

Abstract
Epstein-Barr virus (EBV) maintains a lifelong infection. According to the germinal center model (GCM), latently infected B cells transit the germinal center (GC) to become resting memory cells. Here, the virus resides quiescently, occasionally reactivating to infect new B cells, completing the cycle of infection. The GCM remains the only model that explains EBV biology and the pathogenesis of lymphoma. Recent work suggests modifications to the model notably that the virus contributes only modestly to the GC process and predictions from mathematical models that quiescence within memory B cells shapes the overall structure of viral infection but is not essential for persistence. Rather, it is the cycle of infection which allows viral persistence at the very low levels observed.
Introduction
Epstein-Barr virus is a human herpesvirus with striking biological properties. It persists in a quiescent state in resting memory B lymphocytes[1] for life in virtually every human being, yet it is also a potent transforming virus in vitro for B cells and is associated with several important lymphomas, specifically Burkitt's, Hodgkin's disease and immunoblastic lymphoma[2]. These properties have been satisfactorily explained by a model of viral infection and persistence, the germinal center model (GCM) [3]. The GCM proposes that EBV persists by exploiting normal B cell biology. This involves the virus passing through a cycle of infected stages, each employing a discrete viral gene transcription program. These stages are summarized in Figure 1. Each stage of the cycle has been demonstrated experimentally [1,4,5] and, with the exception of the memory compartment, is potentially regulated by the immune response [6]. Thus, the GCM accounts for all the latent and lytic stages of the virus and thereby provides an explanation for the origin of EBV-associated lymphomas (for a detailed discussion see [2]). It is widely believed that epithelial cell infection amplifies the levels of infectious virus prior to shedding [7].
Figure 1.

The Germinal Center Model (GCM) of EBV persistence.
Infectious virus enters the lymphoid tissue of Waldeyer's ring and then crosses the epithelial barrier where it directly infects naive B cells, activating them into proliferating latently infected Blasts expressing all nine known latent proteins (the growth transcription program). These cells then move into the germinal center (GC) to participate in the GC reaction. Here they express a more restricted pattern of latent proteins, the default program. Eventually these cells leave as latently infected memory B cells that either only express the viral genome tethering protein EBNA1 (the EBNA1 only program) or no viral proteins at all (the latency program). The memory compartment has been considered the site of long-term persistence because the virus is quiescent [42] and therefore invisible to the immune response. At any time a small subset of latently infected memory B cells initiates lytic reactivation in association with terminal differentiation signals [5,43]. Reactivation of the virus is subdivided into three phases; Immediate early when the transcription factors initiating viral replication are expressed, Early when the proteins involved in viral DNA replication are produced, and Late when viral DNA and structural proteins are assembled into virions [44]. This process results in the release of infectious virus that may be shed into saliva for infectious spread or infect new naive B cells, thus completing the cycle.
Since its description 13 years ago, experimental evidence has consistently supported the GCM, however the model now needs updating in light of recent publications.
LMP1, LMP2, and the Germinal Center
The evidence that small numbers of EBV-infected cells reside and participate in the GC is unequivocal [8,9]. What role the virus plays in this process is less clear. Latently infected GC B cells express LMP1 and LMP2, and an abundance of evidence from transgenic mouse and in vitro studies has shown that they have the signaling capacity to allow GC B cells to survive and differentiate in the absence of T cell help and antigen[10,11]. But do they? If true, the resulting latently infected memory B cells should not have undergone antigen selection and this in turn should be reflected in the somatic hypermutation (SHM) patterns of their expressed immunoglobulins. However, the observed patterns are very similar to those of antigen-selected memory B cells, suggesting that the impact of LMP1 and LMP2 on the cells as they traverse the GC is modest[12-14]. Minor differences were noted, the most interesting being that the EBV-infected memory B cells expressed immunoglobulins with a reduced rate of self-reactivity[14]. This is surprising, given suggestions that EBV may play a role in autoimmunity[15], and actually raises the possibility that EBV has evolved to minimize this risk.
