

Abstract
Degradation of heroin to 6-monoacetylmorphine (6-MAM) and then morphine happens rapidly in vivo and in vitro. The rates of heroin and 6-MAM degradation depend on the type of biological samples, and the duration and conditions of storage. In order to optimize conditions for measuring heroin and its metabolites in samples collected for pharmacokinetic studies in rats, we investigated the time course of degradation of heroin, 6-MAM, and morphine in four biological matrices: rat blood, rat brain homogenate, bovine serum, and human plasma under various conditions. Analyte concentrations were measured by LC-MS. The goal was to identify conditions that allow maximum flexibility in scheduling sample collection and analysis, as well as gain more information on the stability of heroin in blood and tissue samples. A solid-phase extraction method with ice-cold solvents, sodium fluoride (NaF) and a low pH (3.0) maintained sample stability. Quality controls were within 94.0–105% of the target value. Variability was 4.0–8.9% for all analytes within the range of 5–200 ng/mL for heroin, 5–1000 ng/mL for 6-MAM, and 10–200 ng/mL for morphine. Heroin degradation to 6-MAM was faster in rat whole blood than in plasma, and faster in rat plasma than in rat brain homogenate. Maintaining NaF at 4 mg/mL throughout processing enhanced stability; higher NaF concentrations added to whole blood caused hemolysis. Samples processed through solid phase extraction and stored as dried pellets at 80°C constituted the most stable environment for heroin, and was superior to the storing of samples in solution prior to or after extraction. Nevertheless, post-extraction heroin and 6-MAM levels declined by 6.7–8.3% over one week in rat plasma under these conditions, and by <1–4.7% in bovine serum or human plasma.
Keywords: Heroin, Assay validation, Morphine, Stability, Rat
1. Introduction
Quantification of heroin in biological specimens can prove challenging due to the rapid deacetylation of heroin (half-life of approximately 5 min in plasma) to its active metabolites 6-monoacetylmorphine (6-MAM) and morphine [1–4]. In humans, heroin degradation is catalyzed by esterases present in blood and tissue [3,5]. However, esterase activity and concentrations differ between species resulting in differential degradation of opioids across species and biological matrices within species [6–8]. Degradation of heroin in vitro is also dependent on pH and temperature [4]. Therefore, measurement of heroin after collection of biological samples is problematic if conditions are not properly controlled.
Analytical methods that include quantification of heroin recognize that heroin and 6-MAM can be unstable in particular matrices and suggest the need to use fresh standards and controls in order to avoid degradation [9,10]. Conditions that favor heroin stability include use of general esterase inhibitor(s), most commonly NaF, low pH, and low temperature with the use of ice-cold reagents and/or immediate freezing of samples in liquid nitrogen [4,10]. While these measures are useful, a wide range of conditions have been studied, and varying degrees of stability have been reported. One method incorporated the freezing of samples with liquid nitrogen and a protein precipitation protocol followed by analysis by LC–MS/MS [10]. The use of liquid nitrogen is not always convenient, and does not appear to prevent significant degradation of heroin in blood samples stored for up to seven days. Analyzing samples immediately after collection to minimize analyte degradation is difficult when scheduling animal or clinical experiments between laboratories, when transportation of samples is required, or in laboratories with shared instrumentation requiring coordination across multiple laboratories and projects. In our study, heroin and 6-MAM stability were investigated under a variety of conditions and an alternative solid phase extraction method was developed that enabled storage of extracted samples collected in several blood matrices (rat plasma, bovine serum, and human plasma) and rat brain homogenate for up to one week, thus enabling flexibility in scheduling of sample analysis and minimizing the degradation of heroin.
2. Methods
2.1. Chemicals
Heroin, 6-monoacetylmorphine, morphine, methadone, codeine, hydrocodone, meperidine, and oxycodone and their deuterated analogs used for analysis were purchased from Cerilliant Analytical Reference Standards (Round Rock, TX). Ammonium formate, formic acid 88%, glacial acetic acid, sodium fluoride, methanol (HPLC grade), and acetonitrile (HPLC grade) were purchased from Fisher Scientific (Fair Lawn, NJ). Human plasma was purchased from Biological Specialty Corporation (Colmar, PA) and bovine serum was purchased from Sigma Chemical Company (St. Louis, MO).
2.2. Preparation of stock standards and quality controls
Stock standards (1 mg/mL) of heroin and 6-MAM were stored in acetonitrile. The stock standard for morphine was stored in methanol (1 mg/mL). All working solutions were stored in 10 mM formate (pH 3.0): methanol (50/50, v/v) at −20°C. Working solutions for calibration samples and quality controls were prepared independently and made fresh every two weeks. Heroin standards were prepared separately. Due to the limited availability of rat plasma, bovine serum was used for preparing calibration standards and quality control samples. Standard solutions were prepared in blank bovine serum to obtain final concentrations of 5, 10, 25, 50, 100, and 200 ng/mL for heroin, 5, 10, 25, 50, 100, 200, 500, 1000 ng/mL for 6-MAM, and 10, 25, 50, 100, 150, and 200 ng/mL for morphine. Quality control samples were prepared in bovine serum at final concentrations of 5.6, 40.0, and 160 ng/mL for heroin, 5.6, 40, 160, and 400 ng/mL for 6-MAM, and 16.0, 40, and 160 ng/mL for morphine. The internal standard working solution was prepared (50 ng/mL) in 10 mM formate buffer pH 3.0, aliquoted into screw-top cryogenic vials, and stored at −80°C.
