

Abstract
Few conventional cytotoxic anticancer therapeutics induce immunogenic cell death (ICD). This means that they induce tumor cells to undergo apoptosis while eliciting the emission of a spatiotemporal-defined combination of damage-associated molecular patterns (DAMPs) decoded by the immune system to activate antitumor immunity effective for long-term therapeutic success. The neurotoxin capsaicin (CPS) can induce both cancer cell apoptosis and immune-mediated tumor regression. In the present study, we investigated whether CPS is capable of eliciting the emission of ICD hallmarks in human bladder cancer cell lines undergoing apoptosis. We demonstrated that CPS induces pre- and early apoptotic cell surface exposure of calreticulin (CRT), HSP90, and HSP70 as well as ATP release. Moreover, CRT exposure was prevented by inhibition of endoplasmic reticulum–Golgi traffic by brefeldin A. Furthermore, high-mobility group box 1, HSP90, and HSP70 were passively released at late apoptotic stages. We provide the first evidence that CPS is an inducer of ICD hallmarks, suggesting CPS as a novel potential immunogenic cytotoxic agent.
Keywords: Capsaicin, Immunogenic cell death, Calreticulin, Heat shock proteins, Adenosine triphosphate, High-mobility group box 1
Introduction
Accumulating evidence exists that a crucial contribution to the long-term success of conventional cytotoxic anticancer therapies is made by their capability to stimulate antitumor immune responses, by acting either on immune cells or on tumor cells (Lake and Robinson 2005). One possibility to stimulate antitumor immunity by acting on tumor cells is based on the induction of cancer cell death with immunogenic properties (Bianchi 2007; Garg et al. 2010; Kepp et al. 2011). The concept that apoptosis, being a physiological cell death, represents always a non-immunogenic cell death model has been invalidated (Albert et al. 1998). Indeed, a restricted panel of radiotherapies (i.e., γ- and UVC-irradiation), chemotherapies (i.e., doxorubicin, mitoxantrone, oxaliplatin, cyclophosphamide, bortezomib), and hypericin-based photodynamic therapy (Hyr-PDT) induce a combination of tumor cell stress and death that result in a functionally peculiar type of immunogenic apoptosis, denominated immunogenic cell death (ICD) (Garg et al. 2010; Vacchelli et al. 2012). ICD relies on the emission by apoptotic cancer cells of a spatiotemporally defined combination of damage-associated molecular patterns (DAMPs) which stimulate immune responses, involving antigen presenting cells (APCs) (i.e., dendritic cells (DCs) and macrophages) and T and natural killer (NK) cells, against dead cell antigens (Garg et al. 2010; Kepp et al. 2011; Multhoff 2009). Distinctive features of ICD (as opposed to non-immunogenic cell death (non-ICD)) are: (1) the cell surface exposure of chaperones including calreticulin (CRT) (Garg et al. 2012a; Obeid et al. 2007) and/or heat shock proteins (HSPs) such as HSP90 and HSP70 (Fucikova et al. 2011; Garg et al. 2012a; Spisek et al. 2007) in pre- or early apoptotic stages, before tolerance to cells dying through apoptosis is induced; (2) the pre- or early apoptotic secretion of ATP (Garg et al. 2012b; Martins et al. 2009); and (3) the late apoptotic passive release of non-histone chromatin protein high-mobility group box 1 (HMGB1) (Bell et al. 2006; Fucikova et al. 2011; Scaffidi et al. 2002) and possibly of HSP70 and HSP90 (Garg et al. 2010; Schmitt et al. 2007; Srivastava et al. 1994). Importantly, these parameters appear to be sufficient to make accurate predictions about the capacity of drugs to induce ICD and, so far, only a few cytotoxic anticancer drugs induce all these phenomena and in the correct spatiotemporal order, thus eliciting bona fide ICD. The ability of cancer therapies to induce ICD depends on their aptitude to induce premortem reactive oxygen species (ROS) production and endoplasmic reticulum (ER) stress, essential to trigger the intracellular danger signaling pathways that govern ICD (Garg et al. 2010; Vacchelli et al. 2012).
