

Abstract
Aberrant activation of the epidermal growth factor receptor (EGFR) is critical to the biology of many common cancers. The molecular events that define how EGFR transmits an extracellular ligand binding event through the membrane are not understood. Here we use a chemical tool, bipartite tetracysteine display, to report on ligand-specific conformational changes that link ligand binding and kinase activation for full-length EGFR on the mammalian cell surface. We discover that EGF binding is communicated to the cytosol through formation of an antiparallel coiled coil within the intracellular juxtamembrane (JM) domain. This interaction is functionally coupled to receptor activation by EGF. In contrast, TGFα binding is communicated to the cytosol through formation of a discrete, alternative helical interface. These findings suggest that the JM region can differentially decode extracellular signals and transmit them to the cell interior, and provide new insight into how EGFR communicates ligand-specific information across the membrane.
Introduction
The epidermal growth factor receptor (EGFR/ErbB1/HER1) receives a stimulus in the form of an extracellular binding event and communicates this information across the cell membrane to effect diverse signaling outcomes.1 When this communication is misregulated via overexpression or mutation, the signaling consequences are associated with a variety of human diseases, including cancer. Therefore, deciphering how EGFR conveys information across the cell membrane is essential to our understanding of its role not only in normal biology, but also in disease progression and therapeutic response.1, 2 Here we apply a novel chemical tool to identify ligand-specific conformational changes that link ligand-induced reorganization of the extracellular domains to kinase domain activation in the context of full-length receptor in mammalian cells. Our findings suggest that the intracellular juxtamembrane segment plays a crucial role not only in receptor activation, but also in relaying the identity of the bound ligand to the cytosol.
Roughly three decades have passed since EGFR was first identified as single-pass transmembrane receptor tyrosine kinase, yet the mechanism through which it conveys extracellular signals through the plasma membrane remains unknown. It is known that extracellular ligand binding induces an intracellular structural transition to result in kinase activation through an asymmetric homodimeric interface (Figure 1A).1, 3-6 However, the nature of this structural transition has remained elusive because of limited structural information describing how the EGFR extracellular and intracellular domains are connected.1, 3-5, 7-12 Recent studies have demonstrated that the intracellular juxtamembrane (JM) segment, which is largely undefined in a structural context, is critical for information transfer by EGFR.9, 11, 13-15 Deletion of the JM segment abrogates receptor activation,15 and crystallographic studies identify contacts with the C-terminal portion of the JM domain that stabilize the active, asymmetric kinase domain interface.9, 14 Furthermore, the intracellular JM domain is required for the observed negative cooperativity in EGF binding.11
Figure 1. Monitoring EGFR Dimerization and Activation Using Bipartite Tetracysteine Display.
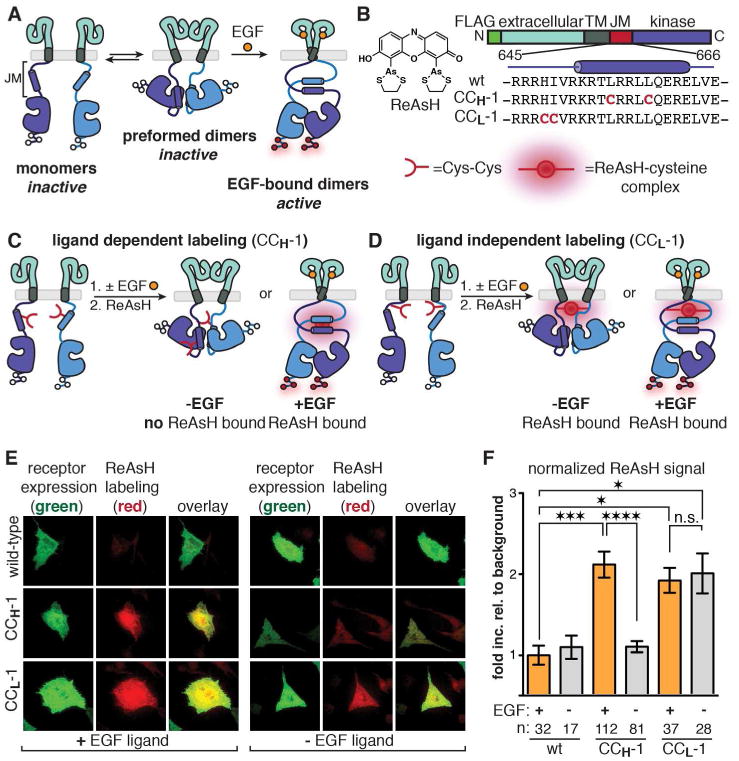
(a) Cartoon depicting the current model for EGFR activation. White circles represent sites where tyrosine becomes phosphorylated (red circles) when the receptor is activated. (b) Chemical structure of ReAsH along with the domain structure of EGFR and the identities of two JM Cys-Cys constructs prepared. (c) Cartoon demonstrating EGF-dependent ReAsH labeling of CCH-1 EGFR. (d) Cartoon depicting ligand independent labeling of CCL-1 EGFR. (e) Representative TIRFM images for monitoring the ReAsH labeling of wild-type, CCH-1, and CCL-1 EGFR in the presence (left) and absence (right) of EGF. (f) Quantification of TIRFM results as a fold increase relative to background that is normalized for receptor expression levels. n is the number of cells quantified. Error bars represent the standard error. *p<0.05, *** p<0.001, **** p<0.0001, one-way ANOVA with Bonferroni post-analysis accounting for multiple comparisons.
ReAsH labeling of CCH-1 EGFR is dependent on the presence of EGF, whereas ReAsH labeling of CCL-1 can occur regardless of ligand. These data support the conclusion that an interhelical JM interaction is present upon EGF binding.
Recent in vitro studies of the N-terminal portion of the JM segment suggest one model for how this domain might potentiate kinase activation.9 NMR studies revealed that a short segment of the N-terminal JM region displays nascent helicity in vitro, and that when two copies of this sequence are fused by a short, flexible linker, the polypeptide folds into an antiparallel coiled coil.9 Notably, no defined interhelical interaction was observed in the absence of the linker, suggesting that the antiparallel interaction may only occur when enforced by an increase in effective concentration and may not represent the exclusive association geometry. Moreover, it has not been established whether this association is the unique allosteric link that couples extracellular ligand binding to intracellular kinase activation for a full-length receptor at the plasma membrane.14, 16
Here we use a novel chemical tool, bipartite tetracysteine display17, 18 to probe structure within the intracellular JM domain of full-length EGFR expressed on the mammalian cell surface. Bipartite tetracysteine display reports on protein conformation and association via a turn-on fluorescent signal that results from coordination of a bis-arsenical fluorophore (ReAsH)19, 20 to an encoded tetracysteine motif that is reconstituted only when the protein is folded and assembled properly (Fig. 1B-D).17 Formation of a bipartite ReAsH binding site is functionally coupled to protein structure and/or association. Therefore, bipartite tetracysteine display is uniquely suited to discern discrete protein interactions within the dynamic environment of a mammalian cell.
