa communities at different sequencing efforts
Although the previous analysis showed that observation of the great majority of OTUs in saliva and mucosal communities would require a great sampling effort, it has been shown that even in under-sampled communities, it is still possible to detect diversity patterns as this will depend on the effect size measured (Kuczynski et al., 2010). Hence, we examined the dissimilarity patterns arising from under-sampled versus exhaustively sampled communities. For this analysis we sequenced the communities of two more subjects using a multiplexing approach. The general characteristics and α-diversity estimates of the salivary and mucosa communities from these two subjects are presented in Tables 2 and 4. In agreement with results for deep-sequenced communities, mucosal communities were less diverse, both in terms of richness and evenness, than salivary communities. Dissimilarity analysis of deep-sequenced communities (Fig. 4A), based on the Jaccard index (takes into account membership only), showed no clustering of samples based on site or subject. In contrast, comparison of community structure in deep-sequenced communities by the θYC measure of dissimilarity (takes into account relative abundance of taxa) clustered communities by site of origin, rather than by subject (P < 0.001).
Table 4. Alpha diversity estimates for microbial communities sequenced by multiplexing.
| Sample | Sobs1 | DInverse Simpson | DNp Shannon | EShannon | Scatchall (lci–uci) | Richness coverage (%) according to catchall2 |
|---|---|---|---|---|---|---|
| 4S | 120 | 14.3 | 3.6 | 0.7 | 145 (135–164) | 82 (73–89) |
| 5S | 160 | 11.7 | 3.4 | 0.7 | 198 (184–221) | 80 (72–87) |
| 4M | 63 | 2.3 | 1.7 | 0.4 | 111 (84–174) | 57 (36–75) |
| 5M | 100 | 2.5 | 1.9 | 0.4 | 166 (136–221) | 60 (45–74) |
Sobs are operational taxonomic units (OTUs) observed at maximum sequencing effort and defined at a 0.3% dissimilarity.
Coverage of richness, calculated as the percentage of OTUs observed (Sobs) from the OTUs predicted to exist by catchall.
Figure 4.

Dissimilarity between salivary (blue) and mucosal (red) communities at different sequencing efforts. Top trees in each panel depict the distance among communities calculated by the Jaccard Index, which takes into account membership only. Lower trees depict the distance among communities calculated by θYC which compares communities based on their structure. (A) Deep-sequenced salivary and mucosal communities from first three subjects. (B) Relationships of all communities sequenced in this study. As a result of great differences in sequencing effort, the number of reads in each community was normalized, by random sampling, to that of the community with fewer reads (4254). (C) Relationships of communities randomly sampled to contain only 40 sequences per community.
To evaluate the efficacy of sequencing efforts, we pooled all the libraries sequenced and normalized by random subsampling so that each community contained the same number of sequences. Even at a sequencing effort of ∼4250, and with the inclusion of two more subjects, we still observed similar clustering to that at a deep-sequencing effort (Fig. 4B). We further decreased sampling (to as few as 40 sequences per library) and observed that even at this very low level of sequencing, the θYC index separated communities based on sites of origin (Fig. 4C). We performed similar tests using a phylogenetic approach to analyse community composition and structure and obtained similar results (data not shown). One of these analyses is shown in Fig. 5A, which depicts principal coordinates analysis of the phylogenetic distance among communities (subsampled to ∼4250), based on the weighted unifrac measure, showing that saliva communities clustered separately from mucosal communities using phylogenetic distance.
Figure 5.
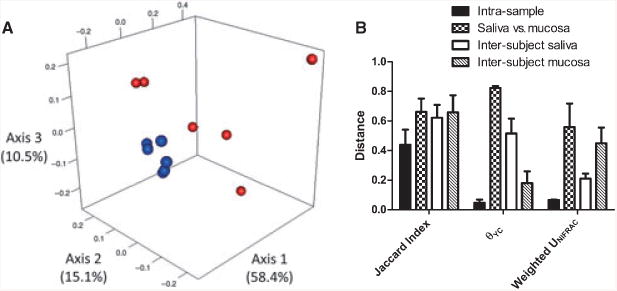
Distance among bacterial communities. (A) Principal coordinates analysis of phylogenetic distance among communities according to the weighted unifrac metric. Salivary communities appear in blue, mucosal communities appear in red. (B) Intra-sample variability, intra-subject (mucosa versus saliva within a subject) and the inter-subject variability at each site. Intra-sample variability was calculated from saliva and mucosal replicate samples of subject 3.
