

Abstract
Heterozygosity for dominant-negative STAT1 mutations underlies autosomal dominant Mendelian susceptibility to mycobacterial diseases. Mutations conferring Mendelian susceptibility to mycobacterial diseases have been identified in the regions of the STAT1 gene encoding the tail segment, DNA-binding domain and SH2 domain. We describe here a new heterozygous mutation, Y701C, in a Japanese two-generation multiplex kindred with autosomal dominant Mendelian susceptibility to mycobacterial diseases. This mutation affects precisely the canonical STAT1 tyrosine phosphorylation site. The Y701C STAT1 protein is produced normally, but its phosphorylation is abolished, resulting in a loss-of-function for STAT1-dependent cellular responses to interferon-γ or interferon-α. In the patients’ cells, the allele is dominant-negative for γ-activated factor-mediated responses to interferon-γ, but not for interferon-stimulated gene factor-3-mediated responses to interferon-α/β, accounting for the clinical phenotype of Mendelian susceptibility to mycobacterial diseases without severe viral diseases. Interestingly, both patients displayed multifocal osteomyelitis, which is often seen in patients with Mendelian susceptibility to mycobacterial diseases with autosomal dominant partial IFN-γR1 deficiency. Multifocal osteomyelitis should thus prompt investigations of both STAT1 and IFN-γR1. This experiment of nature also confirms the essential role of tyrosine 701 in human STAT1 activity in natura.
Introduction
Mendelian susceptibility to mycobacterial diseases (MSMD) (OMIM 209950) is a rare congenital disorder characterized by susceptibility to clinical diseases caused by weakly virulent mycobacteria, such as Mycobacterium bovis Bacille Calmette-Guérin (BCG) and non-tuberculous mycobacteria.1,2 Affected individuals are also susceptible to M. tuberculosis, a more virulent mycobacterial species.3 Nine MSMD-causing genes (IFNGR1, IFNGR2, IL12B, IL12RB1, ISG15, STAT1, IRF8, CYBB and NEMO) defining 17 different genetic etiologies have been identified to date.4–11 Mutations of IL12B, IL12RB1 and NEMO impair the production of interferon (IFN)-γ, whereas mutations of IFNGR1, IFNGR2 and STAT1 impair cellular responses to IFN-γ. Moreover, autosomal recessive (AR) ISG15 deficiency has recently been identified as a genetic cause of MSMD.11 A lack of ISG15 secretion by leukocytes results in impaired IFN-γ production by NK and T lymphocytes, accounting for mycobacterial disease. Thus, single-gene variants disrupting IL-12- or ISG15-dependent, IFN-γ-mediated immunity result in an inherited predisposition to mycobacterial infections.12,13 However, no genetic etiology has yet been established for about half the patients with MSMD.
The first identification of MSMD-causing mutations of STAT1 in 2001 was surprising, because STAT1 is involved in cellular responses mediated by cytokines other than IFN-γ, including IFN-α/β in particular. IFN-γ stimulation results in the phosphorylation of STAT1 on tyrosine 701, inducing its homodimerization to form gamma-activated factor (GAF). GAF binds the gamma-activated sequence (GAS) to induce the transcription of target genes involved in antimycobacterial immunity. On the other hand, IFN-α/β stimulation induces the phosphorylation of both STAT1 and STAT2, resulting in the formation of the heterotrimeric IFN-stimulated gene factor-3 (ISGF3) complex with IRF9. ISGF3 recognizes IFN-stimulated response element (ISRE) motifs in target genes and their expression confers anti-viral immunity. Indeed, heterozygosity for STAT1 dominant-negative alleles is responsible for AD MSMD.14–17 Six mutations, E320Q, Q463H, K637E, M654K, K673R and L706S in STAT1, have been reported (Figure 1A).14–17 The L706S mutation affects the tail segment domain of STAT1, abolishing phosphorylation at Y701.14 The E320Q and Q463H mutations affect the DNA-binding domain.15 They have no effect on STAT1 phosphorylation, but modify the DNA-binding capacity of GAF, impairing STAT1-dependent immunity. The other three mutations affect the SH2 domain. The M654K and K673R mutations impair the tyrosine phosphorylation of STAT1, whereas the K637E mutation impairs both STAT1 phosphorylation and GAF-DNA binding.16,17 These mutations are loss-of-function or hypomorphic and have been shown to exert a dominant-negative effect on wild-type STAT1 for IFN-γ responses.14,15 We report here the molecular and clinical features of a multiplex kindred with MSMD due to a new STAT1 allele, with a mutation of the tyrosine 701 codon.
Figure 1.

(A) Summary of loss-of-function STAT1 mutations. The N-terminal domain, coiled-coil domain, DNA-binding domain, linker domain, SH2 domain, tail segment domain (TS), and transactivation domain (TA) are shown, together with Y701, the site of tyrosine phosphorylation. The dominant-negative mutants (blue) are indicated below the protein and the recessive forms of hypomorphic (green) and loss-of-function mutations (yellow) are shown above the protein. Y701C is shown in red. (B) The heterozygous Y701C mutation was detected in the patient (II.2) and his mother (I.2). Closed symbols indicate an affected individual and open symbols indicate a healthy family member. (C) CD14-positive monocytes were incubated in the presence of lipopolysaccharide (LPS) (100 ng/mL) and various concentrations of IFN-γ for 48 h and TNF production was then analyzed. CD14-positive monocytes from the patients (P1 and P2) produced abnormally small amounts of TNF. Two independent experiments were performed. (D) Flow cytometry analysis of STAT1 phosphorylation upon IFN-γ stimulation, on peripheral monocytes (CD14+ gate). STAT1 phosphorylation levels were lower in the patients’ cells than in control cells. The black line indicates an absence of stimulation and the red line, stimulation with 104 IU/mL IFN-γ. Three independent experiments were performed.
