

Abstract
The BCR-ABL T315I mutation confers resistance to currently licensed tyrosine kinase inhibitors in chronic myelogenous leukemia. However, the impact of this mutation on survival in early stages of disease, in chronic phase, has never been detailed. Using matched pair analysis, a cohort of 64 patients with chronic phase chronic myelogenous leukemia harboring a T315I mutation and resistant to imatinib mesylate was compared to a similar cohort of 53 chronic phase patients resistant to imatinib, but with no detectable T315I mutation, in the pre-ponatinib era. These patients were matched according to age at diagnosis, interval between disease diagnosis and start of imatinib treatment, and duration of imatinib therapy. Kaplan-Meier survival analyses demonstrated the significant negative impact of the presence of the T315I mutation on overall survival (since imatinib-resistance: 48.4 months for T315I+ patients versus not reached for T315I− ones; P=0.006) and failure-free survival (since imatinib-resistance: 34.7 months for T315I+ patients versus not reached for T315I− patients; P=0.003). In addition, Cox proportional hazard models adjusted on overall survival demonstrated the negative influence of the T315I mutation (P=0.02, HR=2.54). These results confirm early assumptions concerning the poor prognosis of chronic phase chronic myelogenous leukemia patients with the T315I mutation who are not eligible for allogeneic transplantation, and demonstrate the need for more therapeutic options.
Introduction
Chronic phase (CP) chronic myelogenous leukemia (CML) originates in a multipotent hematopoietic stem cell and, in the initial stages of disease, is driven by the oncogenic Philadelphia chromosome, the reciprocal translocation t(9;22)(q34;q11),1 its molecular equivalent the BCR-ABL oncogene and its subsequent derivative, the BCR-ABL1 tyrosine kinase. In the absence of specific treatment, the natural evolution of the disease is the inexorable development of a fatal blast crisis. Tyrosine kinase inhibitors (TKI) have revolutionized the overall poor prognosis of this disease, particularly in patients with CP disease, by prolonging, possibly indefinitely, the life of patients.2 However, it has been demonstrated in vitro that none of the agents available is able to eradicate a pool of living, quiescent, BCR-ABL+ undifferentiated cells, which may serve as a reservoir for additional oncogenic events leading to disease progression.3–5 These findings have been confirmed in vivo in the vast majority of patients who remain with persistent molecular residual disease thus forbidding TKI cessation.2 In approximately 50% of cases of imatinib (IM) resistance in CP, patients acquire, under the selective pressure of TKI, recurrent BCR-ABL amino-acid exchanges6–8 which modify the conformation of the ABL tyrosine kinase pocket, and result in impairment, at different levels, of the activity of the three most widely prescribed TKI to date (i.e., IM, dasatinib and nilotinib). The T315I BCR-ABL mutation remains the most problematic since it totally impairs contact between the ABL tyrosine kinase domain and these three TKI, resulting in high IC50 to these TKI, as assessed in engineered murine cell lines.9 These biochemical and cellular in vitro findings are associated with poor survival rates in patients resistant to IM.10–14 However, it remains unclear whether the presence of the T315I mutation within the cells in CP confers a specific virulence to the disease or whether it is simply a co-factor in a cell that is IM-resistant for other unknown reasons.
In this perspective, we further exploited data available from an international registry of patients harboring a T315I mutation previously published in part.14 We did this by restricting our analyses exclusively to chronic phase CML patients (at diagnosis and at the time the T315I mutation was detected) who were not eligible for allogeneic stem cell transplantation (as this option might have rescued a number of them15) and who had not received ponatinib, and updating the data. Using a matched pair analysis in our three European centers, we matched CP CML T315I+ patients with other IM-resistant CP CML patients who did not have a T315I BCR-ABL mutation at resistance, in an effort to demonstrate the negative impact of the presence of this mutation on overall and failure-free survival.
