

Abstract
Simple sequence repeats (SSRs) have been widely used in maize genetics and breeding, because they are co-dominant, easy to score, and highly abundant. In this study, we used whole-genome sequences from 16 maize inbreds and 1 wild relative to determine SSR abundance and to develop a set of high-density polymorphic SSR markers. A total of 264 658 SSRs were identified across the 17 genomes, with an average of 135 693 SSRs per genome. Marker density was one SSR every of 15.48 kb. (C/G)n, (AT)n, (CAG/CTG)n, and (AAAT/ATTT)n were the most frequent motifs for mono, di-, tri-, and tetra-nucleotide SSRs, respectively. SSRs were most abundant in intergenic region and least frequent in untranslated regions, as revealed by comparing SSR distributions of three representative resequenced genomes. Comparing SSR sequences and e-polymerase chain reaction analysis among the 17 tested genomes created a new database, including 111 887 SSRs, that could be develop as polymorphic markers in silico. Among these markers, 58.00, 26.09, 7.20, 3.00, 3.93, and 1.78% of them had mono, di-, tri-, tetra-, penta-, and hexa-nucleotide motifs, respectively. Polymorphic information content for 35 573 polymorphic SSRs out of 111 887 loci varied from 0.05 to 0.83, with an average of 0.31 in the 17 tested genomes. Experimental validation of polymorphic SSR markers showed that over 70% of the primer pairs could generate the target bands with length polymorphism, and these markers would be very powerful when they are used for genetic populations derived from various types of maize germplasms that were sampled for this study.
Keywords: simple sequence repeat, whole-genome sequences, polymorphic SSR markers, teosinte, maize
1. Introduction
Maize (Zea mays L.) is one of the most important food, feed, and industrial crops globally and a model system for the study of genetics, evolution, and domestication. The maize genome is large and complex. The estimated total size of genome draft is 2.3 Gb, with over 80% of repeated sequences of various types.1 The genetic variability in the maize genome can be utilized to enhance biotic and abiotic stress tolerance and to improve agronomic traits such as quality, maturity, and yield potential. Types of variation at the whole-genomic level include microsatellites or simple sequence repeats (SSRs), single-nucleotide polymorphisms (SNPs), insertions and deletions (indels), and various types of structure variation.
SSRs are tandemly repeated mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide sequence motifs flanked by unique sequences.2,3 The unique sequences bordering the SSR motifs provide templates for specific primers to amplify SSR alleles via polymerase chain reaction (PCR), and allelic differences are usually the result of variable numbers of repeat units within a microsatellite structure.4 A larger number of repeated units is generally related to greater genotypic variation, and the shorter motifs such as those with mono-, and di-nucleotides usually possess more repeats than longer motifs such as those with tetra-, penta-, and hexa-nucleotides. However, shorter motifs can produce more slipped-strand mispairing (stuttering) during PCR, which usually lead to genotyping errors.5,6 Based on the repetitive architecture, purity, and complexity of their motifs, SSRs can be classified as perfect (single motif in an uninterrupted array), imperfect, or compound (two or more motifs in interrupted or uninterrupted arrays). As we known, SSR loci with longer or perfect motifs can exhibit a higher level of allelic variability.5
SSRs have been the genetic markers of choice, because they are easy to score, and have multiallelic nature, co-dominant inheritance, and clear advantages over restriction fragment length polymorphism and amplified fragment length polymorphism markers in terms of technical simplicity, throughput level and automation.7 Compared with SNP markers that are generally biallelic,8 SSR markers are more informative because it can detect multiple alleles per locus, so they are still commonly used nowadays.
Thanks to the availability of whole-genome or transcriptome sequences in public databases and in the recent advent of bioinformatics tools, development of genetic markers including SSRs has become much easier and more cost-effective. Genetic markers can be obtained by screening genomic, cDNA sequences, or libraries of clones. To facilitate access to and utilization of SSR markers in Brachypodium, 27 329 SSR markers were successfully designed through genome-wide analysis, but only 398 SSR markers have been developed from its bacterial artificial chromosomes end and expressed sequence tag databases.9 The availability of the completed soybean whole-genome sequence also provided an ideal resource for the genome-wide development of locus-specific SSR markers, and 33 065 high-polymorphic SSRs were developed with the availability of their genome positions and primer sequences.10 Barchi et al.11 combined the recently developed a restriction site-associated DNA approach with Illumina DNA sequencing to rapidly discover a large number of SNP and SSR markers for eggplant. Huang et al.12 identified over 3.6 million SNPs by sequencing 517 rice landraces, which were used in genome-wide association studies for 14 agronomic traits. These results show that genetic markers such as SSRs and SNPs are abundant in different crop genomes and can be easily scored, making it more accessible to the breeders and geneticists.
SSRs are abundant and well distributed throughout the maize genome, which can be employed as a preferred marker system. SSR markers have been utilized extensively in maize to characterize the genetic structure and diversity, to construct phylogenetic trees and to define potential heterotic groups, and to identify unique sources of allelic diversity.13–15 Furthermore, SSR markers have been widely used for genetic map construction, quantitative trait locus (QTL) mapping, map-based cloning, and marker-assisted selection (MAS) because of their ubiquity and high level of polymorphism. Hence, enriching the current maize linkage maps with more SSR markers is of great value for the global maize molecular breeding.
In recent years, many SSR markers have been developed and are publicly available (http://www.maizegdb.org/ssr.php) based on their target sequences among different maize germplasm accessions. However, a relatively low level of polymorphism was observed between cultivated maize and their relatives, and within populations derived from cultivated × teosinte and temperate × tropical maize crosses. The availability of the reference genome sequence and increasingly cost-effective sequencing facilities makes it possible to do whole-genome sequencing for more maize germplasm accessions. We used whole-genome sequence information from 3 typical tropical maize inbreds, 13 typical temperate maize inbreds from different heterotic groups, and 1 teosinte line, to analyse their genetic variation and to develop polymorphic molecular markers that can be used for high-resolution MAS, genomic selection, and QTL mapping. Using germplasm of diverse resources including teosinte and different types of maize lines, we can reveal and utilize unique alleles and loci better. Thus, the objectives of this study were to determine the abundance and characterization of SSRs in the maize genome and to use stringent screening to develop highly polymorphic SSR markers.
2. Materials and methods
2.1. Plant materials
Sequence data were generated for 17 genotypes including 16 improved maize inbred lines and 1 wild relative, Z. mays ssp. mexicana (hereafter Z. mexicana), which were listed in Table 1. Among the 16 improved maize inbreds, CML411 and P1 from International Maize and Wheat Improvement Center (CIMMYT) and 81565 from China were chosen to represent tropical/subtropical germplasm, ES40 was derived from traditional Chinese landrace, and the remaining 12 temperate maize inbreds were chosen to represent different heterotic groups in Chinese temperate maize. Temperate maize lines 178, Huangzao4, Ye478, Zheng22, and B73 representing PB, SPT, PA, LRC, and BSSS heterotic groups, respectively, were widely used for commercial hybrid production. For marker validation, additional six teosinte species obtained from United States Department of Agriculture (USDA) and four maize inbreds were also included (Table 1).
Table 1.
