

Abstract
Binding of bacteria to solid surfaces is complex with many aspects incompletely understood. We investigate Pseudomonas aeruginosa uptake kinetics onto hydrogel surfaces representative of soft-contact lenses made of nonionic poly(2-hydroxyethylmethacrylate) (p-HEMA), anionic poly(methacrylic acid) (p-MAA), and anionic poly(acrylic acid) (p-AA). Using a parallel-plate flow cell under phase-contrast microscopy, we document a kinetic “burst” at the anionic hydrogel surface: dilute aqueous P. aeruginosa first rapidly accumulates and then rapidly depletes. Upon continuing flow, divalent cations in the suspending solution sorb into the hydrogel network causing the previously surface-accumulated bacteria to desorb. The number of bacteria eventually bound to the surface is low compared to the nonionic p-HEMA hydrogel. We propose that the kinetic burst is due to reversible divalent-cation bridging between the anionic bacteria and the negatively charged hydrogel surface. The number of surface bridging sites diminishes as divalent cations impregnate into and collapse the gel. P. aeruginosa association with the surface then falls. Low eventual binding of P. aeruginosa to the anionic hydrogel is ascribed to increased surface hydrophilicity compared to the counterpart nonionic p-HEMA hydrogel.
Keywords: Pseudomonas aeruginosa, Soft-contact lenses, Nonionic and anionic hydrogels, Aqueous divalent cations
1. Introduction
Binding of the common bacterium, Pseudomonas aeruginosa, to solid substrates is important in bioremediation and biomedical devices [1–7]. Of special interest is the attachment of P. aeruginosa to soft-contact lenses because this bacterium causes serious and difficult-to-treat infection [7]. In general, P. aeruginosa does not infect healthy individuals. The majority of time, infection occurs only for those with compromised immune systems [8]. Even though the human eye is particularly resistant to infection [9], P. aeruginosa can infect healthy contact-lens wearers [10,11]. Recent studies also demonstrate that P. aeruginosa bound to soft-contact lenses can initiate infection [6]. Thus, a promising method for alleviating infection is to prevent binding to soft-contact-lens surfaces.
P. aeruginosa attachment to substrates is complex because the bacterium possesses a number of different binding mechanisms arising from a variety of appendages [12]. For example, many strains of wild-type P. aeruginosa have a propelling flagellum that enhances attachment rates to surfaces. Additionally, pili, another appendage of P. aeruginosa, allow both specific and nonspecific binding to surfaces [13–16] (nonspecific binding includes hydrophobic and double-layer electrostatic interactions [17]). Proteins, lipopolysacharides, and exopolysacharides expressed on the surface of the bacterium further modulate both specific and nonspecific binding to surfaces [18–20].
Soft-contact lenses are transparent hydrogels designed to ride closely and comfortably on the cornea [21]. Traditionally, they are composed of poly(2-hydroxyethyl methacrylate) (p-HEMA) sometimes copolymerized with polyeletrolytes, such as poly(methacrylic acid) (p-MAA) or poly(acrylic acid) (p-AA), to enhance water content and wearability [22–24]. Copolymerization with varying amounts of MAA/AAcreates a cross-linked anionic hydrogel with differing properties including matrix charge, mesh size, elastic modulus, ion and solute uptake, and surface wettability [22,25,26]. Possible roles of p-MAA/p-AA in the binding of P. aeruginosa to p-HEMA hydrogels are unknown. Further, incorporation of MAA/AA into hydrogels provides a direct means to investigate the role of matrix charge in bacterial binding.
We investigate P. aeruginosa interaction with the surfaces of traditional p-HEMA-based soft-contact-lens materials and how these interactions change upon co-polymerization with polycarboxylic acids, MAA and AA, to produce anionic-charged hydrogels. Because the bacterial growth medium contains magnesium sulfate, we also investigate the importance of calcium and magnesium cations in bacterial attachment.
2. Experiment
2.1. Materials
2-Hydroxyethylmethacrylate (HEMA, 97%), ethylene glycol dimethacrylate (EGDMA, 98%), methacrylic acid (MAA, 99%), acrylic acid (AA, 99%), 0.01 M phosphate-buffered saline (PBS (0.138-M NaCl), molecular-biology grade), SigmaCote™, ethylene diamine tetra-acetic acid terasodium salt dihydrate (EDTA, 99%), potassium chloride, sodium phosphate monobasic monohydrate, potassium phosphate anhydrous, magnesium sulfate heptahydrate, and sodium nitrate (reagent grade) were purchased from Sigma–Aldrich (St. Louis, MO) and used as received. 2,4,6-Trimethyl-benzoyl-diphenyl-phosphineoxide (Daracure TPO) was kindly supplied by CIBA Specialty Chemicals (Tarrytown, NY). Anhydrous ethanol, acetonitrile (99.9%), and sodium acetate (reagent grade) were purchased from Fisher Scientific. Distilled/deionized water was obtained via a Milli Q system (Millipore, Billerica, MA). Tryptic soy agar and MacConkey agar were purchased from Becton, Dickinson, and Company (Franklin Lakes, NJ).
2.2. Bacteria preparation
Pseudomonas aeruginosa strain PAK was kindly provided by Dr. Stephen Lory (Harvard Medical School) [27]. Frozen stocks were recovered onto Tryptic Soy Agar plates and grown at 37 °C for 14 h. Single colonies were inoculated into 12 mL of modified Mian’s minimal medium (Mian’s) [6,28] and grown while shaken at 250 rpm at 37 °C until the optical density reached 0.15. Modified Mian’s minimal medium consists of sodium phosphate monobasic monohydrate (7.5 mM), potassium phosphate anhydrous (16.8 mM), magnesium sulfate heptahydrate (10 mM), sodium nitrate (23.5 mM), and sodium acetate (10 mM) in ultrapure water and filter sterilized [6,28]. The pH of the Mian’s medium is 7.4. Bacteria were then diluted into fresh modified Mian’s medium to a concentration of ~5 × 106 colony forming units per mL (cfu/mL). In some experiments, bacteria were centrifuged at 6000 rcf (6000g) for 10 min, and washed and resuspended in desired solutions of ~5 × 106 cfu/mL. No detectable viability change was observed in the resuspended solutions. Viable cell counts onto MacConkey agar were performed to validate bacteria concentration. Each bacterial-adhesion study was performed with a separate and freshly prepared culture.