Methods
Study population
The CML patients included in this analysis were in CP, resistant to TKI according to the IRIS study definitions2 or ELN recommendations16,17 and harbored a T315I BCR-ABL mutation detected prospectively or retrospectively on frozen samples, by any validated means, between 1999 and 2010. CP CML patients with a T315I mutation were extracted from a previously described updated international database containing information from 222 T315I+ CML patients (all phases and those with Philadelphia chromosome-positive acute lymphoblastic leukemia).14 In that study,14 the survival of T315I+ patients did not differ between countries (unpublished data). In this study, the comparison group was CML patients in CP at diagnosis and at IM-resistance, previously screened at IM-resistance for BCR-ABL mutations and found to be T315I negative (they could harbor other mutations). These T315− patients were identified in the individual databases from three representative European centers (Lyon and Bordeaux in France and London, UK). All patients who had received an allogeneic transplant were eliminated from this study. Patients with the T315I mutation were pair-matched to T315I− IM-resistant patients according to three different variables: age at CP CML diagnosis (± 5 years), time elapsed between CP CML diagnosis and initiation of IM (± 5 years), and the duration of IM treatment (± 31 months). All patients were in CP at diagnosis and at IM-resistance and none had received ponatinib. These CML patients provided written consent whenever possible. This retrospective analysis was approved by the institutional review board/ethical review committee in each participating site/country whenever necessary.
Data collection
Demographic, clinical, treatment, mutation, and survival data were collected from each of the three European centers for the control group or extracted - and updated whenever possible -from the epidemiological study database previously described.14 Mutation screening was performed using different techniques (predominantly direct sequencing, but also polymerase chain reaction-restriction fragment length polymorphisms and denaturing high performance liquid chromatography in combination with sequencing) and, for some patients, included assaying frozen material. Patients with any T315I clone levels were included. A mutated clone was considered the major clone if it accounted for the highest percentage of cells among all detected clones, when this information was available.
Survival measurement
Survival was analyzed according to Kaplan-Meier methods and log-rank tests calculated from the dates of starting IM, resistance to IM, to death or to the most recent date when the patient was known to be alive. Overall survival was analyzed from the time of diagnosis, IM-initiation, IM-resistance, and the initiation of a second-generation TKI (TKI2). Progression-free survival could not be analyzed precisely since the exact date of progression was not available for all cases; however, it was possible to analyze failure-free survival when considering failure as the time to next treatment, progression or death.
Statistical analysis