In this work we exploit bipartite tetracysteine display to demonstrate that ligand binding to the EGFR extracellular domains is transmitted across the membrane into a defined dimeric helical interface within the JM. Additionally, we discover that ligand identity is communicated to the cell interior through distinct JM conformations. In the presence of EGF and certain other EGFR ligands, our data support formation of the antiparallel helical dimer that assembles in isolation.9 In the presence of transforming growth factor-α (TGFα), however, this antiparallel coiled coil is not formed and an alternative helical interface is present. Formation of these structures is contingent on the ability to assemble an asymmetric kinase interface, providing evidence that the diversity of potential JM interactions imparts a mechanism to transmit ligand-specific information to the kinase domains. Therefore, our findings suggest that the JM segment plays a crucial role not only in receptor activation, but also in decoding and relaying extracellular signals to the cytosol.
Results and Discussion
Design of Bipartite Cys-Cys Variants Within the EGFR Intracellular Juxtamembrane Domain
The JM segment is proposed to associate into an antiparallel coiled coil based on NMR structural information acquired for a tethered peptide dimer in isolation.9 We designed full-length EGFR variants containing judiciously placed Cys-Cys pairs to test the hypothesis that the intracellular JM segment assembles into a coiled coil upon EGF binding in the context of the full-length homodimeric receptor (Figures 1B-D). We used the modeled coordinates generated by Jura et al. to design several EGFR variants that contain Cys-Cys pairs at positions within the interface of the proposed antiparallel helical dimer (Figure 1B,C, Figure 2A-C).9 If the JM coiled coil were present in the EGF-activated receptor, these variants (in particular, CCH-1(Helix)) would be expected to bind ReAsH and fluoresce only upon ligand binding and receptor activation. We also constructed a panel of EGFR variants that contain Cys-Cys pairs in an unstructured section of the JM loop (CCL-1 (Loop); CCL-2—CCL-5 not shown) (Figure 1B,D) and would be expected to provide a ReAsH binding site irrespective of ligand binding or receptor activation. Thus, if ligand binding results in a helical association within the JM, CCH-1 and CCL-1 should perform divergently depending on the receptor activation state when treated with ReAsH, despite differing only minimally in primary sequence (Figure 1B-D).
Figure 2. A JM antiparallel helical dimer is present when EGFR is stimulated with EGF.
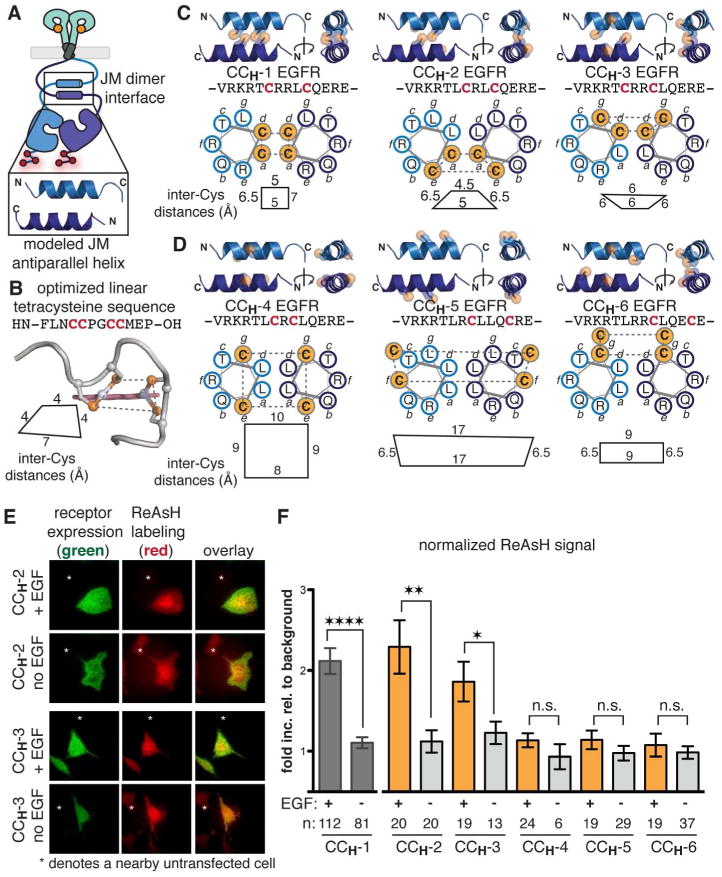
(a) Cartoon representation of the proposed antiparallel coiled coil and the modeled coordinates for this interaction. (b) Structure of an optimized linear tetracysteine peptide in complex with ReAsH.36 Inter-cysteine distances are measured from the sulfur atoms. (c) Bipartite Cys-Cys EGFR variants expected to be labeled with ReAsH if the proposed antiparallel coiled coil is formed (see also Supporting Figure S5). Inter-cysteine distances are measured from the sulfur atoms. (d) Bipartite Cys-Cys EGFR variants not expected to be labeled with ReAsH due to an unfavorable binding site geometry when the coiled coil is present. Inter-cysteine distances are measured from the sulfur atoms. (e) Representative TIRFM images of cells expressing CCH-2 and CCH-3 EGFR treated with ReAsH in the presence and absence of EGF. TIRFM images for CCH-4, CCH-5, and CCH-6 can be found in Supporting Figure S4. (f) Quantification of TIRFM results as a fold increase relative to background that is normalized for receptor expression levels. n is the number of cells quantified. Error bars represent the standard error. * p< 0.05, ** p< 0.01, and **** p< 0.0001, t-test analysis.
The ReAsH labeling results for a series of Cys-Cys variants provide further evidence that the JM interacts through an antiparallel coiled coil when EGFR is stimulated with EGF. Additional analysis (Supporting Figure S5) rules out parallel association in the two most likely registers.
JM Cys-Cys EGFR Variants are Present at the Cell Surface and Activated by EGF
We first set out to confirm that the introduction of Cys-Cys pairs in the EGFR JM segment would not affect ligand-dependent kinase activation. When CCH-1 and CCL-1 were expressed in CHO cells, the levels of EGF-dependent phosphorylation at Y1173 were comparable to those for wild type EGFR. In addition, the basal level of phosphorylation at Y1173 of CCH-1 and CCL-1 in the absence of ligand was also comparable to that of wild-type EGFR. Importantly, when the CCH-1 and CCL-1 variants were expressed in CHO cells and treated with ReAsH, there was no appreciable change in the levels of phosphorylation at Y1173, either in the presence of absence of EGF (Supporting Figure S1). We further confirmed that wild type, CCH-1, and CCL-1 EGFR variants were expressed on the cell surface using a surface biotinylation assay (Supporting Figure S1).21 These experiments revealed that the higher molecular weight species resolvable by SDS-PAGE is the mature, cell surface form of the receptor, and that this species displays an EGF-dependent increase in phosphorylation at Y1173.
