

Abstract
A continuing problem in the area of oligonucleotide-based therapeutics is the poor access of these molecules to their sites of action in the nucleus or cytosol. A number of approaches to this problem have emerged. One of the most interesting is the use of ligand-oligonucleotide conjugates to promote receptor mediated cell uptake and delivery. Here we provide an overview of recent developments regarding targeted conjugates, including use of peptides, carbohydrates and small molecules as ligands. Additionally we discuss our own experience with this approach and point out both advantages and limitations.
INTRODUCTION
The last decade has seen a surge of interest in the therapeutic utilization of various types of oligonucleotides including siRNA, miRNA, single strand antisense oligonucleotides (ASOs) and splice switching oligonucleotides (SSOs) 1-5. While a number of molecules have entered clinical trials, a persistent problem in this field is the poor delivery of oligonucleotides to their sites of pharmacological action in the cytosol or nucleus of tissue cells. Rapid clearance from the circulation, enzymatic degradation, inability to be taken up efficiently by cells, and trapping within endosomes all constrain the pharmacological effectiveness of the different types of oligonucleotides to various degrees 6-9.
Chemical modification has been an important approach to improving the pharmacology of oligonucleotides, providing increased potency, specificity and reduced side effects 10,11. One form of chemical modification, the attachment of ligands designed to improve delivery, has attracted considerable attention recently. Linkage of peptide, lipid, carbohydrate or small molecule moieties at the 5’ or 3’ positions of oligonucleotides has been done either to provide selective binding to cell surface receptors or to alter the physical properties of the oligonucleotide so as to change its clearance kinetics and biodistribution. The ligand conjugation strategy partially contrasts with another major approach to delivery of oligonucleotides, namely the use of nanoparticle carriers 12-14. While nanocarriers can also include targeting ligands, their vastly greater size as compared to molecular scale conjugates implies a far more restricted biodistribution 15. Additionally there are often toxicity issues associated with the cationic lipid or polymer components that are usually included in nanoparticles 16. Thus ligand-oligonucleotide conjugates provide an important alternative to nanocarriers as a delivery strategy.
Several excellent reviews have described many of the issues involved in the conjugation of various ligands with oligonucleotides 11,17-20. One important aspect is the relative merits of solid phase versus solution phase conjugation. Solid phase synthesis is highly efficient and facilitates purification, but the availability of appropriate synthons is often a limitation. Another concern is the need for both ligand and oligonucleotide to be stable under the conditions of synthesis. By contrast, solution phase conjugation first pursues synthesis of each component under the most appropriate and efficient conditions. However, the conjugation reaction itself may be inefficient; further, substantial post-synthesis purification problems can occur. Various groups have utilized both strategies for conjugation, but with the majority preferring solution phase approaches. Recently, some interesting new chemistries have been brought to bear on oligonucleotide conjugation including use of ‘click chemistry’ 21,22 and novel phosphoramidation reactions 23.
Much work has been done on the conjugation of various lipids to siRNA or other oligonucleotides; this can be done at either 3’ or 5’ positions using a variety of linkages 24,25. Lipid conjugation can provide substantial advantages in terms of the blood clearance kinetics, biodistribution and tissue uptake of the oligonucleotide. This was demonstrated early on with cholesterol conjugation of siRNA that causes the molecule to bind to lipoproteins, thus increasing circulation time and promoting uptake into the liver via lipoprotein receptors 26,27. Similar approaches have been undertaken with tocopherol 28 and with a variety of fatty acids and other lipid moieties 29
There has also been extensive work on peptide-oligonucleotide conjugates. For example, a number of laboratories have coupled so-called ‘cell penetrating peptides’ 30-32 to SSOs, particularly to uncharged morpholino or peptide nucleic acid oligomers. These have proven to be promising in correction of defects involved in Duchenne muscular dystrophy and have shown good effects both in muscle cell culture and in dystrophic mice 33-35. Other groups have conjugated siRNAs, ASOs or SSOs with peptides designed to bind to specific receptors. Our laboratory has worked extensively on such targeted conjugates (see below) as have others 36. A variety of linkages have been used to conjugate peptides and oligonucleotides including amide, thioether, thiol-maleimide, ester, and disulfide. An important question is whether use of a bioreversible linkage such as a disulfide is needed to attain biological activity. This does not seem to be the case, however, and both bioreversible and nonreversible linkages can work well, at least for monovalent conjugates 37-39.
Conjugation of carbohydrate moieties to oligonucleotides can provide targeting to the lectin-like proteins that exist on many cell types. Conjugation of monosaccharides to an oligonucleotide can be approached in a simple manner through the preparation of carbohydrate containing phosphoramidites. However, it is far more challenging to prepare oligonucleotides bearing the more complex oligosaccharide structures needed for optimal lectin recognition 18. Recently, ‘click chemistry’ has been used to synthesize complicated oligonucleotide glycoconjugates including branched structures 40,41. An exciting recent application of this approach involves delivery of glycoconjugates of siRNA to liver via the asialoglycoprotein receptor, a cell surface lectin found on hepatocytes 42.
A lesser amount of work has been done on conjugates of small molecules to oligonucleotides. This is somewhat surprising given the plethora of high affinity, receptor-specific ligands for that are known or discoverable 43. While the number of potential small molecule ligands is not unlimited 44, it nonetheless provides a rich field for discovery of oligonucleotide targeting agents. Our laboratory has synthesized and tested mono- and multi-valent conjugates of SSOs with anisamide, a small molecule ligand for the sigma receptor 45. Other groups have synthesized and characterized conjugates with folate 46 or with anandamide 47 that bind respectively to the folate receptor (FOLR1) or a cannabinoid receptor. In all of these cases significant biological activity was seen in the absence of any transfection agent.