Multivariate analysis, according to the Cox proportional hazard model adjusted on overall survival, was performed on the whole population of patients and on the T315I+ subgroup. Covariates, used as continuous variables, included age at IM-resistance, gender, exposure to interferon, intervals between CML diagnosis and IM-initiation, IM-initiation and IM-resistance, and exposure to TKI2. These variables were identified as being significant in an initial univariate analysis. Age was considered as a continuous variable, whereas T315I mutation status, gender, use of interferon-α, TKI2, and different time intervals were analyzed as ordinal variables. P values <0.05 were considered statistically significant. The software package “Survival” (R, version 2.14, www.r-project.org) was used to conduct the statistical analysis.
Results
Demographic information
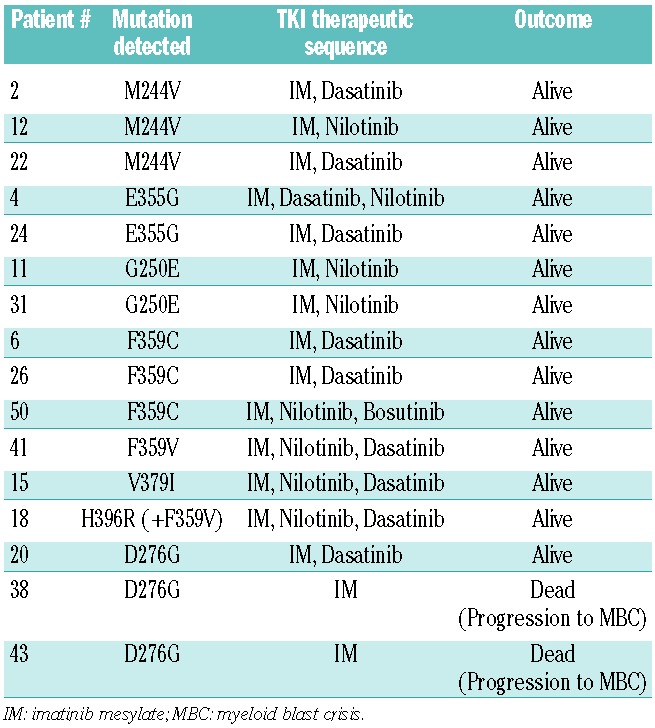
Sixty-four patients with CP CML at diagnosis and at IM-resistance were identified in the initial international T315I+ database and matched with 53 T315I negative counterparts (Table 1). Importantly, among the T315I+ patients, the T315I clone was predominant in 42 patients (78%) out of 53 cases evaluable for quantification, being predominant over wild-type transcripts in 31 patients or other mutated transcripts in 11 patients (21%). In 10 patients (19%) the wild-type clone or another mutated clone was predominant. Significantly more patients had received interferon-α prior to IM in the T315I+ group (64% versus 37.7%; P=0.008), but the duration of interferon-α treatment did not differ between the two groups (10 versus 12 months, P=0.98). The initiation of IM occurred at a median of 33 months after diagnosis in the T315I− group versus 63.8 months in the T315I+ group, which was not a statistically significant difference (P=0.09). Exposure to the selective agent, IM, and the intervals to IM-initiation and IM-resistance were similar. Sixteen patients (30%) in the control group had mutations other than T315I at the time of IM-resistance (3 M244V, 2 G250E, 3 D276G, 2 E355G, 3 F359C, 1 F359V, 1 V379I, 1 H396R+F359V as detailed in Table 2) and 30% in the T315I+ group had additional mutations (2 G250E, 2 Y253H, 2 E255V, 2 E255K, 3 F317L, 1 F359V, 2 F359I, 1 F359C) when the T315I mutation was detected. Forty-seven patients out of 53 had received one or more TKI2 (89%) in the T315I− group and 48/64 patients (75%) in the T315I+ group (P=0.1), after a median of 29 and 36 months, respectively. The median interval between the occurrence of IM-resistance and TKI2 initiation (when given) did not differ being 9 months in the T315I− group and 6 months in the T315I+ group (P=0.35). In the T315I+ group, the mutation was detected in 17/64 (26%) of the cases after the initiation of TKI2, although undetectable at the time of IM-resistance. Importantly, the median follow-up since the diagnosis of CP CML was similar in both groups, being approximately 6 years (P=0.61).
Table 1.
General characteristics of the two paired populations of CP CML patients without and with T315I mutation compared in univariate analysis. Data are presented as median values with the ranges in brackets.
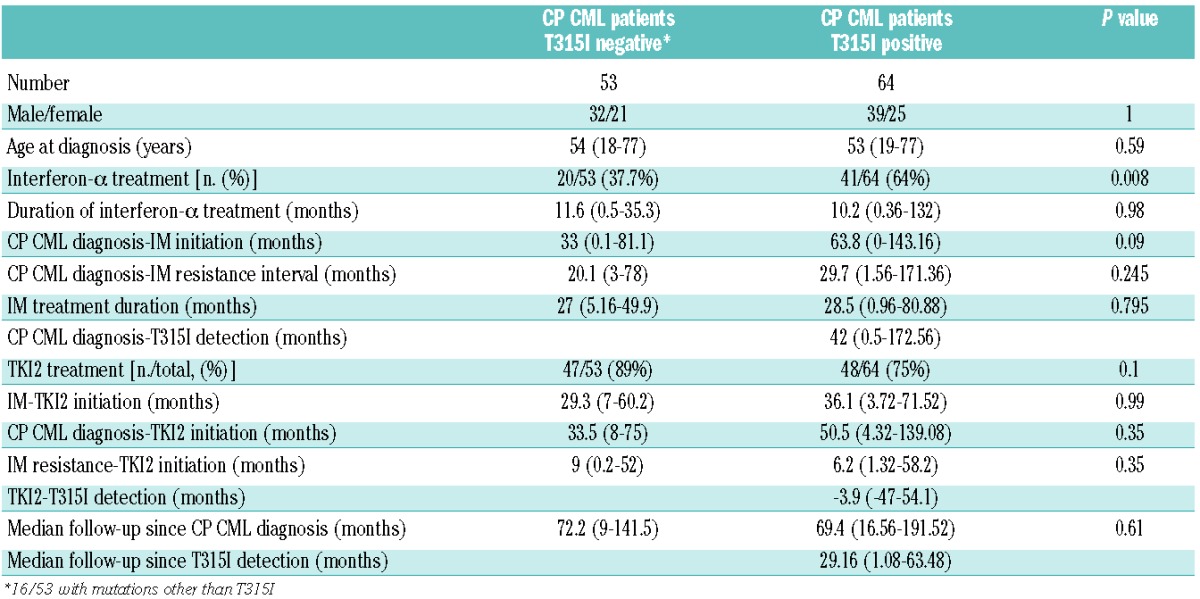
Table 2.