Development of a TIRFM Assay to Monitor ReAsH Labeling of Cys-Cys EGFR Variants
We next developed an experimental protocol that could evaluate ReAsH labeling in a manner uncomplicated by normal receptor internalization upon addition of EGF. We chose to circumvent ligand-stimulated receptor internalization by chemically inhibiting endocytosis preceding stimulation by EGF and labeling with ReAsH. This inhibition protocol did not prevent ligand-dependent phosphorylation of wild type, CCH-1, and CCL-1 EGFR (Supporting Figure S1).4, 22, 23,24 As we were interested primarily in the ReAsH signal at the plasma membrane, we relied on total internal reflectance fluorescence microscopy (TIRFM) to monitor levels of ReAsH fluorescence. We note that this strategy effectively enhanced the signal of interest by diminishing the signal from non-specific cytosolic ReAsH staining25 by restricting fluorophore excitation and emission to a small (100-200 nm) cell surface plane. Receptor expression was monitored using fluorescently labeled antibodies to an N-terminal FLAG epitope (Figure 1B). Taken together, these results and protocols support the viability of using bipartite tetracysteine display to study the EGFR activation mechanism.
Bipartite Tetracysteine Display Can Distinguish Preformed and EGF-bound EGFR Dimers
We used this detection scheme to test the hypothesis that the JM region in full-length EGFR associates into a dimer upon EGF binding. To begin, we incubated mammalian cells expressing CCH-1 EGFR (green) with or without EGF (100 ng/mL, 16.7 nM) in the presence of endocytosis inhibitors and subsequently added ReAsH. Under these conditions, it was possible to discern a significant increase in ReAsH signal (red) only when EGF was present (Figure 1E). As summarized in Figure 1F, cells expressing CCH-1 EGFR displayed roughly 2-fold greater normalized red fluorescence relative to background when stimulated with EGF. This fold increase measurement corrects for differences in expression level (see Methods section). Furthermore, we do not observe a correlation between this normalized fold increase in ReAsH signal and receptor expression levels, suggesting that any effects resulting from receptor density are minimal. This fold increase was comparable to that obtained for cells expressing an EGFR variant containing a linear tetracysteine tag fused to the C-terminus (Supporting Figure S2). Cells expressing wild-type EGFR or a variant containing only one engineered cysteine residue displayed negligible increases in ReAsH signal in the presence and absence of EGF (Figure 1E,F, Supporting Figure S2). We note that wild-type EGFR contains 59 cysteine residues, nine of which are located in the cytoplasmic domains. Consistent with our expectations, cells expressing CCL-1 EGFR treated in this way also displayed a 2-fold increase in normalized ReAsH fluorescence at the cell surface. In the absence of endocytosis inhibitors, ReAsH-labeled endosomes containing EGF were observed in cells expressing CCH-1 and CCL-1 EGFR variants (Supporting Figure S3). In the absence of EGF, however, cells expressing CCL-1 EGFR retained a significant increase in ReAsH fluorescence at their surface, whereas cells expressing CCH-1 EGFR did not (Figure 1E,F). The EGF-dependent increase in ReAsH fluorescence observed for CCH-1 EGFR but not CCL-1 EGFR suggests that EGF binding results in the formation of a discrete dimeric interaction within the JM.
Bipartite Tetracysteine Display Confirms the Antiparallel JM Interaction is Present when EGFR is Activated by EGF
To provide evidence that the dimeric interaction between JM segments induced by the addition of EGF is a discrete, antiparallel coiled coil, we designed a set of additional Cys-Cys EGFR variants based on the modeled structure of the isolated, tethered JM antiparallel coiled coil.9 Two of these variants were expected to respond identically to CCH-1 EGFR, as the Cys-Cys pairs remain proximal on one face of the proposed antiparallel coiled coil: CCH-2 and CCH-3 (Figure 2C). We also designed three constructs (CCH-4, CCH-5, and CCH-6) with substitutions at positions we predicted would not be labeled by ReAsH because the Cys-Cys pairs are displaced axially and are no longer proximal (Figure 2D). Western blot analysis revealed that all variants exhibited a ligand-dependent increase in receptor phosphorylation (Supporting Figure S4). None of these variants (CCH-2, CCH-3, CCH-4, CCH-5, or CCH-6) were labeled by ReAsH in the absence of EGF (Figure 2E,F, Supporting Figure S4). In the presence of EGF, however, mammalian cells expressing CCH-2 and CCH-3 EGFR variants displayed a significant fold increase in ReAsH signal at their surface when stimulated with EGF (2.2 and 1.8, respectively) whereas cells expressing CCH-4, CCH-5, and CCH-6 did not (Figure 2E,F; Supporting Figure S4). These data are consistent with the hypothesis that the dimeric interface assembled within the JM of EGFR in the presence of EGF is an antiparallel coiled coil.
We also used these results to further explore and identify the register and orientation of the helical association (Supporting Figure S5). In particular, the ReAsH labeling results obtained for CCH-1, CCH-2, and CCH-3 are most consistent with an antiparallel orientation in the register proposed by the Jura et al. study (Figure 2C, Supporting Figure S5) and are inconsistent with a parallel arrangement of the two helical regions in the two most probable registers.9 Therefore, these results provide evidence that the JM antiparallel helical interaction observed for the tethered helices in vitro9 is present when EGFR on the cell surface is stimulated with EGF.
Bipartite Tetracysteine Display Links the JM Coiled Coil Interaction to Kinase Domain Activation
Our results thus far demonstrate that the binding of EGF to full-length EGFR leads to formation of an antiparallel coiled coil within the intracellular JM. To explore how this conformation is linked to kinase activation, we made use of two known EGFR mutations that impair ligand-dependent receptor activation.5, 9 We prepared a variant of CCH-1 containing two mutations (R656,657G) within the JM that have been reported to disrupt helicity and attenuate receptor activity.9 We also prepared a CCH-1 variant containing an inactivating point mutation (V924R) that prevents formation of the asymmetric dimer interface (Figure 3A). When cells expressing R656,657G and V924R variants of CCH-1 were exposed to EGF in the presence of endocytosis inhibitors and incubated subsequently with ReAsH, there was no relative increase in ReAsH fluorescence at the cell surface (Figure 3C,D). Western blot analysis confirmed that neither receptor variant was activated by EGF (Figure 3B). The absence of ReAsH labeling in these variants implies a structural linkage between formation of an antiparallel JM coiled coil and the activation state of the receptor: not only does formation of the asymmetric kinase interface depend on helical structure within the JM,9 but helical structure within the JM depends on formation of an asymmetric kinase interface (V924R). In particular, these results demonstrate that the ability of CCH-1 to bind ReAsH is dependent on the formation of helical structure within the JM (R656,657G), and on the ability to form an asymmetric kinase interface (V924R). Furthermore, a classic in vitro disulfide exchange assay26 yielded no appreciable association between JM helices, even at high micromolar concentrations (Supporting Figure S6). Taken together, these results are consistent with a model in which JM association is functionally linked to the global conformation of the intact activated receptor. In other words, the binding of EGF to the EGFR extracellular domain results in a structural change leading to assembly of the intracellular asymmetric kinase interface; this structural signal is transmitted through the formation of an antiparallel coiled coil within the JM segment to which the binding and fluorescence of ReAsH is linked. Consequently, ReAsH binding is positively linked to the global conformation and activation of the homodimeric receptor.