Zeta potential of the P. aeruginosa bacteria was measured with a Brookhaven Zeta Pals (Holtsville, NY). After growth to a concentration of ~5 × 106 cfu/mL, as described above, 10 sets of measurements were done on two separate occasions and averaged. P. aeruginosa strain PAK bacteria suspended in Mian’s minimal medium at pH = 7.4 showed slightly negative zeta potentials of −12 ± 3.3 mV. Thus, P. aeruginosa carries a slight negative charge due to the lipopolysaccarides and organic acids on its surface [29–31].
2.3. Hydrogel synthesis
Hydrogel membranes of p-HEMA were synthesized by UV-initiated free-radical polymerization [32]. 2 mL of HEMA and 2.7 mL of distilled/deionized water were mixed thoroughly with 15 μL of EGDMA. Nitrogen gas was bubbled through the solution to remove oxygen, which may hinder polymerization. 6 mg of the initiator, Daracure TPO, was added, and the solution was mixed while shielded from light. Monomer solution was polymerized immediately between two microscope slides (Fisher Scientific, Waltham, MA) separated by 80-μm-thick plastic shim stock (McMaster Car, Elmhurst, IL) and secured by clamps. Membranes consisting of 30–100% MAA or 30–100% AA by volume were made by substituting the HEMA component with the desired fraction of MAA or AA. Since membranes of MAA or AA are fragile, EGDMA cross-linking volume was increased to 60 μL. Additionally, the initiator was increased to 24 mg. Because the cross-linking agent is very hydrophobic, the amount of water added to the monomer solution was replaced with a solution of 75% water and 25% acetonitrile by volume. Before each experiment, hydrogel membranes were presoaked overnight in the solution of interest and then cut into 10-mm diameter sections.
In some experiments, hydrogel beads ~4 μL in volume were synthesized in a similar fashion by pipetting 2-μL droplets of the monomer solution into a SigmaCote™ treated, glass vial containing 25 mL of Silicone DC 200 fluid (laboratory grade, Accurate Chemical and Scientific Corp, Westbury, NY) and UV polymerized. Similar to the membranes, hydrogel beads were presoaked overnight in the solution under study.
2.4. Membrane wettability
Surface wettability is argued as a modulator of biofouling [33,34]. To assess wettability of the synthesized hydrogel membranes, we measured aqueous contact angles with captive air bubbles. Except for securing the substrates, the procedure was identical to that of Lin and Svitova [35]. Synthesized membranes were placed on a polystyrene plate and needed no further securing. Cover glasses were secured to a polytetrafluoroethylene (PTFE) block with (PTFE) tape.
Fig. 1 reports advancing contact angles of p-HEMA, p-MAA, clean glass, and silanated-glass surfaces measured in PBS (pH = 7.4). Clean and silanated glass surfaces served as controls for hydrophilic and hydrophobic materials, respectively. p-HEMA membranes were pretreated with PBS (pH 7.4), whereas p-MAA membranes were pretreated with PBS (pH 7.4) with 100-mM MgCl2. Clean glass and p-MAA membranes are hydrophilic with advancing contact angles ~20°, the resolution of our optics at low angles. Conversely, p-HEMA and silanated glass are relatively hydrophobic with advancing contact angles of 59 ± 7.6° and 102 ± 4.7°, respectively. Receding angles for all surfaces were 20°, except for silanated glass with a receding angle of 90.6 ± 6°. Thus, as gauged by contact angles, the ionic-homopolymer hydrogel, p-MAA, is considerably more hydrophilic than is p-HEMA. Co-polymerization of methacrylic acid with p-HEMA hydrogels increases the water content of the gel surfaces and in addition to their water wettability [22].
Fig. 1.

Advancing contact angles for surfaces: p-HEMA pretreated with PBS (pH = 7.4), p-MAA pretreated with PBS (pH = 7.4) with 100-mM MgCl2, clean glass, and silanated glass each immersed in aqueous PBS (pH = 7.4) (*denotes statistically significantly different data with comparison to p-HEMA with a p-value <0.05).
2.5. Hydrogel swelling/shrinking
Polyelectrolyte hydrogels respond to changes in pH and salt concentration [36–39] typically manifested by swelling/shrinking due to extension/collapse of polymer chains [38,39]. Thus, to gain information on the hydrogel-matrix charge, we performed swelling experiments. Swelling/shrinking of selected hydrogels was measured by placing a single hydrogel sphere of initial volume ~4 μL (~1-mm diameter) into a glass cuvette containing the aqueous solution of interest. The camera of a Krüss Drop Shape Analysis System 10 (Krüss, Hamburg, Germany) was focused onto the hydrogel sessile bead resting on the bottom of the cuvette. Images were transferred to a PC computer where drop-shape-analysis software (Drop Shape Analysis Version 1.90.0.11, Krüss, Hamburg, Germany) calculated the gel volume every 5 min. Swelling/shrinking results are presented as the average of two or more experiments.
Fig. 2 shows the swelling/shrinkage response of cross-linked p-MAA in terms of the ratio of the transient bead volume, V, to its final volume, Vf, in Fig. 2a, or to its starting volume, Vo, in Fig. 2b. The beads in Fig. 2a were initially saturated for several days in distilled/deionized water (pH = 6.2) and then exposed to PBS at pH = 7.4. A fourfold increase in volume ensued. Conversely, Fig. 2b illustrates that when p-MAA beads initially saturated with PBS (pH = 7.4) were exposed to 100-mM NaCl in PBS (pH = 7.4), they shrank by about 10%. These two results strongly suggest that appended carboxylic side groups in the p-MAA polymeric network are ionized at pH = 7.4 in agreement with the expected pKa of less than 7 [40,41]. Experiments performed on p-MAA and p-AA membranes showed similar results (data not shown). We concluded that p-HEMA membranes copolymerized with p-MAA or p-AA are anionic at pH = 7.4 corresponding to that of Mian’s medium.
Fig. 2.