We then measured inter-site and inter-subject variability using different metrics (Fig. 5B). This analysis also includes intra-sample variability measures from new amplification and deep-sequencing of DNA from saliva and buccal mucosa of subject 3. As this figure illustrates, a comparison of communities based only on membership revealed large differences even within the same sample. For example, of 218 OTUs found in the deep-sequenced salivary communities of subject 3, only 139 (63.8%) were present in both replicates. In contrast, intra-sample variability based on community structure revealed pronounced agreement within the same sample, whereas salivary and mucosal communities differed greatly with inter-site distance within a subject being larger than the inter-subject distance at each site. Interestingly, the θYC metric showed that salivary communities were more variable than mucosal communities, which was the opposite of what was shown when communities were compared based on their phylogenetic relatedness by the weighted unifrac metric.
The large variability in community membership was confirmed by evaluating those OTUs shared among subjects. In total, we found 455 OTUs at the 0.3% level of dissimilarity across all sequenced samples. The number of observed OTUs ranged from 120 to 318 per subject (Scatchall 145–369) in saliva and from 63 to 136 OTUs (Scatchall was 111–377) in mucosa. Of the 455 observed OTUs, only 78 (17.1%) were present in all subjects, whereas 125 (27.5%) were present in four subjects and 182 (40%) were present in three subjects. These results could suggest large inter-individual variability in the oral microbiome, however, this needs to be cautiously interpreted because of large intra-sample variability.
In conclusion, because large biogeographical differences exist in community structure, it is possible to detect these differences even at low sequencing efforts. Comparison of communities based on their membership, however, revealed great variability among all samples. Even within a deep-sequenced replicate sample not all OTUs were shared. These results could be explained by the incomplete coverage of sample richness obtained (36–86%). Hence, it appears that with the current available methods, the determination of the ‘true’ core microbiome, that is those bacterial species present in all humans, is not a feasible endeavor because inter-subject variability in community membership will always prevail, unless the full richness in a sample is surveyed.
OTUs and phylotypes differentially represented in saliva and buccal mucosa that explain biogeographical patterns
Figure 6 depicts the most abundant OTUs in saliva and mucosa samples. Analysis of OTUs differentially represented in saliva or mucosa via metastats (White et al., 2009) found that 79 OTUs were significant, with 66 OTUs more abundant in saliva and 13 in mucosa (see Supplementary material, Tables S1 and S2). Although OTUs identified to species level have to be accepted with caution because of the biological variance within an OTU, it is evident that the different structure in mucosal samples is primarily caused by Streptococcus mitis (Fig. 6). Differentially represented OTUs were also analysed via lefse, which calculates the effect size of each feature after Linear Discriminant Analysis (Segata et al., 2011). Figure 7 shows the results of lefse analysis, which revealed two OTUs more abundant in mucosa and 38 OTUs more abundant in saliva. metastats and lefse analyses largely agreed although the stricter statistical tests used by lefse resulted in a decreased number of significant features compared with metastats. Moreover, lefse analysis confirmed that S. mitis has the greatest effect size discriminating mucosal samples and that Gemella haemolysans also has high affinity for mucosal tissues.
Figure 6.

Relative abundance in saliva and mucosa of 25 most abundant operational taxonomic units (OTUs) across samples. Environment in which the OTU is over-represented (saliva or mucosa, S or M), as calculated by metastats, is indicated by *after each OTU name. OT followed by a number indicates the specific Oral Taxon from the Human Oral Microbiome Database.
Figure 7.