Figure 3. ReAsH Labeling of the JM Antiparallel Helices is Linked to a Global Active Conformation.
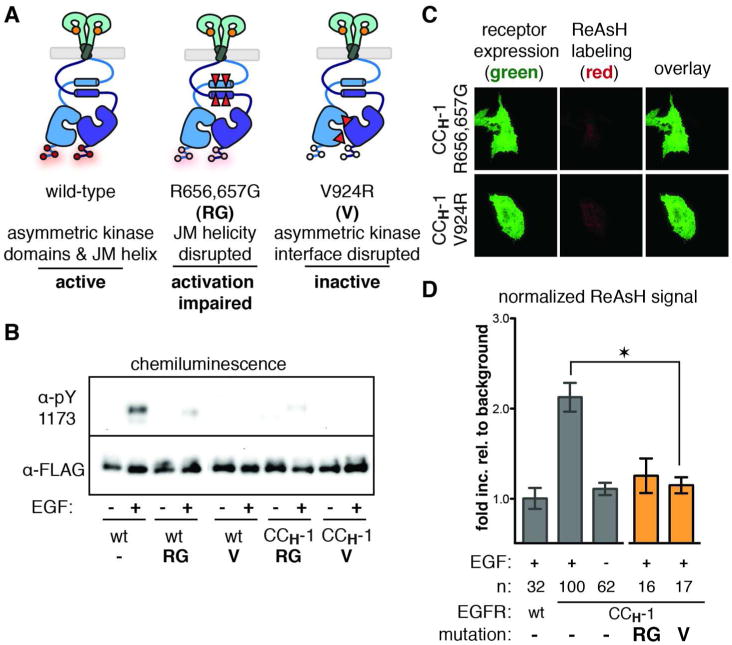
(a) Cartoon depicting the relative positions of the activation-impairing EGFR mutations R656,657G and V924R. (b) Western blots confirm that these mutants are defective in tyrosine autophosphorylation in the context of wild-type and CCH-1 EGFR. (c) Representative TIRFM images of ReAsH-treated cells expressing CCH-1 EGFR variants containing the R656,657G or V924R mutations. (d) Quantification of TIRFM results as a fold increase relative to background that is normalized for receptor expression levels. Error bars represent the standard error. * represents p<0.05 based on ANOVA with Bonferroni post-test.
The ability of CCH-1 to bind ReAsH is dependent on the presence of JM helices (R656,657G) and the global active conformation of kinase domains (V924R). The absence of ReAsH labeling in these variants provides a structural link between the receptor activation and formation of an antiparallel JM coiled coil.
Bipartite Tetracysteine Display Reveals the Antiparallel JM Interaction is Not Present when EGFR is Activated by TGFα
Formation of an antiparallel JM helical interaction in the presence of EGF does not provide a clear model to explain how extracellular ligand identity might be transmitted to the intracellular kinase domains. Is this substructure assembled in the presence of all activating ligands, or might the well-known plasticity of coiled coil domains be exploited to transmit ligand identity? To explore these issues, we treated cells transfected with CCH-1 EGFR with HB or TGFα, along with a related growth factor that does not activate EGFR (heregulin-α, HRG). We confirmed by western blot analysis that both wild-type and CCH-1 EGFR were activated by the addition of HB and TGFα, but not by HRG, as assessed by phosphorylation at Y1173 (Figure 4A). As expected, when mammalian cells expressing CCH-1 EGFR were stimulated by HB (16.7 nM) in the presence of endocytosis inhibitors, we observed a ReAsH signal comparable to that obtained using equimolar concentrations of EGF; when treated with HRG (16.7 nM), there was no relative increase in ReAsH signal at the cell surface (Figure 4B,C). When stimulated with TGFα (16.7 nM), cells expressing CCH-1 EGFR did not exhibit any relative increase in ReAsH signal at their cell surface, even when the TGFα concentration was increased tenfold (Figure 4B,C). These results suggest that the activation of EGFR by HB occurs through formation of an antiparallel coiled coil that resembles that formed upon activation with EGF, whereas activation by TGFα does not. In the case of TGFα, the relative orientation of the JM segments upon ligand binding must be different than the antiparallel coiled coil that assembles upon activation by EGF.
Figure 4. TGFα activates EGFR through an alternative orientation of JM helices.
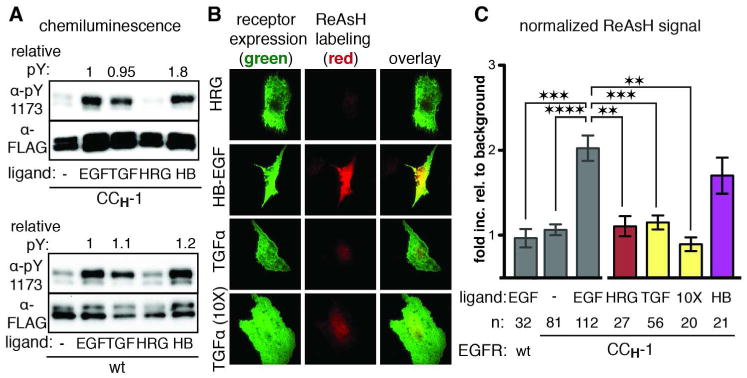
(a) Western blot analysis of wild-type and CCH-1 EGFR stimulated with different growth factor ligands. (b) Representative TIRFM images for ReAsH labeling of CCH-1 EGFR in the presence of HRG, HB-EGF or TGFα. (c) Quantification of TIRFM results as a fold increase relative to background normalized for receptor expression levels. n is the number of cells quantified. Error bars represent the standard error. ** p<0.01, *** p<0.001, **** p<0.0001, one-way ANOVA with Bonferroni post-analysis accounting for multiple comparisons.
The activation of EGFR by TGFα does not involve the JM antiparallel coiled coil that was observed for activation by EGF.
An Alternate and Discrete JM Interaction is Present when EGFR is Activated by TGFα
These results suggest that the activated EGFR homodimer has a unique conformation depending on which ligand is bound. We next sought to understand just how substantial these structural differences might be. When an EGFR point mutant (V924R) that prevents formation of the asymmetric dimer was expressed in CHO cells, there was no detectable phosphorylation at Y1173 regardless of which ligand (EGF or TGFα) was used (Figure 5A). Introduction of the reported helix disrupting R656,657G mutation impaired EGF- and TGFα-dependent activation of wild-type EGFR to the same extent (Figure 5A). These observations suggest that the ability to adopt the asymmetric kinase interface and the presence of intact JM helices is required for activation by both EGF and TGFα. In the context of our studies, this finding is consistent with a model in which ligand identity is transmitted through small structural changes that are distinct from the global conformation required for kinase domain activation.
