

Abstract
Synapse development requires differentiation of presynaptic neurotransmitter release sites and postsynaptic receptive apparatus coordinated by synapse organizing proteins. In addition to the well-characterized neurexins, recent studies identified presynaptic type IIa receptor-type protein tyrosine phosphatases (RPTPs) as mediators of presynaptic differentiation and triggers of postsynaptic differentiation, thus extending the roles of RPTPs from axon outgrowth and guidance. Similarly to neurexins, RPTPs exist in multiple isoforms generated by alternative splicing that interact in a splice-selective code with diverse postsynaptic partners. The parallel RPTP and neurexin hub design facilitates synapse self-assembly through cooperation, pairs presynaptic similarity with postsynaptic diversity, and balances excitation with inhibition. Upon mutation of individual genes in neuropsychiatric disorders, imbalance of this synaptic organizing network may contribute to impaired cognitive function.
Keywords: synaptogenesis, neurexin, NGL-3, TrkC, IL1RAPL1, Slitrk
Introduction
The RPTPs are a large protein family with eight subtypes based on diverse extracellular domains [1,2]. The type IIa RPTPs, composed of three members in vertebrates, leukocyte common antigen-related (LAR), PTPσ, and PTPδ, contain typical cell adhesion immunoglobulin-like (Ig) and fibronectin III (FNIII) domains, suggesting the involvement of RPTPs in cell–cell and cell–matrix interactions [2–4]. Studies on the roles of RPTPs in the central nervous system (CNS) had initially focused on axon outgrowth, guidance, and regeneration [1,2,4]. More recently, however, many cell biology studies have demonstrated novel roles of RPTPs as presynaptic proteins that transsynaptically interact with multiple postsynaptic partners to mediate synaptic adhesion and synapse organization [5–12]. These RPTP-based complexes act in a similar manner and often in parallel with complexes of presynaptic neurexins with postsynaptic neuroligins, leucine-rich repeat transmembrane neuronal proteins (LRRTMs), or cerebellin–glutamate receptor-δ (GluRδ) [13–15]. Furthermore, despite a lack of structural homology or common postsynaptic binding partners, RPTPs and neurexins share a key organizational feature: alternative splicing of each of these presynaptic protein families controls their affinity of interaction with multiple postsynaptic binding partners. In other words, both RPTPs and neurexins display splice-selective binding codes with diverse postsynaptic partners. Accumulating evidence indicates that these two parallel synapse organizing pathways cooperate in the development of many synapses and are linked through presynaptic and postsynaptic intracellular pathways. Further, the genes encoding RPTPs and their postsynaptic partners are associated with neuropsychiatric disorders including autism and schizophrenia [16–20], as are like deleterious mutations in the genes encoding neurexins and partners [13,21], supporting the possibility that aberrant synaptic organization might be a fundamental pathogenesis of neuropsychiatric disorders. We review recent evidence for trans-synaptic interaction between presynaptic RPTPs and their multiple postsynaptic partners and the functions of these complexes in synapse development. Further, we discuss the possible physiological significance of the apparent hub design of RPTP-based as well as neurexin-based synapse organizing complexes.
Structure of RPTPs
LAR, PTPσ, and PTPδ are encoded by three independent genes and share overall 66% amino acid identity [22]. Each contains extracellular Ig and FNIII domains, modified by alternative splicing, which mediate diverse extracellular protein interactions. By contrast, RPTP intracellular protein interactions are less diverse and involve the two intracellular protein tyrosine phosphatase (PTP) domains, the membrane-proximal D1 domain with robust catalytic activity and the membrane-distal D2 domain with residual or no catalytic activity [2–4] (Figure 1). RPTPs undergo constitutive proteolytic processing generating an extracellular subunit that remains noncovalently bound to the phosphatase domain subunit [4], but the functional significance of this modification is not clear. RPTPs can also undergo regulated ectodomain shedding by cleavage at an independent site [23], a mechanism that might be used to curtail the synapse organizing activity of RPTPs.
Figure 1.
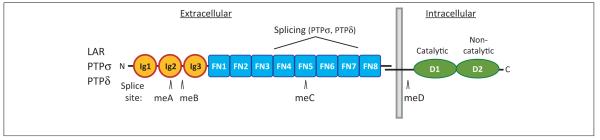
The structure of type IIa receptor-type protein tyrosine phosphatases (RPTPs). Each RPTP contains three extracellular immunoglobulin (Ig)-like domains followed by four or eight fibronectin III (FNIII) domains, depending on alternative splicing, and two intracellular protein tyrosine phosphatase (PTP) domains, the membrane-proximal D1 domain with robust catalytic activity and the membrane-distal D2 domain with residual or no catalytic activity [2–4]. The additional multiple isoforms of RPTPs are generated by alternative splicing of four mini-exons (meA–meD) encoding short amino acid peptides [22,97]. The meA insert with nine or fewer residues is located in the second Ig domain (Ig2), presumably affecting the length of a loop region between the D and E β-strands of Ig2, whereas the meB insert with four residues is located at the end of Ig2 [22,97,98]. The two Drosophila orthologs DLAR and DPTP69D and the single Caenorhabditis elegans ortholog PTP-3 also have two intracellular PTP domains but differ in the number of extracellular Ig and FNIII domains [2,4,31]. The site of constitutive proteolytic processing that generates an extracellular subunit (E-subunit), which remains noncovalently bound to the phosphatase domain subunit (P-subunit) [4], is also indicated.
Hub design of RPTPs: multiple trans-synaptic binding partners
Recent studies demonstrate that presynaptic RPTPs make trans-synaptic adhesion complexes with multiple postsynaptic binding partners to regulate synapse organization. These studies first identified many novel postsynaptic adhesion molecules that can induce presynaptic differentiation in a trans-synaptic manner, here called ‘synaptic organizers’ or ‘synaptogenic’ proteins, through a fibro-blast–neuron co-culture assay [24,25]. In this assay, synaptogenic proteins expressed on the surface of nonneuronal cells trigger formation of functional neurotransmitter release sites in contacting axons of co-cultured neurons. Intriguingly, many of these studies consequently identified RPTPs as presynaptic receptors of the synaptogenic proteins. The postsynaptic binding partners of RPTPs in trans-synaptic complexes have been identified so far as follows (Figure 2): netrin-G ligand-3 (NGL-3) [5,6], neurotrophin receptor tropomyosin-related kinase C (TrkC) [7], interleukin-1-receptor accessory protein-like 1 (IL1RAPL1) [8,9], interleukin-1 receptor accessory protein (IL1RAcP) [10], and Slit and NTRK-like family (Slitrk 1–6) [11,12].
Figure 2.

The selective binding code of receptor-type protein tyrosine phosphatases (RPTPs) with diverse postsynaptic partners. Individual RPTPs bind to overlapping sets of postsynaptic partners, as indicated by the lines; broken lines indicate interactions that can occur in vitro but appear to lack physiological relevance. Importantly, except for netrin-G ligand-3 (NGL-3), these interactions are regulated by alternative splicing of RPTPs at the meA and meB sites. Neurotrophin receptor TrkC can bind all PTPσ forms but insertions at the meA and meB splice sites reduce the apparent interaction [7]. Interleukin-1 receptor accessory protein (IL1RAcP) can bind all forms of PTPδ but insertions at the meA and meB splice sites enhance this interaction [10]. Interleukin-1-receptor accessory protein-like 1 (IL1RAPL1) shows a more complicated selectivity code, binding best to PTPδA9+B+ and PTPδA6+B+, the two most prevalent forms detected in postnatal day 11 (P11) mouse brain, as well as PTPδA9+B− (+ indicates the presence of an insert and the number indicates the length) [8].
Each synaptic organizer displays an individual code in terms of selectivity for RPTP binding (Figure 2). NGL-3 binds to LAR, PTPσ, and PTPδ through their first two FNIII domains [5,6]. TrkC binds selectively to PTPσ, IL1RAPL1 selectively to PTPδ, IL1RAcP to LAR, PTPσ, and PTPδ, and Slitrks selectively to PTPδ and PTPσ, through the Ig domains of the RPTPs [7–12]. Importantly, alternative splicing at the meA and meB sites regulates the affinity of interaction of the RPTPs with all of these partners except for NGL-3. Thus, RPTPs may be considered a presynaptic hub for coupling to diverse postsynaptic partners.
A similar hub design has emerged for neurexins: distinct presynaptic neurexin isoforms generated from different genes, alternative promoters, and alternative splicing bind to distinct postsynaptic partners [13–15]. The parallel design of RPTP and neurexin hubs is striking, very different in nature from cadherin superfamily interactions in which isoform-selective homophilic interactions predominate and contribute to processes such as target recognition and dendritic self-avoidance [26]. However, control of diverse extracellular partnerships by splice-selective binding codes appears to be a feature of Ig superfamily proteins other than RPTPs such as the L1/neuron-glia cell adhesion molecule (NgCAM) family [27,28], and thus may be a more general mechanism. The possible physiological and pathological significance of this hub design of RPTPs as well as neurexins is discussed in Box 1.
Functions of RPTPs in synaptic organization
RPTPs in trans-synaptic complexes have three general functions in synaptic organization (Figure 3). One is to mediate cell–cell adhesion at synapses. The second is to mediate presynaptic differentiation, local recruitment of synaptic vesicles and release and recycling machinery, a form of retrograde synaptogenic signaling triggered by binding of the postsynaptic partner to axonal RPTPs. The third is to trigger postsynaptic differentiation, local recruitment of neurotransmitter receptors, scaffolds, and signaling proteins, a form of anterograde synaptogenic signaling triggered by binding of the presynaptic RPTP to dendritic binding partners. A function of postsynaptic RPTPs has been also reported [29], and the RPTP complexes reviewed here may well participate in regulating axon guidance and target specificity [2]. However, this review focuses on the functions of RPTPs as presynaptic components of synaptic organizing complexes and the associated retrograde and anterograde signaling pathways.
Figure 3.

The roles of receptor-type protein tyrosine phosphatase (RPTP)-based complexes in synapse development. Multiple RPTP complexes have been identified that participate in excitatory synapse development, whereas only PTPδ–Slitrk3 (Slit and NTRK-like family protein 3) is specific for inhibitory synapses. All of the postsynaptic partners can trigger local retrograde differentiation of functional presynaptic sites (red arrows) through the conserved intracellular domains of RPTPs, perhaps through binding liprin-α. RPTP binding to many of the postsynaptic partners can trigger local anterograde recruitment of postsynaptic scaffolds and neurotransmitter (NT) receptors (green arrows), although it is not known whether this is the case for Slitrk1, 2, 4, and 5, and current evidence suggests that Slitrk3 may not mediate postsynaptic differentiation. Individual postsynaptic partners can also directly bind and recruit different scaffolding and signaling proteins as indicated.