Volume swelling/shrinking kinetics of 4-μL p-MAA beads: (a) initially soaked in distilled/deionized water (pH = 6.2) and immersed in PBS (pH 7.4) (b) initially soaked in PBS (pH = 7.4) and immersed in PBS (pH 7.4) with 100-mM NaCl.
2.6. Bacteria-association assays
P. aeruginosa association with transparent surfaces was visualized under phase-contrast, time-lapse microscopy in a parallelplate flow chamber at a surface shear rate of 0.2 s−1 [12]. The flow chamber provided fully-developed laminar flow across the soft contact lens. The chamber frame was constructed of G10 fiberglass epoxy resin. A standard 3 × 1-in. glass microscope slide (Fisher Scientific) served as the chamber top while the bottom was a 60 × 24 mm cover glass slip (Fisher Scientific). The soft contact lens was mounted to the cover glass 35 mm downstream of the chamber entrance and was held in place with a 60 × 24 mm, 0.25-mm thick G10 fiber-glass epoxy resin sheet with a 5-mm diameter aperture allowing for lens exposure to the flow stream. New cover glass and microscope slides were used for each experiment. Flow entered and exited the flow cell via 0.125-cm diameter ports. A 2-mm baffle near the flow-chamber entrance evenly distributed the flow into a parabolic profile. One-dimensional parabolic flow at the location of the soft contact lens was confirmed with Multiphysics simulations (COMSOL) [12].
Before surface assay with phase-contrast microscopy, the flow cell was primed with modified Mian’s minimal medium while the syringe pump (Pharmacia LKB-Pump P-500) and all lines entering the flow cell were primed with the freshly prepared bacterial suspension. During bacterial deposition, a flow rate of 210 mL/h was set corresponding to a surface shear rate of 0.2 s−1. This low shear rate was chosen to provide a well-defined and reproducible mass-transfer rate to the lens surface. In addition, entrained, non-adherent bacteria swam away within seconds. As described below, these bacteria were counted as part of those associated with the surface. Each attachment assay lasted 1–2 h. There was no detectable bacterial growth during that time frame.
Cover glasses (Fisher Scientific, 18 cir) for the assays were cleaned by EtOH saturated with KOH and then washed with distilled/deionized water. To make a cover-glass surface hydrophobic, cleaned cover slips were silanated with a chlorinated organopolysiloxane by dipping into SigmaCote™ (Sigma Aldrich) [42] for 5 s. Treated cover glasses were then baked overnight at 80 °C followed by repeated rinsing first with distilled/deionized water and then with ethanol.
Image processing was accomplished with IMAGE J software (NIH). All bacteria visible were enumerated within the ~4-μm focal depth from the membrane surface. In this region, bacteria were bound and stationary or unbound and moving. Bound bacteria were defined not to move within a 20-s time period. This 20-s interval was chosen for convenience. Experiments with captured frames 0.3 s apart showed no difference in results. The number of bound bacteria was determined via image processing with Image J by subtracting bacteria that moved in between captured frames taken 20 s apart. Unbound bacteria include those that approach the surface but do not bind, bacteria that move on or along the surface, and bacteria that detach between each frame [12].
Bacteria uptake is reported as mean ± the standard deviation of three or more experiments. Student T-test determined the statistical significance when comparing two groups. p-values of less than 0.05 were considered significant. All adhesion measurements were performed at ambient temperature.
3. Results and discussion
3.1. Bacterial uptake
The surface number density of wild-type PAK bacteria in Mian’s medium at pH 7.4 accumulating on the surface versus time is shown in Fig. 3 for a p-HEMA membrane initially soaked in PBS (pH 7.4). Open circles in Fig. 3 correspond to total surface-associated bacteria within the 4-μm depth of focus from the surface and thus represent all bacteria directly adjacent to or on the membrane surface [12]. After a few minutes of suspended bacteria injection into the optical flow cell, PAK bacteria steadily accumulated at an average rate of 230 bacteria/mm2/min, as dictated by convective diffusion [12]. At the surface density of 104 bacteria/mm2, surface coverage is less than 3%. Hence, surface accumulation is an isolated-entity event [12]. Bound bacteria (bacteria stationary for 20 s or more), shown as filled circles in Fig. 3, also accumulated steadily over time. They accounted for 85% of the total number of bacteria at the p-HEMA surface.
Fig. 3.

Surface uptake kinetics of PAK suspended at 5 × 106 cfu/mL in Mian’s medium accumulating on a p-HEMA surface pretreated in PBS (pH = 7.4). Open circles correspond to total bacterial accumulation. Filled circles reflect bound bacteria.
Fig. 4 shows the corresponding surface-association kinetics of PAK bacteria in Mian’s medium at pH 7.4 but now exposed to a 70% p-HEMA/30% p-MAA membrane pretreated with PBS (pH = 7.4). Drastically different binding histories are seen compared to PAK exposed to 100% p-HEMA in Fig. 3. In Fig. 4, the number density of bacteria at the anionic-membrane surface peaks at approximately 20 min. Afterwards, the number of bacteria at the surface diminishes even though bacteria at 5 × 106 cfu/mL continued to be injected.
Fig. 4.
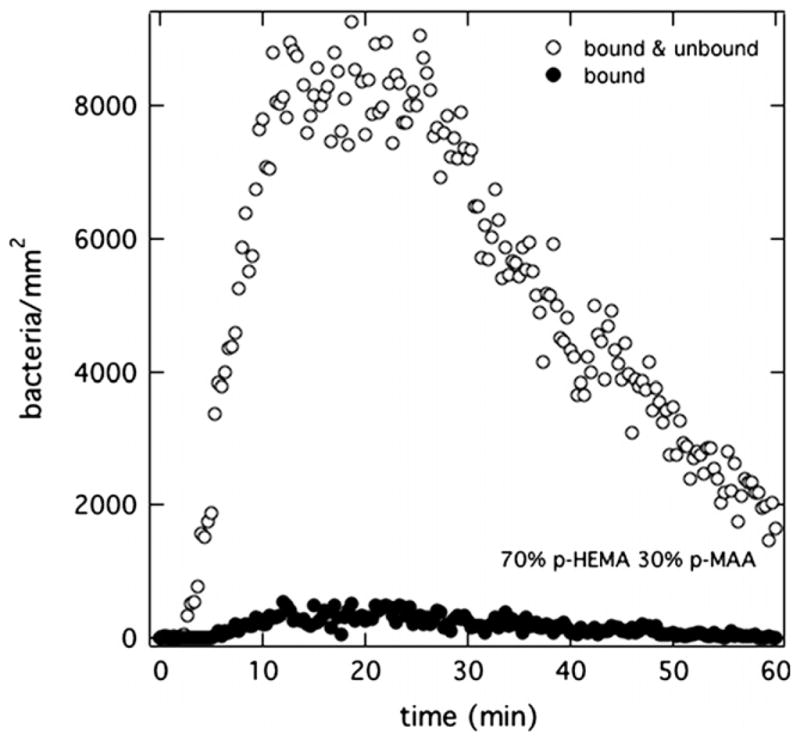
Surface uptake kinetics of PAK at 5 × 106 cfu/mL suspended in Mian’s medium accumulating on a 70% p-HEMA/30% p-MAA surface pretreated in PBS (pH = 7.4). Open circles correspond to total bacterial accumulation. Filled circles reflect bound bacteria.