In contrast, published work in transgenic models has demonstrated potent biological effects for LMP1 and LMP2, including exclusion of B cells from the GC and lymphoma development with LMP1[16] and complete rescue of surface Ig negative B cells and predisposition to autoimmune disease [17] with LMP2 [10]. However, we have criticized these experiments because expression of a viral signaling protein independent of the context of the whole viral genome is equivalent to a proto-oncogene that normally provides a physiologic function and only becomes pathogenic when expression is deregulated. In particular, LMP1 and LMP2 are always expressed together in vivo and their signaling capabilities overlap. Consequently, recent analysis of a double transgenic [18] was important because here LMP1 and LMP2 did not demonstrate their potent activities. Their expression had little or no detectable effect on the GC process and cells transited the GC normally. Thus, in vivo and transgenic experiments now agree that the combined impact of these two proteins is modest.
Together this work supports a counter-intuitive consensus that EBV+ B cells remain subject to antigen selection despite the well-characterized capabilities of LMP1 and LMP2a to circumvent it. If the EBV default program does not function to autonomously drive infected GC B cells into the memory compartment, what advantage does it provide to the virus? It may simply act as insurance to ensure the survival of EBV-infected cells in the competitive environment of the GC. Alternatively, signals from LMP1 and LMP2 may ensure that the infected cells become memory rather than plasma cells. An intriguing possibility is that they provide signals that reduce polyreactivity, thus lowering the risk of developing autoimmune disease associated with acute infection[14]. However, these results may have a wider implication, namely that SHM patterns are not a reliable measure of antigen driven selection and the process of producing EBV-infected memory B cells is antigen independent after all.
Alternate Models
Direct Infection of Naïve B Cells
It has been reported that when EBV infects naïve B cells in vitro it drives them to undergo SHM [19]. This led the authors to speculate that latently infected memory B cells could be produced directly from infected naïve B cells without the need for the GC. However, unlike the latently infected memory B cells seen in vivo, the cells produced in vitro have not undergone class switch recombination and presumably do not express antigen-selected patterns of SHM. Thus, these observations, like many in vitro studies, are of little value unless confirmed in vivo. In this case, they may be an artifact of the known ability of EBV latent proteins to induce AID [20]. The more interesting speculation is that infection of naïve B cells in vivo also initiates the GC process, but the cells need to migrate into and through a GC to emerge as class switched memory B cells with antigen-selected patterns of SHM. In support of this, the authors themselves observed that the in vitro infected cells could undergo class switching. However, T cell help provided by LMP1 seemed inadequate; exogenous T cell help was required – a signal known to arise in the GC in vivo. This work again suggests a modest signaling role for LMP1. In this type of scenario, the role of LMP1 and LMP2 could be to provide the minimal antigen and T cell help necessary to activate the newly infected naïve B cell and push it to the GC, where the default program takes over to ensure survival and differentiation into memory.
Direct Infection of Memory B Cells
Although proposed over 10 years ago by Rajewsky and coworkers [21,22], no further evidence or explanation of the mechanism behind this model has been produced and model predictions were incorrect when tested experimentally, instead supporting the GCM. Thus, infected GC B cells express the viral default transcription program in vivo [4,8] (as predicted by the GCM), not the growth program (as predicted by the direct infection model [23]), and in a transgenic mouse model one of the EBV latent proteins expressed in the GC (LMP2a) was able to drive B cells to form GCs in the absence of antigen [24]. Ironically, evidence of antigen selection in the expressed immunoglobulin genes of latently infected memory B cells[12], not predicted by the GCM, is most easily explained by the direct infection model. Although some minor differences in mutation rates and autoreactivy were noted as compared to normal memory B cells, these could easily reflect variations in repertoire between the targets of infection (limited to the mucosal B cells of Waldeyer's ring for example) and the general memory population.
Infection of Marginal Zone Memory B Cells
One piece of evidence supporting the GCM was the consistent absence of the virus from marginal zone memory B cells (IgD+IgM+CD27+) [13,25]. These are thought to arise through an extrafollicular mechanism and the absence of EBV from this population supports the notion that B cells latently infected with EBV transit the GC. Recently this has been challenged [26]. However, this paper used an indirect method to measure infected cell numbers and also reported levels of infection in GC memory B cells estimated at ∼2 logs too high, suggesting a technical issue with the assay, and it remains unproven that EBV can access this population in either acute or chronic infection. This is not the case in individuals with XLP. These patients fail to produce GC derived memory B cells but harbor latent virus in the IgD+IgM+CD27+ subset ([27] and our laboratory unpublished). This raises the possibility that EBV may access/remain in these cells only if the cells are denied access to GC maturation.
