

Abstract
Multiple myeloma can be categorized into hyperdiploid or non-hyperdiploid myeloma based on the number of chromosomes found in the tumor clone. Among the non-hyperdiploid myelomas, the hypodiploid subtype has the most aggressive clinical phenotype, but the genetic differences between groups are not completely defined. In order to understand the genetic background of hypodiploid multiple myeloma better, we compared the genomic (array-based comparative genomic hybridization) and transcriptomic (gene expression profiling) background of 49 patients with hypodiploid myeloma with 50 other non-hyperdiploid and 125 hyperdiploid myeloma patients. There were significant chromosomal and gene expression differences between hyperdiploid patients and non-hyperdiploid and hypodiploid patients. Non-hyperdiploid and hypodiploid patients shared most of the chromosomal abnormalities; nevertheless a subset of these abnormalities, such as monosomies 13, 14 and 22, was markedly increased in hypodiploid patients. Furthermore, deletions of 1p, 12p, 16q and 17p, all associated with poor outcome or progression in multiple myeloma, were significantly enriched in hypodiploid patients. Molecular risk-stratification indices reinforce the worse prognosis associated with hypodiploid multiple myeloma compared with non-hyperdiploid multiple myeloma. Gene expression profiling clustered hypodiploid and non-hyperdiploid subgroups closer than hyperdiploid myeloma but also highlighted the up-regulation of CCND2, WHSC1/MMSET and FGFR3 in the hypodiploid subtype. In summary, hypodiploid multiple myeloma is genetically similar to non-hyperdiploid multiple myeloma but characterized by a higher prevalence of genetic alterations associated with poor outcome and disease progression. It is provocative to hypothesize that hypodiploid multiple myeloma is an advanced stage of non-hyperdiploid multiple myeloma.
Introduction
Multiple myeloma (MM) is an incurable plasma cell malignancy which can be distinguished into two major subgroups based on the genetic abnormalities that all patients harbor; hyperdiploid MM (≥47 and <75 chromosomes; H-MM) and non-hyperdiploid MM (NH-MM).1 NH-MM is further divided into three subgroups: hypodiploid (≤44 chromosomes), pseudodiploid (45–46 chromosomes) and near tetraploid (>75 chromosomes), where the last is regarded to originate from doubling of the hypodiploid and pseudodiploid karyotypes.2,3 H-MM has been defined by multiple chromosomal gains, preferentially of the odd chromosomes 3, 5, 7, 9, 11, 15, 19 and 21.2,3 NH-MM has a high frequency of IgH translocations (14q32), which are thought to be early events in the disease. The majority of these IgH translocations have as partners one of three groups of proto-oncogenes: (i) the cyclin D family containing D1 (11q13; 15%), D2 (12p13; <1%), and D3 (6p21; 2%); (ii) MMSET/FGFR3 (4p16; 15%); and (iii) the MAF group containing MAF (16q23; 5%), MAFB (20q12; 2%) and MAFA (8q24; <1%).4
Methods for determining DNA content and ultimately ploidy in MM include conventional cytogenetics, flow cytometry, fluorescent in situ hybridization (FISH), array-based comparative genomic hybridization (aCGH) and recently, massively parallel whole genome sequencing. Studies employing high-resolution platforms for the detection of genome-wide copy-number abnormalities in MM have given more insight into the complexity of the disease.5–7 The use of these platforms has clarified the fact that secondary chromosome aberrations are universal in MM but what role they have on tumor progression is still under debate. On the other hand, gene expression profiling (GEP) has been very useful in classifying MM into molecular subgroups mainly based on cyclin D expression, 14q32 translocations and the deregulation of genes on chromosome 1 heavily involved in proliferation.8–10
Overall, NH-MM is associated with worse survival than H-MM.2,3,11,12 The aim of this study was to dissect the hypodiploid MM subgroup in order to identify the genetic relationships with the remaining NH-MM. We used aCGH to search for copy-number abnormalities and GEP to analyze risk-stratification gene signatures as well as differentially expressed genes.
Methods
Patients and sample preparation
Two hundred twenty-four patients with plasma cell neoplasms were analyzed, including 211 with MM, ten with smoldering MM and three with plasma cell leukemia. Of the 224 patients analyzed, 114 were untreated cases and the remaining 110 were previously treated patients. Data were obtained from the Multiple Myeloma Research Consortium genomics initiative (data available at http://www.broadinstitute.org/mmgp/home). All patients provided written informed consent to the use of their samples and the study was approved by the Institutional Review Board. Tumor cells were enriched with anti-CD138+ immunomagnetic beads (Robosep-StemCell Technologies) and stored in TRIzol® reagent (Invitrogen), as we described previously.13 Nucleic acids were isolated from TRIzol following the protocol supplied by the manufacturer. A more detailed description of the methods used can be found in the Online Supplementary Methods section.