Figure 5. TGFα activates EGFR through a distinct orientation of JM helices.
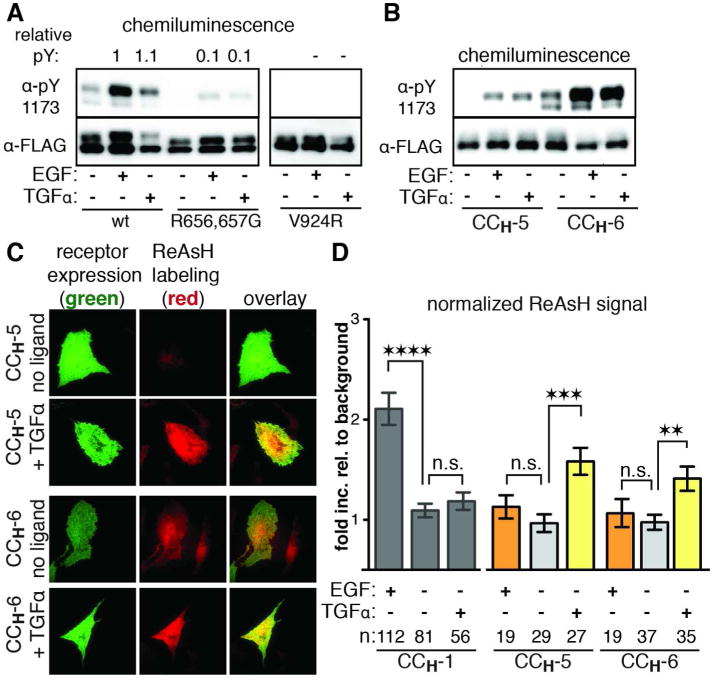
(a) Western blot analysis of wt, R656,657G, and V924R EGFR stimulated with EGF or TGFα. See also Figure 3A. (b) Western blot analysis of CCH-5 and CCH-6 EGFR stimulated with EGF or TGFα. (c) Representative TIRFM images of cells expressing CCH-5 and CCH-6 EGFR that were labeled with ReAsH in the presence or absence of TGFα. See also Supporting Figure S4. (d) Quantification of TIRFM results as a fold increase relative to background that is normalized for receptor expression levels. n is the number of cells quantified. Error bars represent the standard error. ** p<0.01, *** p<0.001, **** p<0.0001, one-way ANOVA with Bonferroni post-analysis accounting for multiple comparisons.
TGFα leads to a structural transition in the JM helices, allowing for CCH-5 and CCH-6 to be labeled with ReAsH. These findings suggest that activation of EGFR by TGFα occurs through a JM helical orientation that is distinct from the antiparallel coiled coil determined for activation by EGF.
Building on the observation that helicity within the JM is important for activation by both EGF and TGFα, we reexamined two constructs, CCH-5 and CCH-6, that were previously not labeled by ReAsH when stimulated with EGF (see above: Figure. 2D,E,F; Supporting Figure S4; Figure 5B). When cells expressing either CCH-5 or CCH-6 EGFR were exposed to TGFα in the presence of endocytosis inhibitors and subsequently incubated with ReAsH, there was a significant increase in normalized ReAsH signal at their cell surface (1.6 and 1.4 fold, respectively), although the magnitude of the increase was not as large as for CCH-1 stimulated with EGF. In the absence of any activating ligand, or in the presence of EGF, cells expressing CCH-5 and CCH-6 did not yield an increase in ReAsH labeling above background (Figure 5C,D). Together, these results indicate that the structural change that occurs in the JM upon activation by TGFα is distinct from that which occurs when the receptor is activated by EGF.
A Structural Explanation for Ligand-Specific Orientations of the Intracellular JM Helices
Structures of the EGF- and TGFα-bound forms of the EGFR ectodomain were reported in 2002; these structures share many commonalities and are often cited synonymously.18, 27-29 We reasoned that any structural differences between EGF- or TGFα- activated intracellular domains must propagate from the extracellular ligand binding domains. Building on a prior analysis,30, 31 we further compared these structures and identified a difference in the orientation of domain IV depending on ligand identity (Figure 6). In the EGF-bound crystal structure, the C-terminal region of domain IV in each monomer forms a homodimeric interface that buries a solvent-exposed surface area of 430 Å2 (Figure 6A,C) However, in all three TGFα- bound homology models there is a substantial steric clash between the two C-termini of domain IV within the EGFR dimer (Figure 6B,C). The structure and orientation of domains III-IV is consistent across all structures analyzed, each of which was obtained using different crystallization conditions and distinct crystal lattices. Within the context of our study, this analysis suggests that the C-terminal region of domain IV in the dimer adopts a different orientation depending on whether TGFα or EGF is bound. This change in orientation would then propagate through the TM domains and affect the relative orientation of the JM helices. At present, we cannot exclude the possibility that the binding of TGFα induces a significant conformational change in the EGFR ectodomain that has not been observed in existing crystal structures; such a change could also affect the relative disposition of the JM helices. Therefore, our analysis of existing structural information is consistent with the finding that activation by TGFα occurs through an orientation of the JM helices that does not correspond to the antiparallel coiled coil elucidated for activation by EGF. In particular, our analysis suggests that differential signaling by TGFα may propagate through a change in the relative orientation of the TM resulting in structural differences in the JM domain.32
Figure 6. The Relative Orientation of Domain IV is Different when TGFα is Bound Instead of EGF.
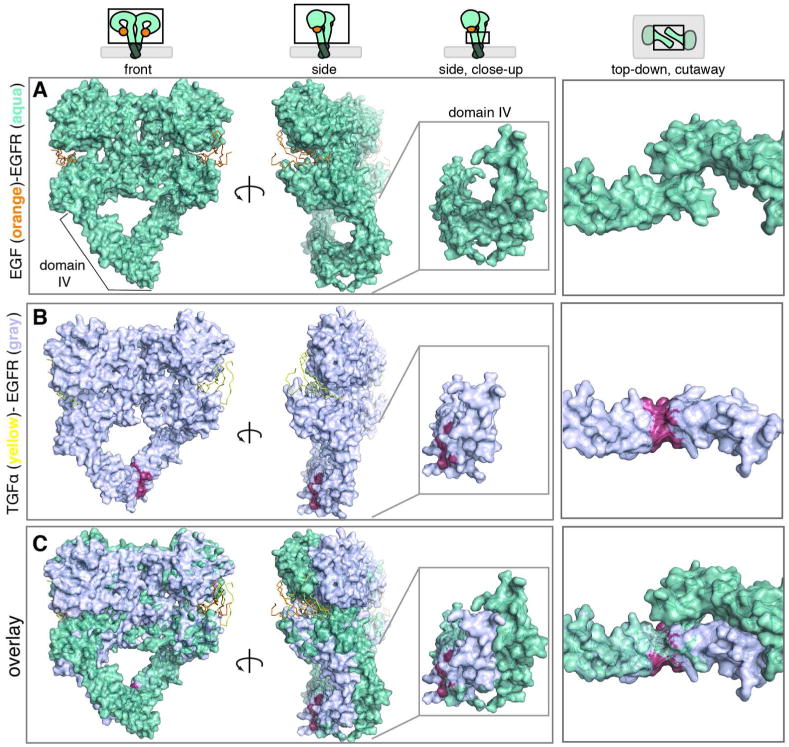
(a) Four views of the crystal structure of the EGFR extracellular domain (aqua) bound to EGF (orange). (b) Four views of the homology model of the EGFR extracellular domain (gray) bound to TGFα (yellow). Red residues represent a steric clash observed in domain IV of the homology model. (c) Overlay of the two structures aligned. Comparison reveals a difference in the orientation of domain IV depending on the ligand identity. This analysis is consistent with a model in which differential signaling by TGFα may propagate through a change in the relative orientation of the TM, which results in structural differences in the JM domain.