RPTP functions in synapse organization were first indicated by genetic studies in invertebrates. Drosophila DLAR and Caenorhabditis elegans PTP-3 mutants show altered presynaptic organization affecting vesicles and active zones [30,31]. Based on expression patterns and phenotypes of mutant mice [4] as well as recent studies of RPTP-based complexes as detailed below, PTPσ and PTPδ may be more important than LAR for synapse organization in vertebrates. In adult brain, LAR shows weak expression whereas PTPσ and PTPδ are highly expressed, PTPσ very broadly, and PTPδ particularly strongly in scattered cortical neurons, presumptive interneurons, and CA2 hippocampal pyramidal cells [32]. Ptprs−/− and Ptprd−/− mice (lacking PTPσ or PTPδ, respectively) exhibit increased paired pulse facilitation, enhanced or reduced long-term potentiation, respectively, and distinct behavioral alterations, supporting distinct synaptic functions [33,34]. However, Ptprs−/− hippocampal neurons also exhibit an increase in synapse density, probably related to enhanced axon sprouting and/or to a role for postsynaptic PTPσ [34]. More generally, Ptprs−/− and Ptprd− mice also exhibit early growth retardation and increased neonatal mortality, and double-mutant mice die at birth due in part to axon targeting defects [33,35–37]. Thus, conditional cell-specific and temporally controlled mutation may be necessary to fully dissect the synaptic organizing roles of PTPσ and PTPδ in mice in vivo.
At least one key downstream effector through which RPTPs signal intracellularly to organize presynaptic terminals has been identified in invertebrates, liprin-α, a scaffolding protein that binds the D2 domains of all RPTPs [22]. Mutation of the orthologs C. elegans SYD-2 and Drosophila Dliprin-α result in synaptic defects similar to, and in cases stronger than, mutation of the RPTP orthologs [30,38]. Additional studies in these systems found that liprin-α dimerization and interaction with liprin-β and with the active zone protein ERC (ELKS/Rab6-interacting/CAST) are important for presynaptic assembly [39–41]. Binding of liprin-α to the active zone proteins RIM and CASK, the Rho effector mDiaphanous, and the Arf GTPase-activating protein GIT1 [42] may also contribute to presynaptic differentiation. The central role of liprin-α/SYD-2 in presynaptic differentiation as established in C. elegans has yet to be tested in mammals, although recent studies demonstrating differential expression and localization of the four α-liprins [43,44] may help fuel further functional studies. RPTP D2 domains also bind other potential effectors including Trio, which contains Rho guanine nucleotide exchange factor (GEF) and Rac GEF domains [45], and the actin-bundling protein MIM [46].
Whether the tyrosine phosphatase activity of the D1 domain is essential for presynaptic differentiation through RPTPs or more modulatory has not yet been determined. Key substrates of DLAR are the Abl tyrosine kinase and Ena, a regulator of actin assembly [2]. Other substrates of RPTPs that may be important for their synaptic functions include N-cadherin, β-catenin, and p250GAP [29,47,48]. The N-cadherin–β-catenin complex stabilizes synaptic connections and controls aspects of transmitter release and spine morphogenesis [49]. Because tyrosine phosphorylation of cadherin and catenin suppresses their association, stabilization of the complex by RPTPs may contribute to synaptic differentiation.
In the anterograde synaptogenic signaling direction, co-culture experiments have shown that RPTPs can trigger excitatory glutamatergic postsynaptic differentiation [5–8]. This anterograde signaling appears to involve multiple postsynaptic partners of RPTPs, likely acting in a synergistic manner and mediating to some degree distinct aspects of postsynaptic differentiation through different pathways (Figure 3).
Next, we will review the evidence for molecular interactions, cellular functions, and signaling pathways of each trans-synaptic complex based on presynaptic RPTPs and their specific postsynaptic binding partners.
LAR/PTPσ–NGL-3 complex
NGL-3 is the first reported synaptic organizer that transsynaptically interacts with RPTPs [5]. NGL-3 is a member of the netrin-G ligand (NGL; LRRC4) family [50]. NGL-1 and NGL-2 but not NGL-3 selectively bind to the glycophosphatidylinositol (GPI)-anchored proteins netrin-G1 and netrin-G2, respectively, and these isoform-dependent interactions are involved in input-dependent dendritic segmentation and laminar-specific excitatory synapse development [50–52]. NGL-3, but not NGL-1 nor NGL-2, binds with high affinity to LAR, PTPσ, and PTPδ [5,6]. NGL-3 binds to the first two FNIII domains of the RPTPs [6], thus is likely to interact with all RPTP splice variants [5].
In the co-culture assay, NGL-3 induces both excitatory and inhibitory presynaptic differentiation in contacting axons, and this activity of NGL-3 is suppressed by addition of soluble LAR ectodomain suggesting that it is mediated by RPTPs [5]. Unlike the co-culture results, however, neuronal overexpression of NGL-3 promotes excitatory, but not inhibitory, presynaptic differentiation, perhaps due to localization of NGL-3 in the excitatory postsynaptic density [5].
Induction-type experiments also indicate that signaling from axonal RPTPs to dendritic NGL-3 can induce excitatory postsynaptic differentiation. Direct aggregation of recombinant NGL-3 on the dendrite surface with antibody-coated beads results in co-clustering of multiple excitatory, but not inhibitory, postsynaptic components including scaffold proteins PSD-95, GKAP, and Shank, AMPA receptors, and NMDA receptors [5]. Notably, the C terminus of NGL-3 binds to the first and second PDZ domains of PSD-95 and all MAGUKs [50]. These data suggest that axonal RPTPs trans-synaptically accumulate NGL-3 on the dendrite surface and consequently promote excitatory postsynaptic differentiation through an NGL-3–PSD-95 interaction.
Although NGL-3 can bind to LAR, PTPσ, and PTPδ in vitro [6], additional evidence indicates that its functional presynaptic partner is LAR and/or PTPσ but not PTPδ. The binding affinity of NGL-3 to PTPδ is much weaker than that of another synaptic organizer IL1RAPL1 (described below) to PTPδ [8]. Moreover, the presynaptic inducing activity of NGL-3 in the co-culture assay is comparable between wild type neurons and Ptprd−/− neurons [8]. Further, mutants of PTPσ or LAR but not PTPδ containing FNIII but not Ig domains, which can still bind to NGL-3 but not to other known synaptic partners such as IL1RAPL1, can induce excitatory postsynaptic differentiation [6,8]. Thus, NGL-3 acts together with PTPσ and/or LAR as a bidirectional excitatory synaptic organizing complex. Based on its high expression in brain, we suggest that PTPσ may be the main presynaptic component in this complex.
NGL-3 is widely expressed in the brain [50], thus is unlikely to control input-selective synapse development as do the other NGL–netrin G complexes. RNAi-mediated knockdown of NGL-3 reduced excitatory synapse density in cultured hippocampal neurons [5]. A critical next step will be to determine the function of NGL-3 in vivo, whether it has unique roles and how it acts synergistically with other synapse organizing complexes.
PTPσ–TrkC complex
The neurotrophin receptor tropomyosin-related kinase (Trk) family is well characterized for its roles in neuronal differentiation, survival, and maintenance through neurotrophin-dependent activation of tyrosine kinase signaling [53,54]. By contrast, it had been predicted that Trks may also have cell adhesion functions given their extracellular domain structures composed of leucine-rich repeat (LRR) and Ig domains [53,54]. Recently, an unbiased expression screen based on the co-culture assay isolated a TrkC noncatalytic isoform as a synaptic organizer [7]. A candidate cell surface binding screen using TrkC ectodomain proteins then isolated PTPσ as a TrkC binding partner. Despite considerable homology, neither TrkA nor TrkB can induce presynaptic differentiation in co-cultured hippocampal or cortical neurons nor bind to RPTPs [7].
In the co-culture assay, both TrkC noncatalytic (TrkCTK−) and catalytic (TrkCTK+) isoforms induce excitatory, but not inhibitory, presynaptic differentiation with almost equal activity [7]. The synaptogenic region of TrkC binds to PTPσ but not PTPδ or LAR (at least not to the splice forms that were tested), and this interaction is regulated by alternative splicing with insertions at the meA and meB splice sites of PTPσ reducing the observed binding [7]. Presynaptic differentiation by TrkC ectodomain-coated beads is abolished by axonal expression of a PTPσ mutant lacking the intracellular region (PTPσΔICD), and forced accumulation of PTPσ, but not PTPσΔICD, on the axon surface is sufficient to induce presynaptic differentiation [7]. This evidence supports the idea that axonal PTPσ mediates presynaptic induction by dendritic TrkC and that signal transduction by the intracellular domain of PTPσ is required.
Trans-synaptic interaction between axonal PTPσ and dendritic TrkC also generates an anterograde synaptogenic signal for excitatory postsynaptic differentiation [7]. Although TrkCTK− and TrkCTK+ lack typical PDZ domain-binding motifs, direct surface aggregation of TrkCTK− or TrkCTK+ on dendrites is sufficient to induce co-aggregation of PSD-95 and NMDA receptors [7]. In addition, TrkCTK− can bind the postsynaptic scaffold protein tamalin to activate Arf6–Rac signaling [55] and TrkCTK+ can bind and activate phospholipase Cγ, Shc, and Frs2 leading to Ras and PI3 kinase [54], critical regulators of excitatory synaptic plasticity. These interactions might be involved in TrkC-mediated excitatory postsynaptic signaling and contribute to the diversity of excitatory postsynaptic composition.