Calculation of copy-number abnormalities using array-based comparative genomic hybridization
aCGH was performed with a 244A microarray (Agilent Technologies). The digestion, labeling and hybridization procedures were done as previously described by us.14 Microarrays were scanned with an Agilent DNA Microarray scanner and data were extracted with Feature Extraction Software. Extracted data were then read into Nexus Software (BioDiscovery). Copy number abnormalities were defined by using the Rank segmentation algorithm with a minimum of three probes per segment. Regions of gain were set at +0.25 for single copy gains and +1.2 for high copy gains. Deletions were set at −0.25 for single copy loss and −1.2 for biallelic loss. The chromosome number was estimated using ideograms. Deviation from diploid count was estimated by accessing the gain or loss of regions bordering the centromere (Online Supplementary Figure S1A,B). Samples estimated to have ≤44 chromosomes were considered to be hypodiploid, those with 45–46 chromosomes were designated NH-MM and samples with ≥47 chromosomes were defined as H-MM. Segmented regions shared by >20% of hypodiploid samples were considered abnormal and listed as an aggregate while the minimal regions of aberrations within the aggregate are listed as peaks only (Online Supplementary Table S1A,B). The most frequent minimal regions found in hypodiploid cases were compared to those in the NH-MM and H-MM groups.
Gene expression profiling
GEP was performed on the U133A Plus_2.0 array (Affymetrix) following the manufacture’s suggested protocol. We transformed the GEP data using a combination of algorithms as follows. First, data were pre-processed using the robust multiarray analysis algorithm. Independently, detection calls (present/marginal/absent) were obtained using the MAS5 algorithm. Raw intensity values obtained from robust multiarray analysis with the detection calls obtained from MAS5 were then merged. Data were filtered on flags, only including cases with present or marginal calls in at least 5% of cases. After filtering on flags, we focused the search on differentially expressed genes (>2-fold change). The analysis was performed for the comparison between ploidy groups, either including all cases or only the untreated MM cases.
Finally, the main gene signatures associated with outcome, including translocation/cyclin D (TC), UAMS 70-gene, proliferation, centrosome and NF-kB indices, were calculated as previously described.8,9,15–17
Statistical analysis
Contingency tables using the χ2 statistic were used for comparisons between groups. Yates’ correction was applied in cases in which at least one group had an expected count smaller than five.
Results
Ploidy and aberrant chromosomal regions
Overall, 224 patients with plasma cell neoplasms were analyzed, including 211 with MM, 10 with smoldering MM and three with plasma cell leukemia. The clinical, demographic and genetic characteristics of the entire cohort are shown in Online Supplementary Table S2.
All abnormalities generated from the segmentation process can be viewed as a genome frequency plot of the entire cohort (Figure 1). Samples were classified into ploidy subgroups as follows: 49 cases (22%) were hypodiploid with a median count of 44 (37–44) chromosomes, 50 (22%) were NH-MM but excluding hypodiploid cases, with a median count of 46 (45–46) chromosomes, and 125 (56%) were H-MM with a median chromosome count of 52 (range, 47–55) (Online Supplementary Figure S2).
Figure 1.
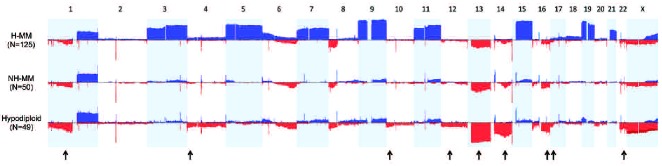
Frequency plots of the copy-number abnormalities per ploidy category. Chromosomes 1 to X are represented from left to right. Gains are represented by upward bars while losses are represented by downward bars. The amplitude in each abnormal region represents the frequency (%) of each copy-number abnormality. The arrows indicate regions that are significantly more affected in hypodiploid MM.
Visual estimation of whole chromosome loss or gain and software generated minimal regions of aberration shared by ≥20% of hypodiploid MM cases are summarized in Table 1 and compared with those of NH-MM and H-MM. Additional information on the recurrent aberrations found in hypodiploid MM, including chromosomal position and genes involved, is shown in Online Supplementary Tables S1 and S3. As previously characterized, multiple copy-number changes, including multiple trisomies in odd chromosomes, differentiate H-MM from the remaining groups. On the other hand, hypodiploid MM and NH-MM share most of the abnormalities (mainly deletions) but there is a clear increase in the prevalence of a subgroup of them in hypodiploid MM (Figure 1). Thus, monosomy 13 was the most frequent abnormality seen in hypodiploid MM, being found in 84% cases compared to 30% in NH-MM (P<0.001). Monosomy 14 was seen in 41% of hypodiploid samples while no NH-MM displayed complete loss (P<0.001). Monosomy 22 was seen in 24% of the hypodiploid samples compared to 4% of cases of NH-MM (P=0.01).
Table 1.
Copy number abnormalities of whole chromosomes and the most frequent minimal region cytobands affected in the hypodiploid group were compared between NH-MM and H-MM groups. A 20% cutoff was used to identify those considered abnormal in hypodiploid cases. “NS” (not significant).