Capsaicin (N-vanillyl-8-methyl-nonenamide; CPS), a homovanillic acid derivative and spicy component of chili pepper, induces the sensation of noxious heat and pain, desensitization, and neurogenic inflammation, by interacting with the vanilloid receptor 1 (VR1) expressed on sensory neurons (Surh and Lee 1995). Apart from its neurological functions, CPS is able to exert cytotoxicity towards cancer cells and immunomodulatory functions, suggesting its potential application in tumor therapy. CPS promotes in vitro and in vivo apoptosis in a wide variety of human cancer cells (derived from urogenital, gastrointestinal, respiratory, and skin tissues) but not in normal cells, by both VR1-dependent- and VR1-independent pathways (Athanasiou et al. 2007; Beltran et al. 2007; Bley et al. 2012; Zhang et al. 2008). Although the exact mechanisms underlying CPS-induced apoptosis remain unclear, ROS generation and ER stress- and mitochondria-mediated death pathways involvement has been reported (Lee et al. 2009; Macho et al. 1999; Yang et al. 2009; Zhang et al. 2008). In addition, CPS exerts anti-inflammatory (Surh 2002) as well as immunostimulatory (Yu et al. 1998) activities and, intriguingly, it can stimulate anticancer immunity. Intratumoral administration of CPS elicits T cell-mediated antitumor immune response, leading to the regression of advanced preexisting solid tumors (Beltran et al. 2007; Flemming 2007). The mechanisms underlying this effect remain unclear. It has been postulated that CPS targets stromal macrophages and T cells, playing a possible immunomodulatory role in the tumor microenvironment (Ghosh and Basu 2012). However, although VR1 expression in macrophages and DCs has been demonstrated, limited and rather controversial data exist on the direct function of CPS on APCs (Basu and Srivastava 2005; Mahmoud et al. 2010; O'Connell et al. 2005; Tóth et al. 2009). To the best of our knowledge, it has not been studied whether the direct action of CPS on tumor cells consists of the induction of a cell death with immunogenic properties, namely whether CPS is an inducer of ICD.
In this study, we investigated whether CPS elicits the emission of ICD-associated DAMPs in two human urinary bladder cancer cell lines. We chose bladder cancer cells for three main reasons. First, the bladder is one anatomic region suitable for local therapy (Patard et al. 1998) and intratumoral administration of CPS should allow this agent to exert its cytotoxic activity directly on tumor cells. Second, the immune system is very likely implicated in the control of bladder cancer cell growth, since bladder cancer is sensitive to immunotherapeutic agents (Patard et al. 1998; Velotti et al. 1991). Finally, intravesical CPS has already been used by clinicians for neurogenic overactive bladder treatment (Sellers and McKay 2007).
Results and discussion
CPS-induced apoptosis in human bladder cancer cells
Before investigating ICD hallmarks, we assessed CPS-induced apoptosis in SD48 and T24 bladder cancer cells. Cells were incubated with different doses of CPS (from 50 to 400 μM) for different periods, and apoptosis was assessed by flow cytometry using the phosphatidylserine (PS)-binding Annexin V-FITC and the vital dye PI staining. As shown in Fig. 1a, CPS induced a dose-dependent increase of apoptosis in SD48 cells, reaching approximately 30 % apoptotic cells using 150 μM CPS for 24 h. The analysis of the kinetic of apoptosis showed early apoptosis (Annexin V+/PI− cells, exposing PS in the presence of membrane integrity) between 3 and 24 h, with progression to late apoptosis (Annexin V+/PI+ and Annexin V−/PI+ cells, with membrane incompetence) after 48 h (Fig. 1b). The analysis of CPS dosage required to induce apoptosis in T24 cells showed a significant percentage of apoptotic cells when 250 μM CPS was used (Fig. 2a). In addition, early apoptotic events (Annexin V+/PI− cells) appeared after 36 h of treatment (Fig. 2b), reaching approximately 30 % apoptotic cells at 60 h. These data illustrate CPS-induced apoptosis in SD48 and T24 cancer cells and show that both different doses and different times are required in the two different cell lines. These observations are consistent with the data in the literature. In fact, different doses of CPS have been used to promote apoptosis in different human tumor cells (Bley et al. 2012), in that different cancer cell lines (deriving from different tumors) represent different populations of cancer cells with different susceptibility to apoptosis. Moreover, a considerable variability in the onset of apoptosis between cells, even in clonal populations, has been reported and a number of different factors have been shown to contribute to it, including differences in the composition of single cells in relation to the mechanisms of apoptosis triggered by specific stimuli (Bhola and Simon 2009).