2.3. Collection of blood and brain tissue samples from rats
The relevant conditions for each experiment are presented in Table 1 and indicated in the section below.
Table 1.
Summary of stability experiments.
| Experiment | Matrix | NaF | Storage temp. | Duration | Figure |
|---|---|---|---|---|---|
| A (whole blood versus plasma) | Rat whole blood | Collected with NaF | 4°C | Processed immediately | 3 |
| Rat plasma | 4 mg/mL | ||||
| B (centrifuge speeds) | Rat whole blood | Collected with NaF | 4°C | Processed immediately | Not shown |
| C (live rat infusion) | Rat plasma | Collected with NaF | 4°C | Processed immediately | 4 |
| Rat brain homogenate (in formate) | |||||
| D (NaF comparison) | Rat plasma | 4 mg/mL NaF added in half samples | 4°C | 10:00 min | 5 |
| E (heroin degradation in brain) | Rat brain homogenate (in water) | 4 mg/mL | 4°C | 30:00 min | 6 |
| Rat brain homogenate (in water) | |||||
| Rat brain homogenate (in formate) | 4 mg/mL | ||||
| Rat brain homogenate (in formate) | |||||
| F (50:50 storage) | Rat dosed with heroin | 4 mg/mL in formate and plasma | −80°C | 24 h | 7, 8 |
| Rat plasma | 4 mg/mL in formate and plasma | ||||
| Rat plasma | 4 mg/mL in plasma | ||||
| Rat brain homogenate (in formate) | 4 mg/mL | Not shown | |||
| G (dried pellet) | Rat dosed with heroin | 4 mg/mL in formate and plasma | −80°C | 7 days | 8 |
| Human plasma | 4 mg/mL | Not shown | |||
| Bovine serum | 4 mg/mL | ||||
| Rat plasma | 4 mg/mL |
2.3.1. Blood collection (experiments A, B, C, D, F, and G, Table 1)
General procedures and processing time are represented in Fig. 1. For in vitro stability studies, male Holtzman Sprague-Dawley rats (300–324 g) were decapitated after being anesthetized with isofluorane and, within 1 min, blood was collected into a vial. Blood was drawn into a syringe containing ice-cold NaF and heparin (final concentration 4 mg/mL NaF) in two 4 mL aliquots (total 8 mL) and transferred to a 15 mL polypropylene tube. Samples were centrifuged at 3100 × g on a Beckman J-6B centrifuge for 3 min at 4°C. Plasma (0.5 mL) was transferred to a 5 mL polypropylene tube and diluted 1:1 with ice cold 10 mM formate buffer (pH 3.0, with 4 mg/mL NaF, unless noted elsewhere).
Fig. 1.
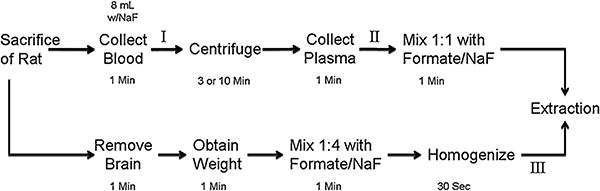
Flowchart describing general method for collecting samples from rats. (I) Point in time where samples were spiked for whole blood experiments (A and B, Table 1). (II) Point in time where samples were spiked for stability experiments (C, E, and F, Table 1). (III) Point in time where samples were spiked for brain homogenate experiments (D and E, Table 1).
2.3.2. Preparation of samples from dosed animals (experiments E and F, Table 1)
Rats were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine both administered i.m. prior to a 1 min infusion of 0.52 mg/kg heroin. Rats were sacrificed 1 min later and blood was collected following the procedure described above.
2.3.3. Preparation of brain samples (experiments C, E, and F, Table 1)
After sacrifice and collection of blood from the rat, the brain was removed, rinsed with ice-cold 10 mM formate pH 3.0 containing 4 mg/mL NaF, and placed in pre-weighed vials. Samples were weighed and then four parts (by mass) of ice-cold 10 mM formate buffer pH 3.0, or distilled water, with or without 4 mg/mL NaF was added. Samples were homogenized for 30–40 s with a Brinkmann homogenizer and placed in a −20°C freezer. Samples were processed within 1 h and then extracted. Before extraction, samples were thawed, swirled to mix (to prevent formation of bubbles), and then 400 μL of this matrix were used for extraction.