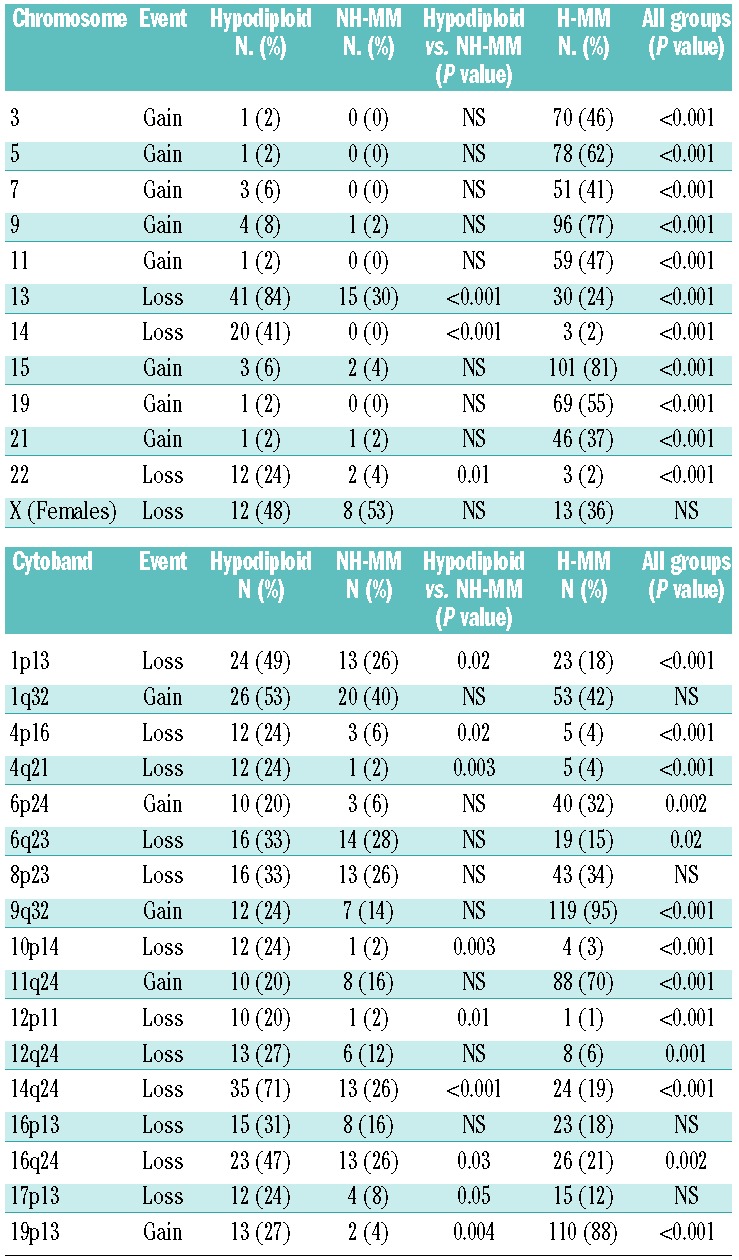
Eight interstitial deletions were significantly enriched in hypodiploid MM compared with NH-MM. The most remarkable regions included deletions of 12p11 (in 20% of hypodiploid MM versus 2% of NH-MM; P=0.01), 16q24 (47% versus 26%; P=0.03) and 17p13 (24% versus 8%; P=0.05), all of which have been previously associated with an adverse prognosis (Table 1 and Figure 1). The remaining five were deletions of 1p13 (49% versus 26%; P=0.02), 4p16 (24% versus 6%; P=0.02), 4q21 (24% versus 2%; P=0.003), 10p14 (24% versus 2%; P=0.003) and 14q24 (71% versus 26%; P<0.001).
Gene expression profiling analysis
GEP data were available for all 224 samples. Differentially expressed genes were analyzed in pairwise comparisons (H-MM versus hypodiploid MM, H-MM versus NH-MM, and NH-MM versus hypodiploid MM). The top 15 genes with the greatest fold change in expression are listed in Table 2 and the remaining can be found in Online Supplementary Table S4A. Overall, 76 genes were differentially expressed (>2-fold change) between H-MM and hypodiploid MM (37 with increased and 39 with decreased expression). Of the 76 differentially expressed genes between H-MM and hypodiploid MM, 64% were also identified in the comparison between H-MM and NH-MM. On the other hand, only 25 genes showed a >2-fold change in expression in the comparison of NH-MM and hypodiploid MM.
Table 2.
The top differentially expressed genes between H-MM vs. hypodiploid MM, H-MM vs. NH-MM and NH-MM vs. hypodiploid MM are listed. The remaining differentially expressed genes (>2-fold change) are listed in Online Supplementary Table S4.