Thus, over the last few years a substantial body of work has appeared regarding the chemistry and biology of oligonucleotide conjugates. In the sections below we will focus on our own experience with these types of molecules and deal with both their potential utility and with possible liabilities.
PEPTIDE-OLIGONUCLEOTIDE CONJUGATES
Our laboratory has worked extensively on peptide conjugates of SSOs and siRNA. The ligands chosen were peptides that are known to have high affinities for particular cell surface receptors that tend to be over-expressed in certain tumor cells. For example, we have extensively used mono- and multi-valent versions of the cyclic RGD peptide that binds selectively to the αvβ3 integrin that is over-expressed in angiogenic vasculature and in certain tumors 48. We have also used bombesin-like peptides that bind to BB2, a G Protein Coupled Receptor over-expressed in certain carcinomas 49. The paragraphs below describe the synthesis of these conjugates.
Solid Phase Synthesis of a Splice Switching Oligonucleotide for Conjugation
The solid phase synthesis of a 2’-O-Me phosphorothioate anionic antisense oligonucleotide with splice switching capability, a 3’ fluorophore, and a 5’ linker for ligand attachment follows our published procedures 39. The sequence we usually employed is termed SSO 623 (5’-GTT ATT CTT TAG AAT GGT GC-3’). The synthesis used a AB3400 DNA synthesizer and was conventional with the exceptions noted below. The synthesis started on solid supports having 2’ -OMe C or Tamra fluorophore at the 3’-end. Detritylation was done using 3% trichloroacetic acid in dichloromethane for 90 seconds followed by an acetonitrile wash. Base condensation was done by adding 0.25M solution of 5-ethylthio-1H-tetrazole in acetonitrile to the next phosphoramidite base to form the activated phosphorous; the free hydroxyl group on the support binds to the activated phosphorous to form the unstable phosphite linkages between the two bases. This coupling takes about 6 min for all ultramild protected 2’-OMe phosphoramidite bases and 10 min for 5’-thiol modifier amidite. The column was then washed with acetonitrile to remove any remaining 5-ethylthio-1H-tetrazole, unreacted base phosphoramidite and by-products. The unstable phosphite linkage was stabilized with a solution of sulfurizing reagent (such as Beaucage reagent) to form the more stable thiophosphate linkage (for phosphorothioates). Remaining unreacted active free hydroxyl group on the solid support (typically less than 2%) was capped to prevent the formation of failure sequences. In the case of regular 2’-OMe-C solid supports, standard acetic anhydride and N-methyl-imidazole solution were used as capping reagents. However, when Tamra fluorophore solid supports and ultramild protected all 2’-O-methyl phosphoramidite bases were used, 5% phenoxyacetic anhydride in tetrahydrofuran/pyridine and 16% N-methyl-imidazole in tetrahydrofuran were used for capping. Then the steps were repeated until the desired length of oligonucleotide sequence has been assembled onto the solid supports. A 5’-thiol linker was introduced at the end of the oligonucleotide synthesis. Normally we obtained a coupling efficiency of 98-100% at each cycle.
After oligonucleotides were synthesized, they were first treated with 10% diethylamine in acetonitrile for 5 min × 2 followed by acetonitrile wash to remove the 2-cyanoethyl group from the phosphate backbone and prevent the elimination of 3’-Tamra fluorophore from the oligonucleotide chain. The oligonucleotides were then reacted with 2 mL of deprotection solution (tert-butylamine: methanol: water = 1:1:2, v/v) per micromole of the oligo bound solid support for 8h at 55 °C. After cooling to room temperature, the supports were filtered off and washed with HPLC grade of water. The combined filtrate was immediately evaporated to dryness. The crude mass was re-suspended in 0.1M TEAA buffer and purified by reverse-phase HPLC using a Zorbax C18 column to obtain 623 SSO disulfide. The structure of the disulfide oligo was confirmed by MALDI-TOF mass spectroscopy.
Bivalent cyclic RGD peptides were synthesized by conventional means and characterized by MALDI-TOF mass spectrometry 50. Peptides of this type have been extensively used in imaging studies 51,52. In this case a maleimide moiety was inserted into the peptide to allow facile conjugation with a thiol group on the 5’ end of the oligonucleotide.
Conjugation of bivalent cRGD peptide to 623 SSO
Conjugation of cRGD peptide to anionic phosphorothioate oligonucleotides is a two-step chemical process as shown in Scheme 1 for the SSO termed 623. It involves generation of a thiol group (-SH) from a disulfide oligonucleotide and reaction of the thiol oligonucleotide to a maleimide bearing bivalent cRGD peptide via Michael addition. Briefly, the disulfide bond of the Tamra-623-disulfide SSO was reduced with 100 mM of aqueous DTT solution at room temperature followed by a Sep-PAK C18 cartridge purification (step I of Scheme 1). Any residual amount of DTT was removed by washing with 5% acetonitrile in a 0.1M TEAA buffer. Thiol oligonucleotide (Tamra-623-SH) was eluted from the Sep-PAK cartridge with 50% aqueous acetonitrile. The eluted thiol oligonucleotides were directly used for the conjugation reaction. When the two species were mixed together in a solution of salt and acetonitrile (400mM KCl, 40% aq. acetonitrile), the reaction was completed within 3h at room temperature (step II of Scheme 1). The progress of the conjugation reaction was monitored by anion exchange HPLC using a pre-pack Resource Q column (1 mL) in 50% of formamide in Tris-chloride buffer. All thiol (>95%) oligonucleotides were shifted to conjugates with bicyclic RGD peptide. We have used 3 equivalents of peptides relative to thiol oligonucleotides in the conjugation step. No aggregation was observed in the reaction mixture. After purification using a Resource Q column, the conjugates were desalted over milli-Q water using slide-A-lyser desalting cassette. The conjugate appeared as a clear solution in water after desalting. After lyophilization, the conjugate was re-suspended in sterile water at a concentration of 50-100 μM for long-term storage. All structures of RGD-623 SSO conjugates were confirmed by MALDI-TOF mass spectroscopy. The chemical structure of the cRGD-623-Tamra conjugate is shown in Figure 1.