EBV and Tumorigenesis
Lymphoma
The GCM provided the first and, to date, only explanation for the origin of the EBV-associated lymphomas and the reason they express restricted patterns of latent proteins [2]. The model predicted that Hodgkin's disease (default program) arises from an EBV-infected GC B cell and Burkitt's lymphoma (EBNA1 only program) from a memory B cell. Recent bioinformatics and genomics analysis however indicate a GC origin also for BL [28] (specifically from centroblasts). However, BL is an extrafollicular tumor, so together with the EBV data we need to amend the interpretation of BL as a GC B cell that has left the follicle to become a resting memory B cell but is unable to do so because of constitutively activated c-myc; thus, it retains a GC cellular phenotype but attains the memory EBV phenotype.
Carcinoma and Epithelial Cell Infection
EBV is also associated with carcinomas, including nasopharyngeal and gastric. Unlike the lymphomas, the GCM has little to say about the origin of the carcinomas. As an extension of the GCM, there is good evidence that the mucosal epithelium of Waldeyer's ring plays an important role in amplifying the virus as it leaves [7], and perhaps also as it enters the body. However, we lack insights into:
what, if anything, viral replication in the epithelium has to do with carcinomagenesis. Specifically, is it a risk factor that this tissue is continuously exposed to high titers of virus throughout lifetime?
whether the tumor is derived from an as yet unidentified reservoir of latent epithelial cell infection that is also critical for persistence.
if NPC, for example, arises because, of all the epithelial tissues in the buccal and nasopharyngeal areas, it alone has a peculiar susceptibility to transformation by EBV.
miRNAs
One enigma of EBV-associated tumors is that they do not express the transcription program associated with viral driven proliferation – the growth program. BL is particularly striking expressing only the EBNA1 protein. The tumors do express the large number of miRNAs encoded in the BART region [29]. Although these miRNAs are dispensable for B cell transformation in vitro, it is clear from recent work that they collectively confer an increased resistance to apoptosis [30,31], as indeed does EBNA1 [32]. Furthermore, a subset of ∼10 BART miRNAs that are restricted to the growth program in vivo are aberrantly expressed in all of the tumors [29] suggesting that the BART miRNAs may confer survival/growth advantages to the tumors. Indeed this has recently been confirmed for both Burkitt's lymphoma [33] and the carcinomas (in preparation). Taken together, these results suggest that the BART miRNAs play a crucial role in vivo for both tumor and normal cell survival and growth.
Mathematical Modeling
Studies on EBV are severely hampered by the lack of an amenable animal model. One alternate approach is to develop mathematical models [34-40]. We have developed a rigorous mathematical description of the GCM which we call the cyclic pathogen model (CPM) (Figure 2) [35]. Solution of the equations describing CPM revealed one and only one solution that was stable and biologically meaningful. We proposed that this describes EBV persistence [41]. Applying biologically credible values for the controlling parameters of CPM, the model successfully predicted stable infection that closely resembles persistent EBV infection. Specifically, the model correctly predicted the observed patterns of cytotoxic T cell regulation and the values for the infected germinal center and memory populations. Crucially, the model predicts that viral quiescence in the memory compartment dictates the pattern of persistence but is not a requirement; it is the cycle of infection that explains persistence and provides the stability that allows EBV to persist at extremely low levels. This shifts the focus away from a single infected stage, the memory B cell, to the whole cycle of infection to explain persistent infection. It will be important now to try and develop experimental approaches that will distinguish between these two interpretations of the mechanism behind the GCM of persistence.
Figure 2.
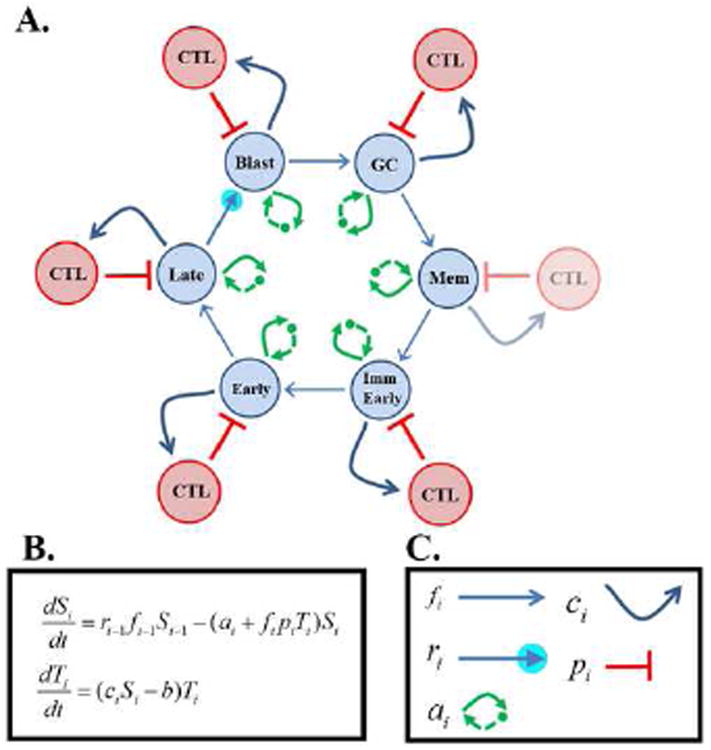
Diagrammatical Representation of the Cyclic Pathogen Model (CPM).