Methods
Case report
The patient (P1) is a 5-year old Japanese boy born to a non-consanguineous family (Figure 1B). At the age of 2 months he presented with a mild fever and rash. Initial laboratory tests demonstrated leukocytosis (28.9×109/L) with eosinophilia (11.1×109/L) and a mild acute-phase inflammatory response. Treatment with cefotaxime was initiated and the patient’s symptoms improved over the first 2 days, but leukocytosis with eosinophilia persisted for 2 weeks. No bacteria could be cultured from blood, the pharynx or stool samples. P1 was vaccinated with BCG at the age of 4 months. At the age of 3 years, he suffered severe back pain and dysbasia. Laboratory tests revealed mild leukocytosis (13.9×109/L) and high levels of C-reactive protein (3.99 mg/dL) and immunoglobulin (IgG; 2070 mg/dL) in the serum. Magnetic resonance imaging and whole-body bone scintigraphy revealed multifocal osteomyelitis in three vertebrae and the cranial, costal, clavicular, bilateral tibial and pelvic bones. Histological findings for the tibial bone were suggestive of tuberculoid granulomas, but no pathogenic bacteria, including Mycobacterium, were detected in the tissues by polymerase chain reaction or culture. The patient’s leukocytes displayed a normal oxidative burst and normal proliferation in response to stimulation with phytohemagglutinin and concanavalin A. STAT1 sequencing revealed a heterozygous nucleotide substitution (2102 A>G) in exon 23, resulting in the substitution of a cysteine for a tyrosine residue at amino-acid position 701 (Y701C). The patient (P1) started treatment with antimycobacterial drugs, including rifampicin, sulfamethoxazole/trimethoprim and clarithromycin. The clinical symptoms and laboratory parameters responded well to the treatment. These treatments have been maintained ever since, with the patient now being 5 years old. The patient has had no episodes of severe viral infection. He has had mumps, chicken pox and flu, but all these diseases followed a normal clinical course. Normal levels of specific antibodies against these viruses were detected in P1 (Online Supplementary Table S1). He has not yet been vaccinated against measles and rubella.
His mother (P2) was vaccinated with BCG in infancy without complications. She had a history of multifocal osteomyelitis in the frontal bone, right maxilla, multiple vertebral bodies and ribs at 18 years of age. Initial laboratory tests showed a moderate acute-phase inflammatory response. Histological findings in the costal bone were consistent with a granulomatous change. No pathogenic bacteria were detected. P2 was treated with levofloxacin hydrate and loxoprofen for 2 years. These treatments improved, but did not cure the symptoms. After this episode, P2 suffered from recurrent cervical and back pain. At the age of 38, confluent changes in the pressure on cervical and lumbar vertebrae were detected on plain X ray, as a sequel of multifocal osteomyelitis. Since the identification of a heterozygous Y701C STAT1 mutation in the family study, P2 has been treated with rifampicin, sulfamethoxazole/trimethoprim and clarithromycin. This treatment appears to be effective, as the recurrent bone pain disappeared after treatment initiation. P2 presented no signs suggestive of immunodeficiency during childhood. She had no history of severe viral infections and normal levels of the specific antibodies against Epstein-Barr, chicken pox, mumps, rubella and measles viruses (Online Supplementary Table S1).
We obtained blood samples from the patients, relatives, and healthy adult controls, after obtaining informed consent. This study was approved by the Ethics Committee/Internal Review Board of Hiroshima University.
The experimental methods are described in detail in the Online Supplementary Methods section.
Results
Identification of a new STAT1 mutation
High-molecular weight genomic DNA was extracted from peripheral blood. The exons and the flanking introns of genes responsible for MSMD, including STAT1, IFNGR1, IFNGR2, IL12B, IL12RB1 and NEMO, were amplified by PCR and analyzed by Sanger sequencing. We identified a new heterozygous mutation, 2102 A>G (Y701C), in exon 23 of STAT1 in P1 (Figure 1B). The Y701C mutation was not found in the National Center for Biotechnology Information, Ensembl or dbSNP databases, or in our own in-house database of 621 exomes. We also sequenced STAT1 in 1,052 controls from 52 ethnic groups from the Centre d’Etude du Polymorphisme Humain and Human Genome Diversity panels; Y701C was not detected in these controls. This mutation was therefore considered to be a rare variant rather than an irrelevant polymorphism. Familial segregation analysis identified the same mutation in the subject’s mother (P2), whereas the father and older brother were both wild-type and healthy. The mother had a history of multiple osteomyelitis of unknown etiology at 18 years of age, revealing an AD pattern of segregation of the MSMD clinical phenotype with heterozygosity for the STAT1 allele. Figure 1A shows previously identified heterozygous or biallelic STAT1 mutations causing AD or AR genetic susceptibility to mycobacterial diseases.14–22 The Y701C mutation affects the Y701 residue, the site of tyrosine phosphorylation in the STAT1 tail segment domain, a residue crucial for the activation of this molecule.23,24
STAT1 phosphorylation and cytokine production by peripheral blood mononuclear cells in response to interferon-γ stimulation
Interferon-γR1 is expressed ubiquitously, at moderate levels, on the cell surface, whereas very little IFN-γR2 is present and the expression of this receptor is tightly regulated, both spatially and temporally. Thus, IFN-γR2 is thought to be the factor determining responsiveness to IFN-γ.25–27 The CD14-positive monocytes in peripheral blood are known to express relatively high levels of IFN-γR2.28 We, therefore, investigated the cellular response to IFN-γ, focusing on CD14-positive monocytes. We purified CD14-positive monocytes by magnetic sorting and incubated them in the presence of lipopolysaccharide and various concentrations of IFN-γ. We then collected the supernatant, in which we determined tumor necrosis factor (TNF) levels. TNF production was severely impaired in the patients (P1 and P2), regardless of the dose of IFN-γ used for stimulation (Figure 1C). We analyzed STAT1 phosphorylation in response to IFN-γ by flow cytometry. STAT1 phosphorylation levels were lower in CD14-positive monocytes from patients than in control cells (Figure 1D). The CD14-positive monocytes of both patients displayed severe impairment of TNF production in the presence of lipopolysaccharide and IFN-γ, probably due to the impairment of Y701 phosphorylation in response to IFN-γ stimulation.