Conclusions
Despite multiple high-resolution views of the EGFR extracellular and intracellular domains, a complete understanding of how the intact receptor transmits information across the plasma membrane has remained elusive. Two regions of poorly defined structure, the transmembrane helix and the juxtamembrane (JM) region, present a genuine obstacle to define the coupled conformational changes that must occur during transmembrane signaling. An additional layer of complexity is provided by the observation that multiple transmembrane helix interfaces are compatible with signaling.33 This finding has led to the conclusion that the extracellular and intracellular domains are “loosely linked”,33-35 which makes it especially difficult to understand how ligand-specific signals are differentially propagated.
Here we have used bipartite tetracysteine display17 along with the bis-arsenical dye ReAsH19 to probe the structural changes that underlie EGFR activation for full-length receptor in mammalian cells. Bipartite tetracysteine display is uniquely suited for this task because the preferred binding site for ReAsH, based on the structurally characterized linear ReAsH-tetracysteine complex,36 is a polygon in which four cysteines occupy vertices spaced 4-7 Å apart. Although some flexibility is tolerated,18, 29 this geometric requirement facilitates the use of ReAsH as a high resolution probe for local structure. In this work, the structural prerequisite for ReAsH binding allowed us to distinguish active EGFR dimers from those that are preformed and inactive. Furthermore, these demanding geometric constraints also permitted us to demonstrate that the antiparallel JM coiled coil, which assembles in vitro and in isolation,9 exists on the surface of mammalian cells when the receptor is activated by EGF.
The spatial requirements for ReAsH binding also enabled the discovery that this same antiparallel coiled coil does not form when EGFR is activated by the related ligand TGFα, and that instead a discrete, alternate (but still helical and dimeric) JM interaction occurs. Three models for the dimeric structure formed in the presence of TGFα are compatible with our result. In the first model, the JM helices remain antiparallel but are displaced axially from their orientation in the EGF-activated dimer; we refer to this arrangement as a “slipped antiparallel dimer.” The second model is a parallel coiled coil, though not necessarily in the registers explored by Jura et al. A third alternative is that TGFα binding eliminates a specific interhelical interaction between the EGFR JM segments, but the helices remain folded and proximal. The observation that ReAsH binds to only a sub-set of bipartite Cys-Cys EGFR variants argues against models in which the JM segments lack helical structure in the presence of ligand. Further work is necessary to discriminate between these three possibilities. Regardless of the model, our results indicate that ligand identity is communicated by EGFR through the formation of multiple, discrete, helical JM conformations.
It has been known for two decades that alternative growth factors ligands such as EGF and TGFα lead to distinct EGFR-mediated signaling outcomes and that EGF- and TGFα-bound receptors are trafficked differently (N.B. heparin-binding EGF (HB) is trafficked similarly to EGF).31, 37-41 Our findings provide, to our knowledge, the first structural evidence that differential signaling by alternative ligands is propagated through unique structures. Coupled with our observations that the asymmetric kinase domain interface and intact JM helices, respectively, are essential for EGFR activation by both EGF and TGFα, our ReAsH-labeling results suggest that interhelical JM interactions may act as a versatile switch through which such signals are propagated. Further work will be necessary to fully characterize this previously unidentified aspect of differential EGFR signaling. Together, the results herein provide new insight into the increasingly complex mechanism through which EGFR transmits signals from the cell surface to the interior.
Methods
Mammalian Cell-Based EGFR Activation Studies
CHO-K1 cells (roughly 1.2 × 106) were seeded into 100 mm dishes (BD Falcon) and incubated at 37 °C in 5% CO2 for 24 h. Transfection of full-length EGFR variants was accomplished using TransIT-CHO (Mirus) according to the manufacturer's instructions. After 8 h, the cells were serum-starved in F-12K with 1% FBS for 16 h. At this time, cells were harvested, washed, and pelleted into two wells of a 96 well plate. Cells were resuspended in either 0.2 mL of unlabeled EGF (100 ng/mL, 16.7 nM) in serum-free media or 0.5 mL of serum-free media. [please note: when other growth factors were used in place of EGF, the concentration was 16.7 nM. ] The plates were incubated at 37 °C for 5 min, and then the cells were pelleted. The supernatent was removed and the cells were washed and then resuspended in 200 μL of lysis buffer (50 mM Tris, 150 mM NaCl, 1 mM EDTA, 1 mM NaF, 1% Triton X-100, pH 7.5) containing 1 mM sodium orthovanadate and a protease inhibitor cocktail (Roche) and were incubated on ice for 1.5 h. The lysates were clarified by centrifugation. Total protein concentrations were determined using the Bradford protein assay (Bio-Rad) in order to normalize the total amount of protein loaded onto the gels (10 μg per lane). SDS-PAGE analysis was accomplished using 10% polyacrylamide gels (BioRad) and was followed by transfer to PVDF membranes (iBlot apparatus, Invitrogen). The membranes were blocked with 5% milk in TBS-T (50 mM Tris, 150 mM NaCl, 0.1 % Tween, pH 7.4) for 2-3 h, followed by incubation with primary (mouse α-FLAG or rabbit α-pY1173) antibodies for 16-18 h at 4 °C. The membranes were washed three times with 5% milk in TBS-T (2× 5 min; 1× 15 min) and exposed to secondary HRP-conjugated α-mouse (FLAG) or α-rabbit (pY) antibodies for 1.5 h. The membranes were washed again using TBS-T, as above, and then were developed using ImmunStar WesternC chemiluminescent reagents (BioRad). Chemiluminescent detection was performed using a ChemiDoc XRS+ (BioRad)
Surface ReAsH Labeling Studies
CHO-K1 cells (75,000) were seeded into glass-bottomed MatTek 35 mm dishes coated with fibronectin and cultured for 24 h. Transient transfection with the EGFR variant of interest was accomplished using TransIT-CHO (Mirus) according to the manufacturer's instructions. After 8 h, the cells were serum-starved using 1% serum in F-12K for 16 h. Receptor endocytosis was inhibited by incubation with an ATP synthesis inhibition cocktail (10 mM NaN3, 2 mM NaF and 5 mM 2-deoxy-D-glucose) for 1 h. At this time, the cells were stimulated with 1 mL of unlabeled EGF (100 ng/mL in serum-free ATP inhibition media) or media alone for 30 min at 8 °C. The EGF solution was removed, and the cells were washed once with ATP inhibition media before incubation with 150 μL ReAsH labeling solution (2 μM ReAsH + 20 μM BAL + 2 μM disperse blue) for 60 min at 37 °C. The ReAsH labeling solution was removed and replaced with 2 mL of ATP inhibition media containing 100 μM BAL. This media was removed immediately and the cells were incubated with 2 mL of ATP inhibition media containing 100 μM BAL for 10 min at 37 °C. The media was removed and the cells were fixed using 4% paraformaldehyde (PFA) for 30 min at rt. The PFA was removed and cells were rinsed once with DPBS and then blocked with 10% BSA in DPBS (PBSB) for 30 min at 37 °C. Cells were labeled with primary antibodies (α-FLAG, 1:1000 dilution in PBSB, 1h, 37 °C) and then washed three times with PBSB (1× immediate, 2× 5 min). The cells were then incubated with FITC-labeled secondary antibodies (α-mouse, 1:200 dilution in PBSB, 1 h, 37 °C) and washed as above using DPBS.