Independent loss-of-function experiments utilizing a TrkC neutralizing antibody that inhibits TrkC–PTPσ interaction or RNAi-mediated knockdown of TrkC support the involvement of the TrkC–PTPσ interaction in excitatory synapse development [7]. Notably, the TrkC neutralizing antibody completely blocks presynaptic induction by TrkC but only partially blocks postsynaptic induction by PTPσ [7], consistent with alternative anterograde pathways from PTPσ such as through NGL-3. TrkC catalytic and noncatalytic isoforms are expressed widely in the postnatal and mature brain [56,57] and localize to excitatory postsynaptic sites [7]. Transgenic overexpression of TrkC results in elevated levels of NMDA receptors and enhanced and longer-lasting long-term potentiation in hippocampus [58], supporting a role of TrkC in excitatory synapse function. However, considering the pleiotropic roles of TrkC in nervous system development and the perinatal lethal phenotype of Ntrk3−/− mice lacking TrkC [59], more selective manipulations will be necessary to test the specific functions of the TrkC–PTPσ synaptic complex in vivo. The TrkC knockdown phenotypes on synaptic markers in culture and spine density in vivo are fully rescued by RNAi-resistant TrkCTK− [7], indicating that the synapse organizing function of TrkC does not require kinase activity. However, the intriguing possibilities remain that TrkC–PTPσ trans-interaction may affect the activity of TrkC tyrosine kinase and/or PTPσ phosphatase to regulate excitatory synapse development and/or plasticity.
PTPδ–IL1RAPL1 and PTPδ/PTPσ/LAR–IL1RAcP complexes
IL1RAPL1 and IL1RAcP belong to the interleukin-1/Toll receptor family, possess Ig-like domains extracellularly and a Toll/IL1R (TIR) domain intracellularly, and share sequence homology [60]. The human gene encoding IL1RAPL1 has been well characterized to be associated with cognitive diseases ranging from nonsyndromic X-linked mental retardation to autism [18,61]. Two independent studies recently revealed that trans-synaptic interaction between postsynaptic IL1RAPL1 and presynaptic PTPδ bidirectionally regulates excitatory synapse development [8,9]. The IL1RAPL1 extracellular region selectively binds to PTPδ but not LAR or PTPσ [8,9]. Furthermore, the IL1RAPL1 extracellular region is sufficient for inducing excitatory presynaptic differentiation in the co-culture assay, and this activity is abolished in co-cultures with Ptprd−/− neurons [8].
Neuronal overexpression of IL1RAPL1 not only enhances excitatory presynaptic inputs but also increases dendritic spine formation and clustering of excitatory postsynaptic proteins such as PSD-95 and Shank2 [8,9,62]. Intracellularly, the IL1RAPL1 TIR domain binds to RhoGAP2, a GTPase-activating protein that inhibits Rac1 activity and regulates dendritic spine number and morphology, and neuronal overexpression of IL1RAPL1 recruits RhoGAP2 to synapses [9]. The IL1RAPL1 C-terminal domain binds to PSD-95 via a noncanonical PDZ binding motif [62]. IL1RAPL1 also activates the JNK pathway and consequently phosphorylates PSD-95 thus regulating synaptic accumulation of PSD-95 [62,63]. Consistent with these molecular pathways, spine induction by IL1RAPL1 requires its TIR domain, whereas Shank2 clustering requires both the TIR and C-terminal domains [8]. Interestingly, postsynaptic protein recruitment and spine induction by IL1RAPL1 require trans-synaptic interaction with PTPδ [8,9]. This finding may reflect a simple requirement for PTPδ binding to localize IL1RAPL1 to excitatory postsynaptic sites. However, it has not been definitively shown whether dendritic recruitment of IL1RAPL1 by PTPδ is sufficient to recruit postsynaptic proteins, for example, by direct aggregation of IL1RAPL1 on dendrites as described above for NGL-3 and TrkC, but this probability is supported by the finding that soluble IL1RAPL1 ectodomain inhibits postsynaptic differentiation by PTPδ [8].
Proteins related to IL1RAPL1 also function in excitatory synapse organization. IL1RAPL2 induces presynaptic differentiation in the co-culture assay, although with less potency than IL1RAPL1, and when overexpressed in neurons IL1RAPL2 also increases excitatory presynaptic inputs and dendritic spine density [9,10]. Other members of the IL1 receptor and accessory protein families do not have synaptogenic activity with one important exception, IL1RAcP [10]. Postsynaptic IL1RAcP and presynaptic PTPδ also function as a bidirectional excitatory synaptic organizing complex [10]. IL1RAcP functions as a co-receptor with interleukin-1 receptor type I (IL1RI) for mediating immune and inflammatory responses to IL1 family cytokines [60], unlike IL1RAPL1 which lacks such function. Thus, reminiscent of TrkC, IL1RAcP appears to serve two distinct functions, one with IL1 and IL1R1 in immune regulation and inflammation and another with PTPδ in synaptic adhesion and synapse organization.
An elegant series of co-culture, ectodomain-bead induction, IL1RAcP neuronal overexpression and RNAi knockdown, recombinant protein interaction, cell surface binding, and cell aggregation assays were used to demonstrate a role for IL1RAcP–PTPδ in excitatory synaptic adhesion and presynaptic and postsynaptic differentiation in cultured neurons [10]. IL1RAcP exists in two isoforms as the more widely expressed IL1RAcP and the CNS-specific IL1RAcPb [64]. IL1RAcPb terminates in a consensus type II PDZ domain binding sequence. Both isoforms of IL1RAcP can induce excitatory presynaptic differentiation using their extracellular regions, but only IL1RAcPb can also promote dendritic spine formation [10]. IL1RAcP-induced presynaptic induction is significantly but partially abolished in co-cultures with Ptprd−/− neurons, indicating that the weaker interaction observed between IL1RAcP and PTPσ and LAR may also be functionally important [10]. Likewise, PTPδ-induced postsynaptic induction is significantly but partially abolished in co-cultures with IL1RAcP−/− neurons [10], indicating that IL1RAcP is one of multiple functional postsynaptic partners for PTPδ.
In vivo knockout data confirm roles for IL1RAPL1 and IL1RAcP in excitatory synapse development. IL1RAPL1−/− mice exhibit deficits in excitatory synapses, reduced asymmetric synapse density, and reduced spine density in hippocampal CA1 stratum radiatum [62]. Long-term potentiation was reduced when elicited by theta burst stimulation but not by high frequency stimulation, thus showing stimulus-dependent effects [62]. Basal transmission assessed by input–output response, minimal stimulation failure rate, and miniature excitatory postsynaptic current (mEPSC) frequency was not significantly impaired. Curiously, mEPSC frequency was substantially reduced in IL1RAPL1−/− hippocampal neurons co-cultured with wild type neurons [62], perhaps reflecting a competitive effect dependent on transcellular differences in IL1RAPL1 level as recently found for neuroligin-1 [65]. Reductions in phosphorylation of JNK and of PSD-95 were also found in IL1RAPL1−/− neurons [62], consistent with the molecular pathways described above. Whether IL1RAPL1 functions exclusively at excitatory postsynaptic sites is not yet clear because differences were observed in synaptic transmission within cerebellar GABAergic networks in IL1RAPL1−/− mice [66] and evidence from zebrafish supports direct presynaptic functions of IL1RAPL1 [67]. Synaptic phenotypes of IL1RAcP−/− mice have not been extensively studied but a significant reduction in spine density also confirms a role in excitatory synapse development in vivo [10].
A major finding from these recent studies on IL1RAPL1 and IL1RAcP as well as TrkC is the revelation of distinct codes for RPTP binding regulated by alternative splicing of RPTPs [7,8,10] (Figure 2). Perhaps related to differences between IL1RAPL1 and IL1RAcP in their RPTP binding codes is the finding that in the co-culture assay, IL1RAPL1 induces only excitatory presynaptic differentiation whereas IL1RAcP induces not only excitatory but also some inhibitory presynaptic differentiation [8,10]. Taken together with additional studies on TrkC and Slitrks (see below), these differences raise the intriguing possibility that glutamatergic and GABAergic axons may have differential expression patterns of RPTP family members and splice forms. Neuron type-specific expression and splicing of RPTP variants remains to be addressed experimentally.
PTPδ/PTPσ–Slitrk1–6 complexes
The Slitrk family consists of six brain-specific transmembrane proteins (Slitrk 1–6) that possess extracellular LRR domains homologous to the axon guidance molecule Slit and intracellular C-terminal tyrosine residues with surrounding sequences homologous to the Trk family [68]. All members of the Slitrk family induce presynaptic differentiation in the co-culture assay [11,69]. Intriguingly, Slitrk3 can induce inhibitory, but not excitatory, presynaptic differentiation whereas other family members induce both excitatory and inhibitory presynapses [11]. Similarly, upon overexpression or knockdown in neurons, Slitrk3 selectively affects GABAergic inputs whereas any of Slitrk1, Slitrk2, Slitrk4, or Slitrk5 affect glutamatergic inputs, and recombinant protein localization for Slitrk1–3 supports these selective phenotypes [11,12].
All Slitrks can interact with PTPδ [11] and Slitrk1–3 at least can also interact with PTPσ [12]. RNAi-mediated knockdown of selective RPTPs in neurons co-cultured with Slitrk-expressing cells suggests that PTPσ mediates the excitatory presynaptic differentiation induced by Slitrks, particularly Slitrk1 and Slitrk2 [12]. Curiously, PTPδ, shown above to function with IL1RAPL1 and IL1RAcP in excitatory synapse development, mediates the inhibitory presynaptic differentiation induced by Slitrks, particularly Slitrk3 [11,12]. Thus, it is puzzling how Slitrk3–PTPδ can function selectively in inhibitory synaptic organization and IL1RAPL1/IL1RacP–PTPδ in excitatory synaptic organization. The PTPδ isoform that contains full meA and meB inserts, a major isoform of PTPδ in the brain [8], can bind to all Slitrks including Slitrk3 [11], IL1RAPL1 [8], and IL1RacP [10], although perhaps with different affinity. Given that the meA and meB splice inserts of PTPδ differentially control its binding to IL1RAPL1 and IL1RAcP [8,10], it is possible that differential splicing of PTPδ in GABAergic versus glutamatergic axons contributes to selectivity in partner binding and function. Such a mechanism would be similar to the role of splicing at the S4 site in neurexins, which along with α/β promoter usage controls their binding to different neuroligins, LRRTM1/2 and cerebellin–GluRδ [14,15]. However, more complicated mechanisms involving axon-selective co-receptors or suppressors to regulate specificity of interactions cannot be ruled out.
Whereas most of the other RPTP complexes have demonstrated bidirectional synaptic organizing activities, the Slitrk–PTPδ/PTPσ interaction has only been shown to induce presynaptic differentiation. A PTPδ isoform that contains full meA and meB inserts and can bind to all Slitrks [11] induces only excitatory not inhibitory postsynaptic protein clustering in co-culture assays [8], suggesting that Slitrk3 may need to cooperate with other synaptogenic complexes such as neurexin–neuroligin-2 for inhibitory synapse development. It remains an open question whether any of the RPTP–Slitrk complexes can directly mediate postsynaptic differentiation.