2.3.4. Whole blood stability experiment (A and B)
To determine the percent degradation occurring from the time of sacrifice of the rat and separation of the plasma, 1.0 mL of heparinized whole blood (containing 4 mg/mL NaF) from sacrificed rats was immediately added to tubes containing 10 μg of heroin. Tubes were then spun at 1200 × g for 10 min at 4°C. Plasma was separated and samples were processed. The time from addition of whole blood to centrifugation was approximately 2 min. For plasma samples, 1 mL of rat plasma (containing NaF) was added to a tube containing 10 μg of heroin and processed. Various centrifuge speeds and times were also examined for optimal recovery of heroin. Samples were collected as described above and centrifuged at 1200 × g for 3 min, 10 min, or at 3100 × g for 3 min.
2.3.5. Time course stability experiment (experiments D, E, F, and G, Table 1)
For time course stability experiments, all samples were prepared by addition of the specific analyte (heroin, 6-MAM, or morphine) directly into freshly collected rat brain homogenate or rat plasma, human plasma, or bovine serum for a final concentration of 100 ng/mL. Unless specified elsewhere, samples were diluted with NaF and formate buffer on ice. The time when an analyte was added to a matrix was considered time zero. Samples were then incubated under different temperature conditions for specified periods of time (up to 30 min for short term experiments, and up to one week for long term stability).
2.4. Sample extraction
Solutions and samples were kept on ice throughout the extraction to promote stability of the analytes. Two hundred microliters of bovine serum was diluted 1:1 with 10 mM formate buffer, pH 3.0, containing 4 mg/mL NaF along with 100 μL of internal standard solution in a 1.5 mL centrifuge tube. For standards and quality control samples, 10 μL of working stock solution was added to the tubes. Four hundred microliters of rat plasma:buffer solution was added to 100 μL of internal standard solution. Brain samples were prepared by adding 400 μL of rat brain homogenate to a 1.5 mL tube with 100 μL of internal standard solution. Brain samples were then centrifuged for 10 min at 9300 × g and the supernatant was used for extraction. All samples were agitated briefly by a vortex mixer before extraction.
Strata-X-RP 1 mL extraction cartridges (Phenomenex, Torrance, CA) were used for extraction. Cartridges were conditioned with 1 mL methanol followed by 1 mL of water. The matrix solution was then loaded onto cartridges and allowed to flow through the cartridge slowly via centrifugation at no more than 55 g for 30 s. Cartridges were washed with 1 mL of cold 1% methanol in acidified water (acidified with formic acid to pH 3.0). In a fresh tube, samples were eluted with 500 μL of cold 1% acetic acid in methanol twice. Both elution volumes were collected in a single tube. Samples were then dried on a Zymark Nitrogen Drier (Hopkinton, MA) at 17 psi and 27°C. Samples were stored as a dry solid until reconstituted in 200 μL of 10 mM formic acid buffer pH 3.0, vortexed for 30 s, and centrifuged for 10 min at 9300 × g. Sixty microliters of sample was then transferred into autosampler vials and analyzed immediately.
2.5. LC-MS conditions and analysis
Fifteen microliters of sample was injected onto a reversed phase Agilent (Santa Clara, CA) Zorbax XDB-C18 (2.1 mm × 50 mm i.d., 3.5 μm) column protected by a reversed phase 4 mm × 2 mm C18 guard column (Phenomenex, Torrance, CA). The LC-MS system consisted of a 2010A Shimadzu (Tokyo, Japan) single-quadrupole LC-MS with a SCL-10Avp controller, dual LC-10ADvp pumps, SIL-10ADvp autoinjector, and DGU 14A degasser. The samples were kept at ambient temperature during injection.
Gradient elution was performed with a mixture of 10 mM formate buffer pH 3.0 (mobile phase A) and acetonitrile (mobile phase B) as follows: 0–0.5 min 7% mobile phase B, 0.5–2.0 min 7 → 20% mobile phase B, 2.0–2.5 min 20% mobile phase B, 2.5–4.0 min 20 → 33% mobile phase B, 4.0–4.5 min 33 → 35% mobile phase B, 4.5–4.75 min 35 → 7% mobile phase B, 4.75–5.0 7% mobile phase B. The flow rate was kept at a constant 0.21 mL/min and the total run time was 5 min. The autosampler needle was washed with 100% methanol following each sample injection.
Ionization was achieved by assisted electrospray in the positive ion mode. Instrument settings were: heat block temperature 200°C, curved desolvation-line temperature 250°C, detector voltage 2.0 kV, and nebulizing gas (N2) flow rate 1.5 mL/min. Data acquisition and peak integration were interfaced to a computer workstation running LCSolutions™ (Tokyo, Japan). LC-MS in the scan mode was used to identify the appropriate ions to monitor heroin, 6-MAM, and morphine. Select ion monitoring for heroin, 6-MAM, and morphine were done at m/z 370 (heroin-d9 m/z 379), m/z 328 (6-MAM-d6 m/z 334), and m/z 286 (morphine-d6 m/z 292), respectively.