Maize genotypes used in the study
| Genotypes | Pedigree | Adaptation |
|---|---|---|
| 178a | Selected from an introduced hybrid | Temperate |
| 18Reda | American hybrid P78599 | Temperate |
| 18Whitea | American hybrid P78599 | Temperate |
| 48-2a | Synthesized population | Temperate |
| A318 | Improved S37 | Tropical |
| B73a | BSSS | Temperate |
| Dan598a | (Dan340 × Danhuang11) × (Danhuang02 × Dan599) | Temperate |
| ES40a | Landrace Linshuidadudu selected from Sichuan | Temperate |
| Han21a | American hybrid P78599 | Temperate |
| Huangzao4a | Improved from Landrace, TangSiPingTou | Temperate |
| Lu9801 | Ye502 × H21 | Temperate |
| Mo17a | C103 × 187-2 | Temperate |
| RP125a | Derived from hybrid Chuandan9 | Temperate |
| Ye478a | U8112 × Shen5003 | Temperate |
| Zheng22a | (Duqing × E28) × Lu Jiu Kuan | Temperate |
| 81565a | (Huobai × Jin03)S2 × Heibai94 | Tropical/subtropical |
| CML411a | P28C7-S4-#-BBBBBBBBBBB | Tropical/subtropical |
| CML206 | [EV7992#/EVPO44SRBC3]#BF37SR-2-3SR-2-4-3-BB | Tropical/subtropical |
| CML85 | P34C5F21-2-#1-2-2-# | Tropical/subtropical |
| P1a | Unknown | Tropical/subtropical |
| Z. mexicanaa | Zea mays ssp. mexicana | Tropical |
| Z. parviglumis | Zea mays ssp. parviglumis | Tropical |
| Z. huehuetenangensis | Zea mays ssp. huehuetenangensis | Tropical |
| Z. nicaraguensis | Zea nicaraguensis | Tropical |
| Z. luxurians | Zea luxurians | Tropical |
| Z. perennis | Zea perennis | Tropical |
| Z. diploperennis | Zea diploperennis | Tropical |
All the materials were used for experimental validation.
aMaterials were only used for SSR identification and markers development. The chromosome number is 2n = 40 for Z. perennis and 2n = 20 for other species.
2.2. Maize genome sequences
For maize lines 81565, 18Red, 18White, 48-2, Dan598, ES40, RP125, and Z. mexicana, sequences were generated, and paired-end libraries were constructed according to the Illumina manufacturer's instructions. An average resequencing depth was 13× and genome coverage was 85% for maize inbreds. One teosinte species Mexican was sequenced with an average of resequencing depth of 9× and genome coverage of 74%. The genome sequences for the remaining maize lines were downloaded from the NCBI Sequence Read Archive (SRA) database (SRA049859 and SRA051245) and NCBI GenBank (JQ886798–JQ887980). All sequence reads were aligned against the maize B73 reference genome (www.maizesequence.org Release 4a.53) using Short Oligonucleotide Alignment Program 2 (http://soap.genomics.org.cn/). Sequencing and reads mapping were carried out at Beijing Genomics Institute (Shenzhen, China).16–18
2.3. SSR identification and primer design
SSR motifs were identified in 17 genomes using MISA (MIcroSAtellite identification tool) program downloaded from the Leibniz Institute of Plant Genetics and Crop Plant Research website (http://pgrc.ipk-gatersleben.de/misa/). Only perfect SSRs including mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide motifs with numbers of uninterrupted repeat units more than 10, 7, 6, 5, 4, and 4, respectively, were targeted. The 5′- and 3′-untranslated regions (UTR), protein coding sequence (CDS), intron, and intergenic regions were determined based on their original annotations of the maize B73 reference genome (www.maizesequence.org Release 4a.53). Promoter sequences were determined at 2 kb upstream of the transcription initiation site.
Any SSR locus to be used to develop genetic markers should include a perfect repeat motif and two unique flanking sequences with 300 bp on each sides of the repeat. In our study, SSR candidate sequences were used for BLASTN search against the genome sequences (e-value cut-off of 1e−10), and filtered with >90% of identity and minimum alignment length with >85% of the flanking sequences. Those with unique hit, together with their specific flanking sequences, were identified as candidate SSR loci. Then, we wrote a Perl script to combine SSRs within 5 kb of different genomes with the same motif and to identify polymorphic SSR loci among 17 genotypes depending on the presence of motifs.
The forward and reverse primers were designed based on unique flanking sequences using Primer 3 (http://primer3.sourceforge.net/). Input parameters for the primer design were as follows: minimum, maximum, and optimal sizes were 18, 27, and 20 nt; minimum and maximum GC were 20 and 80%; and minimum, maximum, and optimal Tm were 57, 63, and 60°C, respectively. The deviation of amplicon size of each SSR primer ranged from 30 to 500 bp based on the expected SSR sequence length.
In addition, electronic polymerase chain reaction (e-PCR) programme (http://www.ncbi.nlm.nih.gov/projects/e-pcr/) was applied to check the uniqueness and specificity of designed primers in the genomes. The parameters were set as following: the word size was 9, the discontiguous word was 1, the maximal allowed deviation of hit product size was 100, the maximum mismatches allowed, and the maximum indels allowed were 1, respectively. On the other hand, the published SSR markers reposited in MaizeGDB (http://www.maizegdb.org/) were downloaded and amplified in silicon through e-PCR programme for further comparison.
2.4. Experimental validation of polymorphic SSR markers
To assess the value of identified SSR markers, 151 primer pairs from 10 chromosomes including all types of SSR were chosen for experimental validation. The samples used in this experiment included 20 improved maize lines and 7 teosinte lines (Table 1). Genomic DNA was extracted from seedlings using the CTAB method. Primers were made by Shanghai DNA Biotechnologies Co., Ltd. PCR was performed in 25 μl reactions containing 2.5 μl buffer, 2.5 μl MgCl2 (25 mM), 4.0 μl dNTP (2.5 mM), 0.2 μl Taq polymerase (5 U/μl), 1 μl template DNA (100 ng/μl),13.8 μl ddH2O, and 0.1 μg primers. The PCR conditions were as follows: 1 cycle at 94°C for 5 min; 35 cycles at 94°C for 30 s, 60°C for 30 s, 72°C for 1 min, and 1 cycle at 72°C for 10 min. PCR products mixed with loading buffer were heated at 95°C for 5 min and quickly chilled on ice. The entire mixture was electrophoresed on 6% denaturing polyacrylamide gel, and the genotype was scored after silver staining. The number of alleles was recorded and the polymorphism information content (PIC) was calculated as described by Smith et al.19
3. Results
3.1. The abundance of SSRs in the maize genome
A large number of perfect SSRs with mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide motifs were identified, but the numbers varied among different genomes (Table 2). The average number of SSRs was 135 693 in 17 genotypes, ranging from 133 346 loci observed in mexicana to 136 723 loci in tropical/subtropical maize inbred 81565. Some reads from Z. mexicana could not be mapped onto the reference genome, which resulted in relatively lower genome coverage and thus, less SSRs identified compared with other maize inbreds. A total of 264 658 unique SSR loci were detected in 17 genomes, of which mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide SSRs were 153 231, 65 236, 25 910, 6572, 8839, and 4870, respectively. The mono-nucleotide motif is the most abundant, accounted for 57.90%. There were 38 971 common SSRs (15% of the total) observed to be the same across 17 genotypes. The SSR density was calculated based on the maize reference genome size of 2.1 Gb, and there was a little difference among 17 genotypes for each nucleotide motif, with an average interval of 15.48 kb between SSR loci for every genome. However, the average intervals for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide SSRs were remarkably different, which were 26.93, 60.88, 150.95, 717.70, 505.05, and 942.55 kb, respectively (Table 2). SSRs were considerably abundant and distributed throughout the maize genome, with a small average marker interval (7.93 kb) for all detected loci.