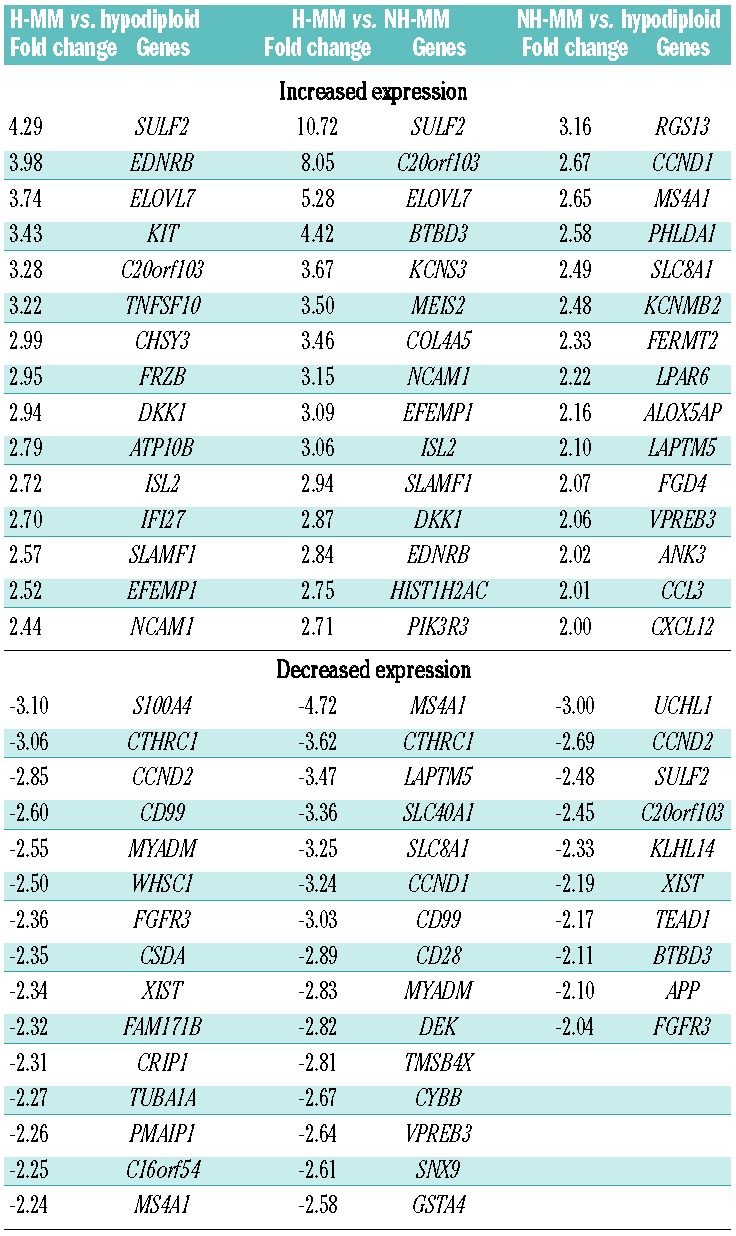
Relevant genes that were found to be differentially expressed in hypodiploid MM compared with NH-MM included increased expression of CCND2 and FGFR3, and reduced expression of CCND1 in the hypodiploid group (Table 2). In order to discard the effects of therapy, we re-ran the analysis only in untreated patients (Online Supplementary Table S4B). By comparing 25 untreated NH-MM with 26 untreated hypodiploid MM, CCND2 and CCND1 remained the top differentially expressed genes between the hypodiploid MM and NH-MM groups. Other relevant genes found in untreated cases were higher expression of IL6R, BIRC3 and CDK6, and lower expression of CD28 in the hypodiploid group.
Gene indices and signatures
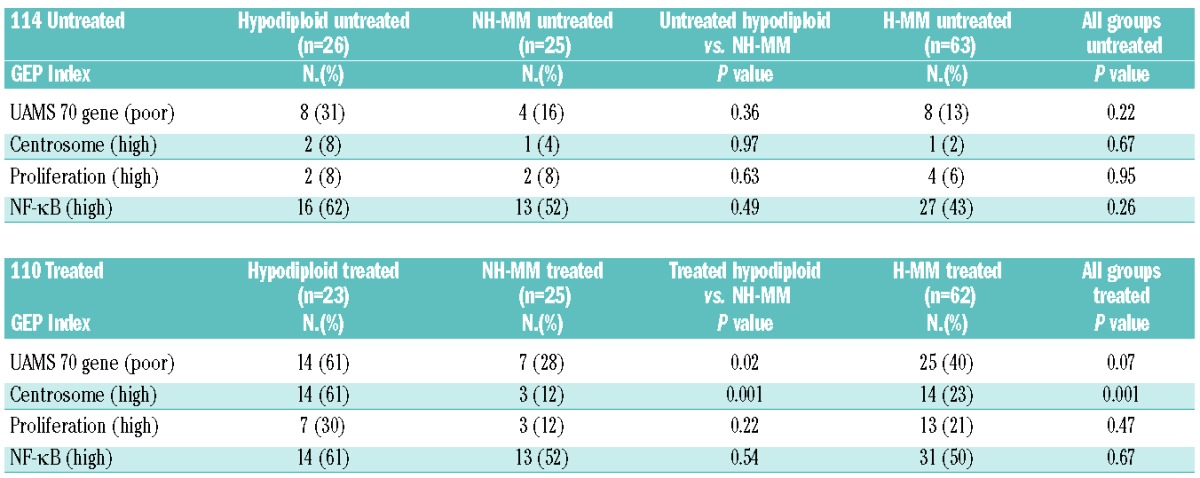
The main gene indices and signatures associated with outcome were calculated for all ploidy subgroups broken down into 114 untreated and 110 previously treated MM (Table 3). Overall, gene indices and signatures confirmed that hypodiploid MM is the higher risk group compared with the remaining NH-MM. In untreated cases, hypodiploid MM showed a higher percentage of cases in the UAMS 70-gene index, although the difference was not statistically significant. The difference did, however, become statistically significant when previously treated cases were considered (61% hypodiploid MM, 28% NH-MM and 40% H-MM; P=0.02). Furthermore, there was a significant increase in cases with high centrosome index in the previously treated hypodiploid MM (61%), compared with NH-MM (12%) and H-MM (23%; P=0.001).