2.6. Validation and statistics
Matrix effects were assessed in rat plasma, rat brain homogenate, bovine serum and human plasma. The matrix effect is found to cause ion enhancement or suppression due to interfering compounds in the matrix that do not appear in blank matrix samples [11,12]. Samples were analyzed in triplicate on three different days. The matrix effect was determined by comparing spiked standard peak areas to unextracted standard peak areas and following the procedure of Matuszewski et al. [11].
Peak area ratios (analyte/internal standard) were used for determination of concentration from extracted matrix. The limit of quantitation (LOQ) was determined to be the lowest standard on the calibration curve that could be reliably measured (≤20% coefficient of variation). The limit of detection (LOD) had a signal to noise ratio of 5. Validation of the assay was performed by preparing standard curves in triplicate (within-run) for each day on five different days (between run, N = 15). Quality control samples were prepared in blank bovine serum and were also run in triplicate. The assay was validated before subsequent stability experiments were conducted. The percentage coefficient of variation (CV) was calculated from the average standard deviation of quality control samples at each level. The estimated variability was determined from calculated concentrations of multiple triplicate weighted (1/x2) standard curves. Accuracy was calculated by dividing the grand mean by the target concentration, and multiplying by 100.
3. Results
3.1. Assay validation
The matrix effect was found to cause ion enhancement in all matrices for the analytes. After correction for the matrix effect, the average extraction recovery for the analytes was found to be within ±20% of the target value.
Precision and accuracy for each analyte's quality controls are shown in Table 2. The LOQ of the assay was determined to be 5 ng/mL for both heroin and 6-MAM and 10 ng/mL for morphine in all matrices. The LOD was 2.5 ng/mL for heroin and 6-MAM, and 5 ng/mL for morphine in each matrix. The within-day and between-day variability ranged from 2.2% to 8.8% and 1.9% to 6.9% respectively. No interference was noted after injection of diluted standards of other common opioids (methadone, codeine, hydrocodone, meperidine, and oxycodone).
Table 2.
Validation results for quality control samples (N = 15).
| Analyte | Concentration (ng/mL) | CVt (%) | Accuracy (%) |
|---|---|---|---|
| Heroin | 5.6 | 5.70 | 104.9 |
| 40 | 7.71 | 100.3 | |
| 160 | 6.69 | 100.2 | |
| 6-MAM | 5.6 | 7.98 | 101.0 |
| 40 | 6.51 | 102.0 | |
| 160 | 4.06 | 94.9 | |
| 400 | 8.88a | 96.3 | |
| Morphine | 16 | 8.59 | 94.6 |
| 40 | 6.13 | 97.8 | |
| 160 | 7.71 | 98.2 |
N = 8.
Fig. 2 shows a sample chromatogram of analytes and internal standards in rat plasma. Under the described chromatographic conditions, the retention times were 3.99 min (heroin), 3.00 min (6-MAM), and 1.27 min (morphine). The retention times of the deuterated internal standards were 3.98 min (heroin-d9), 3.02 min (6-MAM-d6), and 1.26 min (morphine-d6).
Fig. 2.
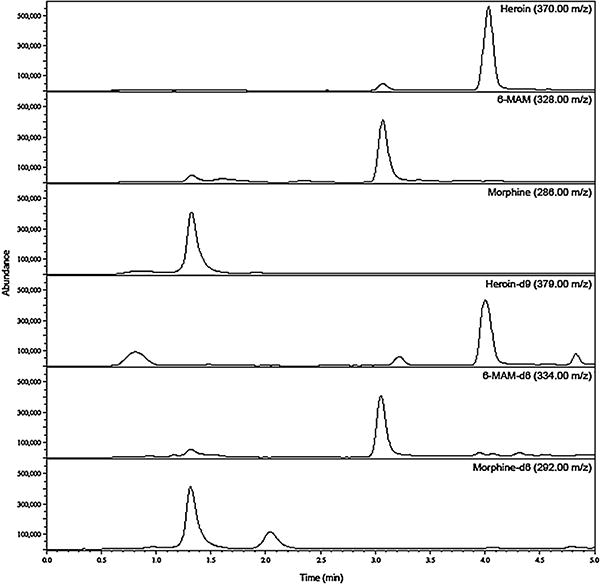
LC/MS chromatogram of heroin, 6-MAM, and morphine with internal standards. 80ng/mL of each analyte was spiked into rat plasma before extraction and analysis.
3.2. Whole blood stability (experiments A and B, Table 1)
Heroin levels decreased by 56.4% after being spiked into fresh rat whole blood with NaF present (Fig. 3) whereas, heroin concentrations decreased by only 5.9% in rat plasma containing NaF. 6-MAM concentrations increased concurrently with the decrease in heroin. Whole blood and plasma samples to which 6-MAM had been added were also compared. No significant difference was seen in 6-MAM concentrations between the two matrices (data not shown) and morphine was not detected. To investigate further the stability of heroin during sample processing, samples were centrifuged under varying conditions. Samples centrifuged at 1200 × g, for 3 min showed a 5.9 ± 5.3% increase in recovery compared to samples spun for 10 min at the same speed (data not shown). Samples spun at 3100 × g for 3 min allowed for a 12 ± 4.5% increase in recovery of heroin compared to 10 min at 1200 × g.