Table 2.
Numbers and density of SSR loci identified in 17 maize genomes
| Genotypes | SSR numbers |
SSR interval (kb) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MNR | DNR | TNR | TTR | PNR | HNR | Total | MNR | DNR | TNR | TTR | PNR | HNR | Total | |
| 178 | 78 367 | 34 604 | 13 929 | 2 964 | 4172 | 2226 | 136 262 | 26.80 | 60.69 | 150.76 | 708.50 | 503.36 | 943.40 | 15.41 |
| 81565 | 79 042 | 34 457 | 13 893 | 2929 | 4178 | 2224 | 136 723 | 26.57 | 60.95 | 151.16 | 716.97 | 502.63 | 944.24 | 15.36 |
| 18White | 77 144 | 34 451 | 13 931 | 2928 | 4166 | 2217 | 134 837 | 27.22 | 60.96 | 150.74 | 717.21 | 504.08 | 947.23 | 15.57 |
| 18Red | 77 049 | 34 495 | 13 919 | 2933 | 4188 | 2221 | 134 805 | 27.26 | 60.88 | 150.87 | 715.99 | 501.43 | 945.52 | 15.58 |
| 48-2 | 76 920 | 34 448 | 13 884 | 2924 | 4123 | 2219 | 134 518 | 27.30 | 60.96 | 151.25 | 718.19 | 509.34 | 946.37 | 15.61 |
| B73 | 77 888 | 34 755 | 14 028 | 2948 | 4181 | 2239 | 136 039 | 26.96 | 60.42 | 149.70 | 712.35 | 502.27 | 937.92 | 15.44 |
| CML411 | 78 591 | 34 546 | 13 900 | 2935 | 4189 | 2225 | 136 386 | 26.72 | 60.79 | 151.08 | 715.50 | 501.31 | 943.82 | 15.40 |
| Dan598 | 78 558 | 34 559 | 13 954 | 2904 | 4159 | 2227 | 136 361 | 26.73 | 60.77 | 150.49 | 723.14 | 504.93 | 942.97 | 15.40 |
| ES40 | 78 978 | 34 372 | 13 894 | 2924 | 4161 | 2221 | 136 550 | 26.59 | 61.10 | 151.14 | 718.19 | 504.69 | 945.52 | 15.38 |
| Han21 | 78 539 | 34 584 | 13 951 | 2898 | 4157 | 2233 | 136 362 | 26.74 | 60.72 | 150.53 | 724.64 | 505.17 | 940.44 | 15.40 |
| Huangzao4 | 76 773 | 34 445 | 13 878 | 2907 | 4165 | 2224 | 134 392 | 27.35 | 60.97 | 151.32 | 722.39 | 504.20 | 944.24 | 15.63 |
| Mo17 | 78 360 | 34 442 | 13 910 | 2902 | 4108 | 2220 | 135 942 | 26.80 | 60.97 | 150.97 | 723.64 | 511.20 | 945.95 | 15.45 |
| P1 | 78 975 | 34 447 | 13 849 | 2961 | 4146 | 2235 | 136 613 | 26.59 | 60.96 | 151.64 | 709.22 | 506.51 | 939.60 | 15.37 |
| RP125 | 76 880 | 34 523 | 13 934 | 2929 | 4198 | 2244 | 134 708 | 27.32 | 60.83 | 150.71 | 716.97 | 500.24 | 935.83 | 15.59 |
| Z. mexicana | 75 997 | 34 339 | 13 806 | 2886 | 4085 | 2233 | 133 346 | 27.63 | 61.15 | 152.11 | 727.65 | 514.08 | 940.44 | 15.75 |
| Ye478 | 78 571 | 34 508 | 13 973 | 2944 | 4159 | 2234 | 136 389 | 26.73 | 60.86 | 150.29 | 713.32 | 504.93 | 940.02 | 15.40 |
| Zheng22 | 78 921 | 34 455 | 13 870 | 2920 | 4156 | 2230 | 136 552 | 26.61 | 60.95 | 151.41 | 719.18 | 505.29 | 941.70 | 15.38 |
| Average | 77 974 | 34 496 | 13 912 | 2926 | 4158 | 2228 | 135 693 | 26.93 | 60.88 | 150.95 | 717.70 | 505.05 | 942.55 | 15.48 |
| Total | 153 231 | 65 236 | 25 910 | 6572 | 8839 | 4870 | 264 658 | 13.70 | 32.19 | 81.05 | 319.54 | 237.58 | 431.21 | 7.93 |
| Common | 22 453 | 9963 | 4603 | 553 | 929 | 470 | 38 971 | 93.53 | 210.78 | 456.22 | 3797.47 | 2260.50 | 4468.09 | 53.89 |
MNR, DNR, TNR, TTR, PNR, and HNR indicate mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide SSRs.
We also examined different SSR repeat types in the genome for all tested genotypes. The frequencies of different nucleotide repeat types in each motif were different, but they showed similar frequency patterns in different genomes. Here, we compared the frequencies of different SSR repeat types by taking the reference line B73 as an example (Table 3). Of mononucleotide motifs, C/G repeats accounted for ∼54.4%, which was slightly higher than A/T repeats. Of the di-nucleotide motifs, (AT)n were most frequent (24.93%), followed by (AG/CT)n (24.36%), (TA)n (21.03%), and (GA/TC) (20.64%), while the (CG)n motif was least frequent (0.80%). Of the tri-nucleotide motifs, (CAG/CTG)n was the most abundant (15.86%), while other nucleotide repeat types had lower frequencies (0.4–6%). Of the tetra-nucleotide SSRs, (AAAT/ATTT)n, was most frequent (10.35%), and the frequencies for the rest nucleotide repeat types were all lower than 7%. There were many types of penta- and hexa-nucleotide SSRs, each with low frequencies, ranging from 0.04 to 4%. The numbers of mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide motifs in different repeat unit classes are also listed in Table 3. The average repeat lengths were different among various motifs ranging from 11.02 for (A/T)n to 58.84 for (AGT/ACT)n.
Table 3.
