

In this issue of Haematologica, Dinner et al. report the outcome of a pilot study of an oral regimen of lenalidomide in combination with dexamethasone and low-dose melphalan in 25 patients with light chain amyloidosis (AL), most of them with cardiac involvement.1 The treatment proved to be toxic and unable to improve survival in patients with advanced amyloid cardiomyopathy and poor performance status. High rates of early cardiac deaths (42%) and of cardiac arrhythmias (33%), probably triggered by high-dose dexamethasone, drug myelotoxicity limiting the duration of treatment to 3 cycles, and a mere 9% cardiac response rate, contributed to a highly disappointing median overall survival of 1.75 months in patients with late-stage heart damage. The outcome of this study highlights the urgent need for novel approaches in order to improve the prospects of AL amyloidosis patients with advanced cardiomyopathy.
Light chain amyloidosis is the most common systemic amyloidosis, with an incidence of 10 patients per million per year, resulting in approximately 5,000 new patients/year in the European Union. It is sustained by a usually small, indolent, plasma cell clone whose biological features, shared by less proliferative plasma cell dyscrasias, make it more sensitive to chemotherapy. The clonal plasma cells synthesize an excess of light chains (LC) with specific mutations and unique structural features causing systemic proteotoxicity. Interactions of the misfolded LCs with cells, extracellular matrix components and other constituents commonly found in amyloid deposits, such as serum amyloid P (SAP), and other molecules, play an important role in the disease process and are potential targets for therapy. AL amyloidosis is also the most severe systemic amyloidosis because over 70% of patients present with cardiac involvement, which results in the development of rapidly progressive heart failure, ventricular arrhythmias and over 50% mortality within one year.2 The incidence of cardiac involvement continues to rise, owing to increasingly effective detection.3 Advanced echocardiographic techniques, such as tissue Doppler and strain imaging, allow the identification of early, subtle changes and provide prognostic information. Cardiac magnetic resonance proves useful in diagnosing and possibly monitoring amyloid deposits. Cardiac scintigraphy with bone tracers helps differentiate AL from non-AL (hereditary, senile) amyloidosis.
The molecular mechanisms involved in amyloid cardiac damage are the subject of intensive investigation. A substantial body of experimental evidence has recently altered the hypothesis underlying the etiology of AL cardiomyopathy, shifting it from a disease of simple passive amyloid infiltration to one in which the direct cardiotoxic effects of amyloidogenic LC proteins play a critical role. Previous and recent research have demonstrated that AL-LC precursor proteins, independent of cardiac fibril formation and passive restriction of cardiac function, trigger a direct cardiotoxic response causing impaired cardiomyocyte function in vitro.4,5 More recently, it has been shown that injection of amyloid LC causes cardiac dysfunction and early death in zebrafish.6 The toxic effect of the LC described in these model systems is supported by clinical observations that cardiac stress, as measured by circulating levels of type B natriuretic peptides (BNP or NT-proBNP) may be reduced in parallel with the reduction in free LC (FLC) levels following chemotherapy and such reductions translate in improved survival.7 The extent of cardiac damage, staged using the cardiac biomarkers NT-proBNP and troponins8 is a powerful predictor of survival, and the burden of the amyloid LC also has a significant impact on outcome.9 It is, therefore, vital to promptly reduce the concentration of cardiotoxic LCs.
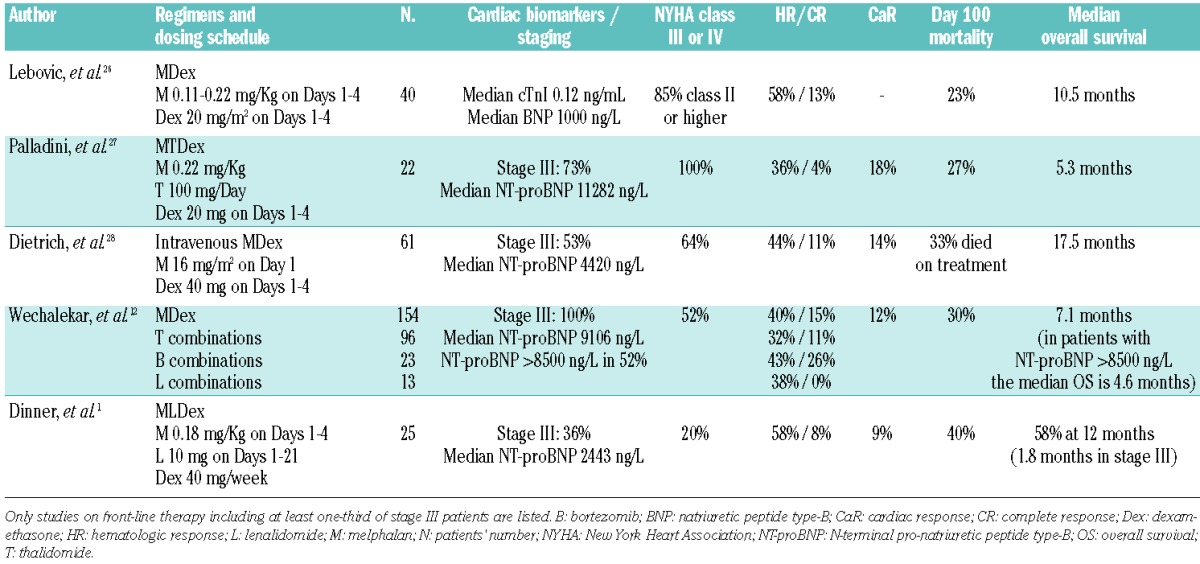