Scheme 1. Conjugation of cRGD peptides with a thiol modified 2’-O-Me phosphorothioate oligonucleotide.
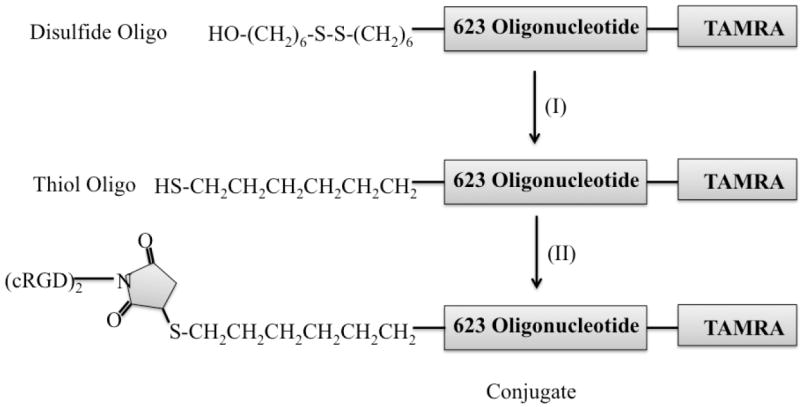
The 5’ disulfide splice switching oligonucleotide is reduced and then conjugated to a maleimide terminated cyclic RGD peptide, as described in the text.
Figure 1. Structure of a bivalent cyclic RGD-oligonucleotide conjugate.
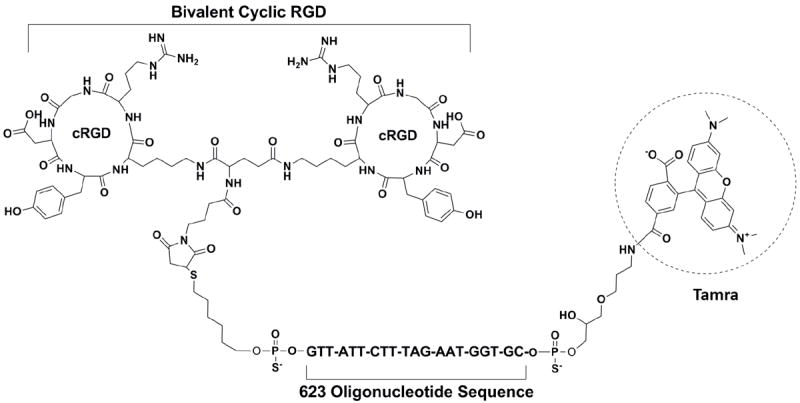
Conjugation of Bombesin (6-14) peptide to 623 SSO
The preparation of bombesin-SSO conjugates 53 was very similar to that used for the cRGD conjugates. The same disulfide version of Tamra-623-SSO as shown in Scheme 1 was used. The chemical structure and synthetic route for bombesin-623-Tamra conjugate is shown in Figure 2. About 5 equivalent of maleimide-bombesin peptide was consumed relative to the thiol oligonucleotide to convert all starting oligo into its conjugated product. The conjugates were analyzed and purified by reverse-phase HPLC method using an XTerra C18 column on a Varian system. The thiol oligos and their bombesin conjugates were clearly separated by the RP-HPLC method. The conjugates were eluted later than thiol oligonucleotide by RP-HPLC analysis and were very soluble in metal free sterile water after lyophilization.
Figure 2. Synthesis and structure of a bombesin-oligonucleotide conjugate.
Conjugation of multivalent cRGD peptides to siRNA
The synthetic route for the chemical conjugation of bi-, tri- and tetravalent cyclic RGD peptides to a firefly luciferase siRNA was extensively discussed in a recent publication 54. Very briefly, a reactive cysteine thiol group (-SH) was introduced into the peptide sequence to allow chemical conjugation with a maleimide functionality of the 3’-sense strand of luciferase siRNA via Michael addition reaction. In other words the opposite strategy was followed as compared to the SSO conjugates. This was necessary to maintain the 5’ position of the antisense strand of the siRNA in an unmodified state. About 2.5 equivalents of reactive thiol bearing peptides were used relative to the maleimide sense strand oligonucleotide. All starting sense strands were consumed and converted to the cRGD conjugated sense strand. None of the conjugates precipitated in the reaction mixture containing 400mM KCl and 40% aq. acetonitrile. After 5-fold of dilution with HPLC grade of water, the crude conjugates were purified by reverse-phase HPLC using an XTerra RP-18 column attached to an AKTA purifier and isolated with an excellent yield (conjugates: bivalent 95%, trivalent 84%, tetravalent 67%). No noticeable aggregation was seen when re-suspended in sterile water after lyophilization. For all cases, the structure of the final conjugate was confirmed by LC-MS analysis. To obtain the desired cRGD-siRNA conjugates, the partner antisense strand was annealed with the cRGD conjugated sense strand in sterile water at elevated temperature for 3 min followed by slow cooling.