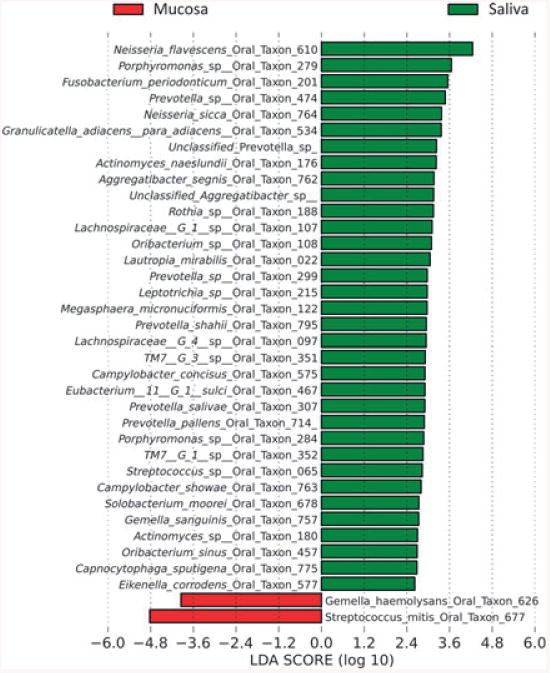
Operational taxonomic units (OTUs) differentially represented in saliva or mucosa as revealed by lefse. Salivary communities appear in green, while mucosal communities appear in red. OTUs are ranked according to their linear discriminant analysis scores.
metastats and lefse can also be used to analyse sequences grouped into phylotypes according to taxonomical classifications. Figure 8 depicts the 25 most abundant genera found across samples. metastats identified two genera, Streptococcus and Gemella, as over-represented in mucosa, whereas 26 genera were over-represented in saliva, largely agreeing with the OTU-based analysis (see Supplementary material, Tables S3 and S4). Figure 9 shows lefse analysis of all taxa, classified from the genus to the phylum levels, differentially represented in either niche and ranked according to the effect size. As Fig. 9 shows, the phyla Proteobacteria, Bacteroidetes and Fusobacteria displayed the least affinity for mucosal surfaces, while the Firmicutes were over-represented in mucosa. Figure 10A shows all phyla across samples and their differential representation according to metastats, and confirms lefse results. According to metastats, only the phylum Firmicutes was over-represented in the communities from buccal mucosa, whereas seven out of nine identified phyla were over-represented in saliva. Although the Firmicutes as a whole appeared more abundant in mucosa, the cladogram for this phylum, shown in Fig. 10B, demonstrates that this difference was largely caused by Streptococcus and Gemella, and other Firmicutes genera do not display predilection for mucosal surfaces.
Figure 8.
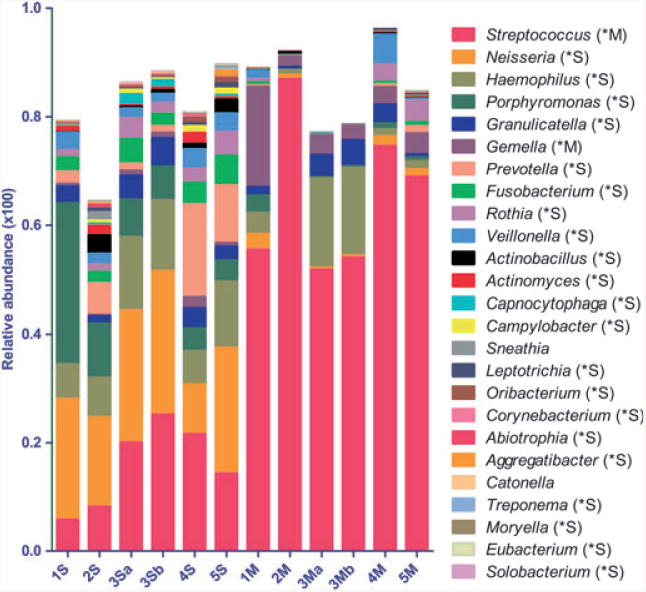
Relative abundance in saliva and mucosa of 25 most abundant genera found across samples. Environment in which the specific genus is over-represented (saliva or mucosa, S or M), as calculated by metastats, is indicated by *after each genus name. Sequences that could not be classified to the genus level were not included in this graph.
Figure 9.

Taxa (classified from the genus to the phylum level) differentially represented in saliva or mucosa as revealed by lefse. Salivary communities appear in green, mucosal communities appear in red. Taxa are ranked according to their linear discriminant analysis scores.
Figure 10.