Slitrks 1–5 are widely expressed in distinct but overlapping patterns in the brain, whereas Slitrk6 is largely restricted to the thalamus, cerebellum, medulla, and is also unique in its high expression in many nonneural tissues [68,70]. Only Slitrk3 and Slitrk5 have been extensively studied with respect to synaptic phenotypes in vivo. Slitrk5−/− mice show reduced levels of AMPA and NMDA receptors in striatum and reduced corticostriatal transmission [71] supporting a role in excitatory synapse development in striatal neurons. Considering the neuron culture and RPTP binding experiments described above, it is likely that Slitrk5 contributes to excitatory synapse development through interaction with PTPσ. Slitrk5−/− mice exhibit excessive self-grooming [71] and may serve as a model for obsessive compulsive disorder (OCD) (Table 1).
Table 1.
Association of RPTPs and postsynaptic partners with neuropsychiatric disorders
| Proteina | Gene | Locus | Mutation | Phenotype | Refs |
|---|---|---|---|---|---|
| PTPδ | PTPRD | 9p23–p24.3 | •Two SNPs in 5′UTR •Four CNVs (hemizygous deletion) •de novo CNV (deletion) •de novo CNV (duplication) |
Restless legs syndrome ADHD ASD Bipolar disorder |
[16,106] [107] [17] [108] |
| PTPσ | PTPRS | 9p13.3 | •de novo CNV (deletion, multigenic) | ASD | [17] |
| IL1RAPL1 | IL1RAPL1 | Xp22.1–p21.3 | •Deletions •Nonsense mutations (Y459X, W487X) •de novo frameshift mutation (I367SfsX6) •Deletions •CNV (duplication) |
Mental retardation ASD Schizophrenia |
[61,109–114] [18,115] [19] |
| Slitrk1 | SLITRK1 | 13q31.1 | •Frameshift mutation (L422fŝ) •3′UTR mutation •Two missense mutations (R584K, S593G) |
Gilles de la Tourette’s syndrome Trichotillomania |
[116,117] [118] |
| Slitrk2 | SLITRK2 | Xq27.3 | •Rare missense mutation (V89M) | Schizophrenia | [115] |
| Slitrk3 | SLITRK3 | 3q26.1 | •Altered expression of miRNAs against Slitrk3 mRNA | Autism | [119] |
| Slitrk6 | SLITRK6 | 13q31.1 | •CNV (deletion) | Epilepsy | [120] |
| TrkC | NTRK3 | 15q25.3 | •Interstitial duplication of 15q24-26 •SNP in 5′UTR •SNP in 3′UTR •SNPs in 3′UTR (two SNPs at miRNA target site) •Five SNPs (intron) •Three SNPs (intron) |
Panic disorder OCD Major depression Schizophrenia |
[121–123] [123,124] [125] [126] |
For LAR encoded by the PTPRF gene at locus 1p34.2, we are not aware of mutations associated with neuropsychiatric disorders.
Slitrk3−/− mice exhibit decreases in GABAergic but not glutamatergic presynaptic markers and decreases in miniature inhibitory postsynaptic current (mIPSC) frequency with no effects on mEPSCs in the hippocampal CA1 region [11]. These data support the cell culture studies, indicating that Slitrk3 mediates functional inhibitory synapse development in vivo, presumably through its interaction with PTPδ. Slitrk3−/− mice also show increased seizure susceptibility and occasional spontaneous seizures, suggesting a widespread impairment of inhibitory synapses [11]. Curiously, within the CA1 region most intensively analyzed, inhibitory terminals in the center of stratum pyramidale were lost whereas those at the edges of stratum pyramidale were spared in Slitrk3−/− mice [11]. This finding may relate to the distinct physiological properties of pyramidal cells residing at different sublayers within stratum pyramidale [72] and presents an interesting cell biological puzzle, perhaps reflecting a selective role of Slitrk3–PTPδ at synapses made by specific classes of basket cells and/or axo-axonic cells. In hippocampal CA1 stratum pyramidale, by immunofluorescence Slitrk3−/− mice show impaired inhibitory presynaptic differentiation ([11]; potential effects on postsynaptic differentiation are not yet known), whereas mice lacking neuroligin-2 show impaired inhibitory postsynaptic differentiation with almost normal inhibitory presynaptic differentiation [73]. Thus, Slitrk3–PTPδ and neuroligin-2–neurexin pathways may cooperatively control inhibitory synapse development by mainly bearing trans-synaptic retrograde and anterograde signals, respectively.
Cooperative mechanisms among RPTP-based and neurexin-based synapse organizing complexes
Overall, the physical and functional trans-interactions of RPTPs as well as neurexins with their multiple postsynaptic binding partners generate a complicated molecular network. Many of these complexes have fairly widespread overlapping expression patterns, consistent with cooperative functions. For example, evidence for function at glutamatergic synapses in hippocampal cultures has been published for RPTPs with NGL-3 [5], TrkC [7], IL1RAPL1 [8], Slitrk1, Slitrk2, Slitrk4, and Slitrk5 [12], and for neurexins with neuroligin-1, neuroligin-3 [74,75], LRRTM1, and LRRTM2 [69,76–78], and about half of these complexes have verified roles at synapses from hippocampal CA3 to CA1 stratum radiatum in vivo. As discussed above, PTPδ–Slitrk3 and neurexin–neuroligin-2 also function together at hippocampal GABAergic synapses, particularly onto pyramidal cell somata. Thus, it appears that multiple RPTP-based and neurexin-based complexes act in parallel to coordinate the organization of individual synapses.
Within the RPTP-based system, the hub design promotes cooperation among complexes with multiple postsynaptic partners, but also competition among postsynaptic partners that bind to individual RPTP isoforms if their amounts are limiting (and similarly within the neurexin-based system). Implications of this hub design are further discussed in Box 1. Within the RPTP-based system, there is also the potential for more direct cooperative interactions by simultaneous binding of multiple postsynaptic partners to an individual RPTP (Figure 4A). Such triple complexes may have enhanced stability and serve to recruit multiple postsynaptic components. Within the presynaptic terminal, intracellular interactions so far appear to be conserved among RPTPs (e.g., all interact with liprin-α), so each RPTP complex may contribute in an additive rather than unique manner. However, individual RPTP postsynaptic partners can recruit unique interacting proteins contributing to the diversity of postsynaptic specializations. Cooperative interactions may also occur within the postsynaptic density, and may extend to neurexin partners as well, by joint binding to scaffold proteins such as PSD-95 (Figure 4B). Thus, scaffolding by PSD-95, which itself forms head-to-head multimers, and other members of the MAGUK family, may stabilize many of these trans-synaptic complexes and function to transduce the anterograde synapse organizing signal by recruiting NMDA receptors, stargazin, and AMPA receptors, other scaffolds such as GKAP/SAPAP, and signaling proteins such as SynGAP [79].
Figure 4.
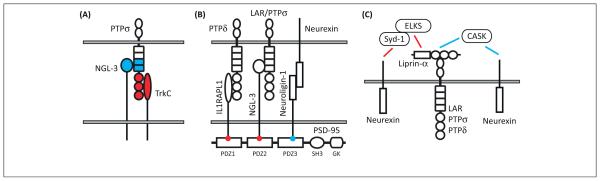
Cooperative mechanisms among receptor-type protein tyrosine phosphatase (RPTP)-based and neurexin-based synapse organizing complexes. (A) Netrin-G ligand-3 (NGL-3) and the neurotrophin receptor TrkC bind to distinct domains of PTPσ [the first two fibronectin III (FNIII) domains for NGL-3 [6] and the Ig domains for TrkC [7]]. Therefore, NGL-3 and TrkC could cooperatively regulate excitatory synapse organization through possible simultaneous binding to PTPσ. Although domain mapping has not been reported for the other RPTP partners, their regulation by meA and meB splicing suggests that interleukin-1-receptor accessory protein-like 1 (IL1RAPL1) and interleukin-1 receptor accessory protein (IL1RAcP) also bind to the Ig domains. Thus, NGL-3, which can bind to all RPTPs, might be rather unique in simultaneously binding to RPTPs along with a variety of other postsynaptic partners. (B) NGL-3 and IL1RAPL1 can each bind to the first two PDZ domains of PSD-95 [50,62], and neuroligins bind to the third PDZ domain of PSD-95 ([127]). Leucine-rich repeat transmembrane proteins (LRRTMs) also bind PSD-95 [69], Thus, scaffolding by PSD-95, which itself forms head-to-head multimers, and other members of the MAGUK family, may stabilize many of these trans-synaptic complexes and function to transduce the anterograde synapse organizing signal by recruiting postsynaptic receptors, other scaffolds, and signaling proteins. (C) The most direct known link from RPTPs to neurexins is through liprin-α binding to CASK, which in turn binds to neurexins [128]. However, cultured cortical neurons lacking CASK exhibit normal evoked glutamatergic and GABAergic transmission [129] and Caenorhabditis elegans liprin-α and CASK orthologs Syd-2 and Lin-2 do not colocalize or genetically interact [39], raising questions about the importance of this link. Another key pathway recently identified in Drosophila [130] links the neurexin DNrx-1 to the liprin-α DSyd-2 through DSyd-1, a Rho GTPase-activating protein that contains PDZ and C2 domains initially identified from a C. elegans screen for genes contributing to synapse development [131]. It is not yet clear whether Syd-1 directly binds liprin-α or whether they interact through simultaneous binding to ERC [132].
Although individual RPTP or neurexin trans-synaptic partners are sufficient to induce presynaptic differentiation in co-culture, it is likely that these multiple systems cooperate for bona fide presynaptic differentiation between neurons. Interestingly, PTPσ as well as neurexins function as receptors for α-latrotoxin, which triggers massive neurotransmitter release [80,81], suggesting shared presynaptic signaling pathways in neurotransmitter release. Based on current limited knowledge about the mechanisms of retrograde synaptogenic signal transduction for the RPTP and neurexin pathways, two major routes of convergence within presynaptic terminals are apparent (Figure 4C), both through liprin-α, which is thought to be a core scaffold protein that governs presynaptic differentiation through RPTPs. It will be important to assess the role of convergent signaling between RPTP and neurexin pathways in presynaptic differentiation and the specific roles of liprin-α, CASK, ERC, and Syd-1 in mammalian systems.