BCR-ABL mutations other than T315I identified in the control group of patients resistant to TKI and individual therapeutic sequence of TKI (only 30% of the patients had detectable levels of various types of mutated cells).
Overall and failure-free survival
We analyzed the overall survival since IM-initiation, IM-resistance and TKI2 initiation in patients with and without the T315I mutation. The results are shown in Figures 1A, 1B, and 1C, respectively. In all three cases, the overall survival of T315I+ patients was impressively and significantly worse than that of T315− patients (P=0.01, 0.006, and 0.0007, respectively) with a median of 79.5 (69.3-not reached] months versus not reached since IM-initiation, 48.4 (44.5–70.5) months versus not reached since IM-resistance and 52.4 (26.8-NR) months versus not reached since TKI2 initiation, in this population of patients who had not been transplanted and had never been treated with ponatinib. There were 26 deaths in the T315I+ group and 14 in the T315I− control group.
Figure 1.
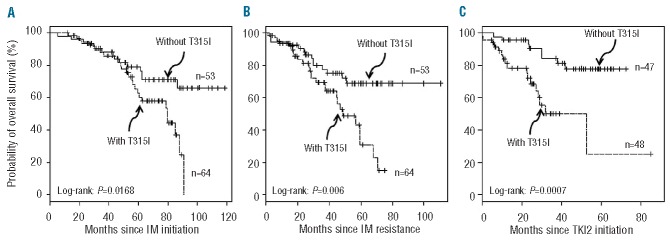
Overall survival of chronic phase CML patients resistant to IM, since IM-initiation (A), since IM-resistance (B), and since TKI2 initiation (C) in months according to T315I status (dashed line patients with T315I mutation, plain line patients without T315I mutation). N: number of patiens.
When we analyzed the failure-free survival (as defined previously) of these two cohorts from the same starting points, survival was unequivocally and significantly worse in patients harboring the T315I for all starting points (P=0.02, P=0.003, P=0.005 respectively, Figure 2A, B, C). Finally, and in order to determine whether the TKI2 might promote the disease by enhancing the selection of T315I cells in vivo, we looked at the survival of patients since TKI2 exposure, according to the moment of detection of the T315I mutation, prior to the exposure to TKI2 (i.e. patients on IM) or after the initiation of TKI2 (Figure 3). Although the overall survival did not seem to differ (P=0.6, data not shown), failure-free survival was significantly worse for patients with a T315I mutation detected after TKI2 initiation (P=0.024), but the number of patients was small. The median interval between the discontinuation of IM and the initiation of the first TKI2 (if any) was 1.04 (0–4.26) months in the T315I+ group and 0.02 (0–2.78) months in the T315− group (P=ns), excluding a deselection phenomenon.18
Figure 2.
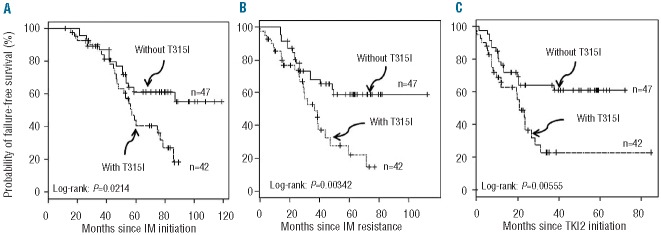
Failure-free survival of chronic phase CML patients resistant to IM, since IM-initiation (A), since IM-resistance (B), and since TKI2 initiation (C) in months according to T315I status (dashed line patients with T315I mutation, plain line patients without T315I mutation). N: number of patients.
Figure 3.