Filled circles taken over the same time interval reveal that less than 10% of the bacteria at the surface were bound to the membrane. This result is in stark contrast to the adhesion kinetics of PAK on a p-HEMA surface. Ultimately in Fig. 4, the total number of bound and unbound bacteria remaining at the surface is less than about 1000 bacteria/mm2 at the end of the 1-h experiment.
Similar uptake experiments for PAK in Mian’s medium were performed with homopolymers of p-MAA and p-AA each pretreated with PBS (pH = 7.4). Fig. 5 graphs the total (bound and unbound) bacterial-accumulation histories for these two hydrogels. Essentially identical results are seen compared to the p-MAA/p-HEMA-copolymer membrane in Fig. 4: rapid accumulation of bacteria, followed by rapid depletion to very low adhesion densities. The fractions of bound bacteria in Fig. 5 are very low, similar to those in Fig. 4.
Fig. 5.

Total surface uptake kinetics of PAK at 5 × 106 cfu/mL suspended in Mian’s medium accumulating on anionic-polycarboxylate homopolymers p-MAA (filled circles) and p-AA (open circles) each pretreated in PBS (pH = 7.4).
We describe the rapid accumulation and depletion of the PAK bacteria at the membrane surface as a “burst”. Three distinct time regimes occur in a burst: (1) fast accumulation of unbound bacteria at the membrane (i.e., faster than for p-HEMA), (2) rapid depletion, and (3) few bacteria eventually binding.
A PAK burst is observed only when p-MAA or p-AA is present in the hydrogel. Apparently, the addition of charged repeat units to the hydrogel matrix plays an important role in the burst phenomenon. To investigate whether membrane charge is necessary for a burst, p-MAA homopolymer membranes were pretreated with deionized/distilled water (pH 6.2) instead of PBS (pH 7.4). Open triangles in Fig. 6 reveal the lack of a bacterial burst when the membrane initially is at pH = 6.2. This result confirms the pre-requisite of membrane charge in initiating a burst.
Fig. 6.

Total surface uptake kinetics for PAK at 5 × 106 cfu/mL suspended in Mian’s medium accumulating on p-MAA membranes with differing pretreatments: aqueous 100-mM NaCl in PBS solution (pH = 7.4) (filled circles) or distilled/deionized water (pH = 6.2) (open triangles).
The importance of membrane charge in PAK bursts suggests that classical double-layer electrostatic forces play a role [43,44]. At high enough indifferent electrolyte concentration, these forces diminish. Accordingly, p-MAA membranes were pretreated with 100-mM NaCl in PBS (pH = 7.4) and were exposed to PAK bacteria in Mian’s medium. At these electrolyte concentrations, the Debye length is less than 1 nm. Far from disappearing, the filled circles in Fig. 6 clearly demonstrate that bursting is unaffected by excess indifferent electrolyte. This result is also consistent with the minor shrinkage observed in Fig. 2b for immersion of p-MAA gel into 100-mM aqueous NaCl at pH = 7.4 Bacterial uptake kinetics at even higher NaCl-pretreatment concentrations was difficult to assess due to rapid changes in membrane conformation during the flow-cell experiments. Nevertheless, we concluded that traditional electrostatic double-layer interaction between the bacterium and the hydrogel surface does not initiate bursting.
Mian’s growth medium contains divalent cations, namely magnesium, that interact specifically with anionic carboxylates [45]. To establish whether magnesium cation interacts specifically with the polycarboxylate gels, we performed swelling/shrinkage experiments on p-MAA gel beads in the presence of magnesium. Results are shown in Fig. 7. When MgCl2 at 10 mM is added to the aqueous PBS solution (pH = 7.4), the beads initially swollen in PBS (pH = 7.4) shrink by 37%. Specific counterion binding of magnesium cations is the most likely explanation for the significant collapse of the anionic p-MAA hydrogel.
Fig. 7.

Volume shrinking kinetics of 4-μL p-MAA beads initially soaked in PBS (pH = 7.4) followed by immersion in PBS (pH 7.4) with 10-mM MgCl2.
Fig. 8 reports the role of magnesium cations in controlling PAK bursting on anionic p-MAA membranes. Uptake kinetics of PAK in Mian’s medium (pH = 7.4) is shown when the membrane is pretreated with various magnesium-salt-containing solutions: Mian’s medium (pH = 7.4), 10-mM MgCl2 in aqueous PBS (pH = 7.4), and 100-mM MgCl2 in aqueous PBS (pH = 7.4). When the presoaking solution contains magnesium cations, PAK bursting is clearly curtailed. The higher is the aqueous magnesium concentration, the more effective is the burst remission. The final number density of bacteria at the membrane surface is less than 2000 bacteria/mm2 even after 1 h of exposure to bacteria.
Fig. 8.

Total surface uptake kinetics of PAK at 5 × 106 cfu/mL accumulating on p-MAA membranes pretreated in Mian’s medium (open triangles), in 10-mM MgCl2 aqueous PBS solution (pH = 7.4) (open squares), and in 100-mM MgCl2 aqueous PBS solution (pH = 7.4) (closed squares). Open triangles are partially hidden.