Possible modulatory mechanisms regulating RPTP-based synaptic organizing complexes
Previous studies have identified additional secreted or extracellular matrix molecules that can bind to the extracellular domains of RPTPs or their trans-synaptic binding partners, raising intriguing possibilities for modulation of the synaptic organizing activity of these complexes (Figure 5). Chondroitin sulfate proteoglycans (CSPGs) and heparan sulfate proteoglycans (HSPGs) bind to RPTPs [82,83]. HSPGs regulate DLAR function in synapse development at the Drosophila neuromuscular junction [84], and several studies demonstrate the involvement of extracellular matrix in synapse formation and/or plasticity in mammalian neurons [85]. Thus, these extracellular matrix molecules might enhance or suppress the synaptogenic activity of RPTP-based synaptic organizing complexes to regulate synapse development and/or plasticity (Figure 5A).
Figure 5.
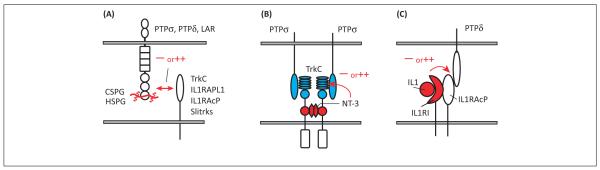
Potential modulatory mechanisms regulating receptor-type protein tyrosine phosphatase (RPTP)-based synapse organizing complexes. (A) Chondroitin sulfate proteoglycans (CSPGs) and heparan sulfate proteoglycans (HSPGs) bind via their glycosaminoglycan chains to the first immunoglobulin-like domain of leukocyte common antigen-related (LAR), PTPσ, and PTPδ [82,83]. CSPGs and HSPGs compete for binding to RPTPs and have opposing PTPσ-dependent effects on neurite outgrowth of dorsal root ganglion neurons, perhaps through differential effects on RPTP oligomerization [133]. The extracellular matrix laminin–nidogen complex also binds to the fifth fibronectin III (FNIII) domain of LAR in a splice-selective manner, only to LAR isoforms lacking an insert at the meC site [134]. These extracellular matrix molecules might modulate the synaptogenic activity of RPTP-based synaptic organizing complexes. (B) The neurotrophin receptor TrkC requires only its second Ig domain to recognize neurotrophin-3 (NT-3) [54,86], whereas only its leucine-rich repeat (LRR) plus first Ig domains are needed to bind PTPσ and trigger synaptogenic signaling [7]. Thus, TrkC could potentially simultaneously bind neurotrophin-3 and PTPσ, raising the possibility for cross-modulation. In general, neurotrophin-3 binding to TrkC induces its dimerization, activation of the kinase, kinase-dependent recruitment of signaling intermediates, and internalization of TrkC [54]. Further, neurotrophin-3 treatment enhances excitatory synaptic transmission and alters synaptic plasticity [87,88]. Some of these effects of neurotrophin-3 may be through modulating the synapse organizing activity of the TrkC–PTPσ complex. (C) In the immune system, interleukin-1 receptor accessory protein (IL1RAcP) forms a heterodimer with the type I interleukin-1 receptor (IL1RI) to form a signaling-competent complex for binding and transducing the effects of IL1 [60]. There is evidence for neuronal expression of IL1R1 [135] and IL1 has rapid effects on NMDA receptor-mediated transmission and synaptic plasticity [136], although whether IL1R1 localizes with IL1RAcP to postsynaptic sites has not been reported. If so, IL1 through IL1RI might modulate the synapse organizing activity of IL1RAcP by altering the binding of IL1RAcP to PTPδ.
Other interactions specific to individual RPTP binding partners may also modulate the function of individual complexes. The isolation of TrkC as a synaptogenic protein was surprising given its well-known role as a neurotrophin receptor. TrkC binds via different domains to neurotrophin-3 and PTPσ [7,54,86], raising the possibility for cross-modulation. Some of the effects of neurotrophin-3 on synaptic efficacy [87,88] may be through modulating the synapse organizing activity of the TrkC–PTPσ complex by its dimerization and/or internalization (Figure 5B). Considering further evidence that neurotrophin-3 undergoes activity-dependent release [89], neurotrophin-3 modulation could represent a way to regulate the TrkC–PTPσ complex by synaptic activity.
Another potential form of modulation specific to IL1RAcP is modulation by the cytokine IL1. In the immune system, IL1RAcP forms a heterodimer with IL1RI to form a signaling-competent complex for binding and transducing the effects of IL1 [60]. If such a heterodimer forms on dendrites, IL1 through IL1RI might modulate the synapse organizing activity of IL1RAcP (Figure 5C). Given that IL1 is a proinflammatory cytokine, such a modulatory mechanism could be important for regulating synaptic function in neuroinflammatory conditions including stroke and brain injury.
Disease association of RPTP and their binding partners
Several neuropsychiatric disorders such as autism spectrum disorders (ASD), schizophrenia, OCD, and attention deficient hyperactivity disorder (ADHD) are highly heritable, motivating large-scale genetic studies for these disorders. Genome-wide single nucleotide polymorphism (SNP) association, copy number variant (CNV) identification, and whole-exome sequencing have uncovered many gene mutations associated with these disorders. A subset of these mutations target genes involved in development and function of synapses. In addition to mutations in genes encoding neurexins and partners neuroligins, LRRTMs, and cerebellin–GluRδ [13,21,90–93], mutations in genes encoding PTPδ, PTPσ, and RPTP postsynaptic binding partners IL1RAPL1, the Slitrk family, and TrkC have been identified in individuals with neuropsychiatric disorders, as summarized in Table 1.
Generally, mutations in a single gene encoding a synaptic organizer are associated with a range of neuropsychiatric disorders. For example, exonic deletions in NRXN1 are associated with ASD, schizophrenia, intellectual disability, and language delay [94,95]. Mutations in RPTP-based synaptic organizing complexes also appear to contribute to multiple neuropsychiatric disorders (Table 1). Conversely, one neuropsychiatric disorder involves mutations of different genes encoding synaptic organizers. Considering the functional similarities of RPTP-based complexes to neurexin-based complexes, dysfunction of either type of synaptic organizing complex by subtly altering the composition and function of synapses could underlie a common fundamental pathogenesis for a range of neuropsychiatric disorders.
Concluding remarks
The RPTP family is an emerging presynaptic hub for synapse organization, molecularly distinct from neurexins but remarkably parallel in function and interaction network organization. A fundamental outstanding question is whether RPTPs and neurexins are interdependent for minimal synapse development or whether synapse assembly may occur largely independently through the two pathways. Identifying the intracellular signal transduction pathways mediating synaptic differentiation through these complexes is an important next step. Although studies so far have identified many postsynaptic RPTP-binding partners, further studies are needed to perform global identification of trans-synaptic binding partners of RPTPs and also neurexins in order to delineate the complete binding codes. In addition, the mechanisms mediating alternative splicing of RPTPs, including cell specificity and activity regulation should be addressed. Given the hub-based design and its possible implications, comprehensive studies of single- and multi-gene mutant mice, including manipulations of alternatively spliced exons, will be necessary in order to understand the specific and cooperative roles of these organizing proteins in synapse development in multiple circuits and in cognitive functions. Screens for small molecule synaptogenic modulators that enhance or suppress activity of these synapse organizing proteins [96] represents a promising therapeutic approach for neuropsychiatric disorders, at least for individuals with mutations in RPTP and neurexin synaptic pathways and perhaps more broadly.
Box 1. Significance of the hub-based design of RPTP and neurexin synapse organizing complexes.
Similarly to the neurexin family, the RPTP family regulates synapse organization through binding to multiple postsynaptic adhesion partners, with relative affinities regulated by gene identity and alternative splicing (and for neurexins also by promoter usage). Thus, RPTPs and neurexins serve as presynaptic hubs (Figure I). This hub-based design could have physiological and pathophysiological significance in light of several aspects of synapse development and function.
Cooperative function: Many individual glutamate synapses contain multiple neurexins and neuroligin and LRRTM partners and multiple RPTPs and NGL-3, TrkC, ILRAPL1, and Slitrk partners, suggesting a highly cooperative function in synapse development. Cooperation may serve to increase both the numbers and diversity of components recruited to developing synapses. Considering the network of protein interactions at synapses, many cooperative actions may be synergistic and not simply additive.
Synapse self-assembly: As indicated by the co-culture assay, sufficient local recruitment of either RPTPs or neurexins can trigger full presynaptic differentiation of functional transmitter release sites in axons, in a sense utilizing the propensity of presynapses for self-assembly [99]. This propensity is likely due in part to a highly interconnected protein interaction network with multiple cross-links among synaptic components, allowing for multiple triggers to nucleate synapses.
Pairing presynaptic similarity with postsynaptic diversity: In general, synapses share many common presynaptic components for regulated neurotransmitter release and recycling, but possess different postsynaptic receptors, scaffolding, and signaling proteins according to chemical type and specific function [100]. Pairing the common neurexin and RPTP presynaptic families with diverse postsynaptic partners could be a means to orchestrate this asymmetry in composition and function.
Balancing excitation and inhibition: Multiple neurexin and PTPδ isoforms interact with excitatory- and inhibitory-specific postsynaptic partners with differential affinity, regulated by alternative splicing, resulting in a mechanism for regulating the balance of excitation and inhibition, the E/I ratio [101].
Combining circuit stability with synaptic plasticity: In such an interconnected system where interactions depend on stoichiometry and relative affinities, altering splicing or levels of one component could alter multiple interactions thus altering the balance of the whole system, generating plasticity [102].
Coordinated regulation: Activity and environmentally regulated alternative splicing of neurexins has been reported [103] and is an ideal way to maintain total levels of neurexins while altering the balance of interactions with diverse postsynaptic partners, perhaps mediating coordinated postsynaptic plasticity. Further coordination could be generated by shared mechanisms regulating the splicing of RPTPs and neurexins.
Specificity of synaptic connections: Although this issue has yet been poorly addressed experimentally, neurexins and RPTPs and partners mediate cell–cell adhesion and may control synaptic partner choice [104,105].
Adaptation to genetic risk: The highly interconnected and partially redundant nature of the design, particularly the utilization of independent neurexin and RPTP hubs, facilitates maintenance of overall function with loss of individual synapse organizing proteins. However, the system is susceptible to shifts in E/I balance and stability/plasticity due to deleterious mutation of individual genes, shifts that might be corrected by enhancing or inhibiting the function of remaining proteins. This issue is clinically relevant given the links of many of these genes to neuropsychiatric disorders.