Fig. 3.
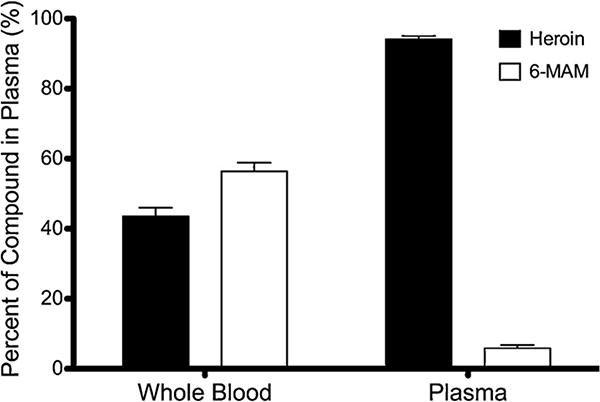
Experiment A. Mean ± SD, n = 3. Measured concentrations of heroin and 6-MAM in rat whole blood and plasma (containing NaF) after the addition of 10 μg/mL of heroin. Heroin degraded 56.4% in whole blood compared to 5.9% degradation in plasma.
3.3. Live rat infusion (experiment C)
Heroin, 6-MAM, and morphine concentrations were measured in rats dosed with 0.52 mg/kg heroin 1 min before sacrifice (Fig. 4). Due to high concentrations of 6-MAM in rat samples a second sample was diluted with bovine serum (1:1) before extraction. Extracted concentrations of all analytes were within the range of the curve. Brain measurements were converted to ng/g. It was found that 6-MAM levels were highest in both plasma and brain, however, brain concentrations of all three analytes was more than 4 fold higher than plasma.
Fig. 4.
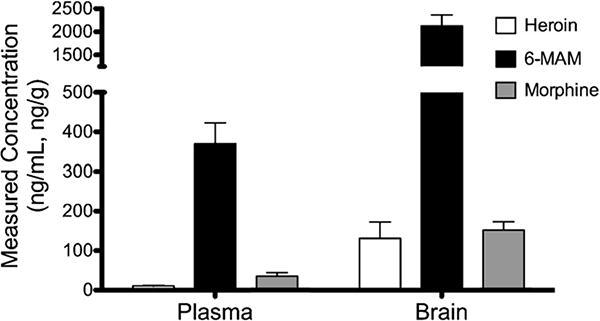
Experiment C. Mean ± SD (n = 6). Measured concentrations of heroin, 6-MAM, and morphine in plasma and brain from rats infused over the course of 1 min with 0.52 mg/kg heroin. Rats were sacrificed 1 min after infusion. Samples were collected with 4 mg/mL NaF, diluted with formate buffer containing 4 mg/mL NaF, and analyzed immediately after extraction.
3.4. Short term stability results (experiments D and E, Table 1)
A 10-min time course of heroin concentrations in rat plasma showed complete degradation of heroin in approximately 2 min when NaF was not present, whereas samples containing 4 mg/mL NaF showed a heroin degradation of 4.2% within 10 min (data not shown). When whole blood was collected with higher concentrations of NaF (9.6 or 20 mg/mL), hemolysis of the sample occurred and more heroin degradation was observed than with 4 mg/mL NaF (data not shown).
Rat brain homogenate (in formate) samples showed a slower degradation of heroin than rat plasma. A 30-min time course with 4 mg/mL NaF added to brain homogenate samples resulted in stable heroin concentrations whereas the absence of NaF resulted in a decline of heroin concentration in rat brain homogenate (Fig. 5, shown up to 10 min). A decline of 35% in final heroin concentrations in the samples containing formate buffer with no NaF was observed. Samples with NaF showed a decrease of only 5.3%. Brain samples homogenized with water containing NaF were equally stable with or without formate buffer. When comparing brain samples homogenized without NaF, it was found that addition of pH 3.0 formate buffer resulted in greater heroin stability compared to water (35% degradation compared to 87%). The instability of heroin in samples without NaF was confirmed by a concomitant increase of 6-MAM observed in samples run under the same conditions for 10 min.
Fig. 5.