Differential representation of all phyla found across samples in saliva and mucosa. (A) Relative abundance of different phyla. Environment in which the specific phylum is over-represented (saliva or mucosa, S or M), as calculated by metastats, is indicated by *after each phylum name. Sequences that could not be classified to any phylum appear as Unclassified Bacteria. (B) A cladogram depicting the phylum Firmicutes and its differentially represented taxa analysed via lefse. Notice that although the phylum appeared over-represented in mucosa according to metastats, only certain clades within the phylum display affinity for mucosa whereas most of the genera within the phylum are over-represented in saliva.
Discussion
This study provides a methodological framework for the analysis of the oral microbiome based on 454-pyrosequencing of 16S rRNA-derived libraries. Although the objective of this study was not to compare PCR primer pairs or DNA isolation protocols, we provide evidence from mock communities of oral microorganisms that DNA isolation, PCR and sequencing bias introduce variability into the results that has to be considered when interpreting data. These results also highlight the importance of using standardized operating protocols by different laboratories to make results comparable across the oral research community. The primers used in this study have been assessed by other investigators for their ability to amplify a vast number of bacterial taxa (Sundquist et al., 2007). Using a mock community with the same number of 16S rRNA copies (mock 1), we confirm that this primer pair detected all species in the community but their relative abundances differed from those expected. After checking the primer pair for mismatches to the 16S rRNA gene from sequences in the RDP, we could not attribute the results obtained to primer mismatches. In fact, the reverse primer had one mismatch to F. nucleatum, an organism over-represented in mock communities, but no sequence mismatches were present with L. casei, an under-represented organism. Hence, other parameters such as different primer binding energies or interferences from DNA flanking the template region may better explain the observed quantitative results (Hansen et al., 1998; Polz & Cavanaugh, 1998).
Moreover, the DNA isolation protocol used in this study was chosen from a group of protocols tested in our laboratory for their efficiency in lysing both gram-positive and gram-negative organisms (data not shown). Despite this, sequence analysis of mock 2 (equal number of cells) did not yield an evenly distributed community, nor a community that resembled the abundances in mock 1. Mocks 2 and 3 showed biases in the same species, which suggests that the actual relative abundance of species in the community does not unduly influence the bias introduced. The species with greater over-representation in mocks 2 and 3 as a consequence of DNA-isolation bias was S. oralis. This finding agrees with previously published results demonstrating over-representation of streptococci after 16S rRNA amplification, cloning and Sanger sequencing (Kroes et al., 1999) and disagrees with our previous findings that streptococci were accurately represented after Sanger methods (Diaz et al., 2006). These discrepancies could be explained by intra-genus variability in lysis efficiency, as exemplified by S. mutans under-representation in mocks 2 and 3. Although such findings of inherent biases may discourage the use of an open-ended molecular method, they do not diminish the great advantages of using powerful high-throughput approaches, which can reveal the breadth and depth of bacterial diversity in a given sample. They do, however, highlight the importance of investigator awareness of limitations, and adoption of standardized protocols when possible to minimize errors.
Our work also presents an improvement in an already highly effective data analysis pipeline (Schloss et al., 2011). By removing singleton OTUs we are able to decrease the number of erroneous OTUs to almost zero. Most singleton OTUs appeared to be chimeric sequences, in agreement with recent results (Schloss et al., 2011). This improvement is of great advantage when using deep-sequenced data to determine richness because the inclusion of amplification and sequencing artifacts could cause rarefaction curves to appear as never leveling. Although application of this correction to real samples could eliminate true taxa, this is preferable to the inclusion of erroneous OTUs, which artifactually increase dissimilarity among datasets. Elimination of singleton OTUs may not be necessary once chimera detection methods improve. It is also noteworthy that mock communities were not deep-sequenced and the number of erroneous OTUs could increase with sequencing effort, as recently reported (Schloss et al., 2011). As a consequence, our deep-sequenced samples could contain more erroneous OTUs than those reported for mock communities. However, this increase is expected to occur in a linear fashion and it may not greatly affect the richness results. It could partly account, however, for the lack of a complete asymptote in rarefaction curves, although it is also expected that not all richness was sampled.