Figure I.
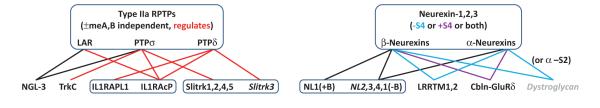
RPTPs and neurexins serve as presynaptic hubs by interacting with diverse postsynaptic partners in an isoform- and splice-selective code. Protein families are boxed; interactions lacking physiological relevance indicated as broken lines in Figure 2 in main text are not drawn here; postsynaptic partners at inhibitory synapses are in italics; nonsynaptogenic postsynaptic partners are in gray whereas all others can trigger presynaptic differentiation.
Acknowledgments
Related work by the authors is supported by National Institutes of Health MH070860, Canadian Institutes of Health Research MOP-125967, and Canada Research Chair awards (to A.M.C.), and a National Alliance for Research on Schizophrenia and Depression (Brain and Behavior Research Fund) Young Investigator award (to H.T.).
References
- 1.Van Vactor D. Protein tyrosine phosphatases in the developing nervous system. Curr. Opin. Cell Biol. 1998;10:174–181. doi: 10.1016/s0955-0674(98)80139-7. [DOI] [PubMed] [Google Scholar]
- 2.Johnson KG, Van Vactor D. Receptor protein tyrosine phosphatases in nervous system development. Physiol. Rev. 2003;83:1–24. doi: 10.1152/physrev.00016.2002. [DOI] [PubMed] [Google Scholar]
- 3.Schaapveld R, et al. Receptor-like protein tyrosine phosphatases: alike and yet so different. Mol. Biol. Rep. 1997;24:247–262. doi: 10.1023/a:1006870016238. [DOI] [PubMed] [Google Scholar]
- 4.Chagnon MJ, et al. Functional significance of the LAR receptor protein tyrosine phosphatase family in development and diseases. Biochem. Cell Biol. 2004;82:664–675. doi: 10.1139/o04-120. [DOI] [PubMed] [Google Scholar]
- 5.Woo J, et al. Trans-synaptic adhesion between NGL-3 and LAR regulates the formation of excitatory synapses. Nat. Neurosci. 2009;12:428–437. doi: 10.1038/nn.2279. [DOI] [PubMed] [Google Scholar]
- 6.Kwon SK, et al. Trans-synaptic adhesions between netrin-G ligand-3 (NGL-3) and receptor tyrosine phosphatases LAR, protein-tyrosine phosphatase delta (PTPdelta), and PTPsigma via specific domains regulate excitatory synapse formation. J. Biol. Chem. 2010;285:13966–13978. doi: 10.1074/jbc.M109.061127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi H, et al. Postsynaptic TrkC and presynaptic PTPsigma function as a bidirectional excitatory synaptic organizing complex. Neuron. 2011;69:287–303. doi: 10.1016/j.neuron.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshida T, et al. IL-1 receptor accessory protein-like 1 associated with mental retardation and autism mediates synapse formation by trans-synaptic interaction with protein tyrosine phosphatase delta. J. Neurosci. 2011;31:13485–13499. doi: 10.1523/JNEUROSCI.2136-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valnegri P, et al. The X-linked intellectual disability protein IL1RAPL1 regulates excitatory synapse formation by binding PTPdelta and RhoGAP2. Hum. Mol. Genet. 2011;20:4797–4809. doi: 10.1093/hmg/ddr418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshida T, et al. Interleukin-1 receptor accessory protein organizes neuronal synaptogenesis as a cell adhesion molecule. J. Neurosci. 2012;32:2588–2600. doi: 10.1523/JNEUROSCI.4637-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takahashi H, et al. Selective control of inhibitory synapse development by Slitrk3-PTPdelta trans-synaptic interaction. Nat. Neurosci. 2012;15:389–398. doi: 10.1038/nn.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yim YS, et al. Slitrks control excitatory and inhibitory synapse formation with LAR receptor protein tyrosine phosphatases. Proc. Natl. Acad. Sci. U.S.A. 2013;110:4057–4062. doi: 10.1073/pnas.1209881110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siddiqui TJ, Craig AM. Synaptic organizing complexes. Curr. Opin. Neurobiol. 2011;21:132–143. doi: 10.1016/j.conb.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuzaki M. Cbln1 and its family proteins in synapse formation and maintenance. Curr. Opin. Neurobiol. 2011;21:215–220. doi: 10.1016/j.conb.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 16.Schormair B, et al. PTPRD (protein tyrosine phosphatase receptor type delta) is associated with restless legs syndrome. Nat. Genet. 2008;40:946–948. doi: 10.1038/ng.190. [DOI] [PubMed] [Google Scholar]
- 17.Pinto D, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piton A, et al. Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum. Mol. Genet. 2008;17:3965–3974. doi: 10.1093/hmg/ddn300. [DOI] [PubMed] [Google Scholar]
- 19.Melhem N, et al. Copy number variants for schizophrenia and related psychotic disorders in Oceanic Palau: risk and transmission in extended pedigrees. Biol. Psychiatry. 2011;70:1115–1121. doi: 10.1016/j.biopsych.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Proenca CC, et al. Slitrks as emerging candidate genes involved in neuropsychiatric disorders. Trends Neurosci. 2011;34:143–153. doi: 10.1016/j.tins.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Francks C, et al. LRRTM1 on chromosome 2p12 is a maternally suppressed gene that is associated paternally with handedness and schizophrenia. Mol. Psychiatry. 2007;12:1129–1139. doi: 10.1038/sj.mp.4002053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pulido R, et al. The LAR/PTP delta/PTP sigma subfamily of transmembrane protein-tyrosine-phosphatases: multiple human LAR, PTP delta, and PTP sigma isoforms are expressed in a tissue-specific manner and associate with the LAR-interacting protein LIP.1. Proc. Natl. Acad. Sci. U.S.A. 1995;92:11686–11690. doi: 10.1073/pnas.92.25.11686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Serra-Pages C, et al. Mutational analysis of proprotein processing, subunit association, and shedding of the LAR transmembrane protein tyrosine phosphatase. J. Biol. Chem. 1994;269:23632–23641. [PubMed] [Google Scholar]
- 24.Craig AM, et al. How to build a central synapse: clues from cell culture. Trends Neurosci. 2006;29:8–20. doi: 10.1016/j.tins.2005.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biederer T, Scheiffele P. Mixed-culture assays for analyzing neuronal synapse formation. Nat. Protoc. 2007;2:670–676. doi: 10.1038/nprot.2007.92. [DOI] [PubMed] [Google Scholar]
- 26.Hirano S, Takeichi M. Cadherins in brain morphogenesis and wiring. Physiol. Rev. 2012;92:597–634. doi: 10.1152/physrev.00014.2011. [DOI] [PubMed] [Google Scholar]
- 27.De Angelis E, et al. Alternative use of a mini exon of the L1 gene affects L1 binding to neural ligands. J. Biol. Chem. 2001;276:32738–32742. doi: 10.1074/jbc.M105156200. [DOI] [PubMed] [Google Scholar]
- 28.Volkmer H, et al. Dissection of complex molecular interactions of neurofascin with axonin-1, F11, and tenascin-R, which promote attachment and neurite formation of tectal cells. J. Cell Biol. 1998;142:1083–1093. doi: 10.1083/jcb.142.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dunah AW, et al. LAR receptor protein tyrosine phosphatases in the development and maintenance of excitatory synapses. Nat. Neurosci. 2005;8:458–467. doi: 10.1038/nn1416. [DOI] [PubMed] [Google Scholar]
- 30.Kaufmann N, et al. Drosophila liprin-alpha and the receptor phosphatase Dlar control synapse morphogenesis. Neuron. 2002;34:27–38. doi: 10.1016/s0896-6273(02)00643-8. [DOI] [PubMed] [Google Scholar]
- 31.Ackley BD, et al. The two isoforms of the Caenorhabditis elegans leukocyte-common antigen related receptor tyrosine phosphatase PTP-3 function independently in axon guidance and synapse formation. J. Neurosci. 2005;25:7517–7528. doi: 10.1523/JNEUROSCI.2010-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaapveld RQ, et al. Developmental expression of the cell adhesion molecule-like protein tyrosine phosphatases LAR, RPTPdelta and RPTPsigma in the mouse. Mech. Dev. 1998;77:59–62. doi: 10.1016/s0925-4773(98)00119-1. [DOI] [PubMed] [Google Scholar]
- 33.Uetani N, et al. Impaired learning with enhanced hippocampal long-term potentiation in PTPdelta-deficient mice. EMBO J. 2000;19:2775–2785. doi: 10.1093/emboj/19.12.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horn KE, et al. Receptor protein tyrosine phosphatase sigma regulates synapse structure, function and plasticity. J. Neurochem. 2012;122:147–161. doi: 10.1111/j.1471-4159.2012.07762.x. [DOI] [PubMed] [Google Scholar]
- 35.Elchebly M, et al. Neuroendocrine dysplasia in mice lacking protein tyrosine phosphatase sigma. Nat. Genet. 1999;21:330–333. doi: 10.1038/6859. [DOI] [PubMed] [Google Scholar]
- 36.Wallace MJ, et al. Neuronal defects and posterior pituitary hypoplasia in mice lacking the receptor tyrosine phosphatase PTPsigma. Nat. Genet. 1999;21:334–338. doi: 10.1038/6866. [DOI] [PubMed] [Google Scholar]
- 37.Uetani N, et al. Mammalian motoneuron axon targeting requires receptor protein tyrosine phosphatases sigma and delta. J. Neurosci. 2006;26:5872–5880. doi: 10.1523/JNEUROSCI.0386-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhen M, Jin Y. The liprin protein SYD-2 regulates the differentiation of presynaptic termini in C. elegans. Nature. 1999;401:371–375. doi: 10.1038/43886. [DOI] [PubMed] [Google Scholar]
- 39.Dai Y, et al. SYD-2 Liprin-alpha organizes presynaptic active zone formation through ELKS. Nat. Neurosci. 2006;9:1479–1487. doi: 10.1038/nn1808. [DOI] [PubMed] [Google Scholar]
- 40.Astigarraga S, et al. Three Drosophila liprins interact to control synapse formation. J. Neurosci. 2010;30:15358–15368. doi: 10.1523/JNEUROSCI.1862-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taru H, Jin Y. The Liprin homology domain is essential for the homomeric interaction of SYD-2/Liprin-alpha protein in presynaptic assembly. J. Neurosci. 2011;31:16261–16268. doi: 10.1523/JNEUROSCI.0002-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sudhof TC. The presynaptic active zone. Neuron. 2012;75:11–25. doi: 10.1016/j.neuron.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spangler SA, et al. Differential expression of liprin-alpha family proteins in the brain suggests functional diversification. J. Comp. Neurol. 2011;519:3040–3060. doi: 10.1002/cne.22665. [DOI] [PubMed] [Google Scholar]