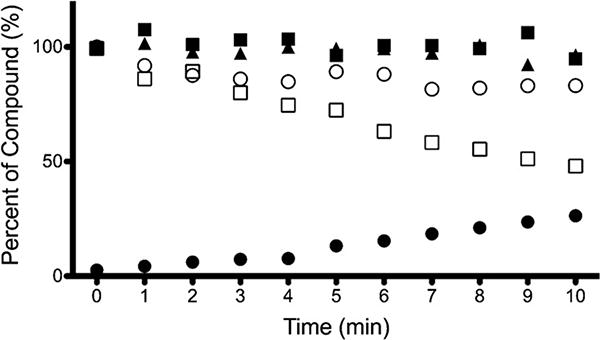
Experiment E. Ten-minute time course of heroin degradation after spiking of 100 ng/mL heroin into rat brain homogenate (brain homogenized with formate containing NaF (
 ) or without NaF (
) or without NaF (
 , run in duplicate) and brain homogenized with water containing NaF (
, run in duplicate) and brain homogenized with water containing NaF (
 ), or without NaF (
), or without NaF (
 ). For samples homogenized with formate buffer and no NaF, corresponding 6-MAM levels are observed (
). For samples homogenized with formate buffer and no NaF, corresponding 6-MAM levels are observed (
 , run in duplicate). Samples were stored on ice during experiment.
, run in duplicate). Samples were stored on ice during experiment.
3.5. Storage condition optimization (experiments F and G, Table 1)
The stability of heroin and 6-MAM in frozen plasma was examined by the addition of NaF to the 10 mM formate buffer (pH 3.0) used to dilute the plasma samples before storage at −80°C. Heroin degraded less over the 24-h storage period when NaF was added with the formate buffer compared to formate buffer alone (13% versus 39% degradation) (data not shown). NaF similarly enhanced the stability of 6-MAM in frozen plasma. Most of the heroin degradation occurred during the freezing process (within the first 3 h) at −80°C with no further degradation noted up to 24 h. Samples spiked with 100 ng/mL of 6-MAM showed minimal degradation regardless of when NaF was added to the sample (4.1% versus 7.6%). Brain homogenate samples stored under similar conditions showed no degradation of any compounds. Post-extraction stability was assessed by storage of samples at −80°C as dried pellets. Heroin and 6-MAM samples remained within 10% of targeted concentrations in bovine serum and human plasma. Analytes were found to be within 12.5% when stored forup to one week. Samples collected 5 min after addition of the analyte (heroin or 6-MAM), < 4.5% degradation was observed. Morphine was stable in all three matrices for up to one week with minimal degradation. Post-collection stability was also assessed by dosing of rats with heroin before sacrifice. Fig. 6 shows a time course stability comparison of samples stored in the 50:50 plasma:buffer solution (Fig. 6a) and in the pellet form after extraction (Fig. 6b). Morphine levels increased throughout the time course in the samples stored pre-extraction whereas the samples stored as dried pellets show less variability.
Fig. 6.
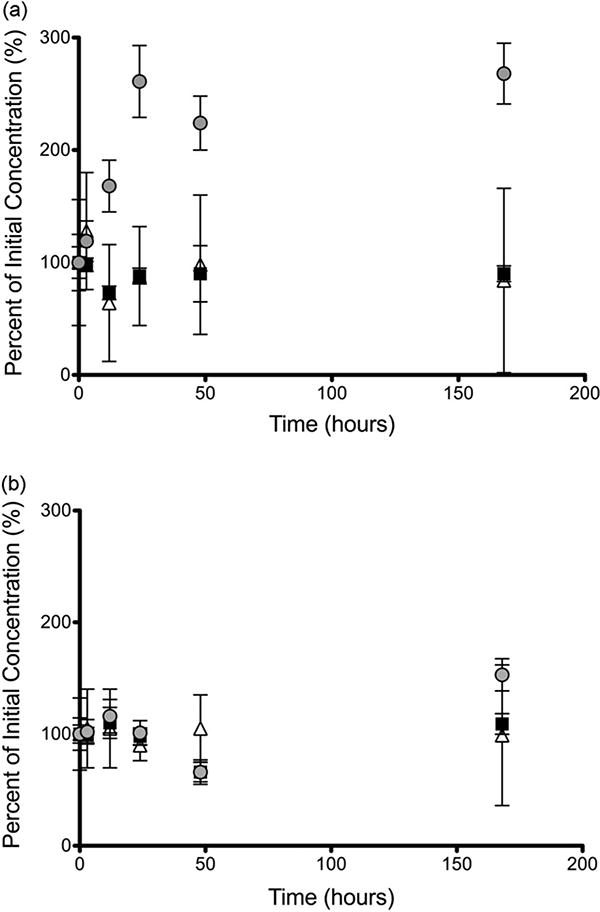
Experiments F and G. Mean ± SD (n = 6). Comparison of heroin (
 ), 6-MAM (
), and morphine (
), 6-MAM (
), and morphine (
 ) stability in plasma post-collection from heroin dosed rats. Samples were stored either as pre-extracted plasma diluted with 10 mM formate buffer containing NaF (a) or in the post-extraction pellet state (b) at −80°C for up to one week.
) stability in plasma post-collection from heroin dosed rats. Samples were stored either as pre-extracted plasma diluted with 10 mM formate buffer containing NaF (a) or in the post-extraction pellet state (b) at −80°C for up to one week.