BIOLOGICAL ACTIONS OF PEPTIDE OLIGONUCLEOTIDE CONJUGATES
In order to examine the specificity of the peptide-oligonucleotide conjugates for membrane receptors, we tested cellular uptake of the conjugates in the tumor cells over-expressing these receptors. Thus, we used integrin αvβ3-expressing A375 melanoma cells 39 and M21+ cells 54 to test the cellular uptake of cRGD-oligonucleotide conjugates, and bombesin (BBN) receptor-expressing PC3 cells to test intracellular delivery of BBN-SSO conjugates 53. In all these cases, peptide-oligonucleotide conjugates showed significantly higher uptake than that of the “free” oligonucleotides, and this increase was reversed by an excess amount of free peptide, confirming receptor specific cellular delivery39,53,54. Enhancement of cellular uptake by peptide conjugation depends on the binding affinity of the peptides to their receptors and the density of the receptors on the cell surface 55. On the other hand, the intrinsic uptake rate of the oligonucleotides also plays a role. “Free” phosphorothioate (PS) oligonucleotides can bind to membrane proteins and enter cells via endocytosis 56, and their cellular uptake is more efficient than “free” siRNA oligonucleotides. Therefore, RGD-siRNA showed about 20-fold higher uptake than “free” siRNA, while RGD-SSO, in which SSO is a PS oligonucleotide, only showed about 2-fold higher uptake than free SSO 39.
Although the enhancement in cellular uptake of RGD-SSO was modest, the functional activity caused by the RGD-SSO conjugate was disproportionately higher than that of “free” SSO 39. The functional delivery was tested in receptor-expressing cells stably transfected with a luciferase gene interrupted by an abnormally spliced intron. Thus, intranuclear delivery of the SSO corrects splicing and allows positive readout of luciferase expression in these cells. At the concentration of 150 nM, RGD-SSO showed about 6-fold higher induction than the SSO in A375 cells 39. Similarly, in BBN receptor-expressing PC3 cells, BBN-SSO showed 2-fold higher cellular uptake, but about 5-fold higher induction than “free” SSO 53. The disconnection between cellular uptake and functional activity was also observed for RGD-siRNA conjugates. In this study, luciferase siRNA was linked with bi-, tri-, and tetravalent cRGD peptides, and their cellular uptake and RNAi activity were then examined using luciferase-expressing and integrin αvβ3 positive M21(+) human melanoma cells 54. The bi-, tri-, and tetravalent cRGD conjugates were taken up by M21(+) cells to approximately the same degree However, only the tri- and tetravalent versions produced RNAi activity, while the bivalent version had little effect 54. One cause of this discord may be the distinct internalization pathways that the peptide-oligonucleotide conjugates undertake. We did a follow-up study to resolve endocytosis pathways of RGD-SSO 57. Using endocytosis inhibitors and a dominant-negative form of dynamin GTPase, we have identified an actin-dependent, dynamin-dependent pathway for uptake of the RGD-SSO conjugate in A375 cells, and an unusual actin-independent, dynamin-independent pathway for uptake of the unconjugated PS oligonucleotide, suggesting that the route of internalization could affect the magnitude of the biological response 57.
The distinct endocytotic process may also cause distinct kinetics of pharmacological action of the peptide-oligonucleotide conjugates. Both RGD-SSO and BBN-SSO conjugates produced a gradual increase followed by a gradual decline in reporter induction, while a cationic lipid-SSO complex caused a rapid increase followed by a monotonic decline 39,53. The peptide-oligonucleotide conjugates need to be transported through multiple endosomal compartments before export from the endosomes, and the release may be slow, as the current conjugate system does not include an endosomal release entity. In order to further enhance the functional activity of the conjugates, the RGD-SSO conjugate was formulated into polyplexes with a cationic polymer, polyethylenimine, which is able to cause endosomal release via the proton sponge effect 58. The resultant polyplexes of RGD-SSO conjugate demonstrated dramatic increases in the pharmacological response of splicing correction compared to free RGD-SSO conjugate or the polyplexes of unconjugated SSO, presumably through integrin-mediated endocytosis and rapid endosomal release 58. This provides a novel method to construct multifunctional nanoparticle-based delivery systems using peptide-oligonucleotide conjugates.
CONJUGATION OF SMALL MOLECULES
As mentioned above, small molecule ligands with high affinity to specific cell surface receptors provide an interesting opportunity for the design of oligonucleotide conjugates. However, there are several caveats involved. First, it is important to understand that much of the structure of the small molecule will be involved in the interaction with its receptor. This limits ones’ ability to introduce chemical modifications that allow conjugation to the oligonucleotide. Second, the presence of the large, highly charged oligonucleotide strand can affect the affinity of binding between the small molecule ligand and its receptor. This latter effect can potentially be ameliorated by introducing an uncharged linker between the oligonucleotide and the ligand. Another consideration is that many of the small molecule ligands developed by the pharmaceutical industry are relatively lipophilic since they are designed for oral administration 59. Lipophilicity can create two types of problems. First, the solubility of the conjugate can be reduced, especially when several ligands are linked to a single oligonucleotide. Second, the presence of lipophilic moieties can lead to non-specific binding to cell membranes; once again this is particularly true when several small molecule ligands are attached.
Our laboratory has had both successful and less than successful experiences with small molecule oligonucleotide conjugates. On the positive side, as mentioned above, we prepared mono-and trivalent conjugates between a SSO and anisamide, a ligand for the sigma receptor 45. Importantly, anisamide had previously been conjugated to liposomes for targeting purposes 60 and thus something was known about how to conjugate the molecule without loss of receptor binding activity. In our study we somewhat modified the coupling approach and then developed a solid phase synthesis of the final ligand-oligonucleotide conjugate. Thus, we synthesized a phosphoramidite version of anisamide that could incorporated using a standard DNA synthesizer as depicted in Scheme 2. The anisamide conjugates, especially the trivalent version, were more effective than an unconjugated SSO in inducing splice correction in a reporter gene stably transfected into human prostate tumor cells. The process seemed to be at least somewhat receptor specific since co-incubation of the cells with haloperidol, a sigma receptor antagonist, partially blocked cellular uptake of the anisamide-SSO conjugates.