Fig. 1.

CPS-induced apoptosis in SD48 bladder cancer cells. a Subconfluent adherent SD48 cells (Chopin et al. 2003) were incubated with either vehicle (Ctr) or the indicated concentrations of CPS (Sigma-Aldrich) for 24 h; b cells were incubated with either vehicle (Ctr) or 150 μM CPS for the indicated periods; then, cells were recovered and apoptosis was assessed by Annexin V-FITC (AV) and propidium iodide (PI) cell staining and cytofluorimetry, as previously described (Merendino et al. 2005). Results are presented as: a mean ± SD of three experiments; b FACS plots, showing the percentage of cells in apoptotic stages; data are representative of at least three independent experiments
Fig. 2.

CPS-induced apoptosis in T24 bladder cancer cells. a Subconfluent adherent T24 cells (ATCC) were incubated with either vehicle (Ctr) or the indicated concentrations of CPS for 48 h; b cells were incubated with either vehicle (Ctr) or 250 μM CPS for the indicated periods. Apoptosis was monitored by cytofluorimetry using annexin V-FITC (AV)/propidium iodide (PI) staining. Results are presented as: a mean ± SD of three experiments; b FACS plots, showing the percentage of cells in apoptotic stages; data are representative of at least three independent experiments
Pre-apoptotic cancer cell surface exposure of CRT, HSP90, and HSP70
Then, we started to investigate the very early event in ICD, such as the pre-apoptotic exposures of CRT, HSP90, and HSP70 on cancer cell surface, by immunoblotting of both the cytoplasmic and the isolated plasma membrane proteins of CPS-treated cells. According to other ICD inducers (Garg et al. 2010), CPS produced increased expression of the three molecules in both cancer cell lines (Fig. 3a) that is very likely to be considered as part of a general response to cellular stress. The analysis of plasma membrane isolated proteins in SD48 cells showed the appearance of a band of 63 kDa corresponding to CRT at 3 h of treatment, which increased at 12 h (Fig. 3a, left panel); on the other hand, HSP90 was considerably enhanced as early as 1 h after treatment (Fig. 3a, left panel). Moreover, surface exposures of CRT and HSP90 were also detectable by flow cytometry after 6 h (Fig. 3b, left panel). These results confirm the ectopic location of both molecules and suggest that their levels might not be high enough on the cell surface to be detected by immunofluorescence earlier. The investigation of CRT and HSP90 externalization in CPS-treated T24 cells revealed the cell surface appearance of both molecules at 30 h (Fig. 3a, right panel) that is well before the apoptosis-associated PS exposure occurring at 36 h (Fig. 2b). According to the results in SD48 cells, CRT and HSP90 surface exposures in T24 cells were also detectable by flow cytometry 36 and 30 h after CPS treatment, respectively (Fig. 3b, right panel). It has been shown that CRT exposure is the result of the relocation on the plasma membrane of the ER-resident CRT (Panaretakis et al. 2009; Garg et al. 2012a, b). Therefore, to exclude a passive leakage of CRT to SD48 and T24 cell plasma membranes, cells were pretreated with 30 μM brefeldin A, an inhibitor of protein transport from ER to Golgi apparatus. As shown in Fig. 3c, the inhibition of ER–Golgi traffic prevented CRT exposure in both cancer cell lines, indicating that CPS-induced CRT externalization depends on anterograde ER–Golgi traffic. This is a crucial point, since the exposure of CRT or HSP90 in pre- and early apoptotic stages (before induction of tolerance by apoptosis-associated PS exposure) is necessary for ICD (Obeid et al. 2007; Spisek et al. 2007). Indeed, the absence of CRT or HSP90 on the cell surface abrogates the immunogenicity of dying cancer cells, thereby constituting one of the major checkpoints determining ICD. In fact, etoposide or cisplatin, although promoting either HSP70 surface exposure and ATP secretion or HMGB1 release, respectively, are non-ICD inducers (Obeid et al. 2007). Surface CRT and HSP90 act both as “eat me” signals, triggering APC-mediated dead cell antigen uptake, a crucial event for priming the innate immune response (Garg et al. 2010; Kepp et al. 2011). Furthermore, taking into account the kinetics of CRT and HSP90 externalization in the two cancer cells, we can speculate that, in contrast to T24 cells where HSP90 and CRT function contemporaneously as “eat me” signals, in SD48 HSP90 might act as primary eat me signal regarding to CRT.