CPM is a mathematical description of the GCM. The CPM consists of a cycle of infected stages (blue circles based on the biological GCM illustrated in Figure 1.) For CPM there are 6 stages: Blast, GC, Memory, Immediate Early, Early and Late infected B cells, each of which is potentially controlled by a distinct CTL response (red circles). Note that the single lytic stage in the GCM is broken down into three discrete stages which are known to be recognized independently by the immune response. Note also that under biological conditions the late [45] and GC [46] stages are not always recognized by CTL and there is never a CTL response against the memory stage however the model allows analysis of theoretical conditions for example where the memory compartment is regulated by CTL. Each stage progresses to the next stage (blue arrows). Late lytic B cells release free virions which produce new infected Blasts. The latter has an amplification factor since loss of a single Late lytic cell produces many infected Blasts (small blue circle). Each infected population may have an inherent rate of proliferation or death (double green arrows). Each stage promotes proliferation of its cognate CTL population (if present) (curved blue arrows) and is in turn controlled by those CTLs (red inhibitory arrows). These CTLs (if present) have an inherent rate of loss in the absence of stimulation (not shown in diagram).
B. Equations of the CPM. For each stage there are two equations governing the rate of change of the infected population Si and the CTL population Ti for a total of 12 equations. Solution of these equations at steady state reveals one and only one solution that is stable and biologically credible. The parameters in these equations give the rates of the processes described above. Except for the rate of loss of CTLs (b), these parameters are stage-specific.
C. Correspondence between the parameters of the equations in B. and processes shown in diagram A
Conclusions
In conclusion, the GCM remains, after 13 years, the only consistent model to explain the diverse biology of EBV and has withstood repeated attempts to disprove it. Currently the major unanswered questions pertain to the exact relationship between EBV and the GC, and the exact role of LMP1 and LMP2 in that process. Is the role of EBV essentially passive as the cells transit the GC or does the virus play an active role? Perhaps the central issue remains determining if the cells are specific for and selected by antigen (consistent with the patterns of SHM but difficult to explain), or is the EBV system challenging the notion that patterns of SHM can be used to predict if a cell population has undergone antigen section. The work on mathematical modeling of persistence reveals the strengths and weaknesses of that approach. The modeling makes a strong case for the cycle of infection being the basis for persistence rather than quiescence in the memory compartment. But an experimental test that distinguishes these possibilities is now required. This is crucial because the predicted targets of anti-viral treatments will be different for the two approaches. Lastly, the outstanding challenge to the modeling is whether it can now be used to make sensible, testable statements about who is and who is not at risk for EBV-associated tumors, particularly PTLD.
Highlights.
We summarize and update the germinal center model of Epstein-Barr virus persistence.
We discuss the weaknesses and strengths of alternate models.
The role of LMP1 and LMP2 in the germinal center process needs further study.
The role of EBV-infected GC B cells in lymphomagenesis needs to be understood.
Mathematical modeling has provided fresh insights into EBV persistence.