STAT1 phosphorylation and DNA-binding ability in Epstein-Barr virus-B cells
We assessed STAT1 production and phosphorylation in Epstein-Barr virus (EBV)-B cells from a healthy control (WT/WT), P1 (Y701C/WT), another patient with AD STAT1 deficiency (L706S/WT) and a patient with complete STAT1 deficiency (1760_1761delAG/1760_1761delAG, −/−), by immunoblotting (Figure 2A).14,20,21 STAT1 protein levels were normal in all EBV-B cells except those from a patient with complete STAT1 deficiency. However, STAT1 phosphorylation in response to stimulation with interferons was weak, but not abolished, in EBV-B cells carrying Y701C or L706S mutations. The DNA-binding ability of the mutant STAT1 proteins was analyzed by electrophoretic mobility shift assay on EBV-B cells. EBV-B cells containing the Y701C or L706S STAT1 proteins displayed a partial impairment of GAF DNA-binding in response to stimulation with interferons (Figure 2C). The GAF-DNA binding complexes were shown, by supershift analysis, to consist of STAT1 homodimers, STAT3 homodimers and STAT1/3 heterodimers, following stimulation with IFN-γ and IFN-α, respectively (data not shown). As previously described, GAF was unable to bind DNA in response to IFN-γ in EBV-B cells from a patient with complete STAT1 deficiency.20 However, a complex identified as STAT3 homodimers was visible following incubation with IFN-α. By contrast, ISRE binding to DNA following IFN-α stimulation was found to be similar in cells from the patients and cells from controls, except for complete STAT1 deficiency. STAT1 phosphorylation and GAF DNA binding in EBV-B cells were impaired to a similar extent in P1 and P2 (Online Supplementary Figure S1B,C). Overall, EBV-B cells from the patients displayed impaired, but not abolished, STAT1 phosphorylation in response to stimulation with interferons, resulting in the partial impairment of GAF-DNA binding. However, ISRE-DNA binding levels in response to IFN-α stimulation were unaffected.
Figure 2.
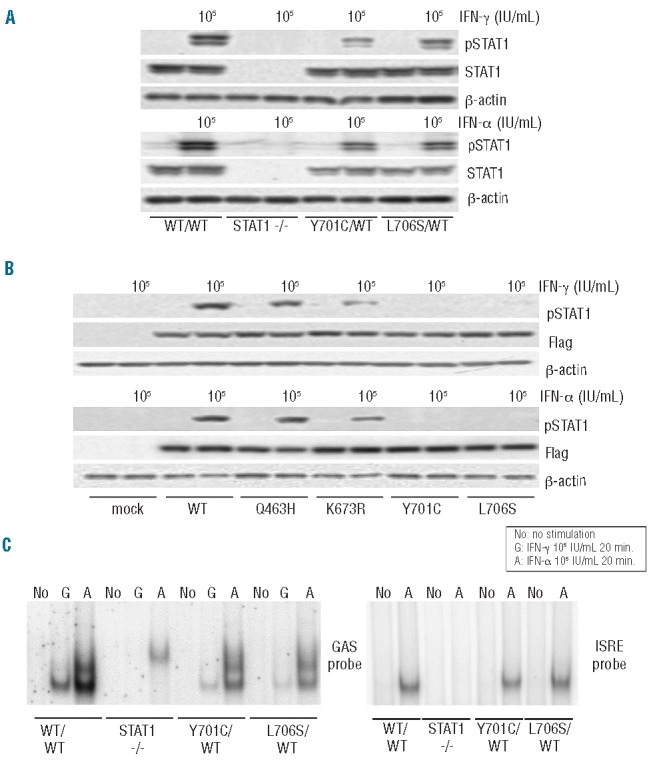
STAT1 phosphorylation and DNA-binding ability in EBV-B cells. STAT1 production and phosphorylation in EBV-B cells (A) and transiently transfected U3C cells (B). The cells were stimulated with 105 IU/mL IFN for 15 min and subjected to immunoblot analysis. (A) EBV-B cells carrying heterozygous Y701C or L706S mutations showed a marked impairment of phosphorylation. The level of pSTAT1 was decreased in Y701C or L706S carrying cells by as much as 50.0% or 57.6% after IFN-γ stimulation and up to 66.3 % or 61.5 % after IFN-α stimulation when compared with healthy controls. (B) The Q463H STAT1 protein was normally phosphorylated, to levels similar to those observed for the WT protein. K673R STAT1 was only weakly phosphorylated. The Y701C STAT1 protein was not phosphorylated. (C) DNA-binding ability in EBV-B cells, as assessed by electrophoretic mobility shift assay with GAS and ISRE probes. EBV-B cells carrying Y701C or L706S mutations displayed a marked impairment of GAF DNA-binding ability. This impairment was particularly strong after IFN-γ stimulation. The ability of ISGF3 to bind DNA upon stimulation with IFN-α was preserved at almost normal levels in the patients’ cells. At least two independent experiments were performed. (G: 105 IU/mL IFN-γ, A: 105 IU/mL IFN-α)
The phosphorylation and nuclear translocation of Y701C STAT1 are impaired
We transiently introduced WT and mutant STAT1 into U3C cells by lipofection, and analyzed, by immunoblotting, the tyrosine 701 phosphorylation of the resulting protein in response to stimulation with IFN-α or IFN-γ (Figure 2B). Both Y701C and L706S STAT1 proteins impaired phosphorylation. As previously reported, the phosphorylation of K673R STAT1 was partially impaired.16 By contrast, the Q463H STAT1 mutant was phosphorylated to almost the same degree as the WT protein. We then analyzed the nuclear translocation of STAT1 in U2OS cells stably expressing flag-tagged WT, Q463H, K673R, L706S and Y701C STAT1 mutant alleles. Unphosphorylated STAT1 was mostly present in the cytoplasm before IFN-γ stimulation (Online Supplementary Figure S2A). After IFN-γ stimulation, STAT1 was found in the nucleus in cells producing the WT and Q463H STAT1 proteins (Online Supplementary Figure S2B). By contrast, STAT1 nuclear translocation was severely impaired in cells producing the Y701C and L706S STAT1 proteins. The K673R mutant STAT1 protein was present in both the nucleus and the cytoplasm, suggesting incomplete nuclear translocation. These results suggest that the Y701C mutation, like L706S, prevents STAT1 phosphorylation and nuclear translocation.