Scheme 2. Preparation of anisamide-oligonucleotide conjugates (reproduced with permission from reference 45).
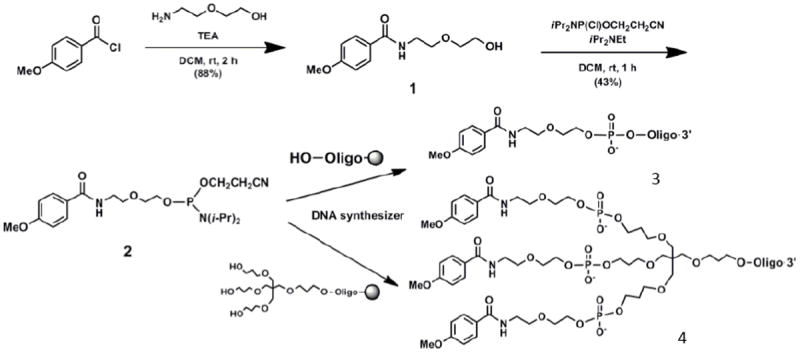
N-[2-(2-hydroxyethoxy)ethyl]-4-methoxybenzamide, a derivative of anisamide with a reactive hydroxyl group (1) is converted to a phosphoramidite (2). Solid phase DNA synthesis is used to prepare monovalent (3) or trivalent (4) anisamide-oligonucleotide conjugates.
We attempted to extend these results to another ligand-receptor system but with less favorable results. DREADDs are G Protein Coupled Receptors (GPCRs) that have been genetically engineered to respond to a unique test ligand rather than to endogenous ligands 61. In this case we used a DREADD that was based on the M3 muscarinic receptor but that responded to clozapine or clozapine-N-oxide (CNO) rather than acetylcholine its normal ligand. Since the M3 receptor and its corresponding DREADD are coupled to Gq/11 G-Proteins, the functional assay for testing potency of ligands in this system is modulation of intracellular calcium levels, which is easily determined via a fluorescent dye assay. Based on structure activity studies of clozapine derivatives in the DREADD system, analogs of clozapine and CNO were synthesized that had a hydroxyl connected via a linker to one of the two nitrogens on the seven-member clozapine ring (Scheme 3). These compounds maintained good potency in the calcium modulation assay (51 nM for the clozapine analog). They were subsequently reacted with 1,1’-carbonyldiimidazole and the product of this reaction was then conjugated to a SSO having a 5’-aminolinker; both mono-and trivalent versions were produced.
Scheme 3. Preparation of clozapine-oligonucleotide conjugates.
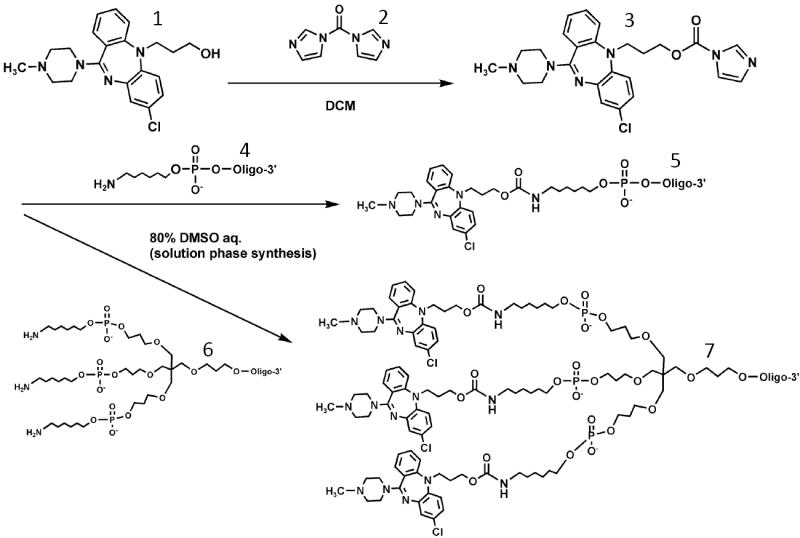
A clozapine derivative with a reactive hydroxyl group (1) was reacted with 1,1’ -carbonyldiimidazole (2) and the product (3) was conjugated to mono- or tri-branched oligonucleotides (4,5) that contained 5’-aminolinkers to produce mono- or trimeric clozapine-oligonucleotide conjugates (6.7).
We then examined the biological activities of these molecules with disappointing results (unpublished observations). For example, using the calcium modulation assay we found that the potency of the conjugates were sharply reduced; thus the monomeric and trimeric clozapine analog-SSO conjugates had ED50s of approximately 1 uM and 0.1uM as compared to approximately 1 nM for clozapine itself. Despite the use of a six-carbon aminolinker, the presence of the charged oligonucleotide seemed to reduce the affinity of the ligand for its receptor. Studies of the cellular uptake of the conjugates were even more disappointing. We used a cell line that had been stably transfected so as to over-express the M3 DREADD. Initially we were pleased to find that the clozapine and CNO analog SSO conjugates displayed substantially higher cellular uptake than the unconjugated parent SSO. However, we then compared uptake in cell lines that did or did not express the DREADD and found little difference. Clozapine and its relatives are quite lipophilic as they must be since they are used to treat disease of the central nervous system; thus the XLogP3 for clozapine is 3.2 (PubChem). We suspect that the rather high lipophilicity of the clozapine analogs caused non-specific binding of the conjugates to cell membranes that overwhelmed the relatively poor specific binding to the receptor. It is interesting to note that anisamide is substantially less lipophilic than clozapine having a XLogP3 of 0.8 which may account for its better success as a ligand for oligonucleotide conjugation.