Number of SSRs in different repeat classes in the maize genome B73
| Motifs | Repeats number |
Total | Average repeat number | Average repeat length (bp) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <5 | 5–7 | 8–10 | 11–15 | 16–20 | 21–25 | 26–30 | 31–40 | >40 | ||||
| G/C | 0 | 0 | 13 085 | 24 297 | 3 832 | 866 | 243 | 45 | 1 | 42 369 | 12.31 | 12.31 |
| A/T | 0 | 0 | 22 410 | 11 687 | 1114 | 192 | 57 | 32 | 27 | 35 519 | 11.02 | 11.02 |
| AT | 0 | 1875 | 3268 | 1379 | 708 | 478 | 378 | 466 | 111 | 8663 | 13.28 | 26.56 |
| CT/AG | 0 | 3615 | 3504 | 740 | 265 | 127 | 72 | 95 | 47 | 8465 | 9.42 | 18.84 |
| TA | 0 | 1725 | 2417 | 1087 | 625 | 465 | 364 | 492 | 134 | 7309 | 14.03 | 28.05 |
| GA/TC | 0 | 3220 | 2938 | 519 | 237 | 103 | 71 | 57 | 28 | 7173 | 9.19 | 18.39 |
| CA/TG | 0 | 666 | 542 | 72 | 15 | 5 | 1 | 5 | 1 | 1307 | 8.24 | 16.48 |
| GT/AC | 0 | 582 | 588 | 73 | 12 | 5 | 4 | 4 | 0 | 1268 | 8.32 | 16.64 |
| GC | 0 | 223 | 66 | 3 | 3 | 0 | 0 | 0 | 0 | 295 | 7.42 | 14.83 |
| CG | 0 | 219 | 55 | 0 | 1 | 0 | 0 | 0 | 0 | 275 | 7.29 | 14.59 |
| CAG/CTG | 0 | 1980 | 242 | 3 | 0 | 0 | 0 | 0 | 0 | 2225 | 6.5 | 19.51 |
| GAT/ATC | 0 | 526 | 264 | 46 | 5 | 1 | 1 | 0 | 0 | 843 | 7.49 | 22.48 |
| GCA/TGC | 0 | 711 | 99 | 0 | 0 | 0 | 0 | 0 | 0 | 810 | 6.42 | 19.25 |
| GAC/GTC | 0 | 587 | 184 | 28 | 1 | 0 | 0 | 0 | 0 | 800 | 7.02 | 21.06 |
| ATT/AAT | 0 | 352 | 124 | 65 | 42 | 28 | 18 | 10 | 4 | 643 | 10.25 | 30.74 |
| TTA/TAA | 0 | 359 | 96 | 66 | 32 | 27 | 24 | 14 | 2 | 620 | 10.31 | 30.94 |
| CGT/ACG | 0 | 429 | 131 | 34 | 0 | 0 | 0 | 0 | 0 | 594 | 7.18 | 21.55 |
| TGA/TCA | 0 | 350 | 167 | 58 | 9 | 3 | 1 | 0 | 0 | 588 | 7.86 | 23.59 |
| CGA/TCG | 0 | 474 | 100 | 4 | 0 | 0 | 0 | 0 | 0 | 578 | 6.69 | 20.06 |
| CGC/GCG | 0 | 505 | 49 | 4 | 1 | 0 | 0 | 0 | 0 | 559 | 6.5 | 19.51 |
| GCC/GGC | 0 | 490 | 42 | 5 | 0 | 0 | 0 | 0 | 0 | 537 | 6.49 | 19.46 |
| TAT/ATA | 0 | 246 | 103 | 43 | 47 | 35 | 13 | 13 | 5 | 505 | 11.39 | 34.16 |
| CGG/CCG | 0 | 416 | 48 | 3 | 0 | 0 | 0 | 0 | 0 | 467 | 6.5 | 19.51 |
| TAC/GTA | 0 | 232 | 50 | 26 | 16 | 6 | 1 | 5 | 1 | 337 | 8.54 | 25.63 |
| TTG/CAA | 0 | 230 | 80 | 16 | 4 | 0 | 0 | 0 | 0 | 330 | 7.32 | 21.96 |
| TTC/GAA | 0 | 288 | 26 | 9 | 0 | 0 | 1 | 2 | 3 | 329 | 7.42 | 22.26 |
| ATG/CAT | 0 | 260 | 45 | 11 | 4 | 0 | 1 | 0 | 0 | 321 | 7.01 | 21.04 |
| GCT/AGC | 0 | 291 | 16 | 1 | 1 | 0 | 0 | 0 | 0 | 309 | 6.36 | 19.09 |
| TGG/CCA | 0 | 280 | 24 | 2 | 0 | 0 | 0 | 0 | 0 | 306 | 6.4 | 19.2 |
| TAG/CTA | 0 | 186 | 49 | 26 | 11 | 10 | 8 | 3 | 3 | 296 | 9.63 | 28.9 |
| CTC/GAG | 0 | 232 | 46 | 6 | 0 | 0 | 0 | 0 | 0 | 284 | 6.82 | 20.45 |
| CTT/AAG | 0 | 245 | 31 | 1 | 1 | 1 | 0 | 1 | 1 | 281 | 7 | 21 |
| TCC/GGA | 0 | 193 | 43 | 8 | 1 | 0 | 0 | 0 | 0 | 245 | 6.91 | 20.74 |
| CCT/AGG | 0 | 187 | 32 | 5 | 2 | 1 | 0 | 0 | 0 | 227 | 6.93 | 20.79 |
| CAC/GTG | 0 | 198 | 18 | 0 | 1 | 0 | 0 | 0 | 0 | 217 | 6.46 | 19.38 |
| ACC/GGT | 0 | 191 | 16 | 1 | 0 | 0 | 0 | 0 | 0 | 208 | 6.39 | 19.17 |
| ACA/TGT | 0 | 125 | 58 | 10 | 0 | 0 | 1 | 0 | 0 | 194 | 7.34 | 22.02 |
| AGA/TCT | 0 | 169 | 21 | 2 | 0 | 1 | 0 | 0 | 0 | 193 | 6.58 | 19.74 |
| GTT/AAC | 0 | 85 | 29 | 4 | 0 | 1 | 1 | 0 | 0 | 120 | 7.45 | 22.35 |
| AAAT/ATTT | 0 | 288 | 15 | 2 | 0 | 0 | 0 | 0 | 0 | 305 | 5.58 | 22.33 |
| AGGC/GCCT | 0 | 153 | 11 | 4 | 0 | 0 | 0 | 0 | 0 | 168 | 5.9 | 23.6 |
| TATT/AATA | 0 | 195 | 10 | 2 | 0 | 0 | 0 | 0 | 0 | 207 | 5.72 | 22.88 |
| TCGT/ACGA | 0 | 126 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 126 | 5.07 | 20.29 |
| TTAT/ATAA | 0 | 111 | 16 | 0 | 0 | 0 | 0 | 0 | 0 | 127 | 5.88 | 23.53 |
| TTTA/TAAA | 0 | 157 | 7 | 2 | 0 | 0 | 0 | 0 | 0 | 166 | 5.67 | 22.67 |
| CGAGC/GCTCG | 153 | 24 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 177 | 4.16 | 24.95 |
| TTTTA/TAAAA | 82 | 28 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 110 | 4.29 | 25.75 |
| ATTTT/AAAAT | 69 | 35 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 105 | 4.42 | 22.1 |
SSR motifs with repeats number >100 in total were listed here.
3.2. Screening of SSR loci and development of maize SSR markers
A total of 2034 SSR markers have been recently developed and reported on MaizeGDB website (www.maizegdb.org). Among the public markers, 1556 SSRs have genomic positions. Through e-PCR programme conducted in B73 genome, 827 SSR markers have specific amplicon, 60 SSR markers have more than one binding sites, and the remaining markers have no proper binding sites on the 10 chromosomes. Here, we developed a new database containing more SSR markers with unique flanking sequences. From the SSRs that could be detected (264 658) across 17 maize genomes, 189 087 (71.45%) of them were identified with unique flanking sequences with an average of 82 741.9 SSR loci for each genome (Table 4). The average numbers of SSR loci with different motifs for each genome were notably different, accounting for 55.19, 74.60, 48.09, 81.94, 80.69, and 68.94% of the total SSRs for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide motifs, respectively (Table 4). It implies that over 80% of tetra- and penta- nucleotide motifs in the maize genome can be used to design SSR markers. A total of 25 437 SSRs with unique flanking sequences were found to be shared across 17 tested genomes, of which 9240 (36.33%) were polymorphic.