Fig. 3.
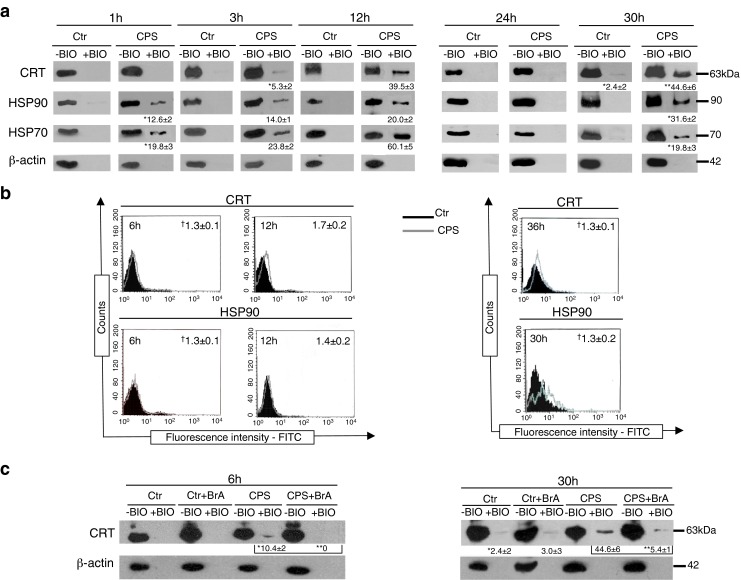
CPS elicits pre- and early apoptotic exposure of CRT, HSP90, and HSP70 on the surface of bladder cancer cells. a–c SD48 (left panels) and T24 (right panels) cells were incubated with either vehicle (Ctr) or 150 and 250 μM CPS, respectively, for the indicated periods; a cell surface proteins were biotinylated and isolated using the Pinpoint Cell Surface Protein Isolation Kit, and Western blot on isolated surface (+BIO) and cytoplasmic (−BIO) proteins was performed as previously described (Molinari et al. 2011), using anti-calreticulin (CRT) (FMC75; MBL International), anti-HSP90 (AC88; StressGen), and anti-HSP70 (C92F3A-5; Santa Cruz Biotechnology) antibodies; β-actin (Ac-40 antibody) was used as loading and intracellular protein control; b immunofluorescence cell staining using anti-CRT, anti-HSP90, or isotype control antibodies and cytofluorimetric analysis while gating on the viable population and excluding dead cells stained with PI; dagger means of the mean fluorescence intensity (MFI) ratio (calculated as the ratio between MFI of positive cells and MFI of control) of three independent experiments ± SD; c cells were pretreated with brefeldin A (BrA; Sigma-Aldrich) and then treated as in a. Asterisk are the mean intensities of bands ± SD of three independent experiments, measured using Quantity One 1-D Analysis software (Bio-Rad) (at the bottom of a and c); statistical comparisons among groups were performed by Student’s t test; differences were considered statistically significant at P < 0.05; **P < 0.05 versus Ctr. Data are representative of three independent experiments
Finally, according to other immunogenic agents (Fucikova et al. 2011; Garg et al. 2012a), we found that CPS induces the pre-apoptotic externalization of HSP70 in both bladder cancer cell lines (Fig. 3a). Ectopic HSP70, by interacting with APCs, has been described to exert multiple activities, including APC migration, tumor antigenic peptide chaperonage, and CD86 and CD40 upregulation (Garg et al. 2010; Schmitt et al. 2007; Srivastava et al. 1994). This last activity is functional to DC-mediated cross-presentation of tumor antigens to CD8+ cytotoxic T lymphocytes. In addition, HSP70 is capable of interacting with NK cells, thereby leading to NK cell activation (Multhoff 2009).