CONCLUSIONS
In summary, it is possible to synthesize a variety of ligand-oligonucleotide conjugates by relatively simple means. In those cases where the ligand binds selectively to a specific cell surface receptor, the conjugates have often shown receptor-dependent, cell type specific uptake and enhanced biological effects. However, important issues remain to be solved. For example, even after receptor dependent uptake, much of the oligonucleotide remains sequestered in pharmacologically inert endosomal compartments. Combining targeted ligand-oligonucleotide conjugates with means to promote endosome escape will be a challenge for the future. We have recently made some progress on this aspect by using a small molecule to enhance escape of anionic antisense and splice switching oligonucleotides from a subset of endosomes resulting in enhanced pharmacological effect 62. A special situation prevails in the case of conjugates involving small molecules. Unlike the situation with peptides or carbohydrates, it is more challenging to link the small molecule ligand to the oligonucleotide in a manner that will preserve specific, high affinity ligand-receptor binding. Despite these concerns, however, it seems likely that ligand-oligonucleotide conjugates will be an important aspect of the future development of oligonucleotides as therapeutic agents.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Spitali P, Aartsma-Rus A. Splice modulating therapies for human disease. Cell. 2012;148:1085–8. doi: 10.1016/j.cell.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 2.Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chem Biol. 2012;19:60–71. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gambari R, Fabbri E, Borgatti M, Lampronti I, Finotti A, Brognara E, Bianchi N, Manicardi A, Marchelli R, Corradini R. Targeting microRNAs involved in human diseases: a novel approach for modification of gene expression and drug development. Biochem Pharmacol. 2011;82:1416–29. doi: 10.1016/j.bcp.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 4.Watts JK, Corey DR. Silencing disease genes in the laboratory and the clinic. J Pathol. 2012;226:365–79. doi: 10.1002/path.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012;11:125–40. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geary RS. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. 2009;5:381–91. doi: 10.1517/17425250902877680. [DOI] [PubMed] [Google Scholar]
- 7.Dominska M, Dykxhoorn DM. Breaking down the barriers: siRNA delivery and endosome escape. Journal of cell science. 2010;123:1183–9. doi: 10.1242/jcs.066399. [DOI] [PubMed] [Google Scholar]
- 8.Juliano RL, Ming X, Nakagawa O. Cellular uptake and intracellular trafficking of antisense and siRNA oligonucleotides. Bioconjug Chem. 2012;23:147–57. doi: 10.1021/bc200377d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010;50:259–93. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 10.Caruthers MH. A brief review of DNA and RNA chemical synthesis. Biochem Soc Trans. 2011;39:575–80. doi: 10.1042/BST0390575. [DOI] [PubMed] [Google Scholar]
- 11.Deleavey GF, Damha MJ. Designing chemically modified oligonucleotides for targeted gene silencing. Chem Biol. 2012;19:937–54. doi: 10.1016/j.chembiol.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 12.Huang L, Liu Y. In vivo delivery of RNAi with lipid-based nanoparticles. Annu Rev Biomed Eng. 2011;13:507–30. doi: 10.1146/annurev-bioeng-071910-124709. [DOI] [PubMed] [Google Scholar]
- 13.Nguyen J, Szoka FC. Nucleic acid delivery: the missing pieces of the puzzle? Acc Chem Res. 2012;45:1153–62. doi: 10.1021/ar3000162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schroeder A, Levins CG, Cortez C, Langer R, Anderson DG. Lipid-based nanotherapeutics for siRNA delivery. J Intern Med. 2010;267:9–21. doi: 10.1111/j.1365-2796.2009.02189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juliano R, Bauman J, Kang H, Ming X. Biological barriers to therapy with antisense and siRNA oligonucleotides. Molecular pharmaceutics. 2009;6:686–95. doi: 10.1021/mp900093r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akhtar S. Cationic nanosystems for the delivery of small interfering ribonucleic acid therapeutics: a focus on toxicogenomics. Expert Opin Drug Metab Toxicol. 2010;6:1347–62. doi: 10.1517/17425255.2010.518611. [DOI] [PubMed] [Google Scholar]
- 17.Singh Y, Murat P, Defrancq E. Recent developments in oligonucleotide conjugation. Chem Soc Rev. 2010;39:2054–70. doi: 10.1039/b911431a. [DOI] [PubMed] [Google Scholar]
- 18.Lonnberg H. Solid-phase synthesis of oligonucleotide conjugates useful for delivery and targeting of potential nucleic acid therapeutics. Bioconjug Chem. 2009;20:1065–94. doi: 10.1021/bc800406a. [DOI] [PubMed] [Google Scholar]
- 19.Phelps K, Morris A, Beal PA. Novel modifications in RNA. ACS Chem Biol. 2012;7:100–9. doi: 10.1021/cb200422t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Juliano RL, Ming X, Nakagawa O. The chemistry and biology of oligonucleotide conjugates. Acc Chem Res. 2012;45:1067–76. doi: 10.1021/ar2002123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer A, Spinelli N, Dumy P, Vasseur JJ, Morvan F, Defrancq E. Oligonucleotide sequential bis-conjugation via click-oxime and click-Huisgen procedures. J Org Chem. 2010;75:3927–30. doi: 10.1021/jo100599m. [DOI] [PubMed] [Google Scholar]