Table 4.
Summary of SSR loci with unique flanking sequences identified in tested maize genomes
| Motifs | Average SSRs |
Total SSRs |
Common SSRs |
Common SSRs with polymorphism |
||||
|---|---|---|---|---|---|---|---|---|
| No. | %a | No. | %b | No. | %c | No. | % | |
| MNR | 43 030.4 | 55.19 | 103 486 | 67.54 | 12 029 | 54.31 | 4588 | 38.14 |
| DNR | 25 733.2 | 74.6 | 52 876 | 81.05 | 8630 | 79.87 | 3776 | 43.75 |
| TNR | 6689.9 | 48.09 | 15 946 | 61.54 | 2755 | 59.53 | 560 | 20.33 |
| TTR | 2397.2 | 81.94 | 5658 | 86.09 | 652 | 83.27 | 142 | 21.78 |
| PNR | 3355.4 | 80.69 | 7481 | 84.64 | 950 | 83.11 | 129 | 13.58 |
| HNR | 1535.8 | 68.94 | 3640 | 74.74 | 422 | 74.17 | 45 | 10.66 |
| Total | 82 741.9 | 60.98 | 189 087 | 71.45 | 25 437 | 63.47 | 9240 | 36.33 |
MNR, DNR, TNR, TTR, PNR, and HNR indicate mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide SSRs.
aPercentage of the average number of SSRs with unique flanking sequences against all for every tested maize genome.
bPercentage of total SSR number with unique flanking sequences against all identified in 17 maize lines.
cPercentage of the common loci against all that are the same in 17 maize lines.
Of 189 087 candidate SSRs, 188 571 loci have specific physical position and would be developed as genetic markers in the study. Primer pairs were then designed for the 188 571 SSR loci, with 13 344 (chromosome 10) to 29 779 (chromosome 1) SSRs on each chromosome, and 173 587 of them were polymorphic with length differences and present-absent variation in 17 genomes. E-PCR programme was further conducted to validate and refine the specificity of new designed SSR markers, and 111 887 primer pairs of them could bind as expected and the others were amplified with multiple binding sites or false match. Through comparing SSR sequences among 17 tested genomes, a new database was developed to include 111 887 SSR markers with specific physical positions, with proportion of 59% of the candidate SSR loci with specific flanking sequences (Table 5 and Supplementary Table S1). Among these markers, SSRs with mono-, di-, tri-, tetra-, penta-, and hexa- nucleotide motifs accounted for 58.00, 26.09, 7.20, 3.00, 3.93, and 1.78%, respectively. A total of 35 573 SSR loci, accounting for 31.8% of the refined SSR markers, showed length polymorphism in the 17 tested genotypes. The PIC for these polymorphic SSRs varied from 0.05 to 0.83, with an average of 0.31 (Supplementary Table S1). SSR markers with mono- and di-nucleotide motifs showed higher levels of polymorphism (33.87 and 37.31%, respectively) than other SSR markers with tetra-, penta-, and hexa-nucleotide motifs (7.44–17.19%). Comparing with the SSR markers in MaizeGDB database, there were 18 606 SSR markers, accounting for 16.6% of the newly developed SSR markers, shared the same loci with the public SSR markers with various motifs. However, only 527 (0.47%) newly developed SSR markers had completely compatible position with public SSR primers. Additionally, the average SSR lengths and number of loci across 10 chromosomes were calculated for three SSR datasets, all SSRs, SSRs with unique flanking sequences, and polymorphic SSRs (Fig. 1). In each of the three SSR datasets, the numbers of loci gradually declined with the increase of SSR lengths, the same as shown in previous studies.20
Table 5.
Numbers of candidate SSR markers, and polymorphic SSR markers detected in 17 maize lines and previously developed SSR in MaizeGDB database
| Chr | Candidate SSR markers |
Poly. (%)a | SSRs in MaizeGDB database | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MNR |
DNR |
TNR |
TTR |
PNR |
HNR |
|||||||||
| Mono- | Poly- | Mono- | Poly- | Mono- | Poly- | Mono- | Poly- | Mono- | Poly- | Mono- | Poly- | |||
| 1 | 6957 | 3422 | 2958 | 1737 | 1129 | 242 | 439 | 84 | 643 | 73 | 304 | 12 | 30.94 | 293 |
| 2 | 4875 | 2488 | 2118 | 1212 | 697 | 157 | 294 | 76 | 418 | 46 | 203 | 17 | 31.71 | 226 |
| 3 | 5079 | 2414 | 2289 | 1217 | 777 | 177 | 330 | 69 | 514 | 49 | 209 | 17 | 30.01 | 224 |
| 4 | 4831 | 2331 | 2246 | 1264 | 774 | 166 | 331 | 73 | 422 | 45 | 192 | 23 | 30.73 | 141 |
| 5 | 4623 | 2498 | 1851 | 1140 | 681 | 164 | 276 | 67 | 461 | 55 | 198 | 18 | 32.76 | 146 |
| 6 | 3501 | 1816 | 1499 | 844 | 579 | 118 | 248 | 44 | 345 | 25 | 158 | 12 | 31.11 | 111 |
| 7 | 3548 | 1870 | 1473 | 929 | 505 | 150 | 271 | 41 | 285 | 44 | 150 | 12 | 32.83 | 112 |
| 8 | 3553 | 1845 | 1391 | 901 | 504 | 118 | 185 | 32 | 315 | 50 | 180 | 13 | 32.56 | 118 |
| 9 | 3059 | 1726 | 1262 | 847 | 451 | 100 | 203 | 39 | 297 | 40 | 137 | 16 | 33.85 | 102 |
| 10 | 2884 | 1572 | 1214 | 803 | 436 | 127 | 203 | 52 | 248 | 26 | 111 | 8 | 33.68 | 83 |
| Total | 42 910 | 21 982 | 18 301 | 10 894 | 6533 | 1519 | 2780 | 577 | 3948 | 453 | 1842 | 148 | 31.79 | 1556 |
MNR, DNR, TNR, TTR, PNR, and HNR indicate mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide SSRs.
Mono: monomorphism; poly: polymorphism; Chr: chromosome.
aPercent of polymorphic SSR markers over all of candidate SSR markers in silicon analysis.
Figure 1.
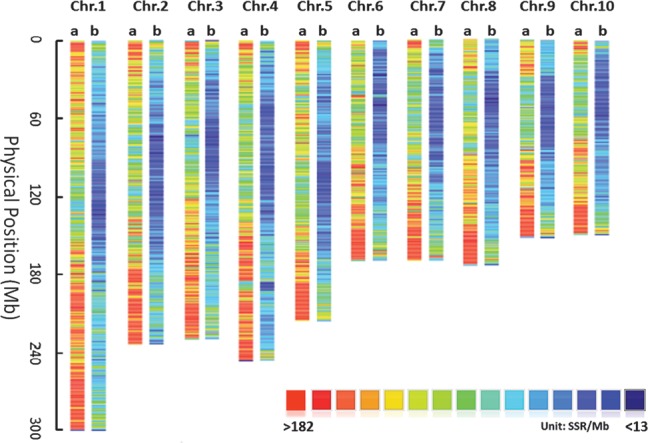
Distributions of 263,423 SSR loci (a) and 111,887 new developed SSR markers (b) with unique physical positions across 10 chromosomes in the B73 reference genome (www.maizesequence.org Release 4a.53). Different colors represent levels of density of SSRs.