Two studies reported the use of the same oral regimen of lenalidomide in combination with dexamethasone and melphalan described in the current study by Dinner et al.1 Moreau et al., in 26 patients (12 of whom were in cardiac stage II or III), reported a 58% hematologic response rate, with 40% cardiac response, and 81% survival at two years.10 Sanchorawala et al. reported a 44% hematologic response rate (no cardiac response) with significant toxicity and a 19% mortality in the first three months in a series including 69% of patients with heart involvement, 81% of whom were in cardiac stage II and III.11 These discordant outcomes depend on the variable percentage and severity of cardiac involvement in the patient populations. A recent European collaborative study analyzed treatment outcomes of 346 patients with cardiac stage III AL amyloidosis from the United Kingdom, Italy, Germany and Greece.12 The results show that cardiac stage III AL amyloidosis is actually a heterogeneous disease, comprising both patients with less advanced cardiac damage, in whom response to therapy conferred survival improvement dependent on the depth of hematologic response, and patients with severe amyloid cardiomyopathy (characterized by extreme elevation of NT-proBNP above 8500 ng/L and systolic blood pressure <100 mm Hg) who die within a few weeks. Several studies evaluated the efficacy of front-line conventional and novel therapies in patients with amyloid cardiomyopathy. However, most of them were retrospective and some were performed before cardiac risk stratification was available, making them hardly comparable and prevented solid conclusions being drawn. Thus, we focused our analysis on the studies reporting cardiac staging and including only patients treated frontline (Table 1). The outcome of these studies is invariably dismal, with 20–40% early mortality rates, median survival ranging from 5 to 17 months depending on the proportion of patients with severe heart failure. The overall response rates seem almost unaffected by the addition of novel agents, although bortezomib combinations probably give a higher rate of complete response that may translate into better cardiac outcome.12 Improvement of cardiac function is infrequent, being observed in less than 20% of cases. Collectively these data indicate that current therapy for patients with amyloid cardiomyopathy is largely unsatisfactory and calls for better approaches. As reported in the European collaborative study, patients with less advanced cardiac damage tolerate chemotherapy better and are significantly more likely to respond to treatment.12 In addition, patients with early heart involvement and only moderately elevated cardiac biomarkers (NT-proBNP < 5000 ng/L and troponin T < 0.06 μg/L) can safely undergo autologous stem cell transplantation (ASCT)13 with satisfactory outcomes.14 These findings highlight the critical importance of early detection of amyloid cardiomyopathy. Furthermore, although the survival of patients with AL amyloidosis has been significantly improved over the last two decades by effective regimens and novel agents,15,16 the 25–30% rate of early deaths (within one year of diagnosis), almost invariably cardiac, still represents the major impediment for further improvements in survival. Investigators involved in the care of patients with AL amyloidosis have deployed several lines of intervention to tackle this vital problem.
Table 1.
Studies on front-line treatment of patients with AL amyloidosis and advanced cardiac involvement.
A) Early diagnosis
The cardiac biomarker NT-proBNP has 100% sensitivity in detecting amyloid cardiac involvement, anticipating echocardiographic abnormalities and preceding the onset of cardiac symptoms by several months.17 The great majority of patients with amyloid cardiomyopathy have an abnormal FLC κ/λ ratio. Therefore, we have recently proposed routine adoption of a simple strategy, i.e. checking NT-proBNP levels during monitoring of patients with monoclonal gammopathy of undetermined significance with abnormal κ/λ ratio, in order to detect heart involvement at a very early stage.18 Increasing knowledge and awareness of these rare diseases, through national and international collaborative networks and patients’ associations, is also fundamental for improving early recognition of these rapidly progressive diseases in which ‘time’ is ‘life’.