Acknowledgments
This work was supported by Public Health Service grants R01 CA65883, R01 AI18757 and RO1 AI062989 to DATL and K25-AI079404 to MS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Babcock GJ, Decker LL, Volk M, Thorley-Lawson DA. EBV persistence in memory B cells in vivo. Immunity. 1998;9(3):395–404. doi: 10.1016/s1074-7613(00)80622-6. [DOI] [PubMed] [Google Scholar]
- 2.Thorley-Lawson DA, Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med. 2004;350(13):1328–1337. doi: 10.1056/NEJMra032015. [DOI] [PubMed] [Google Scholar]
- 3.Thorley-Lawson DA. Epstein-Barr virus: exploiting the immune system. Nat Rev Immunol. 2001;1(1):75–82. doi: 10.1038/35095584. [DOI] [PubMed] [Google Scholar]
- 4.Babcock GJ, Hochberg D, Thorley-Lawson AD. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity. 2000;13(4):497–506. doi: 10.1016/s1074-7613(00)00049-2. [DOI] [PubMed] [Google Scholar]
- 5.Laichalk LL, Thorley-Lawson DA. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J Virol. 2005;79(2):1296–1307. doi: 10.1128/JVI.79.2.1296-1307.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annu Rev Immunol. 2007;25:587–617. doi: 10.1146/annurev.immunol.25.022106.141553. [DOI] [PubMed] [Google Scholar]
- 7.Hadinoto V, Shapiro M, Sun CC, Thorley-Lawson DA. The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog. 2009;5(7):e1000496. doi: 10.1371/journal.ppat.1000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roughan JE, Thorley-Lawson DA. The intersection of Epstein-Barr virus with the germinal center. J Virol. 2009;83(8):3968–3976. doi: 10.1128/JVI.02609-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9*.Roughan JE, Torgbor C, Thorley-Lawson DA. Germinal center B cells latently infected with Epstein-Barr virus proliferate extensively but do not increase in number. J Virol. 2010;84(2):1158–1168. doi: 10.1128/JVI.01780-09. One of two papers demonstrating that B cells latently infected with EBV reside and participate in the germinal center reaction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caldwell RG, Wilson JB, Anderson SJ, Longnecker R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity. 1998;9(3):405–411. doi: 10.1016/s1074-7613(00)80623-8. [DOI] [PubMed] [Google Scholar]
- 11.Gires O, Zimber-Strobl U, Gonnella R, Ueffing M, Marschall G, Zeidler R, Pich D, Hammerschmidt W. Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. Embo J. 1997;16(20):6131–6140. doi: 10.1093/emboj/16.20.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Souza TA, Stollar BD, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. Peripheral B cells latently infected with Epstein-Barr virus display molecular hallmarks of classical antigen-selected memory B cells. Proc Natl Acad Sci U S A. 2005;102(50):18093–18098. doi: 10.1073/pnas.0509311102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Souza TA, Stollar BD, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. Influence of EBV on the peripheral blood memory B cell compartment. J Immunol. 2007;179(5):3153–3160. doi: 10.4049/jimmunol.179.5.3153. [DOI] [PubMed] [Google Scholar]
- 14.Tracy SI, Kakalacheva K, Lunemann JD, Luzuriaga K, Middeldorp J, Thorley-Lawson DA. Persistence of Epstein-Barr virus in self-reactive memory B cells. J Virol. 2012;86(22):12330–12340. doi: 10.1128/JVI.01699-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Posnett DN. Herpesviruses and autoimmunity. Curr Opin Investig Drugs. 2008;9(5):505–514. [PubMed] [Google Scholar]
- 16.Uchida J, Yasui T, Takaoka-Shichijo Y, Muraoka M, Kulwichit W, Raab-Traub N, Kikutani H. Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science. 1999;286(5438):300–303. doi: 10.1126/science.286.5438.300. [DOI] [PubMed] [Google Scholar]