Comparison of the Y701C and L706S mutations
Both the Y701C and L706S mutations affect residues in the tail segment domain of STAT1. We focused on these two mutations and performed further functional characterization. We treated cells carrying these mutations with the tyrosine phosphatase inhibitor pervanadate and then analyzed the phosphorylation of STAT1 upon IFN-γ stimulation.29 STAT1 phosphorylation was restored in the presence of pervanadate in cells carrying the L706S mutation, whereas no such restoration was observed for the Y701C mutant (Figure 3A). Conversely, pervanadate treatment did not rescue GAF-DNA binding for L706S STAT1 (Figure 3A). We investigated the mechanisms underlying these experimental observations by extracting the cytosol and investigating STAT1 phosphorylation by immunoblotting (Figure 3B). The L706S STAT1 protein showed almost normal levels of phosphorylation in the cytosol fraction after pervanadate treatment, whereas phosphorylation was severely impaired in the nuclear fraction. These results suggest that the L706S mutation impairs both phosphorylation and nuclear translocation. This impairment of the nuclear translocation of L706S STAT1 was confirmed by immunostaining (Figure 3C). The L706S mutant protein was, therefore, considered to have at least two molecular defects: severe impairments of both phosphorylation and the nuclear translocation of STAT1. By contrast, Y701C STAT1 phosphorylation was totally abolished and could not be restored by pervanadate treatment.
Figure 3.
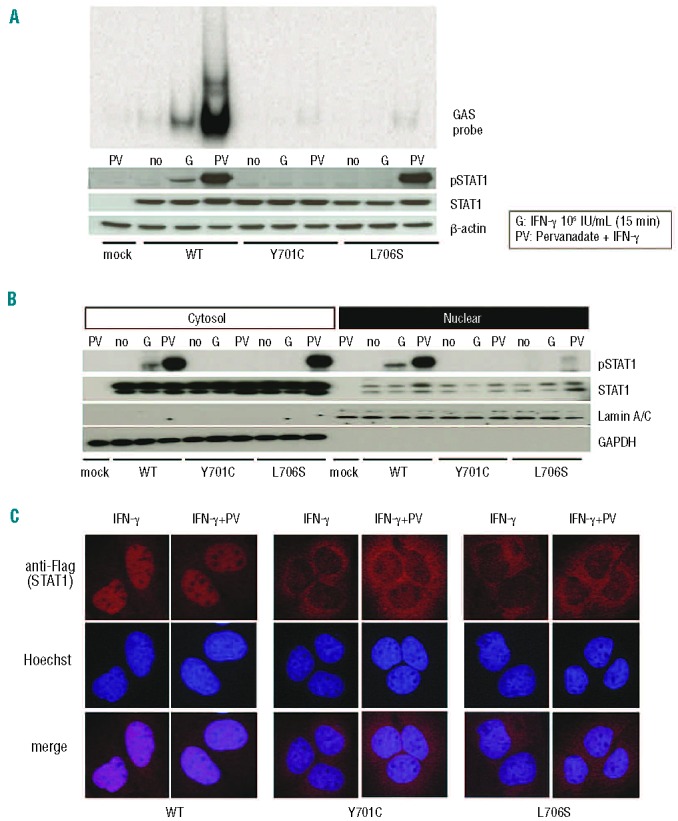
Comparison of the Y701C and L706S mutations. (A) U3C transfectants were stimulated with 105 IU/mL IFN-γ in the presence or absence of pervanadate for 15 min and subjected to immunoblotting and electrophoretic mobility shift assay. The L706S STAT1 protein was phosphorylated to almost normal levels in response to IFN-γ stimulation in the presence of pervanadate, whereas no phosphorylation of the Y701C STAT1 protein was observed. However, L706S STAT1 was still associated with a lack of GAF-DNA binding, even after its phosphorylation had been restored. (B) STAT1 phosphorylation was investigated in the nuclear and cytosolic fractions, by immunoblotting. The phosphorylated L706S STAT1 protein was mostly present in the cytosol fraction. (C) U2OS cells stably expressing Flag-tagged WT, Y701C, or L706S STAT1 were stimulated with IFN-γ for 30 min in the presence or absence of pervanadate and subjected to immunostaining. Nuclear translocation was severely impaired in cells producing either Y701C or L706S STAT1, even after pervanadate treatment. At least two independent experiments were carried out.