Table 3.
Comparisons of untreated and treated ploidy groups considered high-risk based on four published gene signatures.8,9,16,35
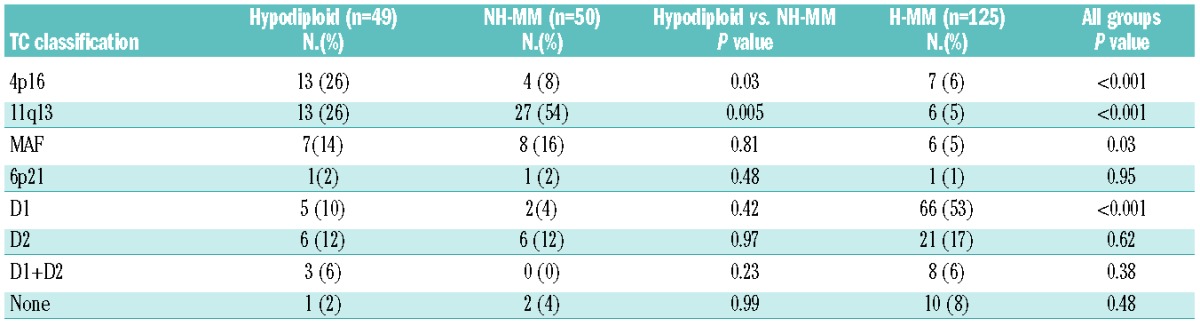
Considering the translocation/cyclin D (TC) classification among the three ploidy groups (Table 4), 4p16 was enriched in hypodiploid MM (26%) in comparison to both NH-MM (8%) and H-MM (6%; P<0.001), 11q13 was enriched in NH-MM (54%) in comparison to hypodiploid MM (26%) and H-MM (5%; P<0.001), MAF showed no statistically significant difference between hypodiploid (14%) and NH-MM (16%) but together showed a slight significant difference in comparison to H-MM (5%; P=0.03), while H-MM was enriched for D1 group (53%) in comparison to both hypodiploid MM (10%) and NH-MM (4%, P<0.001).
Table 4.
Ploidy categories broken down into the eight translocation/cyclin D groups based on the published TC classification.8
When considering the UAMS classification among the three ploidy groups, the MF subgroup was not statistically significantly different between hypodiploid MM (12%) and NH-MM (18%) but together showed a significant enrichment in comparison to H-MM (2%; P=0.002) (Online Supplementary Table S5). The prevalence of the CD1 group was similar in hypodiploid MM (24%) and NH-MM (22%) but together showed significant enrichment in comparison to H-MM (2%; P<0.001). Finally, CD2 was enriched in NH-MM (34%; P<0.001) and HY in H-MM (20%; P<0.001).
Comparing the TC classification to the UAMS classification, 4p16 and MAF groups showed 93% and 88% concordance with the MS and MF groups, respectively. The 11q13 group corresponded with CD1 and CD2 together in 92% of cases and D1 corresponded with HY in 92%. Complete concordance data between the TC and UAMS classifications are shown in Online Supplementary Table S6.
Discussion
There is an accepted dichotomy in MM that separates MM patients into two main ploidy groups, those with H-MM and those with NH-MM. The NH-MM group is further subdivided into three subgroups: hypodiploid, pseudodiploid and near-tetraploid. In this study we analyzed the hypodiploid MM subgroup in order to better characterize the genetic basis and understand the similarities with and differences from the remaining NH-MM cases.