Total Internal Reflectance Fluorescence (TIRF) Microscopy
Total internal reflection fluorescence microscope (TIRFM) imaging was performed using an Olympus I×81 inverted microscope fitted with TIRF optics, a temperature controlled stage, and a 63×/1.45 NA oil immersion TIRF objective. Images were collected on an EMCCD camera (Andor, Belfast). Signal from FITC- labeled antibodies (green) was monitored using the 488 nm line of an Ar/Kr laser for excitation, and emission was collected using a LP500 filter. ReAsH labeling was monitored using the 568 nm line of a He/Ne laser for excitation and a LP585 emission filter. Acquired images were analyzed using ImageJ. The mean red fluorescence was measured for 1) a peripheral region (R1) of a transfected cell 2) a comparable region (R2) of a neighboring untransfected cell and 3) a nearby region (B) of background from the glass. The mean green fluorescence (G1) was also measured for the identical region R1 to account for varying levels of receptor expression. The fold increase (normalized for receptor expression) was assessed as: fold=((R1-B)/(R2-B))/G1. Error bars represent the standard error. Statistical analysis was performed using GraphPad Prism Software.
Supplementary Material
Acknowledgments
We thank J. Kuriyan and N. Jura for providing us with plasmids for full-length FLAG-tagged EGFR and the modeled coordinates for the JM antiparallel helices. We thank R. Baxter for his expertise in creating and analyzing the TGFα-bound homology models. Additionally, we thank D. Toomre and F. Rivera-Molina for access to their Cinema lab facility at Yale Medical School. R.A.S thanks J.P. Miller, J.M. Baskin and P.V. Chang for helpful discussions. This work was funded by NIH 5 R01 GM083257. R.A.S. was supported by an NIH postdoctoral fellowship 5 F32 GM087092-03.
Footnotes
Supporting Information: This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avraham R, Yarden Y. Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nature Reviews Molecular Cell Biology. 2011;12(2):104–117. doi: 10.1038/nrm3048. [DOI] [PubMed] [Google Scholar]
- 3.Lemmon MA. Ligand-induced ErbB receptor dimerization. Experimental Cell Research. 2009;315(4):638–648. doi: 10.1016/j.yexcr.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tao RH, Maruyama IN. All EGF(ErbB) receptors have preformed homo- and heterodimeric structures in living cells. Journal Of Cell Science. 2008;121(19):3207–3217. doi: 10.1242/jcs.033399. [DOI] [PubMed] [Google Scholar]
- 5.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125(6):1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 6.Bessman NJ, Lemmon MA. Finding the missing links in EGFR. Nature Structural & Molecular Biology. 2012;19(1):1–3. doi: 10.1038/nsmb.2221. [DOI] [PubMed] [Google Scholar]
- 7.Bae JH, Boggon TJ, Tome F, Mandiyan V, Lax I, Schlessinger J. Asymmetric receptor contact is required for tyrosine autophosphorylation of fibroblast growth factor receptor in living cells. Proceedings Of The National Academy Of Sciences Of The United States Of America; 2010; pp. 2866–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung I, Akita R, Vandlen R, Toomre D, Schlessinger J, Mellman I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature. 2010;464(7289):783–787. doi: 10.1038/nature08827. [DOI] [PubMed] [Google Scholar]
- 9.Jura N, Endres NF, Engel K, Deindl S, Das R, Lamers MH, Wemmer DE, Zhang X, Kuriyan J. Mechanism for Activation of the EGF Receptor Catalytic Domain by the Juxtamembrane Segment. Cell. 2009;137(7):1293–1307. doi: 10.1016/j.cell.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klein P, Mattoon D, Lemmon M, Schlessinger J. A structure-based model for ligand binding and dimerization of EGF receptors. Proceedings Of The National Academy Of Sciences Of The United States Of America; 2004; pp. 929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Macdonald-Obermann JL, Pike LJ. The Intracellular Juxtamembrane Domain of the Epidermal Growth Factor (EGF) Receptor Is Responsible for the Allosteric Regulation of EGF Binding. Journal Of Biological Chemistry. 2009;284(20):13570–13576. doi: 10.1074/jbc.M109.001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mi LZ, Lu C, Li Z, Nishida N, Walz T, Springer TA. Simultaneous visualization of the extracellular and cytoplasmic domains of the epidermal growth factor receptor. Nature Structural & Molecular Biology. 2011;18(9):984–U1502. doi: 10.1038/nsmb.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adak S, Yang KS, Macdonald-Obermann J, Pike LJ. The Membrane-proximal Intracellular Domain of the Epidermal Growth Factor Receptor Underlies Negative Cooperativity in Ligand Binding. Journal Of Biological Chemistry. 2011;286(52):45146–45155. doi: 10.1074/jbc.M111.274175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brewer MR, Choi SH, Alvarado D, Moravcevic K, Pozzi A, Lemmon MA, Carpenter G. The Juxtamembrane Region of the EGF Receptor Functions as an Activation Domain. Molecular Cell. 2009;34(6):641–651. doi: 10.1016/j.molcel.2009.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thiel KW, Carpenter G. Epidermal growth factor receptor juxtamembrane region regulates allosteric tyrosine kinase activation. Proceedings Of The National Academy Of Sciences Of The United States Of America; 2007; pp. 19238–19243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hubbard SR. The Juxtamembrane Region of EGFR Takes center stage. Cell. 2009;137(7):1181–1183. doi: 10.1016/j.cell.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 17.Luedtke NW, Dexter RJ, Fried DB, Schepartz A. Surveying polypeptide and protein domain conformation and association with FlAsH and ReAsH. Nature Chemical Biology. 2007;3(12):779–784. doi: 10.1038/nchembio.2007.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scheck RA, Schepartz A. Surveying Protein Structure and Function Using Bis-Arsenical Small Molecules. Accounts Of Chemical Research. 2011;44(9):654–665. doi: 10.1021/ar2001028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams S, Campbell R, Gross L, Martin B, Walkup G, Yao Y, Llopis J, Tsien R. New biarsenical Ligands and tetracysteine motifs for protein labeling in vitro and in vivo: Synthesis and biological applications. Journal Of The American Chemical Society. 2002;124(21):6063–6076. doi: 10.1021/ja017687n. [DOI] [PubMed] [Google Scholar]