Transcriptional activity of the mutant STAT1 proteins
We investigated the impact of the STAT1 Y701C mutation on the transcriptional activities of GAS and ISRE, by carrying out reporter assays with GAS or ISRE reporter plasmids. Production of the Y701C and L706S STAT1 proteins was associated with abolition of the transcriptional activities of both GAS and ISRE (Online Supplementary Figure S3A,C). Co-transfection experiments revealed that these mutant proteins had negative effects on the WT protein in GAS transcriptional activity (Online Supplementary Figure S3B). Furthermore, a dose-dependent negative-dominance effect was clearly observed for the Y701C and L706S mutant proteins. By contrast, ISRE transcriptional activity remained at almost normal levels in cells co-transfected with the L706S plasmid (Online Supplementary Figure S3D). The level of ISRE transcriptional activity was repeatedly found to be lower with Y701C STAT1, but no clear dominant-negative effect was detected. These results are consistent with the results we observed in an electrophoretic mobility shift assay using EBV-B cells. Thus, the Y701C STAT1 allele has a dominant-negative effect, decreasing GAS activation but not ISRE activation.
Downstream gene induction in CD14-positive monocytes and Epstein-Barr virus-B cells
The induction of downstream interferon-stimulated genes (ISG) has been investigated with EBV-B cells from patients and in gene expression experiments using U3C cells.15 We investigated the impact of the Y701C mutation further, by studying the induction of ISG in CD14-positive monocytes from patients, comparing the results obtained with those for EBV-B cells. We first purified CD14-positive monocytes, stimulated them with IFN-γ and analyzed the expression of the downstream ISG encoding CXCL9 and IRF1, by real-time quantitative PCR analysis. Strong induction of CXCL9 was observed at late time points (6 hours) in healthy controls, but this induction was severely impaired in the patients’ cells (Figure 4A). In contrast, IRF1 induction was observed at both early and late time points. The patients’ cells displayed a mild but significant impairment of IRF1 induction at late time points, whereas the difference observed at early time points was not statistically significant. We then investigated the induction of CXCL9, IRF1 and ISG15 in EBV-B cells. The induction of these three ISG was STAT1-dependent, as shown by the abolition of induction in STAT1-null EBV-B cells. EBV-B cells carrying heterozygous Y701C or L706S mutations displayed severely impaired induction of CXCL9 and ISG15 in response to interferons (Figure 4B,C). Unlike CD14-positive monocytes, the peak of IRF1 induction was observed at early time points in EBV-B cells. The impairment of IRF1 induction was mild and mostly observed at early time points in EBV-B cells from the patients. Thus, CD14-positive monocytes and EBV-B cells behaved similarly, but not identically, in terms of ISG induction in response to IFN-γ stimulation. These results suggest that the induction of ISG is impaired in the patients’ cells.
Figure 4.

Analysis of gene expression. The induction of ISG was analyzed by real-time quantitative PCR. (A) CD14-positive monocytes from the patients (P1, P2) and two healthy controls were stimulated with 103 IU/mL IFN-γ (for 2 or 6 h) and the induction of CXCL9 and IRF1 was investigated. The induction of CXCL9 was severely impaired in the patients’ cells, whereas the induction of IRF1 was only mildly impaired. *Differences were statistically significant in monocytes from the patients compared with those from healthy individuals (P<0.05). (B, C) The induction of CXCL9, IRF1 and ISG15 in EBV-B cells from the patients, Y701C/WT and L706S/WT cells, and cells from healthy individuals, in response to IFN-γ (B) or IFN-α (C) stimulation. EBV-B cells carrying Y701C or L706S mutations displayed severe impairment of CXCL9 induction in response to IFN stimulation. The induction of IRF1 and ISG15 was also impaired, but to a lesser extent than that of CXCL9. ISG expression was normalized with respect to GAPDH. The results shown are representative of three independent experiments, except for the CD14+ monocyte experiment (which was performed twice). Differences were statistically significant between cells carrying either the Y701C mutation*(P<0.05) or L706S mutation †(P<0.05) and EBV-B cells from healthy individuals.
Discussion
We report here a novel heterozygous STAT1 mutation, 2102 A>G (Y701C), which results in STAT1 deficiency and AD MSMD. The Y701 residue serves as a site of phosphorylation for STAT1, this phosphorylation playing a key role in STAT1-mediated signal transduction. Indeed, AD STAT1 mutations impairing STAT1 phosphorylation have been shown to underlie the pathogenesis of MSMD.14–17 Furthermore, AD mutations that cause gains of phosphorylation because they impair the nuclear dephosphorylation of STAT1 have been identified in patients with chronic mucocutaneous candidiasis.30–34 The Y701C mutant STAT1 protein displayed a complete abolition of Y701 phosphorylation and downstream events, such as the nuclear translocation and transcriptional activities of GAS and ISRE. The Y701C STAT1 allele is dominant for GAF, but recessive for ISGF3. This observation is highly consistent with previously identified STAT1 mutations in patients with AD STAT1 deficiencies and the molecular mechanisms can be explained by differences in the structure of GAS (homodimer of STAT1) and ISGF3 (heterodimer of STAT1/STAT2/IRF9).14–17 The Y701C mutation is thus responsible for MSMD without viral disease. Two heterozygous STAT1 mutations, Y701C and L706S, affect residues located in the same tail segment domain and result in the impairment of Y701 phosphorylation. However, these two mutants responded differently to stimulation in the presence of pervanadate. This treatment rescued Y701 phosphorylation in L706S STAT1, but not in the Y701C protein. The functional defect seemed to be more severe for the Y701C than for the L706S mutant protein, as shown by GAS reporter assays and real-time quantitative PCR analysis. The Y701C mutation may therefore have a stronger negative impact in vitro than L706S STAT1. However, in contrast to the findings of these in vitro studies, the clinical symptoms of the patient and his mother were similar to those of previously identified patients with AD STAT1 deficiency.