- 22.Yamada T, Peng CG, Matsuda S, Addepalli H, Jayaprakash KN, Alam MR, Mills K, Maier MA, Charisse K, Sekine M, Manoharan M, Rajeev KG. Versatile site-specific conjugation of small molecules to siRNA using click chemistry. J Org Chem. 2011;76:1198–211. doi: 10.1021/jo101761g. [DOI] [PubMed] [Google Scholar]
- 23.Wang TP, Chiou YJ, Chen Y, Wang EC, Hwang LC, Chen BH, Chen YH, Ko CH. Versatile phosphoramidation reactions for nucleic acid conjugations with peptides, proteins, chromophores, and biotin derivatives. Bioconjug Chem. 2010;21:1642–55. doi: 10.1021/bc1001505. [DOI] [PubMed] [Google Scholar]
- 24.Raouane M, Desmaele D, Urbinati G, Massaad-Massade L, Couvreur P. Lipid Conjugated Oligonucleotides: A Useful Strategy for Delivery. Bioconjug Chem. 2012 doi: 10.1021/bc200422w. [DOI] [PubMed] [Google Scholar]
- 25.Patwa A, Gissot A, Bestel I, Barthelemy P. Hybrid lipid oligonucleotide conjugates: synthesis, self-assemblies and biomedical applications. Chem Soc Rev. 2011 doi: 10.1039/c1cs15038c. [DOI] [PubMed] [Google Scholar]
- 26.Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, Elbashir S, Geick A, Hadwiger P, Harborth J, John M, Kesavan V, Lavine G, Pandey RK, Racie T, Rajeev KG, Rohl I, Toudjarska I, Wang G, Wuschko S, Bumcrot D, Koteliansky V, Limmer S, Manoharan M, Vornlocher HP. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–8. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 27.Wolfrum C, Shi S, Jayaprakash KN, Jayaraman M, Wang G, Pandey RK, Rajeev KG, Nakayama T, Charrise K, Ndungo EM, Zimmermann T, Koteliansky V, Manoharan M, Stoffel M. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat Biotechnol. 2007;25:1149–57. doi: 10.1038/nbt1339. [DOI] [PubMed] [Google Scholar]
- 28.Nishina K, Unno T, Uno Y, Kubodera T, Kanouchi T, Mizusawa H, Yokota T. Efficient in vivo delivery of siRNA to the liver by conjugation of alpha-tocopherol. Mol Ther. 2008;16:734–40. doi: 10.1038/mt.2008.14. [DOI] [PubMed] [Google Scholar]
- 29.Petrova NS, Chernikov IV, Meschaninova MI, Dovydenko IS, Venyaminova AG, Zenkova MA, Vlassov VV, Chernolovskaya EL. Carrier-free cellular uptake and the gene-silencing activity of the lipophilic siRNAs is strongly affected by the length of the linker between siRNA and lipophilic group. Nucleic Acids Res. 2012;40:2330–44. doi: 10.1093/nar/gkr1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindgren M, Langel U. Classes and prediction of cell-penetrating peptides. Methods Mol Biol. 2011;683:3–19. doi: 10.1007/978-1-60761-919-2_1. [DOI] [PubMed] [Google Scholar]
- 31.van den Berg A, Dowdy SF. Protein transduction domain delivery of therapeutic macromolecules. Curr Opin Biotechnol. 2011;22:888–93. doi: 10.1016/j.copbio.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Abes R, Moulton HM, Clair P, Yang ST, Abes S, Melikov K, Prevot P, Youngblood DS, Iversen PL, Chernomordik LV, Lebleu B. Delivery of steric block morpholino oligomers by (R-X-R)4 peptides: structure-activity studies. Nucleic Acids Res. 2008;36:6343–54. doi: 10.1093/nar/gkn541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ivanova GD, Arzumanov A, Abes R, Yin H, Wood MJ, Lebleu B, Gait MJ. Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res. 2008;36:6418–28. doi: 10.1093/nar/gkn671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yin H, Saleh AF, Betts C, Camelliti P, Seow Y, Ashraf S, Arzumanov A, Hammond S, Merritt T, Gait MJ, Wood MJ. Pip5 Transduction Peptides Direct High Efficiency Oligonucleotide-mediated Dystrophin Exon Skipping in Heart and Phenotypic Correction in mdx Mice. Mol Ther. 2011 doi: 10.1038/mt.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saleh AF, Arzumanov AA, Gait MJ. Overview of alternative oligonucleotide chemistries for exon skipping. Methods Mol Biol. 2012;867:365–78. doi: 10.1007/978-1-61779-767-5_23. [DOI] [PubMed] [Google Scholar]
- 36.Sethi D, Chen CP, Jing RY, Thakur ML, Wickstrom E. Fluorescent peptide-PNA chimeras for imaging monoamine oxidase A mRNA in neuronal cells. Bioconjug Chem. 2012;23:158–63. doi: 10.1021/bc2004507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turner JJ, Arzumanov AA, Gait MJ. Synthesis, cellular uptake and HIV- 1 Tat-dependent trans-activation inhibition activity of oligonucleotide analogues disulphide-conjugated to cell-penetrating peptides. Nucleic Acids Res. 2005;33:27–42. doi: 10.1093/nar/gki142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Astriab-Fisher A, Sergueev D, Fisher M, Shaw BR, Juliano RL. Conjugates of antisense oligonucleotides with the Tat and antennapedia cell-penetrating peptides: effects on cellular uptake, binding to target sequences, and biologic actions. Pharm Res. 2002;19:744–54. doi: 10.1023/a:1016136328329. [DOI] [PubMed] [Google Scholar]
- 39.Alam MR, Dixit V, Kang H, Li ZB, Chen X, Trejo J, Fisher M, Juliano RL. Intracellular delivery of an anionic antisense oligonucleotide via receptor-mediated endocytosis. Nucleic Acids Res. 2008;36:2764–76. doi: 10.1093/nar/gkn115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kiviniemi A, Virta P, Drenichev MS, Mikhailov SN, Lonnberg H. Solid-Supported 2’-O-Glycoconjugation of Oligonucleotides by Azidation and Click Reactions. Bioconjug Chem. 2011;22:1249–1255. doi: 10.1021/bc200097g. [DOI] [PubMed] [Google Scholar]