Figs. 4–8 suggest that anionic PAK bursts at the surface of anionic hydrogels when magnesium is present in the flowing suspension but not initially in the hydrogel. Therefore, removal of magnesium cations from the suspending medium should eliminate bacterial bursts. We performed bacteria-attachment assays with p-AA hydrogels presoaked in PBS (pH = 7.4) but with the suspending Mian’s medium (pH = 7.4) devoid of magnesium salt. To accomplish this, PAK bacteria, initially grown in Mian’s medium, were centrifuged, washed, and resuspended in Mian’s medium but without MgSO4. Removal of magnesium from the suspending medium had no detectable effect on bacteria viability (data not shown). As demonstrated in Fig. 9a, no burst was observed at a p-AA membrane even after 1 h of bacterial exposure. However, once the injected bacteria solution was brought to 10-mM MgCl2, a burst immediately occurred. The results confirm that magnesium in the aqueous suspension is required for PAK burst. The fact that bursts occur either with aqueous MgCl2 or with MgSO4 rules out any influence of salt anions.
Fig. 9.

Burst initiation with divalent cations. Total surface uptake kinetics of PAK at 5 × 106 cfu/mL on a p-AA membrane pretreated in PBS (pH = 7.4). (a) Initially the injected suspension medium is Mian’s without MgSO4 (pH = 7.4). After 1 h, 10-mM MgCl2 was added to the flowing bacterial suspension. (b) Initially, the injected suspension medium is 100-mM KCl and 10-mM CH3COONa at pH = 7.4. After 1 h, 5-mM CaCl2 was added to the flowing bacterial suspension.
Identical experiments were conducted with CaCl2 replacing MgCl2. To prevent calcium phosphate from precipitating in PBS and in Mian’s medium, a KCl solution (100-mM KCl with 10-mM CH3COONa) was used both to pretreat the membrane (instead of PBS) and to suspend the bacteria (instead of Mian’s). Sodium acetate served as a carbon source (same as in Mian’s) for the bacteria, and, thus, maintained bacteria viability. The solution pH was adjusted to 7.4 with small amounts of NaOH. Fig. 9b displays the results. Without calcium ions in the suspending medium, again no burst was observed. Once CaCl2 was added to the suspension medium, a burst appeared. We concluded that calcium cations also specifically ion bind with the anionic polycarboxylate gels.
Another means for removing magnesium cation from the Mian’s suspending medium is via chelation. Fig. 10 demonstrates that ethylene diamine tetraacetic acid (EDTA) successfully inactivates magnesium cations from both the solution and from the anionic gels. p-MAA beads previously shrunken with MgCl2 in Fig. 7 recover their initial swelling when exposed to EDTA in PBS (pH 7.4). Thus, EDTA was used to restore membranes that had previously absorbed divalent cations. A burst should then occur once the divalent cations are removed from the membrane, and the membrane is exposed to more bacteria in the presence of aqueous divalent cations.
Fig. 10.

Volume swelling kinetics of 4-μL p-MAA beads upon exposure to EDTA. p-MAA was initially soaked in PBS (pH = 7.4) followed by immersion in PBS (pH 7.4) with 10-mM MgCl2 (see Fig. 7). At the end of shrinkage in Fig. 7, 10-mM EDTA was added to the MgCl2/PBS solution.
In Fig. 11, PAK bacteria were suspended in Mian’s medium (pH = 7.4) and flowed over a p-AA membrane pretreated in PBS (pH = 7.4). After 1 h, EDTA at an equal concentration to magnesium sulfate (10 mM) was incorporated in the suspending medium for about 50 min. During this period, EDTA chelated the magnesium cations from the bulk solution and from the membrane. After the 50-min chelating period, 10-mM of MgCl2 was then added to the EDTA/Mian’s bacterial suspension and flowed past the membrane. Fig. 11 clearly shows that upon addition of more magnesium cations into the bacterial suspension, another burst occurred.
Fig. 11.

Magnesium burst recovery with EDTA. Total surface uptake kinetics of PAK at 5 × 106 cfu/mL in Mian’s medium (pH = 7.4) for p-AA membranes pretreated in PBS (pH = 7.4). After 1 h, 10-mM EDTA is added to the suspending solution. After about an additional 30 min, 10-mM MgCl2 is added to the suspending solution.
Calcium divalent-cation bridging was also curtailed with addition of EDTA as shown in Fig. 12. In this case, PAK at 5 × 106 cfu/mL in aqueous 100-mM KCl, 10-mM CH3COONa, and 5-mM CaCl2 (pH = 7.4) was exposed to p-AA membranes pretreated in 0.1-M KCl and 10-mM CH3COONa (pH = 7.4). A burst occurs but there is not enough solution Ca2+ to collapse fully the anionic polymer chains. A significant number of bacteria strongly associating with the membrane persists. After 1 h, 5-mM EDTA is added to the suspending solution; essentially all the bacteria leave the hydrogel surface. After an additional 30 min, 5-mM CaCl2 is re-added to the suspending solution and a small burst reappears.
Fig. 12.

Calcium burst recovery with EDTA. Total surface uptake kinetics of PAK at 5 × 106 cfu/mL in aqueous 100-mM KCl, 10-mM CH3COONa, and 5-mM CaCl2 (pH = 7.4) for p-AA membranes pretreated in 100-mM KCl and 10-mM CH3COONa (pH = 7.4).
3.2. Burst etiology
PAK bursts commence when the hydrogel surface is anionic and when the suspending medium contains divalent ions, but the membrane initially does not. Thus, in addition to membrane charge, a gradient in divalent-ion concentration between the suspending solution and the membrane appears requisite. Bacteria migration in concentration gradients towards an attractant or away from a repellent (i.e., chemotaxis) is well documented [46,47]. Specifically, P. aeruginosa migrates in the direction of increasing concentration of organic acids used as a carbon source [47]. The presence of magnesium is apparently also required [48,49]. However, no free acids are present in our experiments. Although bacteria adhered to a surface do respond to chemical stimulae [50], there is no documentation of P. aeruginosa attraction to MAA or AA. Further, such attraction, if present, does not explain the bacterial-depletion sequence of a burst. Chemotaxis driven by divalent-cation concentration gradients also cannot explain the burst phenomenon because such gradients are present in other PAK uptake experiments that do not exhibit bursts, including those for p-HEMA (Fig. 3), O2Optix™ [12], and p-MAA membranes pretreated with distilled/deionized water (see also Fig. 6) Accordingly, we reject chemotaxis as the origin of PAK bursting on anionic hydrogel surfaces.