We also investigated the induction of ISG upon IFN-γ stimulation in CD14-positive monocytes from the patients. ISG induction has been intensively investigated in EBV-B cells,15,17 but this is the first study to investigate the induction of ISG in primary cells from patients. Both EBV-B cell lines and primary monocytes from the patients showed severe impairment of CXCL9 induction. Minor differences in induction patterns were observed, but both types of cell showed mild impairment of IRF1 induction. Thus, the impairment of ISG induction was confirmed not only in EBV-B cells, but also in the patients’ monocytes. IL-12p40 induction is totally dependent on Irf1 in mice.35–37 Macrophages from Irf1-null mice display impaired induction of inducible nitric oxide synthase in response to lipopolysaccharide.38,39 Furthermore, Irf1-null mice develop severe symptoms when infected with Mycobacterium bovis. Thus, the impairment of IRF1 induction observed in CD14-positive monocytes may contribute to host susceptibility to mycobacteria. We also observed an impairment in ISG15 induction in the patients’ EBV-B cells. The recent identification of ISG15 deficiency in some human patients with MSMD has provided evidence of an important role for this molecule in host immunity to mycobacteria.11 Heterozygous STAT1 mutations thus have negative effects on the downstream induction of ISG involved in host defense against mycobacteria, and this in vitro cellular phenotype is commonly observed in the patients’ cells. However, the magnitude of this negative impact may differ between cell types and between the ISG induced. The induction of ISG has also been intensively investigated in patients with AR STAT1 deficiency.22 As in the current study, the severity of impairment differed in the patient’s cells depending on the ISG induced. These observations reflect the complexity of STAT1-mediated signaling.40
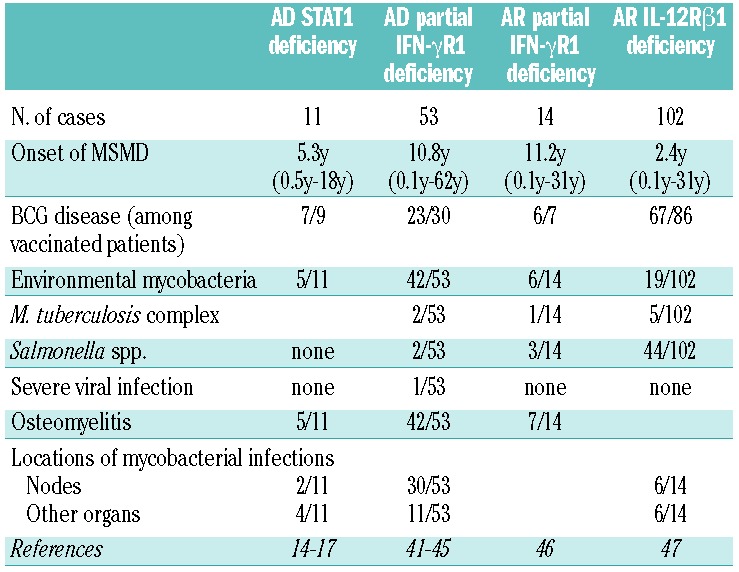
Including the two patients studied here, 11 patients with AD STAT1 deficiency have been identified to date. Five of these 11 patients have developed multifocal osteomyelitis, a typical clinical feature of patients with AD partial IFN-γR1 deficiency.4,41 Our initial patient had multifocal osteomyelitis showing tuberculoid granulomas without the detection of pathogenic mycobacterium by culture and/or PCR amplification. Additionally, his mother had a history of multifocal osteomyelitis. These observations emphasize the importance of multifocal osteomyelitis as one of the representative clinical manifestation even in patients with AD STAT1 deficiency. We have summarized the clinical manifestations of 11 patients with AD STAT1 deficiency,14–17 53 patients with AD partial IFN-γR1 deficiency,41–45 14 patients with AR partial IFN-γR1 deficiency46 and 102 patients with AR IL-12Rβ1 deficiency,47 the most common genetic etiology of MSMD (Table 1). The frequency of multifocal osteomyelitis was high in AD STAT1 deficiency (45.5%), AD partial IFN-γR1 deficiency (79.2%) and AR partial IFN-γR1 deficiency (50.0%), but lower in patients with AR complete IFN-γR1 deficiency (13%).41 Unfortunately, this clinical manifestation has not been investigated in patients with AR IL-12Rβ1 deficiency.47 In many cases, either BCG or environmental mycobacteria have been proven to be present by biopsy of osteomyelitis. By contrast, the frequency of BCG disease, a common sign in patients with MSMD, in patients vaccinated with BCG is similar to that in AD STAT1 deficiency (77.8%), AD partial IFN-γR1 deficiency (76.7%), AR partial IFN-γR1 deficiency (85.7%) and AR IL-12Rβ1 deficiency (77.9%). The clinical signs of AD STAT1 deficiency may, therefore, be considered to be similar to those of partial IFN-γR1 deficiency. There are several possible reasons for this: (i) STAT1 is a non-redundant downstream transcription factor for IFN-γ signaling;4 (ii) STAT1 mutations impair GAF-mediated signaling, but not ISGF3-mediated signaling, and IFN-γ induces GAF, but not ISGF3;48 (iii) IFN-γ signaling is only partially impaired in both disorders.4 The clinical similarities between these two disorders support our hypothesis that the symptoms accompanying AD STAT1 deficiency result from an impairment of cellular responses to IFN-γ. Multifocal osteomyelitis is a characteristic symptom common to three different disorders: AD partial IFN-γR1 deficiency, AR partial IFN-γR1 deficiency and AD STAT1 deficiency. Multifocal osteomyelitis should, therefore, lead to investigations of both STAT1 and IFN-γR1.
Table 1.
Comparison of clinical manifestations.