Detection of ploidy has been historically challenging in MM. Conventional cytogenetics is hindered by the ability of the plasma cell to divide, resulting in an abnormal rate of only 20–30%18–20 and no assurance that a metaphase even originated from the plasma cell clone. FISH on the other hand does not require dividing cells but it is imperative that only the clonal plasma cells be analyzed. Furthermore, FISH is not a discovery tool; it relies on other technologies to steer what to target and is laborious and informative for only a small fraction of the genome at one time. Efforts have been made, with some success, to develop a FISH panel for identifying H-MM;21,22 however, there is no probe combination capable of covering all karyotypic combinations. Using flow cytometry to measure DNA content has been fraught with technical issues and a tendency to underestimate hypodiploid MM.23,24 Like other technologies, aCGH has its own limitations, one of the most important being that only relative and not absolute copy numbers are determined. Since aCGH assay is based on the comparative hybridization of equal amounts of tumor and normal DNA, a tumor with a pure tetraploid (4N) karyotype will have half of the cells analyzed compared with a diploid (2N) control so the number of copies will be compensated and no DNA imbalances will be identified. This likely explains our inability to differentiate near triploid and near tetraploid samples along with the inability of aCGH to distinguish balanced translocations. Using aCGH, however, has proven to be a valuable comprehensive discovery tool not only in genomic characterization but also in sequential analysis focused on clonal evolution and disease progression.25,26 Although the sensitivity of aCGH is limited to abnormalities found in >30% of cells, a combination of aCGH for the initial comprehensive identification of abnormalities followed by the longitudinal screening of such abnormalities by FISH can be viewed as an attractive alternative for establishing clonal variation for a patient.
With the use of aCGH and GEP to characterize hypodiploid MM, we were able to show genetic similarities between NH-MM and hypodiploid MM, yet highlight the markedly higher prevalence of multiple abnormalities, such as monosomies 13, 14, and 22. Furthermore, several abnormalities associated with adverse prognosis such as deletions of 12p, 16q and 17p5,27–29 were also significantly more prevalent in hypodiploid MM than in the remaining NH-MM.
We also used GEP and risk-stratification signatures to differentiate hypodiploid MM better. Risk stratification signatures reinforce the outcome differences between hypodiploid MM and the remaining NH-MM, with aggressive disease markers being more evident in treated hypodiploid patients. In this study we found that CCND2 was one of the top genes with increased expression and CCND1 with decreased expression in hypodiploid MM compared to NH-MM. These differences were found independently of whether the entire cohort or only the untreated MM cases were analyzed. Chromosomal translocations directly leading to the dysregulation of CCND2 are rare in MM, but downstream (trans) expression is common with nearly all (67/68, 98.5%) patients with up-regulated CCND2 also having translocations of c-MAF, MAFB and FGFR3/MMSET.30 Hurt et al. demonstrated transactivation of CCND2 by c-MAF.31CCND2 has been found to be the preferred cyclin D-type gene to inactivate Rb1 when paired with either cdk4 or cdk6 and associated with both proliferation and disease progression.32 Furthermore, the proliferation-inducing ligand (APRIL) stimulates cell cycle progression in CCND2-positive MM cells but has minimal effect in CCND1-positive cells.33 This may suggest a dichotomy with regards to cyclin D activation between hypodiploid MM (cyclin D2) and NH-MM (cyclin D1), giving a reasonable explanation why a higher frequency of t(11;14) is observed in NH-MM and why, overall, these patients tend to do better clinically.29
In summary, we were able to better differentiate and describe the aggressive subtype of hypodiploid MM using a combination of aCGH and GEP. This approach could be used in most cases of MM, considering that only 0.5 μg of DNA are needed for aCGH and 50 ng of RNA for GEP. Hypodiploid patients account for one-fifth of cases of MM, are genetically similar to patients with NH-MM but are characterized by an enrichment of abnormalities associated with poor outcome and progression; we, therefore, hypothesize that hypodiploid MM is a more advanced clonal state of NH-MM (Figure 2). As Darwinian principles of evolution are applied to understanding genome evolution we propose that shedding of redundant and dispensable genomic regions (such as duplicate chromosomes) is ultimately a hallmark of clonal evolution based on natural selection. This is a fundamental observation now that it is clearly recognized that all cancers have a subclonal nature and MM is not the exception. Clonal evolution and disease progression are thought to proceed with the malignant clone acquiring additional genetic events selected based on the clonal advantage they give.34 This hypothesis is provocative and further studies are needed to confirm it, with the ideal approach being longitudinal analyses focusing on the dissection of the karyotypic evolution of this subgroup over time.
Figure 2.
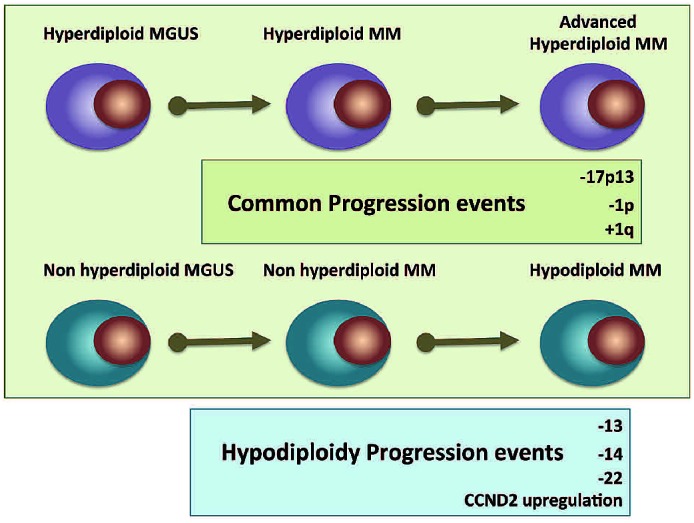
Illustration of the hypothesis suggesting that hypodiploid MM is a late stage of progression in a subgroup of NH-MM. The cartoon shows progression abnormalities shared between ploidy subtypes as well as abnormalities that might be hallmarks of the hypodiploid subtype.