Our study also assessed estimators of richness and coverage. Deep-sequencing of three subjects allowed us to capture a number of OTUs close to those in the sampling universe. Results demonstrated that basing coverage on Good–Turing's estimator greatly underestimates the number of sequences needed to achieve acceptable coverage of richness. catchall provided results that better approximated reality and was more accurate when used in uneven communities than Good–Turing. We also show that catchall rapidly stabilizes and so, after obtaining ∼3000–5000 sequences, it can be used as a reliable estimator of total richness in a sample, even though detection of nearly all OTUs would require 100 times more sequences. Hence, although Good–Turing is widely used by microbial ecologists, these results confirm that this non-parametric estimator is downwardly biased and of lower accuracy. Reduction of sequencing error and the identification of an estimator that reliably predicts total richness allowed us then to answer the question of how many OTUs existed in the microbial communities sampled. This analysis resulted in a range of observed OTUs at each site of 63–318, and total estimated OTUs at each site from 111 to 377. These results are in striking agreement with findings of Zaura et al. (2009), who reported a range of 123–326 OTUs (also defined at 3% dissimilarity) in each oral site they sampled. Richness detected in other oral sites may be higher and may also increase as communities associated with individuals suffering from oral disease are analysed by future studies, because an increased diversity is believed to be associated with the development of conditions such as periodontal disease (Paster et al., 2001; Diaz, 2012).
Another important consideration for large-scale sequencing analysis of communities is the level of sequencing depth needed to answer a specific question or measure an effect. Our analysis demonstrates that a large number of sequences is required to completely cover the richness in salivary or mucosal samples. If communities are compared based on prevalence data, it may be necessary to sample to a greater depth to conclude that a specific phylotype is absent. A similar limitation applies to studies that aim at defining the core oral microbiome, those phylotypes highly prevalent in all human hosts, which will most likely be impacted by under-sampling. We demonstrate, however, that comparing communities based on structure is more feasible than comparisons based on membership. Moreover, depending on the effect size measured, it may not be necessary to achieve great richness coverage. As few as 40 sequences were able to discriminate between saliva and buccal mucosa communities because of considerable differences in structures. Clearly, researchers will benefit from preliminary studies like the one herein to define sequencing depth and associated costs before embarking on large-scale sequencing enterprises.
We demonstrate that patterns in biogeography of oral communities from 454-pyrosequencing of 16S rRNA amplicons largely agree with those previously reported using culturing or other molecular methods, and we further expand the characterization of salivary and buccal mucosa communities. For instance, our results agree with those of Aas et al. (2005), in that S. mitis, S. mitis bv. 2 and G. haemolysans were the predominant species of the buccal epithelium. It is interesting to highlight the intra-genus variability in the affinity of microorganisms for buccal mucosal surfaces because several cultured and uncultured streptococci other than S. mitis, as well as Gemella sanguinis, are over-represented in saliva (see Supplementary material, Table S2). These differences in fine-scale biogeography support the concept of an interplay between environmental selection and microbial traits as a driving force of community assemblage. The attachment of S. mitis to oral epithelial surfaces is mediated by adhesins that bind to sialic acid receptors (Gibbons, 1989; Childs & Gibbons, 1990). The mechanism used by G. haemolysans has not been studied. The abundance of these microorganisms at mucosal surfaces during health suggests a role as prime commensals of the oral cavity of humans. Discerning the mechanisms by which they interact with, and are tolerated by the host as well as their interactions with mucosal pathogens will be important to understand how the dynamics of the host–oral flora cross-talk may promote health or disease.