B) Synergize treatment and improve tolerability
Several lines of evidence indicate that a profound and early reduction of cardiotoxic LC is critical to improving survival. Achieving this aim remains difficult in these frail patients extremely sensitive to the toxicity of therapy. A possible strategy would be to combine agents with synergistic mechanisms of action, thereby allowing lower doses of each individual agent to be used, thus improving tolerance while producing rapid and deep responses.12 Recently, an over 90% survival at two years was reported in a retrospective series in 20 stage III patients receiving cyclophosphamide, bortezomib and dexamethasone. However, this study also included previously treated subjects.19 Prospective trials of regimens based on rapidly acting proteasome inhibitors in association with alkylating agents and low-dose dexamethasone in stage III patients are ongoing to validate this strategy. Silencing the amyloid LC isotype gene could be an alternative and less toxic strategy.20
C) Accelerate recovery of cardiac function
The most advanced stage III patients, with extreme elevations of NT-proBNP and hypotension, pose an extremely difficult challenge. These very ill patients cannot withstand even attenuated regimens and achieving good quality response (≥ VGPR) may not be sufficient to rescue heart function.12 Strategies directed at promoting the resorption of amyloid deposits, such as immunotherapy21 or small molecules like doxycycline22 in combination with effective, attenuated chemotherapy, could potentially accelerate the recovery of cardiac function, improving the quality of life and extending survival. Drugs counteracting the proteotoxicity of the amyloid light chains, such as epigallocatechin-gallate,23 can also improve heart performance. Several trials are ongoing to test these novel strategies.
D) Supporting cardiac function
Careful control of body weight and judicious diuretic use are fundamental. Devices that assist ventricular function have been used in patients with terminally compromised cardiac function, with disappointing results, mostly due to high morbidity.24 Cardiac transplantation represents a life-saving approach, particularly in young patients without significant extra-cardiac organ involvement. Sequential ASCT can warrant extended survival and quality of life.25 However, the feasibility of this approach is limited by the availability of organ donors.
Reduction of morbidity and mortality in patients with cardiac AL amyloidosis requires the development of innovative and more effective remedies. Basic research into the molecular mechanisms involved in cardiac injury caused by amyloid proteins is essential to direct therapeutic efforts. In the near future, new treatment options targeting multiple steps in the process of amyloidogenesis, from reducing the amyloid precursor protein to accelerating the clearance of amyloid deposits and counteracting proteotoxicity, have the potential to improve clinical efficacy while reducing treatment-related complications.
Acknowledgments
This work was supported by grants from “Associazione Italiana per la Ricerca sul Cancro” Special Program Molecular Clinical Oncology 5 per mille n. 9965 “Harnessing tumor cell/microenvironment cross talk to treat mature B cell tumors.”
Footnotes
Financial and other disclosures provided by the author using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are available with the full text of this paper at www.haematologica.org.
References
- 1.Dinner S, Witteles W, Afghahi A, Witteles R, Arai S, Lafayette R, et al. Lenalidomide, melphalan and dexamethasone in a population of patients with immunoglobulin light chain amyloidosis with high rates of advanced cardiac involvement. Haematologica. 2013;98(10): 1593–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenesis and new therapeutic options. J Clin Oncol. 2011;29(14):1924–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falk RH. Cardiac amyloidosis: a treatable disease, often overlooked. Circulation. 2011;124(9):1079–85 [DOI] [PubMed] [Google Scholar]
- 4.Liao R, Jain M, Teller P, Connors LH, Ngoy S, Skinner M, et al. Infusion of light chains from patients with cardiac amyloidosis causes diastolic dysfunction in isolated mouse hearts. Circulation. 2001;104(14):1594–7 [PubMed] [Google Scholar]
- 5.Guan J, Mishra S, Shi J, Plovie E, Qiu Y, Cao X, et al. Stanniocalcin1 is a key mediator of amyloidogenic light chain induced cardiotoxicity. Basic Res Cardiol. 2013;108(5):378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mishra S, Guan J, Plovie E, Seldin DC, Connors LH, Merlini G, et al. Human amyloidogenic light chain proteins result in cardiac dysfunction, cell death, and early mortality in zebrafish. Am J Physiol Heart Circ Physiol. 2013;305(1):H95–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palladini G, Lavatelli F, Russo P, Perlini S, Perfetti V, Bosoni T, et al. Circulating amyloidogenic free light chains and serum N-terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL. Blood. 2006;107(10):3854–8 [DOI] [PubMed] [Google Scholar]