During the burst, the number of bacteria affixed to the membrane surface is very low. In fact, accumulated bacteria appear to diffuse along the membrane surface without strong binding. This observation suggests an attractive force maintaining the bacteria within about a body diameter of the surface. Classical Hamaker forces are sensibly independent of aqueous electrolyte concentration [44,51] and, hence, are inappropriate to bursting. As pointed out earlier, Figs. 4–10 argue for a specific electrostatic attraction. The importance of divalent cations to bursting in Figs. 9, 11, and 12, suggests cationic bridging between negatively charged sites on the anionic bacterium and on the anionic hydrogel surface. Indeed, there is documentation that divalent cations act as a bridge for binding between anionic bacteria and substrate surfaces [52,53]. Fig. 13 illustrates schematically our proposed picture. We suggest that the bridging attraction is local and weak. Nonpermanently bound bacteria may swim away from the surface overcoming the bridging attractive force. They adsorb and desorb reversibly from the membrane on a time scale shorter than our frame speed. Reversible cation bridging explains the initial large accumulation of nonbound PAK on anionic-hydrogel surfaces in Figs. 4 and 5 relative to that on nonionic p-HEMA in Fig. 3. Subsequent bacterial depletion, however, is not accounted for.
Fig. 13.
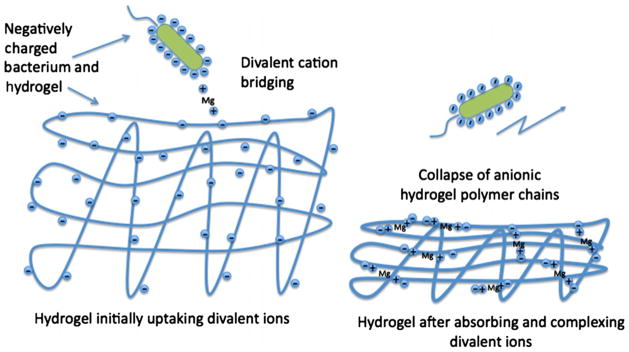
Schematic of proposed bridging/collapse mechanism for PAK bursting. Initially, the hydrogel is expanded allowing divalent-cation bridging between bacteria and substrate surface. Later, the hydrogel collapses destroying surface bridging sites and releasing accumulated bacteria.
On the left of Fig. 13, the membrane surface is immersed in indifferent electrolyte at pH 7.4. Carboxylic-acid side groups are deprotonated causing the polymer chains to repel [38,39] and the gel to expand. Fig. 2a confirms this charge-induced swelling. When exposed to divalent-cation salts, the gel surface ion binds the cations at the anionic charged surface. Some of the externalsurface divalent-ion-bound sites permit cation bridging with PAK and give rise to the enhanced adhesion seen initially in a burst.
Upon longer contact of the membrane with aqueous divalent cations, more absorb into the gel polymer matrix, ion bind, and bridge internally with nearby carboxylated side groups. Nonspecific-bound sodium counterions release in an ion-exchange process [38]. The resulting matrix bridging-charge neutralization contracts the hydrogel, as pictured on the right of Fig. 13 and confirmed in Fig. 7. Gel collapse buries some surface bridging sites making them no longer available to nonpenetrating bacteria. Previously associated bacteria then escape from the surface. Divalent-cation binding within the anionic gel is reversible to chelation with EDTA. Once the aqueous divalent cations are inactivated, the gel again swells, as established in Fig. 10. Gel-surface ion-binding sites re-establish. Hence, as documented in Figs. 11 and 12, re-introduction of aqueous divalent-cation salt permits another PAK burst.
Membranes pretreated with distilled/deionized water at pH 6.2 do not exhibit bacterial bursting because they are not negatively charged enough. In concert with our bridging/collapse model, divalent cations have a much more pronounced effect relative to mono-valent cations on both bursting and gel shrinkage.
The third criterion for a burst is that densities of bound bacteria on the surface are low. Fig. 14 emphasizes this point (see also Fig. 8). Adhesion rates of bound PAK in supernatant Mian’s medium are almost zero for p-MAA pretreated with 100-mM MgCl2 in PBS. In contrast, the comparable bound adhesion rate of PAK in Mian’s medium for p-HEMA is much larger. Fig. 14 also reveals that similar bound adhesion rates for clean glass are small, whereas those for silanated glass are large. This later result suggests that wild-type PAK bacteria do not bind well to hydrophilic materials. Some studies suggest that hydrophilic materials resist fouling [33,34,54,55]. Upon comparing the bound adhesion rates in Fig. 14 with the corresponding advancing contact angles in Fig. 1, we find a definite correlation that low-contact-angle surfaces (more hydrophilic) resist PAK adherence, whereas high-contactangle surfaces (more hydrophobic) do not. Hence, we argue that the reason for the small strong binding of PAK on anionic hydrogels is enhanced water wettability when the gel is charged and of higher water content.
Fig. 14.

PAK total uptake rates at 5 × 106 cfu/mL on p-HEMA pretreated with PBS (pH = 7.4), p-MAA pretreated with PBS (pH = 7.4) and 100-mM MgCl2, glass, and silanated glass. (*denotes statistically significantly different data with comparison to p-HEMA with a p-value <0.05).
However, correlation of bacterial binding with surface wettability is not universal [56,57]. We find that PAK bacteria binds equally well to p-HEMA and silanated glass despite an enormous difference in advancing contact angle. Clearly surface hydrophilicity is not the complete deciding factor for bacterial binding. Perhaps there is a threshold hydrophilicity that must be achieved to prevent strong adhesion.