3.3. Distribution of SSRs in different genomic regions
A total of 264 658 SSRs were detected in 17 genomes, and 263 423 loci of them have specific physical position. The distributions of 263 423 SSR loci and 111 887 newly developed SSR markers refined by e-PCR programme across 17 tested genomes were shown in Fig. 2 a and b , respectively. SSRs were unevenly distributed on chromosome regions, and there were much more loci located in near telomeric regions than near centromeres, which was accordance with the distribution patterns of genes in maize.21 Moreover, we compared SSR distributions across five genomic regions using tested genomes of P1, B73, and Z. mexicana to represent tropical, temperate, and wild maize germplasm, respectively (Table 6). SSR loci were most abundant in intergenic region and least frequent in UTR region. Polymorphism rate and GC content of SSRs in coding regions were higher than other genic regions.
Figure 2.

Correlation between SSR numbers and SSR lengths.
Table 6.
The distribution of SSRs in different genomic regions
| All SSR loci |
SSR loci with unique flanking sequences |
SSR loci with polymorphism |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Count | Interval (kb) | Length (bp) | GC% | Count | Interval (kb) | Length (bp) | GC% | Count | Interval (kb) | Length (bp) | GC% | |
| B73 | ||||||||||||
| 5′-UTR | 3804 | 6.91 | 16.10 | 32.20 | 3601 | 7.30 | 16.19 | 32.60 | 1335 | 19.69 | 16.23 | 26.55 |
| 3′-UTR | 3829 | 6.87 | 16.13 | 33.68 | 3628 | 7.25 | 16.22 | 33.94 | 1341 | 19.62 | 15.90 | 26.70 |
| CDS | 2553 | 17.27 | 19.80 | 74.82 | 2391 | 23.10 | 19.87 | 75.28 | 309 | 178.73 | 21.28 | 56.84 |
| Intron | 11 857 | 10.13 | 16.03 | 29.94 | 11 038 | 10.88 | 16.12 | 29.97 | 4704 | 25.54 | 16.70 | 29.60 |
| Promotors | 10 370 | 6.26 | 17.35 | 27.57 | 9248 | 7.02 | 17.54 | 26.71 | 3344 | 19.42 | 17.26 | 20.82 |
| Intergenic | 107 658 | 21.63 | 16.17 | 50.49 | 65 098 | 28.56 | 17.12 | 46.22 | 21 650 | 85.88 | 17.70 | 42.24 |
| Total/average | 136 039 | 15.15 | 16.33 | 47.01 | 90 441 | 22.79 | 17.11 | 42.62 | 30 900 | 66.70 | 17.49 | 37.61 |
| Z. Mexicana | ||||||||||||
| 5′-UTR | 2520 | 10.25 | 15.55 | 26.23 | 2263 | 11.42 | 15.66 | 24.10 | 1007 | 25.66 | 14.35 | 17.18 |
| 3′-UTR | 3329 | 7.76 | 17.34 | 38.34 | 3092 | 8.35 | 17.42 | 38.10 | 1100 | 23.48 | 15.81 | 23.81 |
| CDS | 3636 | 14.29 | 17.70 | 45.01 | 3252 | 15.98 | 17.93 | 43.96 | 954 | 54.47 | 16.14 | 23.08 |
| Intron | 8102 | 14.23 | 16.24 | 32.41 | 7168 | 16.08 | 16.48 | 30.43 | 3035 | 37.98 | 15.63 | 24.71 |
| Promotors | 10 903 | 5.86 | 17.79 | 34.35 | 10 035 | 6.37 | 17.95 | 34.04 | 3627 | 17.62 | 16.32 | 21.71 |
| Intergenic | 107 785 | 16.50 | 16.18 | 49.95 | 57 813 | 30.76 | 17.51 | 42.07 | 22 413 | 79.33 | 16.64 | 38.84 |
| Total/average | 133 346 | 15.46 | 16.38 | 47.13 | 80 545 | 25.59 | 17.50 | 39.82 | 30 787 | 66.94 | 16.45 | 34.45 |
| P1 | ||||||||||||
| 5′-UTR | 2684 | 9.60 | 15.27 | 24.75 | 2419 | 10.65 | 15.37 | 22.50 | 926 | 27.82 | 15.44 | 20.02 |
| 3’-UTR | 3478 | 7.41 | 17.04 | 37.07 | 3230 | 7.98 | 17.11 | 36.90 | 1044 | 24.68 | 16.96 | 29.53 |
| CDS | 3724 | 13.91 | 17.44 | 44.49 | 3351 | 15.46 | 17.63 | 43.48 | 911 | 56.86 | 17.51 | 28.36 |
| Intron | 8622 | 13.32 | 15.96 | 31.18 | 7659 | 14.99 | 16.14 | 29.13 | 3073 | 37.36 | 16.28 | 28.44 |
| Promotors | 11 218 | 5.68 | 17.56 | 33.82 | 10 316 | 6.18 | 17.67 | 33.50 | 3486 | 18.28 | 17.56 | 24.74 |
| Intergenic | 110 043 | 16.17 | 16.06 | 49.51 | 59 167 | 30.07 | 17.26 | 41.81 | 22 475 | 79.16 | 17.39 | 41.03 |
| Total/average | 136 613 | 15.09 | 16.24 | 46.58 | 82 831 | 24.88 | 17.24 | 39.38 | 30 621 | 67.31 | 17.28 | 37.09 |
The average intervals between SSRs were the longest in intergenic regions, second in CDS regions, and smallest in promotors (Table 6). Distributions for the SSRs with unique flanking sequences and for the polymorphic SSRs across tested genomes also varied among the six genomic regions, but the trend was consistent with that for all the candidate SSR loci. This result is also in full agreement with a previous report in rice.22 In addition, SSR distribution was rather similar among the three representative genotypes.
Furthermore, the repeat types of SSRs in CDS region of B73 were investigated. SSRs with tri-nucleotide repeats were the most (1832) among the six repeat types, with proportion of 71.8% in CDS region. The tri- and hexa-nucleotide SSRs that would not bring the frame shift accounted for 84.1% (2148) of the SSRs in CDS region. Therefore, only 15.9% of the SSRs in CDS region have potential threats to the gene structure.
3.4. SSR markers validated for quality and polymorphism
A total of 151 SSR markers were randomly chosen for experimental validation using 20 maize inbreds and 7 teosinte lines (Fig. 3 and Table 1). Of them, 121 primer pairs (80.1%) generated specific products and distinct bands, while 30 primer pairs failed to produce stable or clear bands due to the lack of sequence specificity in the genomic DNA samples. The majority of the 121 primer pairs (112 primer pairs) revealed high levels of allelic diversity in tested 27 lines, with PIC values of 0.074–0.796 (an average of 0.478). The 112 polymorphic SSR loci contained 329 alleles in total and an average of 2.94 alleles with a range of 2–5 (Supplementary Table S2).
Figure 3.

Experimental validation of six randomly selected SSR markers in 27 genotypes. Lanes 1–27 were PCR products of Zea perennis, Z. diploperennis, Z. mays ssp. parviglumis, Z. mays ssp. huehuetenangensis, Z. nicaraguensis, Z. luxurians, Z. mays ssp. mexicana, RP125, 18Red, 18White, CML206, 81565, A318, P1, Han21, CML85, CML411, Ye478, Mo17, Zheng22, 178, 48-2, B73, Lu9801, ES40, Huangzao4, and Dan598, respectively.