Moreover, our results expand knowledge on the salivary flora of healthy individuals by the deployment of a powerful open-ended molecular method. Mager et al. (2003) used the checkerboard DNA–DNA hybridization technique to evaluate the presence and abundance of 40 bacterial species in healthy individuals. Our results and the latter study agree in finding Veillonella parvula, Prevotella melaninogenica, Fusobacterium periodonticum and S. mitis as predominant microorganisms in the saliva of healthy individuals. Our study broadens this knowledge base by demonstrating that 11 of the 25 most abundant organisms in saliva across individuals were uncultured ‘species-level’ phylotypes, belonging to the genera Prevotella, Porphyromonas, Streptococcus, Haemophilus, Aggregatibacter and Rothia. Of particular interest is the great abundance of Porphyromonas and Prevotella sp., organisms typically thought to favor the subgingival environment because of their oxygen requirements. In a previous study we identified these two genera as present in initial communities formed for 4 or 8 h on the enamel surfaces of healthy individuals (Diaz et al., 2006), a finding in agreement with their pronounced abundance in saliva and their low affinity for soft tissues. Characterization of the properties of these uncultured Prevotella and Porphyromonas species and comparison with cultured species such as Porphyromonas gingivalis or Prevotella nigrescens that are considered pathogenic microorganisms of the subgingival environment (Socransky et al., 1998), remains a question for future investigations. The high abundance of the genus Neisseria in saliva and its low abundance at mucosal surfaces are also interesting findings. Species of Neisseria, in particular Neisseria mucosa, have not been previously demonstrated to differ among oral sites (Mager et al., 2003). Here we show species-level phylotypes identified as Neisseria sicca and Neisseria flavescens present in great abundance in saliva and with low affinity for buccal mucosal surfaces. One speculation is that the great abundance of obligate anaerobes in saliva may depend on the presence of these aerobic, oxygen-metabolizing organisms, as has been suggested by in vitro modeling of oral bacterial consortia (Bradshaw et al., 1996).
In conclusion, 454-pyrosequencing of microbial communities is a powerful method for evaluation of oral biodiversity. However, investigators using this strategy should be aware of limitations and minimize technical error by accounting for it in the design of experimental studies and data analysis. Use of similar operating procedures among oral researchers is highly encouraged. It is also advisable to use a mock community of oral organisms to refine protocols before under-taking large-scale studies. Researchers should also determine the best sequencing effort needed based on their specific study question. Studies directed at determining the existence of a core microbiome in the oral cavity should first assess coverage with a reliable estimator such as catchall before concluding on differences or commonalities. Studies interested in determining drivers of community structure should first determine optimal sequencing depth, depending on the effect size of the expected change. By using these methods we present evidence that fine-scale biogeography variation within the oral cavity is larger than inter-subject variability in the structure of either salivary or mucosal communities. This finding enables the use of 16S rRNA community profiling to understand microbial shifts associated with the development of mucosal disease.
Supplementary Material
Table S1. Operational taxonomic units (OTUs) over-represented in mucosa after adjusting for multiple comparisons (P < 0.0022).
Table S2. Operational taxonomic units (OTUs) over-represented in saliva after adjusting for multiple comparisons (P < 0.0022).
Table S3. Genera over-represented in mucosa after adjusting for multiple comparisons (P < 0.0128).
Table S4. Genera over-represented in saliva after adjusting for multiple comparisons (P < 0.0128).
Acknowledgments
We thank Dr. Patrick D. Schloss for his help and guidance in the use of mothur. This work was supported by Grant 5RO1DE02157 from The National Institutes of Health, National Institute of Dental and Craniofacial Research.
Footnotes
Supporting Information: Additional Supporting Information may be found in the online version of this article:
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author.
References
- Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. Defining the normal bacterial flora of the oral cavity. J Clin Microbiol. 2005;43:5721–5732. doi: 10.1128/JCM.43.11.5721-5732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainamo J, Barmes D, Beagrie G, Cutress T, Martin J, Sardo-Infirri J. Development of the World Health Organization (WHO) community periodontal index of treatment needs (CPITN) Int Dent J. 1982;32:281–291. [PubMed] [Google Scholar]