Pre- and early apoptotic cancer cells release of ATP
Next, we investigated whether CPS induces the pre- and early apoptotic active release of ATP into the extracellular environment. Therefore, we measured intracellular and extracellular ATP levels in CPS-treated SD48 and T24 cells, at their corresponding pre- and early apoptotic stages. According to other immunogenic stimuli (Garg et al. 2012b), we observed that pre-apoptotic intracellular ATP levels increased following CPS treatment (Fig. 4a). Extracellular ATP analysis performed in the conditioned media of SD48 cells showed the onset of measurable levels of ATP at 1 h after treatment (Fig. 4a, left panel), clearly preceding apoptosis-associated PS exposure and under non-permeabilizing plasma membrane conditions (Fig. 1b). According to results in SD48 cells, the analysis of ATP release in CPS-treated T24 cells revealed the onset of measurable levels of extracellular ATP at the pre-apoptotic stage (Fig. 4a, right panel; Fig. 2b). It is known that nucleotides can be released either through active processes (before the plasma membrane becomes permeable) or passively (when cells lose their integrity) (Garg et al. 2012b; Martins et al. 2009). According to Garg et al. (2012b), our results show that CPS induces ATP release before the onset of apoptosis (before plasma membrane permeabilization), thereby suggesting a stress-induced active release of ATP. In addition, while the combination of CRT exposure and optimal ATP secretion induced by some chemotherapeutics (e.g., anthracyclines) requires different doses (Martins et al. 2009), in the case of CPS, as for Hyr-PDT (Garg et al. 2012b), the two events are associated. In fact, surface CRT and extracellular ATP are promoted by the same doses of CPS, thereby integrating the emergence of these two critical immunogenic signals within a possible single therapeutic set-up. This is an important point, since the secretion of ATP is mandatory for the induction of specific antitumor immunity, in that extracellular ATP acts both as “find-me” signal and as activator of the (NOD-like receptor family pyridine domain containing-3) NLRP3 inflammosome, thereby stimulating both the recruitment and the activation of APCs required for the adequate polarization of cytotoxic T lymphocytes (Garg et al. 2012b; Vacchelli et al. 2012).
Fig. 4.

CPS elicits pre- and early apoptotic ATP as well as late apoptotic HMGB1, HSP90, and HSP70 release by bladder cancer cells. a–c (left panels) SD48 and (right panels) T24 cells were incubated with either vehicle (Ctr) or 150 and 250 μM CPS, respectively. At the indicated times, cells and conditioned media were collected and a intracellular and extracellular ATP were measured by lucifern-based ENLITEN® ATP Assay (Promega), following the manufacturer’s instructions. Intracellular ATP was determined after saponin-based cell lysis and extracellular ATP in the conditioned (serum-free) media in the presence of 100 μM ATPase inhibitor 67156 (Sigma-Aldrich); Ctr, vehicle; for assessment of the chemiluminescent signal, the plates were read in a Trilux 1450 Microbeta liquid scintillation and luminescence counter presetted to integrate the amount of light produced over a 10-s interval without an initial delay; standard curves generated from ATP standards (Promega) displayed linearity in the range of 10–12 to 10–7 mol ATP; results are presented as mean ± SD of triplicates; b immunoblotted using anti-HMGB1 (ab18256; Abcam), anti-HSP70, and anti-HSP90 antibodies; β-actin was used as intracellular protein control; Nt, not treated; Ctr, vehicle; Asterisk mean intensities of bands ± SD of three independent experiments (at the bottom of each panel). **P < 0.05 versus Ctr. Data are representative of three independent experiments
Late apoptotic cancer cell release of HMGB1, HSP90, and HSP70
Finally, we investigated whether CPS-induced apoptosis was associated with the late apoptotic extracellular passive release of HMGB1, HSP70, and HSP90, the second line of hallmarks required for ICD. As shown in Fig. 4b, HMGB1 was released in the conditioned media of both SD48 (left panel) and T24 (right panel) cells after 72 h of treatment, that is when approximately 32 and 39 % late apoptotic (PI+, with permeabilized plasma membrane) SD48 (Fig. 1b) and T24 (Fig. 2b) cells were induced, respectively. HMGB1 release is mandatory for ICD because HMGB1, by interacting with APCs, triggers signaling pathways that allow the antigen to traffic toward the antigen presenting compartment, thereby leading to optimal tumor antigen processing and cross-presentation to T cells (Kepp et al. 2011).