Acknowledgments
The authors would like to thank Xiao-Fei Kong, MD, PhD, Vanessa Bryant, PhD, Dusan Bogunovic, PhD, Alexandra Kreins, MD, Marcela Moncada-Velez, BSc, Michael Ciancaneli, PhD, and Avinash Abhyankar, MD, PhD, for helpful discussions and critical reading. We also thank the members of the laboratory, Yelena Nemirovskaya and Eric Anderson for secretarial assistance, and Tiffany Nivare for technical assistance. Sequence analysis support was provided by the Analysis Center of Life Science, Natural Science Center for Basic Research and Development, Hiroshima University.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This study was supported, in part, by Grants in Aid for Scientific Research from the Japan Society for the Promotion of Science [22591161 to M.K.], and by Research on Measures for Intractable Diseases funding from the Japanese Ministry of Health, Labor and Welfare [H22-Nanchi-ippan-078 to MK]. This study was also supported, in part, by the Rockefeller University and grants from the National Center for Research Resources, and the National Center for Advancing Sciences (NCATS), National Institutes of Health (NIH) grant number 8UL1TR000043, NIH grant number R01AI089970, and the St. Giles Foundation. SC was supported by the AXA Research Fund and X-FK by the Stony Wold-Herbert Fund.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Casanova JL, Abel L. Genetic dissection of immunity to mycobacteria: the human model. Annu Rev Immunol. 2002;20:581–620 [DOI] [PubMed] [Google Scholar]
- 2.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledge-base of human genes and genetic disorders. Nucleic Acids Res. 2005;33(Database issue):D514–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alcais A, Fieschi C, Abel L, Casanova JL. Tuberculosis in children and adults: two distinct genetic diseases. J Exp Med. 2005;202(12):1617–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Filipe-Santos O, Bustamante J, Chapgier A, Vogt G, de Beaucoudrey L, Feinberg J, et al. Inborn errors of IL-12/23-and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin Immunol. 2006;18(6):347–61 [DOI] [PubMed] [Google Scholar]
- 5.Vogt G, Bustamante J, Chapgier A, Feinberg J, Boisson Dupuis S, Picard C, et al. Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation. J Exp Med. 2008;205(8): 1729–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bustamante J, Arias AA, Vogt G, Picard C, Galicia LB, Prando C, et al. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol. 2011;12(3):213–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med. 2011;365 (2):127–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al-Muhsen S, Casanova JL. The genetic heterogeneity of mendelian susceptibility to mycobacterial diseases. J Allergy Clin Immunol. 2008;122(6):1043–51; quiz 52-3. [DOI] [PubMed] [Google Scholar]
- 9.Boisson-Dupuis S, Kong XF, Okada S, Cypowyj S, Puel A, Abel L, et al. Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr Opin Immunol. 2012;24(4):364–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity. 2012;36(4):515–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, et al. Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency. Science. 2012;337 (6102):1684–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casanova JL, Abel L. Inborn errors of immunity to infection: the rule rather than the exception. J Exp Med. 2005;202(2):197–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alcais A, Quintana-Murci L, Thaler DS, Schurr E, Abel L, Casanova JL. Life-threatening infectious diseases of childhood: single-gene inborn errors of immunity? Ann NY Acad Sci. 2010;1214:18–33 [DOI] [PubMed] [Google Scholar]
- 14.Dupuis S, Dargemont C, Fieschi C, Thomassin N, Rosenzweig S, Harris J, et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science. 2001;293(5528):300–3 [DOI] [PubMed] [Google Scholar]
- 15.Chapgier A, Boisson-Dupuis S, Jouanguy E, Vogt G, Feinberg J, Prochnicka-Chalufour A, et al. Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet. 2006;2(8):e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsumura M, Okada S, Sakai H, Yasunaga S, Ohtsubo M, Murata T, et al. Dominant-negative STAT1 SH2 domain mutations in unrelated patients with Mendelian susceptibility to mycobacterial disease. Hum Mutat. 2012;33(9):1377–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sampaio EP, Bax HI, Hsu AP, Kristosturyan E, Pechacek J, Chandrasekaran P, et al. A novel STAT1 mutation associated with disseminated mycobacterial disease. J Clin Immunol. 2012;32(4):681–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kristensen IA, Veirum JE, Moller BK, Christiansen M. Novel STAT1 alleles in a patient with impaired resistance to mycobacteria. J Clin Immunol. 2011;31(2): 265–71 [DOI] [PubMed] [Google Scholar]
- 19.Vairo D, Tassone L, Tabellini G, Tamassia N, Gasperini S, Bazzoni F, et al. Severe impairment of IFN-gamma and IFN-alpha responses in cells of a patient with a novel STAT1 splicing mutation. Blood. 2011;118 (7):1806–17 [DOI] [PubMed] [Google Scholar]
- 20.Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003;33(3):388–91 [DOI] [PubMed] [Google Scholar]
- 21.Chapgier A, Wynn RF, Jouanguy E, Filipe-Santos O, Zhang S, Feinberg J, et al. Human complete Stat-1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo. J Immunol. 2006;176 (8):5078–83 [DOI] [PubMed] [Google Scholar]