- Belda-Ferre P, Alcaraz LD, Cabrera-Rubio R, et al. The oral metagenome in health and disease. ISME J. 2011;30:85. doi: 10.1038/ismej.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bik EM, Long CD, Armitage GC, et al. Bacterial diversity in the oral cavity of 10 healthy individuals. ISME J. 2010;4:962–974. doi: 10.1038/ismej.2010.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw DJ, Marsh PD, Allison C, Schilling KM. Effect of oxygen, inoculum composition and flow rate on development of mixed-culture oral biofilms. Microbiology. 1996;142:623–629. doi: 10.1099/13500872-142-3-623. [DOI] [PubMed] [Google Scholar]
- Bunge J. Estimating the number of species with catchall. Pacific Symposium on Biocomputing; 2011. pp. 121–130. [DOI] [PubMed] [Google Scholar]
- Chang JY, Antonopoulos DA, Kalra A, et al. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J Infect Dis. 2008;197:435–438. doi: 10.1086/525047. [DOI] [PubMed] [Google Scholar]
- Chao A, Shen TJ. Nonparametric estimation of Shannon's index of diversity when there are unseen species in sample. Environ Ecol Statistics. 2003;10:429–443. [Google Scholar]
- Childs WC, 3rd, Gibbons RJ. Selective modulation of bacterial attachment to oral epithelial cells by enzyme activities associated with poor oral hygiene. J Periodontal Res. 1990;25:172–178. doi: 10.1111/j.1600-0765.1990.tb01040.x. [DOI] [PubMed] [Google Scholar]
- Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhirst FE, Chen T, Izard J, et al. The human oral microbiome. J Bacteriol. 2010;192:5002–5017. doi: 10.1128/JB.00542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz PI. Microbial diversity and interactions in subgingival communities. In: Kinane DF, Mombelli A, editors. Periodontal Disease Front Oral Biol. Vol. 15. Basel: Karger; 2012. pp. 17–40. [DOI] [PubMed] [Google Scholar]
- Diaz PI, Chalmers NI, Rickard AH, et al. Molecular characterization of subject-specific oral microflora during initial colonization of enamel. Appl Environ Microbiol. 2006;72:2837–2848. doi: 10.1128/AEM.72.4.2837-2848.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg PB, Relman DA. The role of microbes in Crohn's disease. Clin Infect Dis. 2007;44:256–262. doi: 10.1086/510385. [DOI] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J, Sheneman L, Foster J. Relaxed neighbor joining: a fast distance-based phylogenetic tree construction method. J Mol Evol. 2006;62:785–792. doi: 10.1007/s00239-005-0176-2. [DOI] [PubMed] [Google Scholar]
- Gibbons RJ. Bacterial adhesion to oral tissues: a model for infectious diseases. J Dent Res. 1989;68:750–760. doi: 10.1177/00220345890680050101. [DOI] [PubMed] [Google Scholar]
- Good IJ. The population frequencies of species and the estimation of population parameters. Biometrika. 1953;40:237–264. [Google Scholar]
- Hansen MC, Tolker-Nielsen T, Givskov M, Molin S. Biased 16S rDNA PCR amplification caused by interference from DNA flanking the template region. FEMS Microbiol Ecol. 1998;26:141–149. [Google Scholar]
- Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol. 2010;12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail AS, Behrendt CL, Hooper LV. Reciprocal interactions between commensal bacteria and gamma delta intraepithelial lymphocytes during mucosal injury. J Immunol. 2009;182:3047–3054. doi: 10.4049/jimmunol.0802705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474:327–336. doi: 10.1038/nature10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klappenbach JA, Saxman PR, Cole JR, Schmidt TM. rrndb: the Ribosomal RNA Operon Copy Number Database. Nucleic Acids Res. 2001;29:181–184. doi: 10.1093/nar/29.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroes I, Lepp PW, Relman DA. Bacterial diversity within the human subgingival crevice. Proc Natl Acad Sci USA. 1999;96:14547–14552. doi: 10.1073/pnas.96.25.14547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczynski J, Liu Z, Lozupone C, McDonald D, Fierer N, Knight R. Microbial community resemblance methods differ in their ability to detect biologically relevant patterns. Nat Methods. 2010;7:813–819. doi: 10.1038/nmeth.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar PS, Brooker MR, Dowd SE, Camerlengo T. Target region selection is a critical determinant of community fingerprints generated by 16S pyrosequencing. PLoS ONE. 2011;6:e20956. doi: 10.1371/journal.pone.0020956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunin V, Engelbrektson A, Ochman H, Hugenholtz P. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol. 2010;12:118–123. doi: 10.1111/j.1462-2920.2009.02051.x. [DOI] [PubMed] [Google Scholar]