Moreover, at late apoptotic stages, we also observed extracellular release of HSP70 and HSP90 in both cancer cell lines (Fig. 4b). As mentioned above, extracellular HSPs promote the formation of tumor antigen–HSP complexes that are processed by APCs for T cell cross-priming more efficiently than tumor antigen alone (Garg et al. 2010; Schmitt et al. 2007; Srivastava et al. 1994; Vacchelli et al. 2012).
Conclusions
Our findings provide the first evidence that CPS is an inducer of ICD hallmarks. Moreover, they lay down a novel and solid foundation for both future investigations aiming to characterize the mechanisms underlying the induction of ICD-associated molecules by CPS and functional experiments confirming the capacity of CPS to stimulate ICD-dependent anticancer immune responses in vivo. Since most anticancer drugs fail to elicit one or more of the specific ICD-associated DAMPs (being thus intrinsically unable to induce ICD), our studies might lead to the expansion of the number of ICD inducers, thereby contributing to design innovative therapeutic strategies that harness the immunogenicity of cancer cells.
Acknowledgments
We thank Rocco Fraioli for the skillful technical assistance. The research was financially supported by grant Programmi di Ricerca di Rilevante Interesse Nazionale 2009 from Italian Ministero dell’Istruzione, Università e Ricerca.
References
- Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86–89. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- Athanasiou A, Smith PA, Vakilpour S, et al. Vanilloid receptor agonists and antagonists are mitochondrial inhibitors: how vanilloids cause non-vanilloid receptor mediated cell death. Biochem Biophys Res Commun. 2007;354:50–55. doi: 10.1016/j.bbrc.2006.12.179. [DOI] [PubMed] [Google Scholar]
- Basu S, Srivastava P. Immunological role of neuronal receptor vanilloid receptor 1 expressed on dendritic cells. Proc Natl Acad Sci U S A. 2005;102:5120–5125. doi: 10.1073/pnas.0407780102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell CW, Jiang W, Reich CF, 3rd, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol. 2006;291:C1318–C1325. doi: 10.1152/ajpcell.00616.2005. [DOI] [PubMed] [Google Scholar]
- Beltran J, Ghosh AK, Basu S. Immunotherapy of tumors with neuroimmune ligand capsaicin. J Immunol. 2007;178:3260–3264. doi: 10.4049/jimmunol.178.5.3260. [DOI] [PubMed] [Google Scholar]
- Bhola PD, Simon SM. Determinism and divergence of apoptosis susceptibility in mammalian cells. J Cell Sci. 2009;122:4296–4302. doi: 10.1242/jcs.055590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- Bley K, Boorman G, Mohammad B, McKenzie D, Babbar S. A comprehensive review of the carcinogenic and anticarcinogenic potential of capsaicin. Toxicol Pathol. 2012;40:847–873. doi: 10.1177/0192623312444471. [DOI] [PubMed] [Google Scholar]
- Chopin D, Barei-Moniri R, Maillé P, Le Frère-Belda MA, Muscatelli-Groux B, Merendino N, Lecerf L, Stoppacciaro A, Velotti F. Human urinary bladder transitional cell carcinomas acquire the functional Fas ligand during tumor progression. Am J Pathol. 2003;162:1139–1149. doi: 10.1016/S0002-9440(10)63910-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemming A. Red chillies tackle tumours. Nat Rev Drug Discov. 2007;6:269. doi: 10.1038/nrd2299. [DOI] [Google Scholar]
- Fucikova J, Kralikova P, Fialova A, Brtnicky T, Rob L, Bartunkova J, Spísek R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011;71:4821–4833. doi: 10.1158/0008-5472.CAN-11-0950. [DOI] [PubMed] [Google Scholar]
- Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta. 2010;1805:53–71. doi: 10.1016/j.bbcan.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Garg AD, Krysko DV, Vandenabeele P, Agostinis P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol Immunother. 2012;61:215–221. doi: 10.1007/s00262-011-1184-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg AD, Krysko DV, Verfaillie T, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012;31:1062–1079. doi: 10.1038/emboj.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Basu S. Tumor macrophages as a target for capsaicin mediated immunotherapy. Cancer Lett. 2012;324:91–97. doi: 10.1016/j.canlet.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Kepp O, Galluzzi L, Martins I, Schlemmer F, Adjemian S, Michaud M, Sukkurwala AQ, Menger L, Zitvogel L, Kroemer G. Molecular determinants of immunogenic cell death elicited by anticancer chemotherapy. Cancer Metastasis Rev. 2011;1:61–69. doi: 10.1007/s10555-011-9273-4. [DOI] [PubMed] [Google Scholar]
- Lake RA, Robinson WS. Immunotherapy and chemotherapy—a practical partnership. Nat Rev Cancer. 2005;5:397–405. doi: 10.1038/nrc1613. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Kee KH, Suh CH, Lim SC, Oh SH. Capsaicin-induced apoptosis is regulated by endoplasmic reticulum stress- and calpain-mediated mitochondrial cell death pathways. Toxicology. 2009;264:205–214. doi: 10.1016/j.tox.2009.08.012. [DOI] [PubMed] [Google Scholar]
- Macho A, Calzado MA, Muñoz-Blanco J, Gómez-Díaz C, Gajate C, Mollinedo F, Navas P, Muñoz E. Selective induction of apoptosis by capsaicin in transformed cells: the role of reactive oxygen species and calcium. Cell Death Differ. 1999;6:155–165. doi: 10.1038/sj.cdd.4400465. [DOI] [PubMed] [Google Scholar]
- Mahmoud ME, Nikami H, Shiina T, Takewaki T, Shimizu Y. Capsaicin inhibits IFN-gamma-induced MHC class II expression by suppressing transcription of class II transactivator gene in murine peritoneal macrophages. Int Immunopharmacol. 2010;10:86–90. doi: 10.1016/j.intimp.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, Séror C, Métivier D, Perfettini JL, Zitvogel L, Kroemer G. Chemotherapy induces ATP release from tumor cells. Cell Cycle. 2009;8:3723–3728. doi: 10.4161/cc.8.22.10026. [DOI] [PubMed] [Google Scholar]
- Merendino N, Loppi B, D'Aquino M, Molinari R, Pessina G, Romano C, Velotti F. Docosahexaenoic acid induces apoptosis in the human PaCa-44 pancreatic cancer cell line by active reduced glutathione extrusion and lipid peroxidation. Nutr Cancer. 2005;52:225–233. doi: 10.1207/s15327914nc5202_12. [DOI] [PubMed] [Google Scholar]
- Molinari R, D'Eliseo D, Manzi L, Zolla L, Velotti F, Merendino N. The n3-polyunsaturated fatty acid docosahexaenoic acid induces immunogenic cell death in human cancer cell lines via pre-apoptotic calreticulin exposure. Cancer Immunol Immunother. 2011;60:1503–1507. doi: 10.1007/s00262-011-1074-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Multhoff G. Activation of natural killer cells by heat shock protein 70. Int J Hyperth. 2009;25:169–175. doi: 10.1080/02656730902902001. [DOI] [PubMed] [Google Scholar]
- Obeid M, Tesniere A, Ghiringhelli F, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- O'Connell PJ, Pingle SC, Ahern GP. Dendritic cells do not transduce inflammatory stimuli via the capsaicin receptor TRPV1. FEBS Lett. 2005;579:5135–5139. doi: 10.1016/j.febslet.2005.08.023. [DOI] [PubMed] [Google Scholar]