- 20.Griffin B, Adams S, Tsien R. Specific covalent labeling of recombinant protein molecules inside live cells. Science. 1998;281(5374):269–272. doi: 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- 21.Adak S, DeAndrade D, Pike LJ. The tethering arm of the EGF receptor is required for negative cooperativity and signal transduction. Journal Of Biological Chemistry. 2011;286(2):1545–1555. doi: 10.1074/jbc.M110.182899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hertel C, Coulter SJ, Perkins JP. A Comparison of Catecholamine-Induced Internalization of Beta-Adrenergic Receptors and Receptor-Mediated Endocytosis of Epidermal Growth-Factor in Human Astrocytoma-Cells - Inhibition by Phenylarsine Oxide. Journal Of Biological Chemistry. 1985;260(23):2547–2553. [PubMed] [Google Scholar]
- 23.Schlessinger J, Shechter Y, Willingham MC, Pastan I. Direct Visualization of Binding, Aggregation, and Internalization of Insulin and Epidermal Growth-Factor on Living Fibroblastic Cells. Proceedings Of The National Academy Of Sciences Of The United States Of America; 1978; pp. 2659–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.We note that low temperature incubation23 and phenylarsine oxide (PAO)22 are frequently used to inhibit EGFR endocytosis. We found that low temperature incubation was not compatible with subsequent ReAsH labeling. We avoided the use of PAO to minimize any potential competition with the bis-arsenical fluorophore. The ATP inhibition cocktail we chose (2-DG/NaN3/NaF)23 has been used previously to inhibit RGFR endocytosis. We acknowledge that this protocol likely dampens the phosphorylation event that follows ligand binding by EGFR, but will have no effect on our assay, which detects events upstream of phosphorylation.
- 25.Uttamapinant C, White KA, Baruah H, Thompson S, Fernández-Suárez M, Puthenveetil S, Ting AY. A fluorophore ligase for site-specific protein labeling inside living cells. Proceedings of the National Academy of Sciences; 2010; pp. 10914–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Shea E, Rutkowski R, Stafford W, Kim P. Preferential Heterodimer Formation by Isolated Leucine Zippers From Fos and Jun. Science. 1989;245(4918):646–648. doi: 10.1126/science.2503872. [DOI] [PubMed] [Google Scholar]
- 27.Garrett T, McKern N, Lou M, Elleman T, Adams T, Lovrecz G, Zhu H, Walker F, Frenkel M, Hoyne P, Jorissen R, Nice E, Burgess A, Ward C. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell. 2002;110(6):763–773. doi: 10.1016/s0092-8674(02)00940-6. [DOI] [PubMed] [Google Scholar]
- 28.Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, Kim J, Saito K, Sakamoto A, Inoue M, Shirouzu M, Yokoyama S. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110(6):775–787. doi: 10.1016/s0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- 29.Goodman JL, Fried DB, Schepartz A. Bipartite Tetracysteine Display Requires Site Flexibility for ReAsH Coordination. Chembiochem. 2009;10(10):1644–1647. doi: 10.1002/cbic.200900207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dawson JP, Berger MB, Lin CC, Schlessinger J, Lemmon MA, Ferguson KM. Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface. Molecular And Cellular Biology. 2005;25(17):7734–7742. doi: 10.1128/MCB.25.17.7734-7742.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson KJ, Gilmore JL, Foley J, Lemmon MA, Riese DJ. Functional selectivity of EGF family peptide growth factors: implications for cancer. Pharmacology & Therapeutics. 2009;122(1):1–8. doi: 10.1016/j.pharmthera.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jura N, Zhang X, Endres NF, Seeliger MA, Schindler T, Kuriyan J. Catalytic Control in the EGF Receptor and Its Connection to General Kinase Regulatory Mechanisms. Molecular Cell. 2011;42(1):9–22. doi: 10.1016/j.molcel.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu C, Mi LZ, Grey MJ, Zhu J, Graef E, Yokoyama S, Springer TA. Structural Evidence for Loose Linkage between Ligand Binding and Kinase Activation in the Epidermal Growth Factor Receptor. Molecular And Cellular Biology. 2010;30(22):5432–5443. doi: 10.1128/MCB.00742-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Escher C, Cymer F, Schneider D. Two GxxxG-like motifs facilitate promiscuous interactions of the human ErbB transmembrane domains. Journal Of Molecular Biology. 2009;389(1):10–16. doi: 10.1016/j.jmb.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 35.Stanley A. The Transmembrane Domains of ErbB Receptors do not Dimerize Strongly in Micelles. Journal Of Molecular Biology. 2005 doi: 10.1016/j.jmb.2005.01.059. [DOI] [PubMed] [Google Scholar]
- 36.Madani F, Lind J, Damberg P, Adams SR, Tsien RY, Graslund AO. Hairpin Structure of a Biarsenical-Tetracysteine Motif Determined by NMR Spectroscopy. Journal Of The American Chemical Society. 2009;131(13):4613. doi: 10.1021/ja809315x. + [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carpenter G. Receptors for Epidermal Growth Factor and Other Polypeptide Mitogens. Annual Review Of Biochemistry. 1987;56(1):881–914. doi: 10.1146/annurev.bi.56.070187.004313. [DOI] [PubMed] [Google Scholar]
- 38.Ebner R, Derynck R. Epidermal Growth-Factor And Transforming Growth Factor-Alpha -Differential Intracellular Routing And Processing Of Ligand-Receptor Complexes. Cell Regulation. 1991;2(8):599–612. doi: 10.1091/mbc.2.8.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roepstorff K, Grandal MV, Henriksen L, Knudsen SLJ, Lerdrup M, Grovdal L, Willumsen BM, van Deurs B. Differential Effects of EGFR Ligands on Endocytic Sorting of the Receptor. Traffic. 2009;10(8):1115–1127. doi: 10.1111/j.1600-0854.2009.00943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roepstorff K, Grovdal L, Grandal M, Lerdrup M, van Deurs B. Endocytic downregulation of ErbB receptors: mechanisms and relevance in cancer. Histochemistry And Cell Biology. 2008;129(5):563–578. doi: 10.1007/s00418-008-0401-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yarden Y, Sliwkowski M. Untangling the ErbB signalling network. Nature Reviews Molecular Cell Biology. 2001;2(2):127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.