4. Conclusions
We report that wild-type PAK strain of P. aeruginosa exhibits a burst-attachment phenomenon on anionic hydrogel membranes when the suspending medium contains divalent cations and when the membrane initially does not. Early-time accumulation rates are larger than on counterpart nonionic hydrogels. The initial high accumulation rate is followed by spontaneous bacterial detachment to a low number of strongly bound bacteria. We explain the bursting phenomenon as arising from reversible divalent-cation bridging between the anionic bacterium and anionic charged sites on the hydrogel surface. As divalent cations penetrate the hydrogel lattice, the gel collapses destroying surface binding sites and releasing most of the initial surface accumulation of bacteria. Few strongly bound PAK bacteria remain on the gel surface, most likely due to increased hydrophilicity compared to the nonionic hydrogels. This study emphasizes the delicate balance of physical and chemical forces that play important roles in the interaction of bacteria with solid surfaces. In the experiments presented here, the hydrogel ionic content was at least 30% by monomer volume. In comparison, soft-contact lenses that incorporate charged copolymers may do so at only a few percent [58]. At these low charged copolymer fractions, bursting may not be as evident.
Acknowledgments
We thank Dr. Tatiana Svitova for helpful guidance. V.T. acknowledges funding from the National Science Foundation Graduate Fellowship. Unrestricted funds from Alcon Laboratories, Fort Worth, TX are gratefully appreciated. We thank Alcon Laboratories for use of the Brookhaven Zeta Pals instrument. This work was also partially supported by NIH Grant EY011221 to SMJF.
References
- 1.Blasco MD, Esteve C, Alcaide E. J Appl Microbiol. 2008;105:469. doi: 10.1111/j.1365-2672.2008.03765.x. [DOI] [PubMed] [Google Scholar]
- 2.Das K, Bose S, Bandyopadhyay A, Karandikar B, Gibbins BL. J Biomed Mater Res Part B. 2008;87B:455. doi: 10.1002/jbm.b.31125. [DOI] [PubMed] [Google Scholar]
- 3.Liu C, Zhao Q, Liu Y, Wang S, Abel EW. Colloids Surf B. 2008;61:182. doi: 10.1016/j.colsurfb.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 4.Moritz MM, Flemming HC, Wingender J. Int J Hyg Environ Health. 2010;213:190. doi: 10.1016/j.ijheh.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Stubblefield BA, Howery KE, Islam BN, Santiago AJ, Cardenas WE, Gilbert ES. Appl Microbiol Biotechnol. 2010;86:1941. doi: 10.1007/s00253-010-2473-y. [DOI] [PubMed] [Google Scholar]
- 6.Tam C, Mun JJ, Evans DJ, Fleiszig SMJ. Invest Ophthalmol Vis Sci. 2010;51:3100. doi: 10.1167/iovs.09-4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garg P, Sharma S, Rao GN. Ophthalmology. 1999;106:1319. doi: 10.1016/S0161-6420(99)00717-4. [DOI] [PubMed] [Google Scholar]
- 8.Lyczak JB, Cannon CL, Pier GB. Microbes Infect. 2000;2:1051. doi: 10.1016/s1286-4579(00)01259-4. [DOI] [PubMed] [Google Scholar]
- 9.Selinger DS, Selinger RC, Reed WP. Surv Ophthalmol. 1979;24:33. doi: 10.1016/0039-6257(79)90145-0. [DOI] [PubMed] [Google Scholar]
- 10.Morgan PB, Efron N, Brennan NA, Hill EA, Raynor MK, Tullo AB. Invest Ophthalmol Vis Sci. 2005;46:3136. doi: 10.1167/iovs.05-0133. [DOI] [PubMed] [Google Scholar]
- 11.Keay L, Edwards K, Naduvilath T, Taylor HR, Snibson GR, Forde K, Stapleton F. Ophthalmology. 2006;113:109. doi: 10.1016/j.ophtha.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 12.Tran V, Fleiszig S, Evans D, Radke C. Appl Environ Microbiol. 2011;77:3644. doi: 10.1128/AEM.02656-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lillehoj EP, Kim BT, Kim KC. Am J Physiol – Lung Cell Mol Physiol. 2002;282:L751. doi: 10.1152/ajplung.00383.2001. [DOI] [PubMed] [Google Scholar]
- 14.Giltner CL, van Schaik EJ, Audette GF, Kao D, Hodges RS, Hassett DJ, Irvin RT. Mol Microbiol. 2006;59:1083. doi: 10.1111/j.1365-2958.2005.05002.x. [DOI] [PubMed] [Google Scholar]
- 15.Drake D, Montie TC. J Gen Microbiol. 1988;134:43. doi: 10.1099/00221287-134-1-43. [DOI] [PubMed] [Google Scholar]
- 16.Saiman L, Prince A. J Clin Invest. 1993;92:1875. doi: 10.1172/JCI116779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Busscher HJ, Weerkamp AH. FEMS Microbiol Rev. 1987;46:165. [Google Scholar]
- 18.Myszka K, Czaczyk K. Curr Microbiol. 2009;58:541. doi: 10.1007/s00284-009-9365-3. [DOI] [PubMed] [Google Scholar]
- 19.Atabek A, Camesano TA. J Bacteriol. 2007;189:8503. doi: 10.1128/JB.00769-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blanchard B, Nurisso A, Hollville E, Tetaud C, Wiels J, Pokorna M, Wimmerova M, Varrot A, Imberty A. J Mol Biol. 2008;383:837. doi: 10.1016/j.jmb.2008.08.028. [DOI] [PubMed] [Google Scholar]
- 21.Epstein A. In: Textbook of Opthalmology. Agarwal S, Agarwal A, Apple DJ, Buratto L, Alio JL, Pandey SK, Agarwal A, editors. Vol. 1. Jaypee Brothers Medical Publishers; New Delhi, India: 2002. pp. 205–218. [Google Scholar]