Acknowledgments
The authors would like to thank the The Henry Predolin Foundation and the Multiple Myeloma Research Foundation for their support. EB is a recipient of a Marriott Specialized Workforce Development Award in Individualized Medicine, The Henry Predolin Foundation Career Development Award and the George Haub Family Career Development Award Fund in Cancer Research. AB is supported by the Multiple Myeloma Research Consortium. RF is a Clinical Investigator of the Damon Runyon Cancer Research Fund.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work is supported by grants SPORE CA90297052, P01 CA62242, R01 CA83724, ECOG CA 21115T, Predolin Foundation, Mayo Clinic Cancer Center and the Mayo Foundation.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Fonseca R, Barlogie B, Bataille R, Bastard C, Bergsagel PL, Chesi M, et al. Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer Res. 2004;64(4):1546–58 [DOI] [PubMed] [Google Scholar]
- 2.Debes-Marun CS, Dewald GW, Bryant S, Picken E, Santana-Davila R, Gonzalez-Paz N, et al. Chromosome abnormalities clustering and its implications for pathogenesis and prognosis in myeloma. Leukemia. 2003;17(2):427–36 [DOI] [PubMed] [Google Scholar]
- 3.Smadja NV, Bastard C, Brigaudeau C, Leroux D, Fruchart C. Hypodiploidy is a major prognostic factor in multiple myeloma. Blood. 2001;98(7):2229–38 [DOI] [PubMed] [Google Scholar]
- 4.Kuehl WM, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nat Rev Cancer. 2002;2(3):175–87 [DOI] [PubMed] [Google Scholar]
- 5.Avet-Loiseau H, Li C, Magrangeas F, Gouraud W, Charbonnel C, Harousseau JL, et al. Prognostic significance of copy-number alterations in multiple myeloma. J Clin Oncol. 2009;27(27):4585–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrasco DR, Tonon G, Huang Y, Zhang Y, Sinha R, Feng B, et al. High-resolution genomic profiles define distinct clinicopathogenetic subgroups of multiple myeloma patients. Cancer Cell. 2006;9(4):313–25 [DOI] [PubMed] [Google Scholar]
- 7.Walker BA, Leone PE, Chiecchio L, Dickens NJ, Jenner MW, Boyd KD, et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood. 2010;116(15): e56–65 [DOI] [PubMed] [Google Scholar]
- 8.Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J., Jr Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood. 2005;106(1):296–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaughnessy JD, Jr, Zhan F, Burington BE, Huang Y, Colla S, Hanamura I, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109(6):2276–84 [DOI] [PubMed] [Google Scholar]
- 10.Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, et al. The molecular classification of multiple myeloma. Blood. 2006;108(6):2020–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calasanz MJ, Cigudosa JC, Odero MD, Garcia-Foncillas J, Marin J, Ardanaz MT, et al. Hypodiploidy and 22q11 rearrangements at diagnosis are associated with poor prognosis in patients with multiple myeloma. Br J Haematol. 1997;98(2):418–25 [DOI] [PubMed] [Google Scholar]
- 12.Fassas AB, Spencer T, Sawyer J, Zangari M, Lee CK, Anaissie E, et al. Both hypodiploidy and deletion of chromosome 13 independently confer poor prognosis in multiple myeloma. Br J Haematol. 2002; 118(4):1041–7 [DOI] [PubMed] [Google Scholar]
- 13.Ahmann GJ, Chng WJ, Henderson KJ, Price-Troska TL, DeGoey RW, Timm MM, et al. Effect of tissue shipping on plasma cell isolation, viability, and RNA integrity in the context of a centralized good laboratory practice-certified tissue banking facility. Cancer Epidemiol Biomarkers Prev. 2008;17(3):666–73 [DOI] [PubMed] [Google Scholar]
- 14.Braggio E, Keats JJ, Leleu X, Van Wier S, Jimenez-Zepeda VH, Valdez R, et al. Identification of copy number abnormalities and inactivating mutations in two negative regulators of nuclear factor-kappaB signaling pathways in Waldenstrom’s macroglobulinemia. Cancer Res. 2009;69(8): 3579–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12(2):115–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12(2):131–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chng WJ, Ahmann GJ, Henderson K, Santana-Davila R, Greipp PR, Gertz MA, et al. Clinical implication of centrosome amplification in plasma cell neoplasm. Blood. 2006;107(9):3669–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calasanz MJ, Cigudosa JC, Odero MD, Ferreira C, Ardanaz MT, Fraile A, et al. Cytogenetic analysis of 280 patients with multiple myeloma and related disorders: primary breakpoints and clinical correlations. Genes Chromosomes Cancer. 1997;18(2):84–93 [PubMed] [Google Scholar]