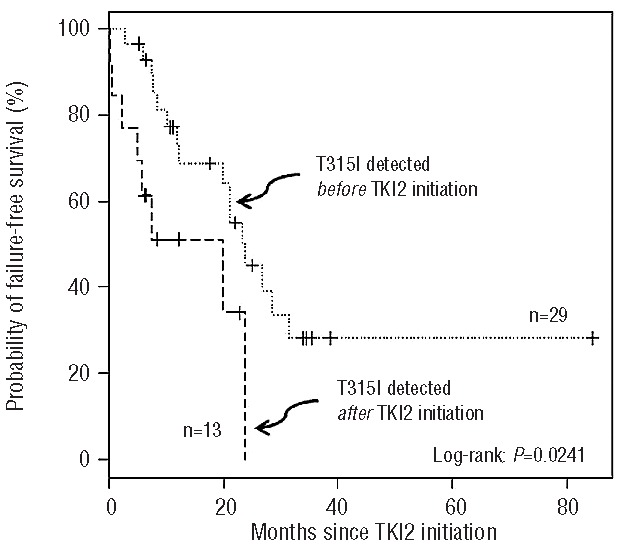
Failure-free survival of CP CML patients resistant to IM harboring a T315I mutation, in months, according to the time of detection of the mutation before (dotted line) of after (dashed line) exposure to the TKI2. N: number of patients.
Cox proportional hazard model on overall survival
The Cox proportional hazard model on overall survival since IM-resistance was performed in patients analyzing the impact of the seven variables detailed in the Methods section (Figure 4A). The negative impact of the presence of the T315I mutation [P=0.02, hazard ratio (HR) = 2.4], suggested by the overall and failure-free survival curves, was confirmed in this model, and we were able to identify some factors with a positive impact on overall survival such as an exposure to interferon-α (P=0.01, HR=0.36), and exposure to TKI2, probably influenced by its beneficial effects in the T315I− patients (P=0.0002, HR=0.2). The interval between the initiation of IM and the resistance to these agents did not have a statistically significant influence on overall survival (P=0.43, HR=0.99). A younger age at IM-resistance and shorter interval between CML diagnosis and IM-initiation were significant factors (P=0.02 and P=0.01, respectively) having favorable effects on overall survival, with HR of 1.03 and 1.01, respectively.
Figure 4.
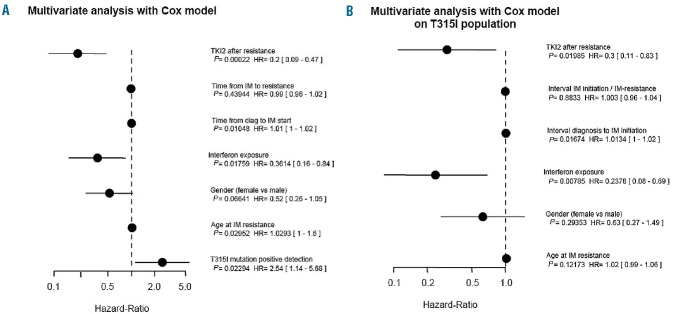
(A) Forest plot (log scale) showing the results of the multivariate analysis with Cox model, adjusted on overall survival since IM-resistance for the whole population of CP CML patients resistant to IM studied here. Horizontal bars represent the 95% confidence intervals. P values for each variable are indicated, the hazard ratio (HR) is stated for the each variable “Hazard Ratios” and numbers in brackets indicate the exact 95% confidence intervals. (B) Forest plot (log scale) showing the results of the multivariate analysis with Cox model adjusted on overall survival since IM-resistance for the T315I+ population of CP CML studied here. Horizontal bars represent the 95% confidence intervals. P values for each variable are indicated. The hazard ratio (HR) is stated for each variable and the numbers in brackets are the exact 95% confidence intervals.
In a Cox proportional hazard model we further analyzed the impact of the same variables (except the presence of the T315I mutation) on overall survival in the population of T315I+ patients (n=64, Figure 4B), in an effort to identify favorable or unfavorable factors in this selected subpopulation. Again, treatment with interferon-α had a positive impact on overall survival (P=0.007, HR=0.24), and, unexpectedly, exposure to TKI2 was found to have a positive impact on overall survival in these T315I+ patients (P=0.0199, HR=0.3). Age, gender, interval between IM-initiation and resistance did not influence overall survival in this model.