- 22.Nicolson PC, Vogt J. Biomaterials. 2001;22:3273. doi: 10.1016/s0142-9612(01)00165-x. [DOI] [PubMed] [Google Scholar]
- 23.Tonge S, Jones L, Goodall S, Tighe B. Curr Eye Res. 2001;23:51. doi: 10.1076/ceyr.23.1.51.5418. [DOI] [PubMed] [Google Scholar]
- 24.Moradi O, Modarress H, Noroozi M. J Colloid Interface Sci. 2004;271:16. doi: 10.1016/j.jcis.2003.11.031. [DOI] [PubMed] [Google Scholar]
- 25.Amende MT, Hariharan D, Peppas NA. React Polym. 1995;25:127. [Google Scholar]
- 26.Johnson BD, Beebe DJ, Crone W. Mater Sci Eng C – Biomimetic Supramol Syst. 2004;24:575. [Google Scholar]
- 27.Strom MS, Lory S. J Bacteriol. 1986;165:367. doi: 10.1128/jb.165.2.367-372.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lakkis C, Fleiszig SMJ. J Clin Microbiol. 2001;39:1477. doi: 10.1128/JCM.39.4.1477-1486.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Makin SA, Beveridge TJ. Microbiology – UK. 1996;142:299. doi: 10.1099/13500872-142-2-299. [DOI] [PubMed] [Google Scholar]
- 30.Gacesa P. Microbiology – UK. 1998;144:1133. doi: 10.1099/00221287-144-5-1133. [DOI] [PubMed] [Google Scholar]
- 31.Bruinsma GM, Rustema-Abbing M, van der Mei HC, Busscher HJ. J Microbiol Methods. 2001;45:95. doi: 10.1016/s0167-7012(01)00238-x. [DOI] [PubMed] [Google Scholar]
- 32.Kapoor Y, Chauhan A. J Colloid Interface Sci. 2008;322:624. doi: 10.1016/j.jcis.2008.02.028. [DOI] [PubMed] [Google Scholar]
- 33.Aye Aye M, Lee W, Mun S, Ahn C, Lee S, Yoon J. Biofouling. 2010;26:313. doi: 10.1080/08927010903576389. [DOI] [PubMed] [Google Scholar]
- 34.Bennett SM, Finlay JA, Gunari N, Wells DD, Meyer AE, Walker GC, Callow ME, Callow JA, Bright FV, Detty MR. Biofouling. 2010;26:235. doi: 10.1080/08927010903469676. [DOI] [PubMed] [Google Scholar]
- 35.Lin MC, Svitova TF. Optom Vis Sci. 2010;87:440. doi: 10.1097/OPX.0b013e3181dc9a1a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chu Y, Varanasi PP, McGlade MJ, Varanasi S. J Appl Polym Sci. 1995;58:2161. [Google Scholar]
- 37.De-Wei Y, Olvera de la Cruz M, de Pablo JJ. J Chem Phys. 2009;131:194907. doi: 10.1063/1.3264950. [DOI] [PubMed] [Google Scholar]
- 38.Toomey R, Tirrell M. Annu Rev Phys Chem. 2008;59:493. doi: 10.1146/annurev.physchem.59.032607.093623. [DOI] [PubMed] [Google Scholar]
- 39.Zhang J, Peppas NA. Macromolecules. 2000;33:102. [Google Scholar]
- 40.Chao GT, Deng HX, Huang Q, Jia WJ, Huang WX, Gu YC, Tan HP, Fan LY, Liu CB, Huang AL, Lei K, Gong CY, Tu MJ, Qian ZY. J Polym Res. 2006;13:349. [Google Scholar]
- 41.Izumrudov V, Sukhishvili SA. Langmuir. 2003;19:5188. [Google Scholar]
- 42.van der Heiden AP, Willems GM, Lindhout T, Pijpers AP, Koole LH. J Biomed Mater Res. 1998;40:195. doi: 10.1002/(sici)1097-4636(199805)40:2<195::aid-jbm4>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 43.Hermansson M. Colloids Surf B. 1999;14:105. [Google Scholar]
- 44.Overbeek JTG. In: Colloid Science I: Irreversible Systems. Kruyt HR, editor. Chapters IV and VI Elsevier; Amsterdam: 1952. [Google Scholar]
- 45.Cannan RK, Kibrick A. J Am Chem Soc. 1938;60:2314. [Google Scholar]
- 46.Berg HC. Annu Rev Biophys Bioeng. 1975;4:119. doi: 10.1146/annurev.bb.04.060175.001003. [DOI] [PubMed] [Google Scholar]
- 47.Kato J, Kim HE, Takiguchi N, Kuroda A, Ohtake H. J Biosci Bioeng. 2008;106:1. doi: 10.1263/jbb.106.1. [DOI] [PubMed] [Google Scholar]
- 48.Ferrandez A, Hawkins AC, Summerfield DT, Harwood CS. J Bacteriol. 2002;184:4374. doi: 10.1128/JB.184.16.4374-4383.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moench TT, Konetzka WA. J Bacteriol. 1978;133:427. doi: 10.1128/jb.133.1.427-429.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deziel E, Lepine F, Milot S, Villemur R. Microbiology – Sgm. 2003;149:2005. doi: 10.1099/mic.0.26154-0. [DOI] [PubMed] [Google Scholar]
- 51.Hamaker HC. Physica. 1937;4:1058. [Google Scholar]
- 52.De Kerchove AJ, Elimelech M. Langmuir. 2008;24:3392. doi: 10.1021/la7036229. [DOI] [PubMed] [Google Scholar]
- 53.Simoni SF, Bosma TNP, Harms H, Zehnder AJB. Environ Sci Technol. 2000;34:1011. [Google Scholar]
- 54.Kodjikian L, Casoli-Bergeron E, Malet F, Janin-Manificat H, Freney J, Burillon C, Colin J, Steghens JP. Graefes Arch Clin Exp Ophthalmol. 2008;246:267. doi: 10.1007/s00417-007-0703-5. [DOI] [PubMed] [Google Scholar]
- 55.Henriques M, Sousa C, Lira M, Elisabete M, Oliveira R, Azeredo J. Optom Vis Sci. 2005;82:446. doi: 10.1097/01.opx.0000168585.53845.64. [DOI] [PubMed] [Google Scholar]
- 56.Salerno MB, Logan BE, Velegol D. Langmuir. 2004;20:10625. doi: 10.1021/la048372z. [DOI] [PubMed] [Google Scholar]
- 57.Bruinsma GM, van der Mei HC, Busscher HJ. Biomaterials. 2001;22:3217. doi: 10.1016/s0142-9612(01)00159-4. [DOI] [PubMed] [Google Scholar]
- 58.Soltys-Robitaille CE, Ammon DM, Valint PL, Grobe GL. Biomaterials. 2001;22:3257. doi: 10.1016/s0142-9612(01)00163-6. [DOI] [PubMed] [Google Scholar]