4. Discussion
Heroin is susceptible to degradation at several points during sample processing. Initial degradation can happen during the time necessary for separation of plasma from erythrocytes due to esterases present in erythrocyte membranes or in plasma. Degradation can also occur during the extraction process. Optimization of all methods used in sample collection, processing, and analysis are critical in order to minimize degradation and to avoid underestimation of heroin in biological specimens. When separating plasma from red blood cells the use of high centrifuge speeds for even a short amount of time (3 min) still allows for up to 40–50% degradation of heroin to occur. Significant degradation of heroin also takes place in post-separation conditions during sample extraction and while in storage before analysis. Differences in the time course of heroin degradation were seen across matrices with rat brain being the most stable, followed by bovine serum and human plasma, with rat plasma being the least stable. Optimization of pH, NaF concentration, and temperature in collected samples for storage alone was found to be insufficient for stability of heroin in bovine serum, rat brain homogenate, and rat plasma. Optimal stability for at least one week before analysis was attained by storing samples as post-extracted pellets at −80°C.
Stability of heroin in rat whole blood compared to rat plasma showed that plasma is a more stable environment for heroin than whole blood. This supports previous studies that show various esterase-catalyzed compounds to be less stable in whole blood than in plasma [8,13] and also supports the use of quantitative assays that use plasma instead of whole blood. Morphine was not observed in any samples incubated with heroin alone. The absence of morphine could be attributed to the slower rate of degradation of 6-MAM into morphine, and the short amount of time 6-MAM was in whole blood (centrifugation occurred within 2 min of collection). In human blood, esterases catalyzing heroin act much more rapidly than esterases involved in 6-MAM degradation [5].
It is normal practice to collect samples containing heroin into tubes containing NaF as a general esterase inhibitor [10,14,15]. When working with tissue samples, NaF can be diluted when buffers are added to the sample. Maintaining a constant NaF concentration by directly adding NaF to the 10 mM formate buffer (pH 3.0) aided in the stabilization of heroin.
Rat brain homogenate was a more stable matrix for heroin samples than the blood matrices. This could be due to greater esterase activity in blood matrices than in brain and the difference in the type of esterases present in brain [5]. These results are supported by findings in the mouse by Karinen et al. [10] that show brain samples are a more stable matrix than acidified whole blood samples for heroin.
Sample storage in solution, even when optimized for NaF, pH, and temperature conditions was not possible for longer than 24 h without significant degradation of heroin (13% degradation after 24 h). When it is difficult to coordinate schedules for instrument time or transport samples between laboratories, sample stability is essential. In order to find a stable stopping point in the sample preparation procedure, we examined the stability of heroin in samples that were extracted immediately after sample collection and stored in a dried pellet form. These analyte samples were more stable than when stored in dilute pre-extraction solutions containing NaF. Morphine showed minimal degradation when stored as a pellet. Heroin and 6-MAM degradation occurred, 7.1% and <1% at 3 h and 12.3% and 11.4% at one week, respectively. Previous studies show 50% heroin degradation over a period of 3 h to one week of storage [10]. Our results show a degradation of 5.6% between 3 h and one week after extraction for heroin.
Faster degradation rates (lower recoveries) of heroin were observed in rat plasma compared to bovine serum or human plasma and seem to occur primarily during the time between addition of plasma to NaF and dilution with formate buffer. All rat plasma samples had a lower heroin level at the earliest time of measurement, perhaps due to greater esterase activity in rat plasma compared to other matrices [6–8,16] as well as the rapid half-life of heroin (approximately 5 min in human plasma) [1].
5. Conclusion
Optimization of assay conditions that result in minimal degradation of heroin is imperative for pharmacokinetic studies involving quantification of heroin from blood. It is of particular importance to examine stability of samples and to identify points within the methodology that can provide the most stable environment for sample transport between laboratories. In addition, knowledge of the extent and time course of heroin degradation is important when translating results from pharmacokinetic studies. Systematic evaluation of various techniques that are known to decrease heroin degradation were incorporated into this assay in order to provide a method that is sensitive, reliable, and allows for sample storage up to one week before analysis by LC–MS.
Acknowledgments
We would like to thank Michaela Roslawski for her help in revision of this manuscript. This work was supported by NIH/NIDA R01 DA030715, NIH/NIDA R01 DA026300, and T32-DA07097 from the National Institute on Drug Abuse (NIDA). The content is solely the responsibility of the authors and does not necessarily represent the official views of NIDA or the National Institutes of Health.
Footnotes
Conflict of interest: The authors have no conflict of interest to disclose.