- 44.Zurner M, et al. Analyses of the spatiotemporal expression and subcellular localization of liprin-alpha proteins. J. Comp. Neurol. 2011;519:3019–3039. doi: 10.1002/cne.22664. [DOI] [PubMed] [Google Scholar]
- 45.Debant A, et al. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc. Natl. Acad. Sci. U.S.A. 1996;93:5466–5471. doi: 10.1073/pnas.93.11.5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gonzalez-Quevedo R, et al. Receptor tyrosine phosphatase-dependent cytoskeletal remodeling by the hedgehog-responsive gene MIM/BEG4. J. Cell Biol. 2005;168:453–463. doi: 10.1083/jcb.200409078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siu R, et al. N-cadherin is an in vivo substrate for protein tyrosine phosphatase sigma (PTPsigma) and participates in PTPsigma-mediated inhibition of axon growth. Mol. Cell. Biol. 2007;27:208–219. doi: 10.1128/MCB.00707-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chagnon MJ, et al. Receptor tyrosine phosphatase sigma (RPTPsigma) regulates, p250GAP, a novel substrate that attenuates Rac signaling. Cell. Signal. 2010;22:1626–1633. doi: 10.1016/j.cellsig.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 49.Brigidi GS, Bamji SX. Cadherin-catenin adhesion complexes at the synapse. Curr. Opin. Neurobiol. 2011;21:208–214. doi: 10.1016/j.conb.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 50.Kim S, et al. NGL family PSD-95-interacting adhesion molecules regulate excitatory synapse formation. Nat. Neurosci. 2006;9:1294–1301. doi: 10.1038/nn1763. [DOI] [PubMed] [Google Scholar]
- 51.Nishimura-Akiyoshi S, et al. Axonal netrin-Gs transneuronally determine lamina-specific subdendritic segments. Proc. Natl. Acad. Sci. U.S.A. 2007;104:14801–14806. doi: 10.1073/pnas.0706919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DeNardo LA, et al. NGL-2 regulates input-specific synapse development in CA1 pyramidal neurons. Neuron. 2012;76:762–775. doi: 10.1016/j.neuron.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barbacid M. Structural and functional properties of the TRK family of neurotrophin receptors. Ann. N. Y. Acad. Sci. 1995;766:442–458. doi: 10.1111/j.1749-6632.1995.tb26693.x. [DOI] [PubMed] [Google Scholar]
- 54.Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- 55.Esteban PF, et al. A kinase-deficient TrkC receptor isoform activates Arf6-Rac1 signaling through the scaffold protein tamalin. J. Cell Biol. 2006;173:291–299. doi: 10.1083/jcb.200512013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barbacid M. The Trk family of neurotrophin receptors. J. Neurobiol. 1994;25:1386–1403. doi: 10.1002/neu.480251107. [DOI] [PubMed] [Google Scholar]
- 57.Valenzuela DM, et al. Alternative forms of rat TrkC with different functional capabilities. Neuron. 1993;10:963–974. doi: 10.1016/0896-6273(93)90211-9. [DOI] [PubMed] [Google Scholar]
- 58.Sahun I, et al. Dissociation between CA3-CA1 synaptic plasticity and associative learning in TgNTRK3 transgenic mice. J. Neurosci. 2007;27:2253–2260. doi: 10.1523/JNEUROSCI.4055-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tessarollo L, et al. Targeted deletion of all isoforms of the trkC gene suggests the use of alternate receptors by its ligand neurotrophin-3 in neuronal development and implicates trkC in normal cardiogenesis. Proc. Natl. Acad. Sci. U.S.A. 1997;94:14776–14781. doi: 10.1073/pnas.94.26.14776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dunne A, O’Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci. STKE. 2003;2003:re3. doi: 10.1126/stke.2003.171.re3. [DOI] [PubMed] [Google Scholar]
- 61.Carrie A, et al. A new member of the IL-1 receptor family highly expressed in hippocampus and involved in X-linked mental retardation. Nat. Genet. 1999;23:25–31. doi: 10.1038/12623. [DOI] [PubMed] [Google Scholar]
- 62.Pavlowsky A, et al. A postsynaptic signaling pathway that may account for the cognitive defect due to IL1RAPL1 mutation. Curr. Biol. 2010;20:103–115. doi: 10.1016/j.cub.2009.12.030. [DOI] [PubMed] [Google Scholar]
- 63.Pavlowsky A, et al. Neuronal JNK pathway activation by IL-1 is mediated through IL1RAPL1, a protein required for development of cognitive functions. Commun. Integr. Biol. 2010;3:245–247. doi: 10.4161/cib.3.3.11414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith DE, et al. A central nervous system-restricted isoform of the interleukin-1 receptor accessory protein modulates neuronal responses to interleukin-1. Immunity. 2009;30:817–831. doi: 10.1016/j.immuni.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kwon HB, et al. Neuroligin-1-dependent competition regulates cortical synaptogenesis and synapse number. Nat. Neurosci. 2012;15:1667–1674. doi: 10.1038/nn.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gambino F, et al. IL1RAPL1 controls inhibitory networks during cerebellar development in mice. Eur. J. Neurosci. 2009;30:1476–1486. doi: 10.1111/j.1460-9568.2009.06975.x. [DOI] [PubMed] [Google Scholar]
- 67.Yoshida T, Mishina M. Zebrafish orthologue of mental retardation protein IL1RAPL1 regulates presynaptic differentiation. Mol. Cell. Neurosci. 2008;39:218–228. doi: 10.1016/j.mcn.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 68.Aruga J, Mikoshiba K. Identification and characterization of Slitrk, a novel neuronal transmembrane protein family controlling neurite outgrowth. Mol. Cell. Neurosci. 2003;24:117–129. doi: 10.1016/s1044-7431(03)00129-5. [DOI] [PubMed] [Google Scholar]
- 69.Linhoff MW, et al. An unbiased expression screen for synaptogenic proteins identifies the LRRTM protein family as synaptic organizers. Neuron. 2009;61:734–749. doi: 10.1016/j.neuron.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beaubien F, Cloutier JF. Differential expression of Slitrk family members in the mouse nervous system. Dev. Dyn. 2009;238:3285–3296. doi: 10.1002/dvdy.22160. [DOI] [PubMed] [Google Scholar]
- 71.Shmelkov SV, et al. Slitrk5 deficiency impairs corticostriatal circuitry and leads to obsessive-compulsive-like behaviors in mice. Nat. Med. 2010;16:598–602. doi: 10.1038/nm.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mizuseki K, et al. Hippocampal CA1 pyramidal cells form functionally distinct sublayers. Nat. Neurosci. 2011;14:1174–1181. doi: 10.1038/nn.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Poulopoulos A, et al. Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron. 2009;63:628–642. doi: 10.1016/j.neuron.2009.08.023. [DOI] [PubMed] [Google Scholar]
- 74.Graf ER, et al. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004;119:1013–1026. doi: 10.1016/j.cell.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chih B, et al. Control of excitatory and inhibitory synapse formation by neuroligins. Science. 2005;307:1324–1328. doi: 10.1126/science.1107470. [DOI] [PubMed] [Google Scholar]
- 76.de Wit J, et al. LRRTM2 interacts with Neurexin1 and regulates excitatory synapse formation. Neuron. 2009;64:799–806. doi: 10.1016/j.neuron.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ko J, et al. LRRTM2 functions as a neurexin ligand in promoting excitatory synapse formation. Neuron. 2009;64:791–798. doi: 10.1016/j.neuron.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Siddiqui TJ, et al. LRRTMs and neuroligins bind neurexins with a differential code to cooperate in glutamate synapse development. J. Neurosci. 2010;30:7495–7506. doi: 10.1523/JNEUROSCI.0470-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Elias GM, Nicoll RA. Synaptic trafficking of glutamate receptors by MAGUK scaffolding proteins. Trends Cell Biol. 2007;17:343–352. doi: 10.1016/j.tcb.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 80.Krasnoperov V, et al. Protein-tyrosine phosphatase-sigma is a novel member of the functional family of alpha-latrotoxin receptors. J. Biol. Chem. 2002;277:35887–35895. doi: 10.1074/jbc.M205478200. [DOI] [PubMed] [Google Scholar]
- 81.Ushkaryov YA, et al. Neurexins: synaptic cell surface proteins related to the alpha-latrotoxin receptor and laminin. Science. 1992;257:50–56. doi: 10.1126/science.1621094. [DOI] [PubMed] [Google Scholar]
- 82.Aricescu AR, et al. Heparan sulfate proteoglycans are ligands for receptor protein tyrosine phosphatase sigma. Mol. Cell. Biol. 2002;22:1881–1892. doi: 10.1128/MCB.22.6.1881-1892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shen Y, et al. PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science. 2009;326:592–596. doi: 10.1126/science.1178310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Johnson KG, et al. The HSPGs Syndecan and Dallylike bind the receptor phosphatase LAR and exert distinct effects on synaptic development. Neuron. 2006;49:517–531. doi: 10.1016/j.neuron.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 85.Dityatev A, Schachner M. The extracellular matrix and synapses. Cell Tissue Res. 2006;326:647–654. doi: 10.1007/s00441-006-0217-1. [DOI] [PubMed] [Google Scholar]
- 86.Urfer R, et al. An immunoglobulin-like domain determines the specificity of neurotrophin receptors. EMBO J. 1995;14:2795–2805. doi: 10.1002/j.1460-2075.1995.tb07279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim HG, et al. Neurotrophin 3 potentiates neuronal activity and inhibits gamma-aminobutyratergic synaptic transmission in cortical neurons. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12341–12345. doi: 10.1073/pnas.91.25.12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 89.Kolarow R, et al. Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium calmodulin kinase II signaling and proceeds via delayed fusion pore opening. J. Neurosci. 2007;27:10350–10364. doi: 10.1523/JNEUROSCI.0692-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Betancur C, et al. The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 2009;32:402–412. doi: 10.1016/j.tins.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ludwig KU, et al. Supporting evidence for LRRTM1 imprinting effects in schizophrenia. Mol. Psychiatry. 2009;14:743–745. doi: 10.1038/mp.2009.28. [DOI] [PubMed] [Google Scholar]