- 17.Swanson-Mungerson MA, Caldwell RG, Bultema R, Longnecker R. Epstein-Barr virus LMP2A alters in vivo and in vitro models of B-cell anergy, but not deletion, in response to autoantigen. J Virol. 2005;79(12):7355–7362. doi: 10.1128/JVI.79.12.7355-7362.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18**.Vrazo AC, Chauchard M, Raab-Traub N, Longnecker R. Epstein-Barr virus LMP2A reduces hyperactivation induced by LMP1 to restore normal B cell phenotype in transgenic mice. PLoS Pathog. 2012;8(4):e1002662. doi: 10.1371/journal.ppat.1002662. Transgenic mouse model demonstrating that when co-expressed LMP1 and LMP2a do not have the drastic phenotypic effects observed when express separately. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19*.Heath E, Begue-Pastor N, Chaganti S, Croom-Carter D, Shannon-Lowe C, Kube D, Feederle R, Delecluse HJ, Rickinson AB, Bell AI. Epstein-Barr virus infection of naive B cells in vitro frequently selects clones with mutated immunoglobulin genotypes: implications for virus biology. PLoS Pathog. 2012;8(5):e1002697. doi: 10.1371/journal.ppat.1002697. Demonstration that EBV infection of B cells in vitro has the capacity to drive the somatic hypermutation characteristic of the germinal center process but not class switch recombination. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He B, Raab-Traub N, Casali P, Cerutti A. EBV-encoded latent membrane protein 1 cooperates with BAFF/BLyS and APRIL to induce T cell-independent Ig heavy chain class switching. J Immunol. 2003;171(10):5215–5224. doi: 10.4049/jimmunol.171.10.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurth J, Hansmann ML, Rajewsky K, Kuppers R. Epstein-Barr virus-infected B cells expanding in germinal centers of infectious mononucleosis patients do not participate in the germinal center reaction. Proc Natl Acad Sci U S A. 2003;100(8):4730–4735. doi: 10.1073/pnas.2627966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurth J, Spieker T, Wustrow J, Strickler GJ, Hansmann LM, Rajewsky K, Kuppers R. EBV-infected B cells in infectious mononucleosis: viral strategies for spreading in the B cell compartment and establishing latency. Immunity. 2000;13(4):485–495. doi: 10.1016/s1074-7613(00)00048-0. [DOI] [PubMed] [Google Scholar]
- 23.Siemer D, Kurth J, Lang S, Lehnerdt G, Stanelle J, Kuppers R. EBV transformation overrides gene expression patterns of B cell differentiation stages. Mol Immunol. 2008;45(11):3133–3141. doi: 10.1016/j.molimm.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 24.Casola S, Otipoby KL, Alimzhanov M, Humme S, Uyttersprot N, Kutok JL, Carroll MC, Rajewsky K. B cell receptor signal strength determines B cell fate. Nat Immunol. 2004;5(3):317–327. doi: 10.1038/ni1036. [DOI] [PubMed] [Google Scholar]
- 25.Joseph AM, Babcock GJ, Thorley-Lawson DA. EBV persistence involves strict selection of latently infected B cells. J Immunol. 2000;165(6):2975–2981. doi: 10.4049/jimmunol.165.6.2975. [DOI] [PubMed] [Google Scholar]
- 26.Chaganti S, Heath EM, Bergler W, Kuo M, Buettner M, Niedobitek G, Rickinson AB, Bell AI. Epstein-Barr virus colonization of tonsillar and peripheral blood B-cell subsets in primary infection and persistence. Blood. 2009;113(25):6372–6381. doi: 10.1182/blood-2008-08-175828. [DOI] [PubMed] [Google Scholar]
- 27.Chaganti S, Ma CS, Bell AI, Croom-Carter D, Hislop AD, Tangye SG, Rickinson AB. Epstein-Barr virus persistence in the absence of conventional memory B cells: IgM+IgD+CD27+ B cells harbor the virus in X-linked lymphoproliferative disease patients. Blood. 2008;112(3):672–679. doi: 10.1182/blood-2007-10-116269. [DOI] [PubMed] [Google Scholar]
- 28*.Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R, Nussenzweig MC. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood. 2012;120(11):2240–2248. doi: 10.1182/blood-2012-03-415380. A genomic study demonstrating phenotypes for germinal center subsets and the germinal center origin of Bukitt's lymphoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29*.Qiu J, Cosmopoulos K, Pegtel M, Hopmans E, Murray P, Middeldorp J, Shapiro M, Thorley-Lawson DA. A novel persistence associated EBV miRNA expression profile is disrupted in neoplasia. PLoS Pathog. 2011;7(8):e1002193. doi: 10.1371/journal.ppat.1002193. Demonstration that the expression of the EBV BART miRNAs is deregulated in EBV cancers suggesting a role in cancer development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30*.Marquitz AR, Mathur A, Nam CS, Raab-Traub N. The Epstein-Barr Virus BART microRNAs target the pro-apoptotic protein Bim. Virology. 2011;412(2):392–400. doi: 10.1016/j.virol.2011.01.028. Demonstration with Seto et al (reference 31) that the EBV BART miRNAs have an anti-apoptotic function in vitro. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31*.Seto E, Moosmann A, Gromminger S, Walz N, Grundhoff A, Hammerschmidt W. Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog. 2010;6(8):e1001063. doi: 10.1371/journal.ppat.1001063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kennedy G, Komano J, Sugden B. Epstein-Barr virus provides a survival factor to Burkitt' lymphomas. Proc Natl Acad Sci U S A. 2003;100(24):14269–14274. doi: 10.1073/pnas.2336099100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33**.Vereide DT, Seto E, Chiu YF, Hayes M, Tagawa T, Grundhoff A, Hammerschmidt W, Sugden B. Epstein-Barr virus maintains lymphomas via its miRNAs. Oncogene. 2013 doi: 10.1038/onc.2013.71. Demonstration that the EBV miRNAs promote the survival of Burkitt lymphoma tumor cells in vitro. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castiglione F, Duca K, Jarrah A, Laubenbacher R, Hochberg D, Thorley-Lawson D. Simulating Epstein-Barr virus infection with C-ImmSim. Bioinformatics. 2007;23(11):1371–1377. doi: 10.1093/bioinformatics/btm044. [DOI] [PubMed] [Google Scholar]
- 35*.Delgado-Eckert E, Shapiro M. A model of host response to a multi-stage pathogen. J Math Biol. 2011;63(2):201–227. doi: 10.1007/s00285-010-0365-5. The first mathematical description of a pathogen like EBV that undergoes a cycle of infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duca KA, Shapiro M, Delgado-Eckert E, Hadinoto V, Jarrah AS, Laubenbacher R, Lee K, Luzuriaga K, Polys NF, Thorley-Lawson DA. A virtual look at Epstein-Barr virus infection: biological interpretations. PLoS Pathog. 2007;3(10):1388–1400. doi: 10.1371/journal.ppat.0030137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huynh G, Rong L. Modeling the dynamics of virus shedding into the saliva of Epstein-Barr virus positive individuals. J Theor Biol. 2012;310C:105–114. doi: 10.1016/j.jtbi.2012.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huynh GT, Adler FR. Alternating host cell tropism shapes the persistence, evolution and coexistence of epstein-barr virus infections in human. Bull Math Biol. 2011;73(8):1754–1773. doi: 10.1007/s11538-010-9590-8. [DOI] [PubMed] [Google Scholar]
- 39.Huynh GT, Adler FR. Mathematical modelling the age dependence of Epstein-Barr virus associated infectious mononucleosis. Math Med Biol. 2011 doi: 10.1093/imammb/dqr007. [DOI] [PubMed] [Google Scholar]
- 40.Shapiro M, Duca KA, Lee K, Delgado-Eckert E, Hawkins J, Jarrah AS, Laubenbacher R, Polys NF, Hadinoto V, Thorley-Lawson DA. A virtual look at Epstein-Barr virus infection: simulation mechanism. J Theor Biol. 2008;252(4):633–648. doi: 10.1016/j.jtbi.2008.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41**.Hawkins JB, Delgado-Eckert E, Thorley-Lawson DA, Shapiro M. A re-evaluation of the model of Epstein-Barr virus persistent infection. Plos Pathog (in revision) 2013 Characterization and validation of the first comprehensive mathematical model of EBV persistent infection and the demonstration that persistence is dependent upon the cycle of infection. [Google Scholar]
- 42.Hochberg D, Middeldorp JM, Catalina M, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. Demonstration of the Burkitt's lymphoma Epstein-Barr virus phenotype in dividing latently infected memory cells in vivo. Proc Natl Acad Sci U S A. 2004;101(1):239–244. doi: 10.1073/pnas.2237267100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kraus RJ, Mirocha SJ, Stephany HM, Puchalski JR, Mertz JE. Identification of a novel element involved in regulation of the lytic switch BZLF1 gene promoter of Epstein-Barr virus. J Virol. 2001;75(2):867–877. doi: 10.1128/JVI.75.2.867-877.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kieff E, Rickinson AB. Epstein-Barr Virus and Its Replication. In: Knipe DM, Howley PM, editors. Fields Virology. Vol. 2. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 2603–2654. [Google Scholar]
- 45.Pudney VA, Leese AM, Rickinson AB, Hislop AD. CD8+ immunodominance among Epstein-Barr virus lytic cycle antigens directly reflects the efficiency of antigen presentation in lytically infected cells. J Exp Med. 2005;201(3):349–360. doi: 10.1084/jem.20041542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murray RJ, Kurilla MG, Brooks JM, Thomas WA, Rowe M, Kieff E, Rickinson AB. Identification of target antigens for the human cytotoxic T cell response to Epstein-Barr virus (EBV): implications for the immune control of EBV-positive malignancies. J Exp Med. 1992;176(1):157–168. doi: 10.1084/jem.176.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]