References
- 1.Nakamura GR, Thornton JI, Noguchi TT. Kinetics of heroin deacetylation in aqueous alkaline solution and in human serum and whole blood. J Chromatogr. 1975;110:81–89. doi: 10.1016/s0021-9673(00)91213-5. [DOI] [PubMed] [Google Scholar]
- 2.Garrett ER, Gurkan T. Pharmacokinetics of morphine and its surrogates II: methods of separation of stabilized heroin and its metabolites from hydrolyzing biological fluids and applications to protein binding and red blood cell partition studies. J Pharm Sci. 1979;68:26–32. doi: 10.1002/jps.2600680111. [DOI] [PubMed] [Google Scholar]
- 3.Lockridge O, Mottershaw-Jackson N, Eckerson HW, La Du BN. Hydrolysis of diacetylmorphine (heroin) by human serum cholinesterase. J Pharmacol Exp Ther. 1980;215:1–8. [PubMed] [Google Scholar]
- 4.Barrett DA, Dyssegaard AL, Shaw PN. The effect of temperature and pH on the deacetylation of diamorphine in aqueous solution and in human plasma. J Pharm Pharmacol. 1992;44:606–608. doi: 10.1111/j.2042-7158.1992.tb05474.x. [DOI] [PubMed] [Google Scholar]
- 5.Salmon AY, Goren Z, Avissar Y, Soreq H. Human erythrocyte but not brain acetylcholinesterase hydrolyses heroin to morphine. Clin Exp Pharmacol Physiol. 1999;26:596–600. doi: 10.1046/j.1440-1681.1999.03090.x. [DOI] [PubMed] [Google Scholar]
- 6.Koitka M, Hochel J, Gieschen H, Borchert HH. Improving the ex vivo stability of drug ester compounds in rat and dog serum: inhibition of the specific esterases and implications on their identity. J Pharm Biomed Anal. 2010;51:664–678. doi: 10.1016/j.jpba.2009.09.023. [DOI] [PubMed] [Google Scholar]
- 7.Liederer BM, Phan KT, Ouyang H, Borchardt RT. Significant differences in the disposition of cyclic prodrugs of opioid peptides in rats and guinea pigs following IV administration. J Pharm Sci. 2005;94:2676–2687. doi: 10.1002/jps.20476. [DOI] [PubMed] [Google Scholar]
- 8.Minagawa T, Kohno Y, Suwa T, Tsuji A. Species differences in hydrolysis of iso-carbacyclin methyl ester (TEI-9090) by blood esterases. Biochem Pharmacol. 1995;49:1361–1365. doi: 10.1016/0006-2952(95)00071-7. [DOI] [PubMed] [Google Scholar]
- 9.Zuccaro P, Ricciarello R, Pichini S, Pacifici R, Altieri I, Pellegrini M, Dascenzo G. Simultaneous determination of heroin, 6-monoacetylmorphine, morphine, and its glucuronides by liquid chromatography atmospheric pressure ionspray mass spectrometry. J Anal Toxicol. 1997;21:268–277. doi: 10.1093/jat/21.4.268. [DOI] [PubMed] [Google Scholar]
- 10.Karinen R, Andersen JM, Ripel A, Hasvold A, Hopen AB, Morland J, Christophersen AS. Determination of heroin and its main metabolites in small sample volumes of whole blood and brain tissue by reversed-phase liquid chromatography-tandem mass spectrometry. J Anal Toxicol. 2009;33:345–350. doi: 10.1093/jat/33.7.345. [DOI] [PubMed] [Google Scholar]
- 11.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem. 2003;75:3019–3030. doi: 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- 12.Srinivas NR. Dodging matrix effects in liquid chromatography tandem mass spectrometric assays-compilation of key learnings and perspectives. Biomed Chromatogr. 2009;23:451–454. doi: 10.1002/bmc.1152. [DOI] [PubMed] [Google Scholar]
- 13.Skopp G, Klingmann A, Potsch L, Mattern R. In vitro stability of cocaine in whole blood and plasma including ecgonine as a target analyte. Ther Drug Monit. 2001;23:174–181. doi: 10.1097/00007691-200104000-00014. [DOI] [PubMed] [Google Scholar]
- 14.Rook EJ, Hillebrand MJX, Rosing H, van Ree JM, Beijnen JH. The quantitative analysis of heroin, methadone and their metabolites and the simultaneous detection of cocaine, acetylcodeine and their metabolites in human plasma by high-performance liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B: Analyt Technol Biomed Life Sci. 2005;824:213–221. doi: 10.1016/j.jchromb.2005.05.048. [DOI] [PubMed] [Google Scholar]
- 15.Rentsch KM, Kullak-Ublick GA, Reichel C, Meier PJ, Fattinger K. Arterial and venous pharmacokinetics of intravenous heroin in subjects who are addicted to narcotics. Clin Pharm Ther. 2001;70:237–246. doi: 10.1067/mcp.2001.117981. [DOI] [PubMed] [Google Scholar]
- 16.Welch RM, Brown A, Ravitch J, Dahl R. The in vitro degradation of cisatracurium, the R, cis-R'-isomer of atracurium, in human and rat plasma. Clin Pharmacol Ther. 1995;58:132–142. doi: 10.1016/0009-9236(95)90190-6. [DOI] [PubMed] [Google Scholar]