- Panaretakis T, Kepp O, Brockmeier U, et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28:578–590. doi: 10.1038/emboj.2009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patard JJ, Saint F, Velotti F, Abbou CC, Chopin DK. Immune response following intravesical bacillus Calmette–Guerin instillations in superficial bladder cancer: a review. Urol Res. 1998;26:155–159. doi: 10.1007/s002400050039. [DOI] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, Bianchi ME (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418:191–195 [DOI] [PubMed]
- Schmitt E, Gehrmann M, Brunet M, Multhoff G, Garrido C. Intracellular and extracellular functions of heat shock proteins: repercussions in cancer therapy. J Leukoc Biol. 2007;81:15–27. doi: 10.1189/jlb.0306167. [DOI] [PubMed] [Google Scholar]
- Sellers DJ, McKay N. Developments in the pharmacotherapy of the overactive bladder. Curr Opin Urol. 2007;17:223–230. doi: 10.1097/MOU.0b013e3281299033. [DOI] [PubMed] [Google Scholar]
- Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood. 2007;109:4839–4845. doi: 10.1182/blood-2006-10-054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava PK, Udono H, Blachere NE, Li Z. Heat shock proteins transfer peptides during antigen processing and CTL priming. Immunogenetics. 1994;39:93–98. doi: 10.1007/BF00188611. [DOI] [PubMed] [Google Scholar]
- Surh YJ. Anti-tumor promoting potential of selected spice ingredients with antioxidative and anti-inflammatory activities: a short review. Food Chem Toxicol. 2002;40:1091–1097. doi: 10.1016/S0278-6915(02)00037-6. [DOI] [PubMed] [Google Scholar]
- Surh YJ, Lee SS. Capsaicin, a double-edged sword: toxicity, metabolism, and chemopreventive potential. Life Sci. 1995;56:1845–1855. doi: 10.1016/0024-3205(95)00159-4. [DOI] [PubMed] [Google Scholar]
- Tóth BI, Benko S, Szöllosi AG, Kovács L, Rajnavölgyi E, Bíró T. Transient receptor potential vanilloid-1 signaling inhibits differentiation and activation of human dendritic cells. FEBS Lett. 2009;583:1619–1624. doi: 10.1016/j.febslet.2009.04.031. [DOI] [PubMed] [Google Scholar]
- Vacchelli E, Galluzzi L, Fridman WH, Galon J, Sautès-Fridman C, Tartour E, Kroemer G. Trial watch: chemotherapy with immunogenic cell death inducers. Oncoimmunology. 2012;1:179–188. doi: 10.4161/onci.1.2.19026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velotti F, Stoppacciaro A, Ruco L, et al. A Local activation of immune response in bladder cancer patients treated with intra-arterial infusion of recombinant interleukin-2. Cancer Res. 1991;51:2456–2462. [PubMed] [Google Scholar]
- Yang KM, Pyo JO, Kim GY, Yu R, Han IS, Ju SA, Kim WH, Kim BS. Capsaicin induces apoptosis by generating reactive oxygen species and disrupting mitochondrial transmembrane potential in human colon cancer cell lines. Cell Mol Biol Lett. 2009;14:497–510. doi: 10.2478/s11658-009-0016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R, Park JW, Kurata T, Erickson KL. Modulation of select immune responses by dietary capsaicin. Int J Vitam Nutr Res. 1998;68:114–119. [PubMed] [Google Scholar]
- Zhang R, Humphreys I, Sahu RP, Shi Y, Srivastava SK. In vitro and in vivo induction of apoptosis by capsaicin in pancreatic cancer cells is mediated through ROS generation and mitochondrial death pathway. Apoptosis. 2008;13:1465–1478. doi: 10.1007/s10495-008-0278-6. [DOI] [PubMed] [Google Scholar]