In addition, we made a detailed comparison of the allele number and PIC value in silicon analysis and in maker validation in 17 tested genotypes. Forty-one of the 121 primers possessed the practical alleles in accordance with the expected alleles, 38 primer pairs had more allele number, and 42 primer pairs had less allele number in silicon analysis than in maker validation (Supplementary Table S2). Additionally, comparing polymorphism in silicon analysis using 17 tested genomes and in maker validation using 27 genotypes, we found that 51 primer pairs showed more alleles and higher PIC values in validation experiment (Supplementary Table S2). Interestingly, 26 of 151 SSR markers with no polymorphism in silicon analysis showed more than one alleles in validation experiment. We also found that the length of PCR products in silicon analysis almost consist with those in marker validation (Supplementary Tables S1 and S2). The results indicate that newly developed SSR markers are informative and useful, and 70% of the SSR markers in our database are valid and polymorphic.
4. Discussion
SSRs are co-dominant, abundant, high polymorphic, and dispersed throughout plant genomes. Based on the survey across genomes, on average one SSR was found every 1.14 kb in Arabidopsis,23 3.6 kb in rice,22 4 kb in Brassica oleracea,24 4.5 kb in soybean,10 220 kb in sorghum,25 and 578 kb in wheat.26 In this study, average SSR density was one SSR every 15.48 kb. These may reflect real genetic differences existing among plant genomes at DNA level, and also the differences involved in sequencing methods and procedures. We used maize inbred B73 as the reference genome, some reads from maize wild relative, Z. mexicana, could not be mapped onto the reference, resulting in a relatively lower genome coverage and thus, less SSRs identified compared with other maize inbreds. Therefore, the number of SSR loci identified from Z. Mexicana may be underestimated.
In general, a small difference in SSR distribution was found for different populations or ecotypes in the same species. For instance, a very similar SSR distribution was found between indica and japonica rice, and the SSR density (interval between two SSRs) varied from one SSR every 2.0–8.1 kb, which was higher in 5′-UTR (one SSR every 2.1 and 2.0 kb, respectively) but low in CDS regions (one SSR every 8.1 and 7.7 kb, respectively).22 Our study revealed a similar SSR distribution pattern across the tested temperate, tropical, and wild maize lines. However, SSRs are not evenly distributed in different genomic regions with much lower SSR density in CDS region than in UTR and intronic regions. Intriguingly, we found that majority of SSRs resided in CDS region were tri-nucleotide repeats, which was consistent with other report and implied the specific selection against frame shift mutations in coding regions.27 Comparing with rice genome, the average SSR length was approximately identical (16–17 bp), but the average GC content in maize SSR sequences was much higher (47%) than rice (27%). In the maize genome, the proportions of mono-, di-, and tri-nucleotide SSR motifs were ∼60, 20, and 10%, respectively. Tetra-, penta-, and hexa-nucleotide SSR motifs were less abundant, together accounting for 10%, which was accordance with the report in rice.22 SSR densities for different motifs were also unbalanced and the average interval varied from 26 to 950 kb. Moreover, we found that C/G, AT, and CAG/CTG repeats were the most common for mono-, di-, and tri-nucleotide SSRs, respectively, in maize, while A/T, AG, and AGG/CCT repeats are the most common in rice.22 Meanwhile, AT repeats were also the most common dinucleotide motifs in sorghum.25
Short-read data from next-generation sequencing technologies are now being generated across a range of research projects. The fidelity of this data can be affected by several factors, and mapping errors and gaps still exist to a certain extent.28 However, the availability of the maize genome sequence still affords us a simple and economical way to survey and identify markers, thus enabling us to develop more convenient molecular markers for breeding applications. Several sets of maize germplasm including temperate, tropical, and their wild relatives were resequenced using next-generation sequencing technology.18,29–31 There are two major advantages in using currently available data for the analysis of SSR distribution and marker development. The maize germplasms from different ecological regions and heterotic groups (PB, SPT, PA, LRC, and BSSS) are highly diverse and host rare and unique alleles, providing an opportunity of using these types of genetic variation in hybrid maize breeding. On the other hand, whole-genome sequence data provide an ideal resource and the most complete picture of genetic variation for developing high-density genetic markers.
SSR markers of highly polymorphic among diverse germplasms provide some advantages in genetics and breeding applications. In spite of considerable efforts in developing molecular markers in maize, the number of SSRs publicly available is still limited. From >260 000 SSRs identified from 17 tested genomes, we detected 111 887 SSR loci with unique flanking sequence and single binding site through genome sequence blast and e-PCR analysis. These SSR loci can be developed as polymorphic markers in silico and public on the MaizeGDB database, which are ∼60 times more than those deposited in the MaizeGDB database so far. A total of 1556 SSR markers from the MaizeGDB database have specific location, and 16.6% of the newly developed SSR markers shared the same loci with public SSR markers. For some of the public SSR markers, the amplicon size was too large and it contained several newly developed SSR primers with different motifs. Therefore, only 0.47% (527) of newly developed SSR markers had completely compatible position with public SSR markers. Another reason for a few common SSRs shared with the two datasets maybe the traditional method for SSR marker development was based on screening of small-insert or microsatellite-enriched genomic libraries by hybridization in different materials,32 which was different from our analyses based on B73 reference genome and other resequenced genomes. Furthermore, the second-generation sequencing was different from the Sanger sequencing, which also lead to the differences. The experimental validation also proved to detect more alleles than the expected in silicon analysis due to diverse materials used in the study, but some SSR loci with little length differences were also hard to distinguish. The average marker density for the newly developed dataset reached one SSR per 14.7 kb in the B73 reference genome, indicating that maize is a highly polymorphic species.33 The availability of abundant SSR markers allows dramatic improvement in the efficiency of marker-assisted selection and fine mapping of QTL regions.
Previous studies have mainly focused on di-, tri-, and tetra-nucleotide SSRs, whereas mono-, penta-, and hexa-nucleotide SSRs have not drawn enough attention for marker development. We found that mono-nucleotide SSRs had much higher polymorphism rates than others, and penta- and hexa-nucleotide SSRs had relatively longer repeat units. Intron and UTR SSRs were more polymorphic than CDS SSRs due to low selective pressure in non-coding regions, which were consistent with previous reports.22,34–36 Experimental validation using 20 maize inbreds and 7 teosinte species showed that over 70% of the primer pairs could generate the target bands with length polymorphism, promising a great potential for the application of these SSR markers. In practice, it would be very powerful when they are used for genetic populations derived from various types of maize germplasm that were sampled for this study.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This work was supported by the National High Technology Research and Development Program of China (2012AA101104) to Y.L. and Y.X., Sichuan Youth Science and Technology Foundation of China (2012JQ0003), and the National Natural Science Foundation of China (31101162) to Y.L.