- 22.Kong XF, Ciancanelli M, Al-Hajjar S, Alsina L, Zumwalt T, Bustamante J, et al. A novel form of human STAT1 deficiency impairing early but not late responses to interferons. Blood. 2010;116(26):5895–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shuai K, Ziemiecki A, Wilks AF, Harpur AG, Sadowski HB, Gilman MZ, et al. Polypeptide signalling to the nucleus through tyrosine phosphorylation of Jak and Stat proteins. Nature. 1993;366(6455): 580–3 [DOI] [PubMed] [Google Scholar]
- 24.Chen X, Vinkemeier U, Zhao Y, Jeruzalmi D, Darnell JE, Jr, Kuriyan J. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93(5):827–39 [DOI] [PubMed] [Google Scholar]
- 25.Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol. 1997;15:563–91 [DOI] [PubMed] [Google Scholar]
- 26.van Boxel-Dezaire AH, Stark GR. Cell type-specific signaling in response to interferon-gamma. Curr Top Microbiol Immunol. 2007;316:119–54 [DOI] [PubMed] [Google Scholar]
- 27.Kong XF, Vogt G, Itan Y, Macura-Biegun A, Szaflarska A, Kowalczyk D, et al. Haploinsufficiency at the human IFNGR2 locus contributes to mycobacterial disease. Hum Mol Genet. 2013;22(4):769–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernabei P, Coccia EM, Rigamonti L, Bosticardo M, Forni G, Pestka S, et al. Interferon-gamma receptor 2 expression as the deciding factor in human T, B, and myeloid cell proliferation or death. J Leukoc Biol. 2001;70(6):950–60 [PubMed] [Google Scholar]
- 29.Tourkine N, Schindler C, Larose M, Houdebine LM. Activation of STAT factors by prolactin, interferon-gamma, growth hormones, and a tyrosine phosphatase inhibitor in rabbit primary mammary epithelial cells. J Biol Chem. 1995;270 (36):20952–61 [DOI] [PubMed] [Google Scholar]
- 30.Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. 2011;208(8):1635–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med. 2011;365(1):54–61 [DOI] [PubMed] [Google Scholar]
- 32.Smeekens SP, Plantinga TS, van de Veerdonk FL, Heinhuis B, Hoischen A, Joosten LA, et al. STAT1 hyperphosphorylation and defective IL12R/IL23R signaling underlie defective immunity in autosomal dominant chronic mucocutaneous candidiasis. PLoS One. 2011;6(12):e29248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takezaki S, Yamada M, Kato M, Park MJ, Maruyama K, Yamazaki Y, et al. Chronic mucocutaneous candidiasis caused by a gain-of-function mutation in the STAT1 DNA-binding domain. J Immunol. 2012; 189(3):1521–6 [DOI] [PubMed] [Google Scholar]
- 34.Toth B, Mehes L, Tasko S, Szalai Z, Tulassay Z, Cypowyj S, et al. Herpes in STAT1 gain-of-function mutation [corrected]. Lancet. 2012;379(9835):2500. [DOI] [PubMed] [Google Scholar]
- 35.Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001;19:623–55 [DOI] [PubMed] [Google Scholar]
- 36.Taki S, Sato T, Ogasawara K, Fukuda T, Sato M, Hida S, et al. Multistage regulation of Th1-type immune responses by the transcription factor IRF1. Immunity. 1997;6(6): 673–9 [DOI] [PubMed] [Google Scholar]
- 37.Lohoff M, Ferrick D, Mittrucker HW, Duncan GS, Bischof S, Rollinghoff M, et al. Interferon regulatory factor-1 is required for a T helper 1 immune response in vivo. Immunity. 1997;6(6):681–9 [DOI] [PubMed] [Google Scholar]
- 38.Kamijo R, Harada H, Matsuyama T, Bosland M, Gerecitano J, Shapiro D, et al. Requirement for transcription factor IRF1 in NO synthase induction in macrophages. Science. 1994;263(5153):1612–5 [DOI] [PubMed] [Google Scholar]
- 39.Martin E, Nathan C, Xie QW. Role of interferon regulatory factor 1 in induction of nitric oxide synthase. J Exp Med. 1994;180 (3):977–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gough DJ, Levy DE, Johnstone RW, Clarke CJ. IFNgamma signaling - does it mean JAK-STAT? Cytokine Growth Factor Rev. 2008;19(5–6):383–94 [DOI] [PubMed] [Google Scholar]
- 41.Dorman SE, Picard C, Lammas D, Heyne K, van Dissel JT, Baretto R, et al. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet. 2004;364(9451):2113–21 [DOI] [PubMed] [Google Scholar]
- 42.Hoshina T, Takada H, Sasaki-Mihara Y, Kusuhara K, Ohshima K, Okada S, et al. Clinical and host genetic characteristics of mendelian susceptibility to mycobacterial diseases in Japan. J Clin Immunol. 2011;31 (3):309–14 [DOI] [PubMed] [Google Scholar]
- 43.Lee WI, Huang JL, Lin TY, Hsueh C, Wong AM, Hsieh MY, et al. Chinese patients with defective IL-12/23-interferon-gamma circuit in Taiwan: partial dominant interferon-gamma receptor 1 mutation presenting as cutaneous granuloma and IL-12 receptor beta1 mutation as pneumatocele. J Clin Immunol. 2009;29(2):238–45 [DOI] [PubMed] [Google Scholar]
- 44.Glosli H, Stray-Pedersen A, Brun AC, Holtmon LW, Tonjum T, Chapgier A, et al. Infections due to various atypical mycobacteria in a Norwegian multiplex family with dominant interferon-gamma receptor deficiency. Clin Infect Dis. 2008;46(3):e23–7 [DOI] [PubMed] [Google Scholar]
- 45.Okada S, Ishikawa N, Shirao K, Kawaguchi H, Tsumura M, Ohno Y, et al. The novel IFNGR1 mutation 774del4 produces a truncated form of interferon-gamma receptor 1 and has a dominant-negative effect on interferon-gamma signal transduction. J Med Genet. 2007;44(8):485–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sologuren I, Boisson-Dupuis S, Pestano J, Vincent QB, Fernandez-Perez L, Chapgier A, et al. Partial recessive IFN-{gamma}R1 deficiency: genetic, immunological and clinical features of 14 patients from 11 kindreds. Hum Mol Genet. 2011;20(8):1509–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, et al. Revisiting human IL-12Rbeta1 deficiency: a survey of 141 patients from 30 countries. Medicine (Baltimore). 2010;89(6):381–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ginter T, Bier C, Knauer SK, Sughra K, Hildebrand D, Munz T, et al. Histone deacetylase inhibitors block IFNgamma-induced STAT1 phosphorylation. Cell Signal. 2012;24(7):1453–60 [DOI] [PubMed] [Google Scholar]