Discussion
Several studies have suggested that CML patients harboring a T315I mutation have poor prognosis7,10,12,13 because this mutation confers universal resistance in vitro to all currently licensed TKI8 except the very recently available ponatinib. The T315I mutation is the most frequently identified mutation in CML TKI-resistant and mutated patients19 particularly in those in advanced phases of disease. It is not clear whether the poor prognosis observed is related to the pleiotropic resistance to TKI (which may efficiently select the mutated clone and subsequently worsen the prognosis), or whether the mutation itself confers a gain-of-function to Philadelphia chromosome-positive leukemic cells, thereby increasing oncogenicity and promoting progression (as suggested in vitro in engineered models20,21). In retrospective studies the T315I mutation has been identified in ~50% of cases in IM-resistant CML patients14 in advanced phases, but these studies often mix T315I+ patients from all CML phases together and it remains difficult to understand what happens at earlier stages.
We analyzed here a cohort of patients with detectable predominant T315I mutated clones (more likely to drive disease resistance), and matched them to a multicenter cohort of CP CML patients resistant to TKI, screened for BCR-ABL mutations, but without detectable T315I mutation. The two groups were grossly equally balanced according to the different characteristics liable to affect selection (IM treatment duration, proportion of TKI2 treated patients in both groups, interval between IM and TKI2, interval between IM-resistance and TKI2). Age was comparable to that observed in previous academic studies of TKI-resistant patients,10,12,14 and should not have influenced the genetic background of CML cells in one group or the other, or, introduced additional co-morbidities that, in older patients, can affect survival. It is essential to obtain two comparable groups of patients in whom T315I mutated clones may be detected at diagnosis of CML by sensitive techniques,22,23 but it remains unknown why some patients experience IM-resistance with detectable levels of T315I clones while others do not.24 Exposure to interferon-α treatment (for a similar duration) was more frequent in the T315+ group than in the T315I− group, although the interval between diagnosis and IM-initiation was longer in the latter group. In the Cox models adjusted on overall survival for the two groups and for the T315I+ group (Figure 4), interferon-α had a significant positive impact. In the unmutated IM-resistant group, 20/53 patients received interferon-α prior to IM-resistance but never in combination with IM or alone once IM-resistance had occurred. In the T315I+ group, five patients had received interferon-α after the identification of the T315I mutation, and 36 before (30 alone prior to IM, 6 in combination with IM, data not shown). We hypothesize that the fact that some patients received interferon-α after the identification of the T315I mutation, a possible therapeutic option25 along with omacetaxine mepesuccinate25,26 in patients not eligible for allogeneic stem cell transplantation, might explain this positive effect. However, it remains unclear whether interferon-α exerts a specific activity on the T315I mutated clone 25 or whether it is simply the withdrawal of the selective agent, the TKI, that improves the outcome of these patients.18
In this matched pair analysis, the presence of the T315I mutation in CP undoubtedly impaired overall and failure-free survival (Figures 1A,B,C and 2A,B,C). The median overall survival and failure-free survival were never reached in any case for T315I− patients, and were always reached for T315I+ patients. The multivariate analysis adjusted on overall survival (Figure 4A) strengthened such results, in which the presence of the T315I negatively influenced survival (P=0.023, HR=2.54). These findings suggest that the presence of the mutation, whatever the therapeutic options used in these patients, confers a competitive growth advantage to the CP CML cells. A previous study27 demonstrated that kinase domain mutations confer a poor prognosis to CML patients in CP. In the present study, 30% of the CP patients in the T315I− control group harbored diverse mutations concurring to IM-resistance, however, these patients’ overall survival and failure-free survival remained better than those of the T315I+ patients, suggesting that the T315I mutation by itself specifically worsens their prognosis.
TKI2, as more selective agents, might affect survival although it is difficult to analyze this in our study since not all the patients received TKI2, and some of them received one, two and even three TKI2 (nilotinib, dasatinib, bosutinib). In the comparison group, 30% of the patients had mutations and were rescued, in the majority of the cases, by the use of one or two TKI2. The multivariate analyses performed on the whole population studied demonstrated the positive impact of TKI2 administration on overall survival, which is in line with several reports in the literature,8,28 but more surprisingly, TKI2, administered after resistance [median duration = 6.9 (0.5–33.4) months] appeared to improve the overall survival significantly within the T315I+ population (P=0.01, HR=0.3) as well, possibly by limiting the progression of the unmutated tumor cell mass. The failure-free survival, analyzed from the initiation of TKI2, was significantly worse in the group of patients in whom the T315I mutation was detected after starting TKI2 (P=0.024, Figure 3), suggesting that these inhibitors, by selecting the T315I+ cells even more efficiently than IM would do, might promote a rapid failure in such a situation. The general assumption in this regard would be not to use TKI2 once a T315I mutation has been detected in IM-resistant CP CML patients, as recommended.29
This study, despite the obvious caveats, confirms that the presence of the T315I mutation in IM-resistant CP CML patients impairs both overall survival and failure-free survival. It also suggests that the use of TKI2 after the identification of the mutation in IM-resistant patients might selectively impair the failure-free survival of such patients. We believe that the introduction of third-generation TKI for patients resistant to first and second-generation TKI, including T315I+ patients, might help to improve survival for patients remaining ineligible for allogeneic stem cell transplantation. One such third-generation TKI is ponatinib, which is currently being tested in a prospective clinical trial30 and has been approved for use in the USA.