Supplementary Material
Footnotes
Edited by Dr Masahiro Yano
References
- 1.Schnable P.S., Ware D., Fulton R.S., et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–5. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 2.Chambers G.K., MacAvoy E.S. Microsatellites: consensus and controversy. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2000;126:455–76. doi: 10.1016/s0305-0491(00)00233-9. [DOI] [PubMed] [Google Scholar]
- 3.Ellegren H. Microsatellites: simple sequences with complex evolution. Nat. Rev. Genet. 2004;5:435–45. doi: 10.1038/nrg1348. [DOI] [PubMed] [Google Scholar]
- 4.McCouch S.R., Chen X., Panaud O., et al. Microsatellite marker development, mapping and applications in rice genetics and breeding. Plant Mol. Biol. 1997;35:89–99. [PubMed] [Google Scholar]
- 5.Zalapa J.E., Cuevas H., Zhu H., et al. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Botany. 2012;99:193–208. doi: 10.3732/ajb.1100394. [DOI] [PubMed] [Google Scholar]
- 6.Kelkar Y.D., Tyekucheva S., Chiaromonte F., Makova K.D. The genome-wide determinants of human and chimpanzee microsatellite evolution. Genome Res. 2008;18:30–8. doi: 10.1101/gr.7113408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varshney R.K., Graner A., Sorrells M.E. Genic microsatellite markers in plants: features and applications. Trends Biotechnol. 2005;23:48–55. doi: 10.1016/j.tibtech.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Lu Y., Yan J., Guimaraes C.T., et al. Molecular characterization of global maize breeding germplasm based on genome-wide single nucleotide polymorphisms. Theor. Appl. Genet. 2009;120:93–115. doi: 10.1007/s00122-009-1162-7. [DOI] [PubMed] [Google Scholar]
- 9.Sonah H., Deshmukh R.K., Sharma A., et al. Genome-wide distribution and organization of microsatellites in plants: an insight into marker development in Brachypodium. PloS One. 2011;6:e21298. doi: 10.1371/journal.pone.0021298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song Q., Jia G., Zhu Y., et al. Abundance of SSR motifs and development of candidate polymorphic SSR markers (BARCSOYSSR_1. 0) in soybean. Crop Sci. 2010;50:1950–60. [Google Scholar]
- 11.Barchi L., Lanteri S., Portis E., et al. Identification of SNP and SSR markers in eggplant using RAD tag sequencing. BMC Genomics. 2011;12:304. doi: 10.1186/1471-2164-12-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang X., Wei X., Sang T., et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010;42:961–7. doi: 10.1038/ng.695. [DOI] [PubMed] [Google Scholar]
- 13.Liu K., Goodman M., Muse S., Smith J.S., Buckler E., Doebley J. Genetic structure and diversity among maize inbred lines as inferred from DNA microsatellites. Genetics. 2003;165:2117–28. doi: 10.1093/genetics/165.4.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xia X., Reif J., Melchinger A., et al. Genetic diversity among CIMMYT maize inbred lines investigated with SSR markers. Crop Sci. 2005;45:2573–82. [Google Scholar]
- 15.Warburton M., Reif J., Frisch M., et al. Genetic diversity in CIMMYT nontemperate maize germplasm: landraces, open pollinated varieties, and inbred lines. Crop Sci. 2008;48:617–24. [Google Scholar]
- 16.Li R., Yu C., Li Y., et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–7. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 17.Huang J., Lu X., Yan H., et al. Transcriptome characterization and sequencing-based identification of salt-responsive genes in Millettia pinnata, a semi-mangrove plant. DNA Res. 2012;19:195–207. doi: 10.1093/dnares/dss004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chia J.M., Song C., Bradbury P.J., et al. Maize HapMap2 identifies extant variation from a genome in flux. Nat. Genet. 2012;44:803–7. doi: 10.1038/ng.2313. [DOI] [PubMed] [Google Scholar]
- 19.Smith J., Chin E., Shu H., et al. An evaluation of the utility of SSR loci as molecular markers in maize (Zea mays L.): comparisons with data from RFLPs and pedigree. Theor. Appl. Genet. 1997;95:163–73. [Google Scholar]
- 20.Temnykh S., DeClerck G., Lukashova A., Lipovich L., Cartinhour S., McCouch S. Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 2001;11:1441–52. doi: 10.1101/gr.184001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soderlund C., Descour A., Kudrna D., et al. Sequencing, mapping, and analysis of 27,455 maize full-length cDNAs. PLoS Genet. 2009;5:e1000740. doi: 10.1371/journal.pgen.1000740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z., Deng Y., Tan J., Hu S., Yu J., Xue Q. A genome-wide microsatellite polymorphism database for the indica and japonica rice. DNA Res. 2007;14:37–45. doi: 10.1093/dnares/dsm005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawson M.J., Zhang L. Distinct patterns of SSR distribution in the Arabidopsis thaliana and rice genomes. Genome Biol. 2006;7:R14. doi: 10.1186/gb-2006-7-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iniguez-Luy F.L., Voort A.V., Osborn T.C. Development of a set of public SSR markers derived from genomic sequence of a rapid cycling Brassica oleracea L. genotype. Theor. Appl. Genet. 2008;117:977–85. doi: 10.1007/s00122-008-0837-9. [DOI] [PubMed] [Google Scholar]
- 25.Yonemaru J., Ando T., Mizubayashi T., Kasuga S., Matsumoto T., Yano M. Development of genome-wide simple sequence repeat markers using whole-genome shotgun sequences of sorghum (Sorghum bicolor (L.) moench) DNA Res. 2009;16:187–93. doi: 10.1093/dnares/dsp005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morgante M., Hanafey M., Powell W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 2002;30:194–200. doi: 10.1038/ng822. [DOI] [PubMed] [Google Scholar]
- 27.Metzgar D., Bytof J., Wills C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000;10:72–80. [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X.V., Blades N., Ding J., Sultana R., Parmigiani G. Estimation of sequencing error rates in short reads. BMC Bioinformatics. 2012;13:185. doi: 10.1186/1471-2105-13-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hufford M.B., Xu X., Van Heerwaarden J., et al. Comparative population genomics of maize domestication and improvement. Nat. Genet. 2012;44:808–11. doi: 10.1038/ng.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lai J., Li R., Xu X., et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat. Genet. 2010;42:1027–30. doi: 10.1038/ng.684. [DOI] [PubMed] [Google Scholar]
- 31.Jiao Y., Zhao H., Ren L., et al. Genome-wide genetic changes during modern breeding of maize. Nat. Genet. 2012;44:812–5. doi: 10.1038/ng.2312. [DOI] [PubMed] [Google Scholar]
- 32.Sharopova N., McMullen M.D., Schultz L., et al. Development and mapping of SSR markers for maize. Plant Mol. Biol. 2002;48:463–81. doi: 10.1023/a:1014868625533. [DOI] [PubMed] [Google Scholar]
- 33.Buckler E.S., Gaut B.S., McMullen M.D. Molecular and functional diversity of maize. Curr. Opin. Plant Biol. 2006;9:172–6. doi: 10.1016/j.pbi.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 34.Lu Y., Shah T., Hao Z., et al. Comparative SNP and haplotype analysis reveals a higher genetic diversity and rapider LD decay in tropical than temperate germplasm in maize. PloS One. 2011;6:e24861. doi: 10.1371/journal.pone.0024861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu H., Lin Y., Chen G., Shen Y., Liu J., Zhang S. Genome-scale identification of resistance gene analogs and the development of their intron length polymorphism markers in maize. Mol. Breeding. 2012;29:437–47. [Google Scholar]
- 36.Tian X., Strassmann J.E., Queller D.C. Genome nucleotide composition shapes variation in simple sequence repeats. Mol. Biol. Evol. 2011;28:899–909. doi: 10.1093/molbev/msq266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.