- 19.Dewald GW, Kyle RA, Hicks GA, Greipp PR. The clinical significance of cytogenetic studies in 100 patients with multiple myeloma, plasma cell leukemia, or amyloidosis. Blood. 1985;66(2):380–90 [PubMed] [Google Scholar]
- 20.Sawyer JR, Waldron JA, Jagannath S, Barlogie B. Cytogenetic findings in 200 patients with multiple myeloma. Cancer Genet Cytogenet. 1995;82(1):41–9 [DOI] [PubMed] [Google Scholar]
- 21.Wuilleme S, Robillard N, Lode L, Magrangeas F, Beris H, Harousseau JL, et al. Ploidy, as detected by fluorescence in situ hybridization, defines different subgroups in multiple myeloma. Leukemia. 2005;19 (2):275–8 [DOI] [PubMed] [Google Scholar]
- 22.Chng WJ, Van Wier SA, Ahmann GJ, Winkler JM, Jalal SM, Bergsagel PL, et al. A validated FISH trisomy index demonstrates the hyperdiploid and nonhyperdiploid dichotomy in MGUS. Blood. 2005;106(6): 2156–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greipp PR, Trendle MC, Leong T, Oken MM, Kay NE, Van Ness B, et al. Is flow cytometric DNA content hypodiploidy prognostic in multiple myeloma? Leuk Lymphoma. 1999;35(1–2):83–9 [DOI] [PubMed] [Google Scholar]
- 24.Duque RE, Andreeff M, Braylan RC, Diamond LW, Peiper SC. Consensus review of the clinical utility of DNA flow cytometry in neoplastic hematopathology. Cytometry. 1993;14(5):492–6 [DOI] [PubMed] [Google Scholar]
- 25.Braggio E, Kay NE, VanWier S, Tschumper RC, Smoley S, Eckel-Passow JE, et al. Longitudinal genome-wide analysis of patients with chronic lymphocytic leukemia reveals complex evolution of clonal architecture at disease progression and at the time of relapse. Leukemia. 2012;26(7):1698–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E, et al. Clonal competition with alternating dominance in multiple myeloma. Blood. 2012;120(5):1067–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drach J, Ackermann J, Fritz E, Kromer E, Schuster R, Gisslinger H, et al. Presence of a p53 gene deletion in patients with multiple myeloma predicts for short survival after conventional-dose chemotherapy. Blood. 1998;92(3):802–9 [PubMed] [Google Scholar]
- 28.Fonseca R, Blood E, Rue M, Harrington D, Oken MM, Kyle RA, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003;101(11):4569–75 [DOI] [PubMed] [Google Scholar]
- 29.Gertz MA, Lacy MQ, Dispenzieri A, Greipp PR, Litzow MR, Henderson KJ, et al. Clinical implications of t(11;14)(q13;q32), t(4;14)(p16.3;q32), and -17p13 in myeloma patients treated with high-dose therapy. Blood. 2005;106(8):2837–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanamura I, Huang Y, Zhan F, Barlogie B, Shaughnessy J. Prognostic value of cyclin D2 mRNA expression in newly diagnosed multiple myeloma treated with high-dose chemotherapy and tandem autologous stem cell transplantations. Leukemia. 2006;20(7):1288–90 [DOI] [PubMed] [Google Scholar]
- 31.Hurt EM, Wiestner A, Rosenwald A, Shaffer AL, Campo E, Grogan T, et al. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell. 2004;5(2):191–9 [DOI] [PubMed] [Google Scholar]
- 32.Ely S, Di Liberto M, Niesvizky R, Baughn LB, Cho HJ, Hatada EN, et al. Mutually exclusive cyclin-dependent kinase 4/cyclin D1 and cyclin-dependent kinase 6/cyclin D2 pairing inactivates retinoblastoma protein and promotes cell cycle dysregulation in multiple myeloma. Cancer Res. 2005;65(24):11345–53 [DOI] [PubMed] [Google Scholar]
- 33.Quinn J, Glassford J, Percy L, Munson P, Marafioti T, Rodriguez-Justo M, et al. APRIL promotes cell-cycle progression in primary multiple myeloma cells: influence of D-type cyclin group and translocation status. Blood. 2011;117(3):890–901 [DOI] [PubMed] [Google Scholar]
- 34.Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12(5):335–48 [DOI] [PubMed] [Google Scholar]
- 35.Chng WJ, Braggio E, Mulligan G, Bryant B, Remstein E, Valdez R, et al. The centrosome index is a powerful prognostic marker in myeloma and identifies a cohort of patients that might benefit from aurora kinase inhibition. Blood. 2008;111(3):1603–9 [DOI] [PubMed] [Google Scholar]