Acknowledgments
F-EN thanks Mrs M. Etienne, CRA, Pierre-Bénite, France, for help with the data collection; Mr Mohamad Sobh, PharmD, CRA, Pierre Bénite, France for editorial assistance, the association “Regarde un jour le monde” for its constant support, and Mrs Barbara White, for her critical proof-reading of this manuscript. We also thank the hematolog tumor bank of the CHU of Bordeaux, France.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243(5405):290–3 [DOI] [PubMed] [Google Scholar]
- 2.Hughes TP, Hochhaus A, Branford S, Muller MC, Kaeda JS, Foroni L, et al. Long-term prognostic significance of early molecular response to imatinib in newly diagnosed chronic myeloid leukemia: an analysis from the International Randomized Study of Interferon and STI571 (IRIS). Blood. 2010;116(19):3758–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319–25 [DOI] [PubMed] [Google Scholar]
- 4.Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, Jordanides N, et al. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006;107(11):4532–9 [DOI] [PubMed] [Google Scholar]
- 5.Jorgensen HG, Allan EK, Jordanides NE, Mountford JC, Holyoake TL. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood. 2007;109(9): 4016–9 [DOI] [PubMed] [Google Scholar]
- 6.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2(2):117–25 [DOI] [PubMed] [Google Scholar]
- 7.Muller MC, Cortes JE, Kim DW, Druker BJ, Erben P, Pasquini R, et al. Dasatinib treatment of chronic-phase chronic myeloid leukemia: analysis of responses according to preexisting BCR-ABL mutations. Blood. 2009;114(24):4944–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hughes T, Saglio G, Branford S, Soverini S, Kim DW, Muller MC, et al. Impact of baseline BCR-ABL mutations on response to nilotinib in patients with chronic myeloid leukemia in chronic phase. J Clin Oncol. 2009;27(25):4204–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O'Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan J, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65(11):4500–5 [DOI] [PubMed] [Google Scholar]
- 10.Soverini S, Colarossi S, Gnani A, Rosti G, Castagnetti F, Poerio A, et al. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res. 2006; 12(24):7374–9 [DOI] [PubMed] [Google Scholar]
- 11.Soverini S, Iacobucci I, Baccarani M, Martinelli G. Targeted therapy and the T315I mutation in Philadelphia-positive leukemias. Haematologica. 2007;92(4): 437–9 [DOI] [PubMed] [Google Scholar]
- 12.Nicolini FE, Corm S, Le QH, Sorel N, Hayette S, Bories D, et al. Mutation status and clinical outcome of 89 imatinib mesylate-resistant chronic myelogenous leukemia patients: a retrospective analysis from the French intergroup of CML (Fi(φ)-LMC group). Leukemia. 2006; 20(6):1061–6 [DOI] [PubMed] [Google Scholar]
- 13.Nicolini FE, Hayette S, Corm S, Bachy E, Bories D, Tulliez M, et al. Clinical outcome of 27 imatinib mesylate-resistant chronic myelogenous leukemia patients harboring a T315I BCR-ABL mutation. Haematologica. 2007; 92(9):1238–41 [DOI] [PubMed] [Google Scholar]
- 14.Nicolini FE, Mauro MJ, Martinelli G, Kim DW, Soverini S, Muller MC, et al. Epidemiologic study on survival of chronic myeloid leukemia and Ph(+) acute lymphoblastic leukemia patients with BCR-ABL T315I mutation. Blood. 2009;114(26): 5271–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicolini FE, Basak GW, Soverini S, Martinelli G, Mauro MJ, Muller MC, et al. Allogeneic stem cell transplantation for patients harboring T315I BCR-ABL mutated leukemias. Blood. 2011;118(20):5697–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baccarani M, Saglio G, Goldman J, Hochhaus A, Simonsson B, Appelbaum F, et al. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European Leukemia Net. Blood. 2006; 108(6):1809–20 [DOI] [PubMed] [Google Scholar]