- 8.Dispenzieri A, Gertz M, Kyle R, Lacy M, Burritt M, Therneau T, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. 2004;22(18):3751–7 [DOI] [PubMed] [Google Scholar]
- 9.Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30(9):989–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moreau P, Jaccard A, Benboubker L, Royer B, Leleu X, Bridoux F, et al. Lenalidomide in combination with melphalan and dexamethasone in patients with newly diagnosed AL amyloidosis: a multicenter phase 1/2 dose-escalation study. Blood. 2010;116(23):4777–82 [DOI] [PubMed] [Google Scholar]
- 11.Sanchorawala V, Patel JM, Sloan JM, Shelton AC, Zeldis JB, Seldin DC. Melphalan, lenalidomide and dexamethasone for the treatment of immunoglobulin light chain amyloidosis: results of a phase II trial. Haematologica. 2013;98(5):789–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wechalekar AD, Schonland SO, Kastritis E, Gillmore JD, Dimopoulos MA, Lane T, et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood. 2013;121(17):3420–7 [DOI] [PubMed] [Google Scholar]
- 13.Gertz MA, Lacy MQ, Dispenzieri A, Kumar SK, Dingli D, Leung N, et al. Refinement in patient selection to reduce treatment-related mortality from autologous stem cell transplantation in amyloidosis. Bone Marrow Transplant. 2013;48(4):557–61 [DOI] [PubMed] [Google Scholar]
- 14.Madan S, Kumar SK, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, et al. High-dose melphalan and peripheral blood stem cell transplantation for light-chain amyloidosis with cardiac involvement. Blood. 2012;119(5):1117–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar SK, Gertz MA, Lacy MQ, Dingli D, Hayman SR, Buadi FK, et al. Recent improvements in survival in primary systemic amyloidosis and the importance of an early mortality risk score. Mayo Clin Proc. 2011;86(1):12–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merlini G. CyBorD: stellar response rates in AL amyloidosis. Blood. 2012;119(19):4343–5 [DOI] [PubMed] [Google Scholar]
- 17.Palladini G, Campana C, Klersy C, Balduini A, Vadacca G, Perfetti V, et al. Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation. 2003;107(19):2440–5 [DOI] [PubMed] [Google Scholar]
- 18.Merlini G, Wechalekar AD, Palladini G. Systemic light chain amyloidosis: an update for treating physicians. Blood. 2013;121(26):5124–30 [DOI] [PubMed] [Google Scholar]
- 19.Venner CP, Lane T, Foard D, Rannigan L, Gibbs SD, Pinney JH, et al. Cyclophosphamide, bortezomib, and dexamethasone therapy in AL amyloidosis is associated with high clonal response rates and prolonged progression-free survival. Blood. 2012;119(19):4387–90 [DOI] [PubMed] [Google Scholar]
- 20.Hovey BM, Ward JE, Soo Hoo P, O’Hara CJ, Connors LH, Seldin DC. Preclinical development of siRNA therapeutics for AL amyloidosis. Gene Ther. 2011;18(12):1150–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature. 2010;468(7320):93–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ward JE, Ren R, Toraldo G, Soohoo P, Guan J, O’Hara C, et al. Doxycycline reduces fibril formation in a transgenic mouse model of AL amyloidosis. Blood. 2011;118(25):6610–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mereles D, Buss SJ, Hardt SE, Hunstein W, Katus HA. Effects of the main green tea polyphenol epigallocatechin-3-gallate on cardiac involvement in patients with AL amyloidosis. Clin Res Cardiol. 2010;99(8):483–90 [DOI] [PubMed] [Google Scholar]
- 24.Swiecicki PL, Edwards BS, Kushwaha SS, Dispenzieri A, Park SJ, Gertz MA. Left ventricular device implantation for advanced cardiac amyloidosis. J Heart Lung Transplant. 2013;32(5):563–8 [DOI] [PubMed] [Google Scholar]
- 25.Dey BR, Chung SS, Spitzer TR, Zheng H, Macgillivray TE, Seldin DC, et al. Cardiac transplantation followed by dose-intensive melphalan and autologous stem-cell transplantation for light chain amyloidosis and heart failure. Transplantation. 2010;90(8):905–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lebovic D, Hoffman J, Levine BM, Hassoun H, Landau H, Goldsmith Y, et al. Predictors of survival in patients with systemic light-chain amyloidosis and cardiac involvement initially ineligible for stem cell transplantation and treated with oral melphalan and dexamethasone. Br J Haematol. 2008;143(3):369–73 [DOI] [PubMed] [Google Scholar]
- 27.Palladini G, Russo P, Lavatelli F, Nuvolone M, Albertini R, Bosoni T, et al. Treatment of patients with advanced cardiac AL amyloidosis with oral melphalan, dexamethasone, and thalidomide. Ann Hematol. 2009;88(4):347–50 [DOI] [PubMed] [Google Scholar]
- 28.Dietrich S, Schonland SO, Benner A, Bochtler T, Kristen AV, Beimler J, et al. Treatment with intravenous melphalan and dexamethasone is not able to overcome the poor prognosis of patients with newly diagnosed systemic light chain amyloidosis and severe cardiac involvement. Blood. 2010;116(4):522–8 [DOI] [PubMed] [Google Scholar]