- 92.Fallin MD, et al. Bipolar I disorder and schizophrenia: a 440-single-nucleotide polymorphism screen of 64 candidate genes among Ashkenazi Jewish case-parent trios. Am. J. Hum. Genet. 2005;77:918–936. doi: 10.1086/497703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Treutlein J, et al. Dissection of phenotype reveals possible association between schizophrenia and Glutamate Receptor Delta 1 (GRID1) gene promoter. Schizophr. Res. 2009;111:123–130. doi: 10.1016/j.schres.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 94.Rujescu D, et al. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum. Mol. Genet. 2009;18:988–996. doi: 10.1093/hmg/ddn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ching MS, et al. Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am. J. Med. Genet. B: Neuropsychiatr. Genet. 2010;153B:937–947. doi: 10.1002/ajmg.b.31063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shi P, et al. Synapse microarray identification of small molecules that enhance synaptogenesis. Nat. Commun. 2011;2:510. doi: 10.1038/ncomms1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pulido R, et al. Molecular characterization of the human transmembrane protein-tyrosine phosphatase delta. Evidence for tissue-specific expression of alternative human transmembrane protein-tyrosine phosphatase delta isoforms. J. Biol. Chem. 1995;270:6722–6728. doi: 10.1074/jbc.270.12.6722. [DOI] [PubMed] [Google Scholar]
- 98.Williams AF, Barclay AN. The immunoglobulin superfamily–domains for cell surface recognition. Annu. Rev. Immunol. 1988;6:381–405. doi: 10.1146/annurev.iy.06.040188.002121. [DOI] [PubMed] [Google Scholar]
- 99.Garner CC, et al. Molecular mechanisms of CNS synaptogenesis. Trends Neurosci. 2002;25:243–251. doi: 10.1016/s0166-2236(02)02152-5. [DOI] [PubMed] [Google Scholar]
- 100.Craig AM, Boudin H. Molecular heterogeneity of central synapses: afferent and target regulation. Nat. Neurosci. 2001;4:569–578. doi: 10.1038/88388. [DOI] [PubMed] [Google Scholar]
- 101.Levinson JN, El-Husseini A. New players tip the scales in the balance between excitatory and inhibitory synapses. Mol. Pain. 2005;1:12. doi: 10.1186/1744-8069-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Abraham WC, Robins A. Memory retention–the synaptic stability versus plasticity dilemma. Trends Neurosci. 2005;28:73–78. doi: 10.1016/j.tins.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 103.Iijima T, et al. SAM68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell. 2011;147:1601–1614. doi: 10.1016/j.cell.2011.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Missler M, Sudhof TC. Neurexins: three genes and 1001 products. Trends Genet. 1998;14:20–26. doi: 10.1016/S0168-9525(97)01324-3. [DOI] [PubMed] [Google Scholar]
- 105.Shen K, Scheiffele P. Genetics and cell biology of building specific synaptic connectivity. Annu. Rev. Neurosci. 2010;33:473–507. doi: 10.1146/annurev.neuro.051508.135302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yang Q, et al. Family-based and population-based association studies validate PTPRD as a risk factor for restless legs syndrome. Mov. Disord. 2011;26:516–519. doi: 10.1002/mds.23459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Elia J, et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol. Psychiatry. 2010;15:637–646. doi: 10.1038/mp.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Malhotra D, et al. High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron. 2011;72:951–963. doi: 10.1016/j.neuron.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jin H, et al. Two novel members of the interleukin-1 receptor gene family, one deleted in Xp22.1-Xp21.3 mental retardation. Eur. J. Hum. Genet. 2000;8:87–94. doi: 10.1038/sj.ejhg.5200415. [DOI] [PubMed] [Google Scholar]
- 110.Zhang YH, et al. IL1RAPL1 is associated with mental retardation in patients with complex glycerol kinase deficiency who have deletions extending telomeric of DAX1. Hum. Mutat. 2004;24:273. doi: 10.1002/humu.9269. [DOI] [PubMed] [Google Scholar]
- 111.Tabolacci E, et al. A truncating mutation in the IL1RAPL1 gene is responsible for X-linked mental retardation in the MRX21 family. Am. J. Med. Genet. A. 2006;140:482–487. doi: 10.1002/ajmg.a.31107. [DOI] [PubMed] [Google Scholar]
- 112.Bhat SS, et al. Disruption of the IL1RAPL1 gene associated with a pericentromeric inversion of the X chromosome in a patient with mental retardation and autism. Clin. Genet. 2008;73:94–96. doi: 10.1111/j.1399-0004.2007.00920.x. [DOI] [PubMed] [Google Scholar]
- 113.Franek KJ, et al. Deletion of the immunoglobulin domain of IL1RAPL1 results in nonsyndromic X-linked intellectual disability associated with behavioral problems and mild dysmorphism. Am. J. Med. Genet. A. 2011;155A:1109–1114. doi: 10.1002/ajmg.a.33833. [DOI] [PubMed] [Google Scholar]
- 114.Behnecke A, et al. Intragenic deletions of IL1RAPL1: report of two cases and review of the literature. Am. J. Med. Genet. A. 2011;155A:372–379. doi: 10.1002/ajmg.a.33656. [DOI] [PubMed] [Google Scholar]
- 115.Piton A, et al. Systematic resequencing of X-chromosome synaptic genes in autism spectrum disorder and schizophrenia. Mol. Psychiatry. 2011;16:867–880. doi: 10.1038/mp.2010.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Abelson JF, et al. Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science. 2005;310:317–320. doi: 10.1126/science.1116502. [DOI] [PubMed] [Google Scholar]
- 117.O’Roak BJ, et al. Additional support for the association of SLITRK1 var321 and Tourette syndrome. Mol. Psychiatry. 2010;15:447–450. doi: 10.1038/mp.2009.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zuchner S, et al. SLITRK1 mutations in trichotillomania. Mol. Psychiatry. 2006;11:887–889. doi: 10.1038/sj.mp.4001898. [DOI] [PubMed] [Google Scholar]
- 119.Talebizadeh Z, et al. Feasibility and relevance of examining lymphoblastoid cell lines to study role of microRNAs in autism. Autism Res. 2008;1:240–250. doi: 10.1002/aur.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mefford HC, et al. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010;6:e1000962. doi: 10.1371/journal.pgen.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gratacos M, et al. A polymorphic genomic duplication on human chromosome 15 is a susceptibility factor for panic and phobic disorders. Cell. 2001;106:367–379. doi: 10.1016/s0092-8674(01)00447-0. [DOI] [PubMed] [Google Scholar]
- 122.Armengol L, et al. 5′ UTR-region SNP in the NTRK3 gene is associated with panic disorder. Mol. Psychiatry. 2002;7:928–930. doi: 10.1038/sj.mp.4001134. [DOI] [PubMed] [Google Scholar]
- 123.Muinos-Gimeno M, et al. Allele variants in functional MicroRNA target sites of the neurotrophin-3 receptor gene (NTRK3) as susceptibility factors for anxiety disorders. Hum. Mutat. 2009;30:1062–1071. doi: 10.1002/humu.21005. [DOI] [PubMed] [Google Scholar]
- 124.Alonso P, et al. Genetic susceptibility to obsessive-compulsive hoarding: the contribution of neurotrophic tyrosine kinase receptor type 3 gene. Genes Brain Behav. 2008;7:778–785. doi: 10.1111/j.1601-183X.2008.00418.x. [DOI] [PubMed] [Google Scholar]
- 125.Verma R, et al. Linkage disequilibrium mapping of a chromosome 15q25-26 major depression linkage region and sequencing of NTRK3. Biol. Psychiatry. 2008;63:1185–1189. doi: 10.1016/j.biopsych.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Otnaess MK, et al. Evidence for a possible association of neurotrophin receptor (NTRK-3) gene polymorphisms with hippocampal function and schizophrenia. Neurobiol. Dis. 2009;34:518–524. doi: 10.1016/j.nbd.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 127.Irie M, et al. Binding of neuroligins to PSD-95. Science. 1997;277:1511–1515. doi: 10.1126/science.277.5331.1511. [DOI] [PubMed] [Google Scholar]
- 128.Wei Z, et al. Liprin-mediated large signaling complex organization revealed by the liprin-alpha/CASK and liprin-alpha/liprin-beta complex structures. Mol. Cell. 2011;43:586–598. doi: 10.1016/j.molcel.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 129.Atasoy D, et al. Deletion of CASK in mice is lethal and impairs synaptic function. Proc. Natl. Acad. Sci. U.S.A. 2007;104:2525–2530. doi: 10.1073/pnas.0611003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Owald D, et al. Cooperation of Syd-1 with Neurexin synchronizes pre- with postsynaptic assembly. Nat. Neurosci. 2012;15:1219–1226. doi: 10.1038/nn.3183. [DOI] [PubMed] [Google Scholar]
- 131.Hallam SJ, et al. SYD-1, a presynaptic protein with PDZ, C2 and rhoGAP-like domains, specifies axon identity in C. elegans. Nat. Neurosci. 2002;5:1137–1146. doi: 10.1038/nn959. [DOI] [PubMed] [Google Scholar]
- 132.Patel MR, Shen K. RSY-1 is a local inhibitor of presynaptic assembly in C. elegans. Science. 2009;323:1500–1503. doi: 10.1126/science.1169025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Coles CH, et al. Proteoglycan-specific molecular switch for RPTPsigma clustering and neuronal extension. Science. 2011;332:484–488. doi: 10.1126/science.1200840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.O’Grady P, et al. The laminin-nidogen complex is a ligand for a specific splice isoform of the transmembrane protein tyrosine phosphatase LAR. J. Cell Biol. 1998;141:1675–1684. doi: 10.1083/jcb.141.7.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Friedman WJ. Cytokines regulate expression of the type 1 interleukin-1 receptor in rat hippocampal neurons and glia. Exp. Neurol. 2001;168:23–31. doi: 10.1006/exnr.2000.7595. [DOI] [PubMed] [Google Scholar]
- 136.Pickering M, O’Connor JJ. Pro-inflammatory cytokines and their effects in the dentate gyrus. Prog. Brain Res. 2007;163:339–354. doi: 10.1016/S0079-6123(07)63020-9. [DOI] [PubMed] [Google Scholar]