- 41.Pourceau G, Meyer A, Vasseur JJ, Morvan F. Synthesis of a Glycomimetic Oligonucleotide Conjugate by 1,3-Dipolar Cycloaddition. Methods Mol Biol. 2011;751:167–193. doi: 10.1007/978-1-61779-151-2_11. [DOI] [PubMed] [Google Scholar]
- 42.Akinc A, Querbes W, De S, Qin J, Frank-Kamenetsky M, Jayaprakash KN, Jayaraman M, Rajeev KG, Cantley WL, Dorkin JR, Butler JS, Qin L, Racie T, Sprague A, Fava E, Zeigerer A, Hope MJ, Zerial M, Sah DW, Fitzgerald K, Tracy MA, Manoharan M, Koteliansky V, Fougerolles A, Maier MA. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol Ther. 2010;18:1357–64. doi: 10.1038/mt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allen JA, Roth BL. Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2011;51:117–44. doi: 10.1146/annurev-pharmtox-010510-100553. [DOI] [PubMed] [Google Scholar]
- 44.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–30. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 45.Nakagawa O, Ming X, Huang L, Juliano RL. Targeted intracellular delivery of antisense oligonucleotides via conjugation with small-molecule ligands. J Am Chem Soc. 2010;132:8848–9. doi: 10.1021/ja102635c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dohmen C, Frohlich T, Lachelt U, Rohl I, Vornlocher HP, Hadwiger P, Wagner E. Defined Folate-PEG-siRNA Conjugates for Receptor-specific Gene Silencing. Mol Ther Nucleic Acids. 2012;1:e7. doi: 10.1038/mtna.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willibald J, Harder J, Sparrer K, Conzelmann KK, Carell T. Click-modified anandamide siRNA enables delivery and gene silencing in neuronal and immune cells. J Am Chem Soc. 2012;134:12330–3. doi: 10.1021/ja303251f. [DOI] [PubMed] [Google Scholar]
- 48.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jensen RT, Battey JF, Spindel ER, Benya RV. International Union of Pharmacology. LXVIII. Mammalian bombesin receptors: nomenclature, distribution, pharmacology, signaling, and functions in normal and disease states. Pharmacol Rev. 2008;60:1–42. doi: 10.1124/pr.107.07108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen X, Plasencia C, Hou Y, Neamati N. Synthesis and biological evaluation of dimeric RGD peptide-paclitaxel conjugate as a model for integrin-targeted drug delivery. Journal of medicinal chemistry. 2005;48:1098–106. doi: 10.1021/jm049165z. [DOI] [PubMed] [Google Scholar]
- 51.Chen X. Multimodality imaging of tumor integrin alphavbeta3 expression. Mini reviews in medicinal chemistry. 2006;6:227–34. doi: 10.2174/138955706775475975. [DOI] [PubMed] [Google Scholar]
- 52.Shi J, Kim YS, Zhai S, Liu Z, Chen X, Liu S. Improving tumor uptake and pharmacokinetics of (64)Cu-labeled cyclic RGD peptide dimers with Gly(3) and PEG(4) linkers. Bioconjug Chem. 2009;20:750–9. doi: 10.1021/bc800455p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ming X, Alam MR, Fisher M, Yan Y, Chen X, Juliano RL. Intracellular delivery of an antisense oligonucleotide via endocytosis of a G protein-coupled receptor. Nucleic Acids Res. 2010;38:6567–76. doi: 10.1093/nar/gkq534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alam MR, Ming X, Fisher M, Lackey JG, Rajeev KG, Manoharan M, Juliano RL. Multivalent cyclic RGD conjugates for targeted delivery of small interfering RNA. Bioconjug Chem. 2011;22:1673–81. doi: 10.1021/bc200235q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Juliano RL, Carver K, Cao C, Ming X. Receptors, endocytosis, and trafficking: the biological basis of targeted delivery of antisense and siRNA oligonucleotides. Journal of drug targeting. 2012 doi: 10.3109/1061186X.2012.740674. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ming X. Cellular delivery of siRNA and antisense oligonucleotides via receptor-mediated endocytosis. Expert Opin Drug Deliv. 2011;8:435–49. doi: 10.1517/17425247.2011.561313. [DOI] [PubMed] [Google Scholar]
- 57.Alam MR, Ming X, Dixit V, Fisher M, Chen X, Juliano RL. The biological effect of an antisense oligonucleotide depends on its route of endocytosis and trafficking. Oligonucleotides. 2010;20:103–9. doi: 10.1089/oli.2009.0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ming X, Feng L. Targeted delivery of a splice-switching oligonucleotide by cationic polyplexes of RGD-oligonucleotide conjugate. Molecular pharmaceutics. 2012;9:1502–10. doi: 10.1021/mp300113c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 60.Li SD, Chen YC, Hackett MJ, Huang L. Tumor-targeted delivery of siRNA by self-assembled nanoparticles. Mol Ther. 2008;16:163–9. doi: 10.1038/sj.mt.6300323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A. 2007;104:5163–8. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ming X, Carver K, Fisher M, Noel R, Cintrat JC, Gillet D, Barbier J, Cao C, Bauman J, Juliano RL. The small molecule Retro-1 enhances the pharmacological actions of antisense and splice switching oligonucleotides. Nucleic Acids Res. 2013;41:3673–87. doi: 10.1093/nar/gkt066. [DOI] [PMC free article] [PubMed] [Google Scholar]