- 17.Baccarani M, Cortes J, Pane F, Niederwieser D, Saglio G, Apperley J, et al. Chronic myeloid leukemia: an update of concepts and management recommendations of European Leukemia Net. J Clin Oncol. 2009; 27(35):6041–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanfstein B, Muller MC, Kreil S, Ernst T, Schenk T, Lorentz C, et al. Dynamics of mutant BCR-ABL-positive clones after cessation of tyrosine kinase inhibitor therapy. Haematologica. 2011;96(3):360–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Apperley JF. Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007;8(11):1018–29 [DOI] [PubMed] [Google Scholar]
- 20.Skaggs BJ, Gorre ME, Ryvkin A, Burgess MR, Xie Y, Han Y, et al. Phosphorylation of the ATP-binding loop directs oncogenicity of drug-resistant BCR-ABL mutants. Proc Natl Acad Sci USA. 2006;103(51):19466–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 2008;15(10):1109–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roche-Lestienne C, Lai JL, Darre S, Facon T, Preudhomme C. A mutation conferring resistance to imatinib at the time of diagnosis of chronic myelogenous leukemia. N Engl J Med. 2003;348(22):2265–6 [DOI] [PubMed] [Google Scholar]
- 23.Ernst T, Erben P, Muller MC, Paschka P, Schenk T, Hoffmann J, et al. Dynamics of BCR-ABL mutated clones prior to hematologic or cytogenetic resistance to imatinib. Haematologica. 2008;93(2):186–92 [DOI] [PubMed] [Google Scholar]
- 24.Willis SG, Lange T, Demehri S, Otto S, Crossman L, Niederwieser D, et al. High-sensitivity detection of BCR-ABL kinase domain mutations in imatinib-naive patients: correlation with clonal cytogenetic evolution but not response to therapy. Blood. 2005;106(6):2128–37 [DOI] [PubMed] [Google Scholar]
- 25.de Lavallade H, Khorashad JS, Davis HP, Milojkovic D, Kaeda JS, Goldman JM, et al. Interferon-alpha or homoharringtonine as salvage treatment for chronic myeloid leukemia patients who acquire the T315I BCR-ABL mutation. Blood. 2007;110(7): 2779–80 [DOI] [PubMed] [Google Scholar]
- 26.Nicolini FE, Chomel JC, Roy L, Legros L, Chabane K, Ducastelle S, et al. The durable clearance of the T315I BCR-ABL mutated clone in chronic phase chronic myelogenous leukemia patients on omacetaxine allows tyrosine kinase inhibitor rechallenge. Clin Lymphoma Myeloma Leuk. 2010;10(5):394–9 [DOI] [PubMed] [Google Scholar]
- 27.Khorashad JS, de Lavallade H, Apperley JF, Milojkovic D, Reid AG, Bua M, et al. Finding of kinase domain mutations in patients with chronic phase chronic myeloid leukemia responding to imatinib may identify those at high risk of disease progression. J Clin Oncol. 2008;26(29): 4806–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hochhaus A, Kantarjian HM, Baccarani M, Lipton JH, Apperley JF, Druker BJ, et al. Dasatinib induces notable hematologic and cytogenetic responses in chronic-phase chronic myeloid leukemia after failure of imatinib therapy. Blood. 2007;109(6):2303–9 [DOI] [PubMed] [Google Scholar]
- 29.Soverini S, Hochhaus A, Nicolini FE, Gruber F, Lange T, Saglio G, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European Leukemia Net. Blood. 2011; 118(5):1208–15 [DOI] [PubMed] [Google Scholar]
- 30.Cortes J, Kim D, Pinilla-Ibarz J. Initial findings from the PACE trial: a pivotal phase 2 study of Ponatinib in patients with CML and Ph+ ALL resistant or intolerant to dasatinib or nilotinib, or with the T315I mutation. Blood. 2011;118(21):52 Abstract 109. [Google Scholar]