- Lazarevic V, Whiteson K, Hernandez D, Francois P, Schrenzel J. Study of inter- and intra-individual variations in the salivary microbiota. BMC Genomics. 2010;11:523. doi: 10.1186/1471-2164-11-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos LN, Fulthorpe RR, Triplett EW, Roesch LF. Rethinking microbial diversity analysis in the high throughput sequencing era. J Microbiol Methods. 2011;86:42–51. doi: 10.1016/j.mimet.2011.03.014. [DOI] [PubMed] [Google Scholar]
- Li L, Hsiao WW, Nandakumar R, et al. Analyzing endodontic infections by deep coverage pyrosequencing. J Dent Res. 2010;89:980–984. doi: 10.1177/0022034510370026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljemark WF, Gibbons RJ. Ability of Veillonella and Neisseria species to attach to oral surfaces and their proportions present indigenously. Infect Immun. 1971;4:264–268. doi: 10.1128/iai.4.3.264-268.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljemark WF, Gibbons RJ. Proportional distribution and relative adherence of Streptococcus miteor (mitis) on various surfaces in the human oral cavity. Infect Immun. 1972;6:852–859. doi: 10.1128/iai.6.5.852-859.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager DL, Ximenez-Fyvie LA, Haffajee AD, Socransky SS. Distribution of selected bacterial species on intraoral surfaces. J Clin Periodontol. 2003;30:644–654. doi: 10.1034/j.1600-051x.2003.00376.x. [DOI] [PubMed] [Google Scholar]
- Marrugan AE, McGill BJ. Biological Diversity: Frontiers in Measurement and Assessment. Oxford; Oxford University Press; 2011. [Google Scholar]
- Marsh PD. Are dental diseases examples of ecological catastrophes? Microbiology. 2003;149:279–294. doi: 10.1099/mic.0.26082-0. [DOI] [PubMed] [Google Scholar]
- Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- Morgan JL, Darling AE, Eisen JA. Metagenomic sequencing of an in vitro-simulated microbial community. PLoS ONE. 2010;5:e10209. doi: 10.1371/journal.pone.0010209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paster BJ, Boches SK, Galvin JL, et al. Bacterial diversity in human subgingival plaque. J Bacteriol. 2001;183:3770–3783. doi: 10.1128/JB.183.12.3770-3783.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polz MF, Cavanaugh CM. Bias in template-to-product ratios in multitemplate PCR. Appl Environ Microbiol. 1998;64:3724–3730. doi: 10.1128/aem.64.10.3724-3730.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushalkar S, Mane SP, Ji X, et al. Microbial diversity in saliva of oral squamous cell carcinoma. FEMS Immunol Med Microbiol. 2011;61:269–277. doi: 10.1111/j.1574-695X.2010.00773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravel J, Gajer P, Abdo Z, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeder J, Knight R. The ‘rare biosphere’: a reality check. Nat Methods. 2009;6:636–637. doi: 10.1038/nmeth0909-636. [DOI] [PubMed] [Google Scholar]
- Schloss PD. The effects of alignment quality, distance calculation method, sequence filtering, and region on the analysis of 16S rRNA gene-based studies. PLoS Comput Biol. 2010;6:e1000844. doi: 10.1371/journal.pcbi.1000844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL. Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl Environ Microbiol. 2011;77:3219–3226. doi: 10.1128/AEM.02810-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE. 2011;6:e27310. doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson EH. Measurement of diversity. Nature. 1949;163:688. [Google Scholar]
- Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–144. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- Sundquist A, Bigdeli S, Jalili R, et al. Bacterial flora-typing with targeted, chip-based Pyrosequencing. BMC Microbiol. 2007;7:108. doi: 10.1186/1471-2180-7-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Gordon JI. The core gut microbiome, energy balance and obesity. J Physiol. 2009;587:4153–4158. doi: 10.1113/jphysiol.2009.174136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol. 2009;5:e1000352. doi: 10.1371/journal.pcbi.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue JC, Clayton MK. A similarity measure based on species proportions. Commun Statistics Theory Meth. 2005;34:2123–2131. [Google Scholar]
- Zaura E, Keijser BJ, Huse SM, Crielaard W. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 2009;9:259. doi: 10.1186/1471-2180-9-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Operational taxonomic units (OTUs) over-represented in mucosa after adjusting for multiple comparisons (P < 0.0022).
Table S2. Operational taxonomic units (OTUs) over-represented in saliva after adjusting for multiple comparisons (P < 0.0022).
Table S3. Genera over-represented in mucosa after adjusting for multiple comparisons (P < 0.0128).
Table S4. Genera over-represented in saliva after adjusting for multiple comparisons (P < 0.0128).